
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurol. , 08 July 2021
Sec. Movement Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.701351
This article is part of the Research Topic Genetics of Paroxysmal Movement Disorders View all 9 articles
 Josua Kegele1*
Josua Kegele1* Johanna Krüger1
Johanna Krüger1 Mahmoud Koko1
Mahmoud Koko1 Lara Lange2Ana Victoria Marco Hernandez3Francisco Martinez3,4
Lara Lange2Ana Victoria Marco Hernandez3Francisco Martinez3,4 Alexander Münchau5Holger Lerche1
Alexander Münchau5Holger Lerche1 Stephan Lauxmann1
Stephan Lauxmann1Paroxysmal dyskinesias (PxD) are rare movement disorders with characteristic episodes of involuntary mixed hyperkinetic movements. Scientific efforts and technical advances in molecular genetics have led to the discovery of a variety of genes associated with PxD; however, clinical and genetic information of rarely affected genes or infrequent variants is often limited. In our case series, we present two individuals with PxD including one with classical paroxysmal kinesigenic dyskinesia, who carry new likely pathogenic de novo variants in KCNA1 (p.Gly396Val and p.Gly396Arg). The gene has only recently been discovered to be causative for familial paroxysmal kinesigenic dyskinesia. We also provide genetic evidence for pathogenicity of two newly identified disease-causing variants in SLC2A1 (p.Met96Thr and p.Leu231Pro) leading to paroxysmal exercise-induced dyskinesia. Since clinical information of carriers of variants in known disease-causing genes is often scarce, we encourage to share clinical data of individuals with rare or novel (likely) pathogenic variants to improve disease understanding.
Paroxysmal dyskinesias (PxD) are rare movement disorders, characterized by provoked or spontaneous episodes of hyperkinetic involuntary mixed movements such as choreoathetosis, dystonia, or ballism (1, 2). There are three main PxD subtypes: paroxysmal kinesigenic dyskinesia (PKD), paroxysmal exercise-induced dyskinesia (PED), and paroxysmal non-kinesigenic dyskinesia (PNKD). Clinically, they differ from each other mainly in their duration, provocation factors, and frequency of dyskinetic attacks (Table 1) but also regarding their genetic pathology and underlying mechanism: PKD mainly results from mutations in PRRT2 (Proline-rich transmembrane protein 2), mutations in PNKD (metallo-beta-lactamase domain-containing protein, formerly known as MR-1) may induce PNKD, and SLC2A1 (Solute carrier family 2, member 1; known as glucose transporter 1 gene or GLUT1) is known to cause PED, among other diseases. Further genes have been linked to PxD, albeit less frequently than the already mentioned ones. Described PKD-associated genes are SCN8A, PNKD, SLC2A1, KCNMA1, DEPDC5, KCNA1, and CHRNA4 (6–11). Genes associated with PNKD are KCNMA1 (12) and SLC2A1 (10), and the following genes have been linked to PED: GCH1 (13, 14), ECHS (15, 16), and PRRT2 (10). The aforementioned list of genes refers to isolated/predominant PxD. However, PxD can also be part of the clinical presentation in a variety of complex neurogenetic disorders (17).
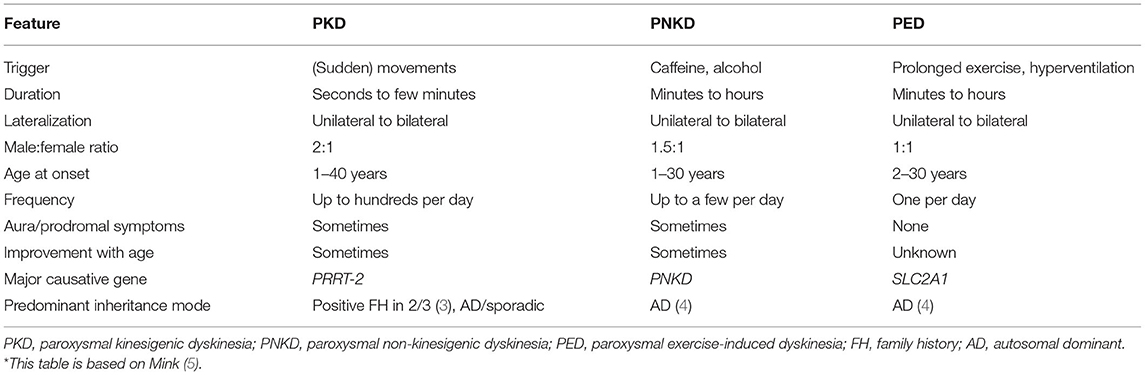
Table 1. Phenotypic characteristics of paroxysmal dyskinesias*.
On the one hand, there is an astonishing cumulation of a few recurrent variants in PxD. For example, up to 78.5% of PRRT-2-positive individuals with PKD carry the frameshift mutation c.649dupC (18). On the other hand, there are reports of disease-causing variants that have occurred only in one family or only in a single individual and whose genetic evidence underpinning the pathogenicity is often limited.
In contrast, there are also secondary causes that lead to PxD such as stroke, trauma, central nervous infection, and multiple sclerosis (19), but these will not be the focus of this report.
Recently, KCNA1 has been linked to familial PKD and PxD (7, 8, 11). In this case series, we strengthen the evidence for KCNA1 as a causative gene in sporadic PKD and other PxD with a detailed description of the clinical phenotype of patients with two newly identified de novo missense mutations in KCNA1. We further describe a novel missense mutation in SLC2A1 resulting in autosomal dominant PED as well as another SLC2A1 missense mutation resulting in PED which has not been associated with this phenotype until now but has been previously published once in a different context as a de novo mutation in a patient with glucose transporter 1 deficiency syndrome (GLUT1-DS).
The study was performed according to local regulations and was approved by the ethics committees involved. Informed consent for publication of the clinical and genetic information was provided by all index individuals and living tested relatives. Consent for the publication of genetic information of deceased individuals was obtained from the next of kin. All study-related procedures are in accordance with the ethical standards laid down in the Declaration of Helsinki and its later amendments.
Clinical data were acquired by the investigators directly from the patient, from medical charts, or from the responsible physician. EDTA samples were taken from each available family member and DNA was extracted using standard procedures.
We performed whole-exome sequencing (WES) in all index individuals. The exomes have been sequenced within previous research projects (individual 1: IonNeuroNet/EuroEPINOMICS, individual 3: EuroEPINOMICS) or done on clinical diagnostic basis (individuals 2 and 4). All discovered variants were validated by Sanger sequencing. The genetic variants were interpreted according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants. The fulfilled criteria are listed in brackets with the abbreviations used in the reference literature (20).
This 49-year-old man has been followed up from 2003 to 2020. He started having attacks of PKD at the age of 10 years. The paroxysmal events were usually provoked by sudden movements, for example, at the start of a 100-m run, or when he was suddenly called while waiting in the waiting room. He reported having twisting and cramping movements of the right arm and leg which rarely spread to the left side of the body. Sometimes, the symptoms were so severe that he could no longer hold himself upright. The attacks lasted between 20 and 60 s and there were no prodromes, for example, sensory phenomena and no alteration of consciousness. He also did not notice any vertigo or the feeling of being drunk during the episodes. Other triggers such as consumption of alcohol, caffeinated drinks, or certain food or the feeling of being cold or fatigue were denied. In case of high fever, the attacks were more frequent, and on rare occasions, they could also occur at rest. The family history was negative for neurological disorders.
Carbamazepine had reduced attacks but was not tolerated because of side effects. Gabapentin was not helpful. The patient did not want to try other medications. The neurological examination revealed an asymmetrical right accentuated arm tremor but was otherwise normal (in particular no ataxia or myokymia, electromyography was not performed). EEG and MRI findings were normal. Genetic findings will be presented with the ones of individual #2.
This 17-year-old woman suffers from idiopathic generalized epilepsy and PxD. She had her first epileptic seizure at the age of 18 months when she suffered from atonic seizures with a sudden loss of tone for a few seconds with falling. She started to have generalized tonic–clonic seizures (GTCS) at the age of 6 years. At the same age, she had a febrile convulsive status epilepticus requiring intubation. Her first episodes with abnormal involuntary choreatiform movements started at the age of 12 years. Both legs and arms and facial muscles were affected. The paroxysmal events lasted 3 to 5 s, occurred two to three times per day, and were triggered by emotional stress, not by sudden movements or exercise.
Development was described as normal, yet when she was 6 years old, she was diagnosed with attention deficit hyperactivity disorder and with a mild difficulty in expressive language. There have been no neurologic disorders in the family.
She was first treated with valproate which was withdrawn due to lack of seizure control. Her second antiseizure drug was topiramate which was stopped due to anhidrosis. Levetiracetam led to an increase in seizure activity so the medication was exchanged with oxcarbazepine which was very effective. When she suffered from a febrile convulsive status epilepticus at the age of 6 years, valproate was added again and taken together with oxcarbazepine until the age of 15. Since paroxysmal dyskinesia did continue, she additionally received brivaracetam from the age of 14 years on. Only when lacosamide, a third anticonvulsive drug, was added 1 year later (in exchange for valproate), she became free of paroxysmal dyskinetic events. Her current treatment (last update: February 2021) is oxcarbazepine 2 × 600 mg (19.05 mg/kg body weight per day), brivaracetam 2 × 50 mg (1.59 mg/kg body weight per day), and lacosamide 2 × 150 mg (4.76 mg/kg body weight per day).
Physical examination did not reveal any signs of ataxia, impaired coordination, or tremor. Myokymia was observed clinically. Further significant findings were generalized hyperhidrosis and acne vulgaris.
A 6-h EEG recording after sleep deprivation (10–20 standard EEG recording, age: 15 years and 9 months) revealed intermittent generalized theta activity (4–6/s) and high amplitude delta activity (3–4/s) predominantly in stage I sleep as well as generalized 3/s spike-wave activity of up to 1 s duration during wakefulness without any clinical correlate. A small subcortical area of gliosis/demyelination right frontobasally was found in the MRI of the brain and interpreted as an unspecific finding.
Whole-exome sequencing for individual #1 was performed in 2012. At that time, no disease-causing variant could be identified (including analysis of PRRT2 and SLC2A1). Exome data reanalysis of six exome-negative patients with paroxysmal movement disorders from our local patient database in 2020 revealed the variant p.Gly396Val in KCNA1 (Figure 1). In the remaining five exomes (three samples of individuals with PKD and two samples of individuals with PED), we did not find any significant variants. Individual #2 received whole-exome sequencing on a diagnostic basis in 2020 where the variant p.Gly396Arg was detected.
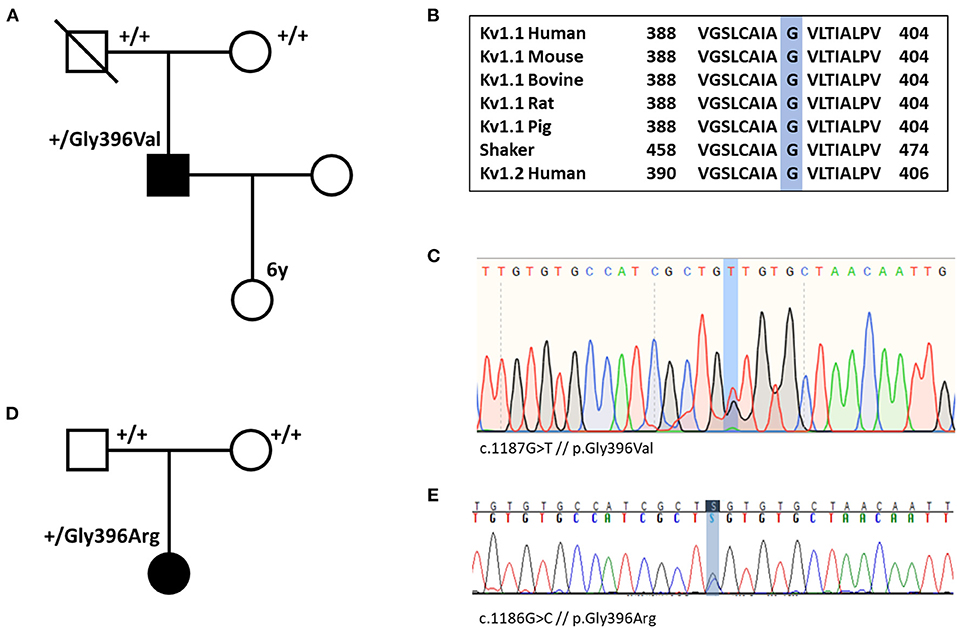
Figure 1. (A) Pedigree of individual #1 shows that the variant p.Gly396Val (p.G396V) is de novo. (B) Amino acid sequence alignment of Kv1.1 subunits across several species and human Kv1.2 subunit demonstrates conservation of the Gly396 residue. (C) Nucleotide sequence with heterozygote substitution of G with T (c.1187G>T) in individual #1. (D) Pedigree of individual #2 demonstrates de novo status of the variant. (E) Nucleotide sequence with heterozygote substitution of G with C in individual #2 (c.1186G>C).
Both variants are confirmed to be de novo and they were not listed in gnomAD (https://gnomad.broadinstitute.org/) (21). The amino acid change is located in the pore-forming transmembrane segment S6. Alignment of Kv1 channels across species, shaker, and Kv1.2 (Figure 1) indicates that the Gly396 residue is highly conserved among all species and tested channels. Only two familial variants in KCNA1 causing PKD have been described so far. While p.Asn255Lys sits in the S3-helix, p.Leu319Arg is located in an intracellular linker that connects the voltage-sensing domain to the pore (between S4 and S5) (22, 23). Both variants are in a completely different region compared with our variants. According to the ACMG criteria, the variants of individual #1 and #2 are predicted to be likely pathogenic [PM1, PM2, PM6, PP2, PP3; the abbreviations refer to the ACMG reference publication (20)]. All 21 in silico prediction tools listed on VarSome (free version) predict a deleterious effect. Furthermore, electrophysiological studies in Xenopus oocytes at homolog positions in the KCNA2 gene (Gly398Cys) and in the well-studied shaker channel (Gly466Ala and Gly466Trp) revealed loss-of-function defects for all three variants (24–26).
This 26-year-old woman has suffered from PED since childhood. Symptoms emerged when she began to be able to do prolonged exercise like hiking with the family or playing table tennis. Predominant symptoms of PED were dystonia of the legs that occurred after or during prolonged exercise and up to three to four times per week. After the interruption of physical activity and intake of glucose (dextrose), she was able to move on after 10–15 min. Regular intake of carbamazepine retard 15 mg/kg per day led to a reduction of the attack frequency to three to four episodes per 6 months.
At the age of 10 years, she presented with an episode which was classified as an attack of hemiplegic migraine: in school, she suffered from a sudden headache with subsequent paresis of the left leg, followed by twitches of the left leg and stiffness of the left arm, which was rotated inwards. Consciousness and awareness have been normal for the whole episode; there was no tongue bite, no defecation, and no enuresis. The speech was blurred. Her symptoms were regressing during the transport to the hospital. EEG revealed delta slowing of the right hemisphere. The paresis faded after 1 to 2 h. A hemiplegic migraine attack had been suggested. Such an episode did not recur. Since she did not feel the episodes of PED to be disturbing, she stopped taking carbamazepine between 11 and 13 years of age.
Mild coordination impairment was observed during development and she needed logopedic support to improve her speech skills. She had a learning disability and physical examination revealed mildly reduced fine motor skills. EEG repeatedly showed increased non-lateralized theta rhythms.
Family history was positive for PED (for pedigree, see Figure 2). Her mother and grandmother also suffered from the disease with PED of the legs after prolonged walks or hiking. All affected family members started to suffer from PED with onset in childhood. The brother of the index patient has early-onset absence epilepsy (with onset in the third year of life) in addition to PED, and the grandmother suffered from Parkinson's disease and idiopathic/genetic generalized epilepsy with GTCS.
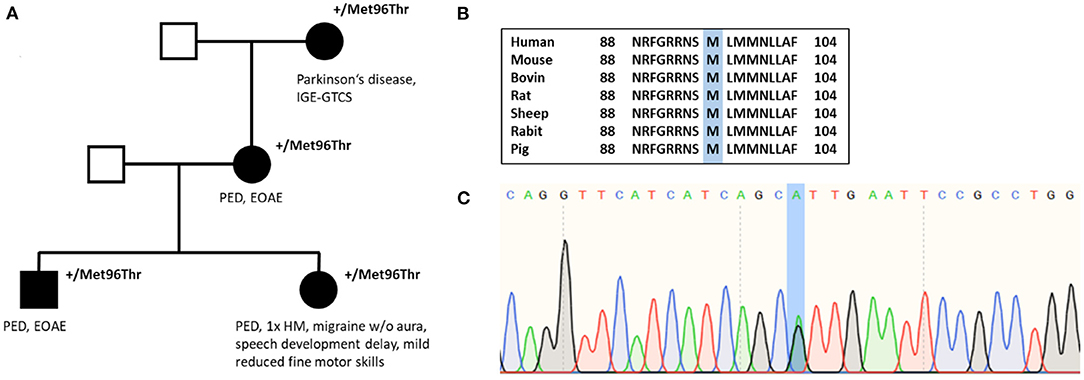
Figure 2. (A) Pedigree of individual #3 shows autosomal dominant inheritance. (B) Amino acid sequence alignment of SLC2A1 across several species demonstrates conservation of the Met96 residue. (C) Nucleotide sequence with substitution of A with G (c.286A>G).
In 2008, the first genetic test performing Sanger sequencing of PNKD and SLC2A1 was negative. In December 2011, whole-exome sequencing revealed the missense variant c.287T>C in SLC2A1 leading to an amino acid change from methionine to threonine (p.Met96Thr), which was reconfirmed by Sanger sequencing. The variant has been classified as pathogenic by MutationTaster and as probably damaging by PolyPhen-2. The amino acid position is located in the S3 helix and is conserved among several species (Figure 2). Evidence details have recently been uploaded to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/847673/). The variant p.Met96Val (c.286A>G), which has been described as a de novo variant in a patient with GLUT1-DS (27), has been investigated recently showing a significant reduction of glucose uptake compared with average protein function (28). However, our variant p.Met96Thr has not been published or functionally analyzed before. According to ACMG criteria, the variant has been classified as a variant of uncertain significance until recently. We suggest that, with our additional evidence [cosegregation with disease in multiple affected family members in a gene definitely known to cause the disease (PP1 and PM5)], the variant can be reclassified as likely pathogenic (PM2, PM5, PP1, PP2, PP3).
A 21-year-old man presented at our outpatient clinic for clarification of intermittent pain associated with cramps in the legs due to exertion, which he experienced the first time 1 year ago. In the beginning, symmetrical cramping pains in the legs had occurred in the afternoon, at the end of or after working as a warehouse keeper. When the symptoms were getting worse, the legs would turn inwards or forwards. Sometimes he noticed an indefinite feeling as a prodromal. When he sat down at the end of work, the symptoms eased after a certain time. Initially, L-dopa-sensitive dystonia was suspected by a practicing neurologist, but the symptoms did not improve by taking L-dopa. About 1 week after starting L-dopa treatment, he had his first and only GTCS. Immediately after the seizure, a cerebral CT scan and an EEG were performed, which were unremarkable. The individual had a history of early-onset absence epilepsy, which is why an antiepileptic treatment was started with valproic acid. There were no further seizures during the year until presentation, and also, the painful involuntary movements of the legs occurred less frequently.
Motor development was normal except for a circumscribed mild impairment of gross motor skills. He had mild psychomotor slowing and experienced difficulties in school with lack of concentration, but with support, he could complete secondary school. His maternal great-grandmother had a child with epilepsy (no further details available). The family history was otherwise negative for neurological diseases. On physical examination, mild gait insecurity was observed. The remaining neurological examination was normal.
At the age of 12 years, he underwent a 24-h EEG where generalized irregular 3/s spike-wave discharges with a maximal duration of 4 s had been recorded during sleep. There were no clinical absences during wakefulness. An MRI has never been performed.
Whole-exome sequencing revealed the variant c.692T>C which results in a substitution of leucine with proline (p.Leu231Pro). There has been no contact with the father, which is why a segregation analysis was not possible. Amino acid alignment shows conservation of the amino acid among several species (human, mouse, rat, bovine, sheep, pig, rabbit, chicken). There is one publication of an individual with suspected GLUT1-DS which carried the same variant. Segregation analysis in this individual confirmed de novo status. She had predominant eyelid myoclonus with onset at the age of 3 months and mild intellectual disability, so that the phenotype of this individual differs significantly from ours, also indicating phenotypic heterogenicity (29). According to ACMG criteria, the variant is likely pathogenic (PM2, PM6, PP2, PP3, PP5), whereas criterion PM6 is based on the already published individual.
We here describe four individuals with PxD, three of whom bear newly identified likely pathogenic variants in known PxD genes (individual #1: KCNA1, p.Gly396Val; individual #2: KCNA1, p.Gly396Arg; and individual #3: SLC2A1, p.Met96Thr). The clinical presentation of the fourth individual expands the phenotypic spectrum of an SLC2A1 variant previously associated only with GLUT1-DS (individual #4: SLC2A1, p.Leu231Pro).
The missense mutations of individual #1 and #2 (KCNA1; p.Gly396Val and p.Gly396Arg) further corroborate the recent findings of KCNA1 as a causative gene for PxD since only two PKD families with an underlying KCNA1 variant have been described so far (7, 8, 11). Moreover, these patients carry the first described KCNA1 variants with PxD phenotype occurring de novo.
This evidence is urgently needed since only the index patient of the family carrying the variant p.Leu319Arg in KCNA1 already published by Yin and colleagues (8) fulfilled the diagnostic criteria for PKD (3). Four other affected individuals with KCNA1-related PxD suffered from episodic dystonic attacks not fulfilling the criteria of PKD (3) because they were too long and not triggered by movement. The episodes were therefore termed “paroxysmal dyskinesia”—without further classification. Similar to our individual #2, the episodes were triggered by stress and anger (in our individual triggered by emotional stress). Given the long duration and/or the aforementioned triggering factors, one may suggest that there is a phenotypic overlap between PNKD in KCNA1-associated PxD. However, data on KCNA1-related PxD are only available from four different variants occurring in single families, so that closer assertions cannot be made yet.
KCNA1 is commonly associated with episodic ataxia 1 (EA1) but shows a broad phenotypic spectrum including myokymia and hypomagnesemia without EA1, developmental and epileptic encephalopathy, and neuromyotonia (23). Up to 10% of patients with EA1 suffer from epilepsy (30), and myokymia and hyperthermia have been reported to co-occur in KCNA1 mutation carriers as well. Therefore, it is not surprising that epilepsy has also been described to be a phenotypic feature of the known PxD-related KCNA1 variants p.Leu319Arg and p.Asn255Lys (7, 8). Accordingly, individual #2 suffered also from epilepsy and myokymia, representing the diverse phenotypic spectrum of KCNA1-related disorders.
Currently, there is no targeted treatment available for KCNA1-associated disorders, which are mainly caused by a biophysical loss-of-function effect (23). The carbonic anhydrase inhibitor acetazolamide, which is often used in EA1 patients, seems to be effective in some patients, but there are still many patients suffering from uncontrolled disease course (31). Our individuals and the individuals of the affected family carrying the p.Leu319Arg variant responded well to the sodium channel blocker (SCB) oxcarbazepine, although seizure freedom was not reached. Unfortunately, treatment data of individuals with the p.Asn255Lys variant were not available, but individual #2 became free of attacks when another SCB—lacosamide—was added to oxcarbazepine. Consequently, we consider that the usage of SCBs in combination or as monotherapy (and in analogy with EA1) might be a good choice for acetazolamide-resistant patients or even as a first-line therapy in KCNA1-associated PxD. There have been first in silico approaches for a specific, causative treatment for KCNA1-related disorders with new substances (32) and different SCBs using a targeted “drug repurposing” approach (33). The biophysical ratio of a broadened shape of the action potential (AP) due to a reduced function of voltage-gated potassium tetramers, such as KV1.1 subunits, and a consequently affected repolarization phase (34) seems plausible, given the fact that SCBs could “correct” this broadened AP by very well-described effects, such as enhancing the inactivated state (35, 36), blocking the persistent sodium current [e.g., riluzole (37)], and reducing repetitive firing (38). Nevertheless, further studies in heterologous expression systems and neurons are necessary to enable individualized treatment.
The two families in our series with a variant in SLC2A1 have PED and early-onset absence epilepsy or a positive family history of both. SLC2A1 is known to be the major causative gene of the two diseases (39–41). We therefore think that the combination of the manifestations is highly suggestive for SLC2A1 to be a candidate gene. The variant p.Met96Thr of individual #3 has not been described to date, but the evident cosegregation (Figure 2) increases the likelihood of pathogenicity of this variant. There have been functional measurements on a different variant at the same amino acid position (p.Met96Val), which has once been observed in a patient with GLUT1-DS (27, 28). A Xenopus laevis oocyte glucose uptake assay showed a significantly marked reduction of glucose uptake compared with the average protein function indicating pathogenicity. The variant with a valine substitution though has been found among 3 of 12,332 individuals of Finnish ancestry so that the likelihood of pathogenicity of this variant (p.Met96Val) becomes more disputable (21, 28).
Our case series has major limitations as we do only have single individuals or families carrying the presented variants and we do not have functional data to support our hypotheses. We know that the variants can only be classified as likely pathogenic and therefore leave room for some uncertainty. Also, we could not reliably retrace why the SLC2A1 variant of individual #3 was missed in the first sequencing approach via single-gene sequencing. However, we believe that in conjunction with the literature on related variants and their phenotypes, we increase the evidence of pathogenicity of the detected variants and give an overview of the current knowledge about them. Furthermore, functional characterizations of three other variants in homologous channels at the same position but different amino acids revealed a clear loss of function (see results). We want to highlight that two of our four described variants (KCNA1 p.Gly396Val; SLC2A1 p.Met96Thr) have been identified by reanalysis of existing genetic information. Therefore, reanalysis of existing genomic data may be considered, especially if the phenotype is highly suggestive for a specific gene or genetic phenotype. We encourage clinicians and researchers to share clinical information of individuals carrying rare or novel likely pathogenic or pathogenic variants, which might lead to a better understanding of the disease mechanism.
The original contributions presented in the study are publicly available. This data can be found at ClinVar with the following accession numbers: NM_000217.3(KCNA1):c.1187G>T SCV001623013; NM_000217.3(KCNA1):c.1186G>C SCV001623014; NM_006516.4(SLC2A1):c.287T>C SCV001623015 and NM_006516.4(SLC2A1):c.692T>C SCV001623016.
The studies involving human participants were reviewed and approved by Ethics Committee at the Medical Faculty of the University of Tuebingen and the Comite de Etica de Investicacion con medicamentos La Fe. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
JKe, LL, AM, AVM, and FM recruited and phenotyped the patients. JKr, MK, JKe, AVM, FM, and SL analyzed and interpreted the genetic data. JKe, JKr, and SL wrote the manuscript. All authors read, revised, and approved the manuscript.
The research was supported by the German Federal Ministry for Education and Research (BMBF, IonNeurONet: 01GM1105A, TreatION 01GM1907A), the EuroEPINOMICS Programme, and the European Reference Network—Rare Neurological Diseases (ERN—RND; Project ID No. 739510). AVM holds a grant CM19/00181 (Instituto de Salud Carlos III-Río Hortega).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patients and family members that have contributed samples for the purpose of this study. C. Hengsbach and A. Maisch are thanked for data organization and sample logistics.
1. Berkovic SF. Paroxysmal movement disorders and epilepsy Links across the channel. Neurology. (2000) 55:169–70. doi: 10.1212/WNL.55.2.169
2. Bhatia KP. Familial (idiopathic) paroxysmal dyskinesias: an update. Semin Neurol. (2001) 21:69–74. doi: 10.1055/s-2001-13121
3. Bruno MK, Hallett M, Gwinn-Hardy K, Sorensen B, Considine E, Tucker S, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. (2004) 63:2280–7. doi: 10.1212/01.WNL.0000147298.05983.50
4. Erro R, Sheerin U-M, Bhatia KP. Paroxysmal dyskinesias revisited: a review of 500 genetically proven cases and a new classification. Mov Disord. (2014) 29:1108–16. doi: 10.1002/mds.25933
5. Mink JW. Treatment of paroxysmal dyskinesias in children. Curr Treat Options Neurol. (2015) 17:23. doi: 10.1007/s11940-015-0350-9
6. Gardella E, Becker F, Møller RS, Schubert J, Lemke JR, Larsen LHG, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. (2016) 79:428–36. doi: 10.1002/ana.24580
7. Tian W-T, Huang X-J, Mao X, Liu Q, Liu X-L, Zeng S, et al. Proline-rich transmembrane protein 2–negative paroxysmal kinesigenic dyskinesia: Clinical and genetic analyses of 163 patients. Mov Disord. (2018) 33:459–67. doi: 10.1002/mds.27274
8. Yin X-M, Lin J-H, Cao L, Zhang T-M, Zeng S, Zhang K-L, et al. Familial paroxysmal kinesigenic dyskinesia is associated with mutations in the KCNA1 gene. Hum Mol Genet. (2018) 27:625–37. doi: 10.1093/hmg/ddx430
9. Jiang Y, Yuan F, Yang Y, Sun X, Song L, Jiang W. CHRNA4 variant causes paroxysmal kinesigenic dyskinesia and genetic epilepsy with febrile seizures plus? Seizure. (2018) 56:88–91. doi: 10.1016/j.seizure.2018.02.005
10. Gardiner AR, Jaffer F, Dale RC, Labrum R, Erro R, Meyer E, et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain. (2015) 138:3567–80. doi: 10.1093/brain/awv310
11. Huang X-J, Wang S-G, Guo X-N, Tian W-T, Zhan F-X, Zhu Z-Y, et al. The Phenotypic and genetic spectrum of paroxysmal kinesigenic dyskinesia in China. Mov Disord. (2020) 35:1428–37. doi: 10.1002/mds.28061
12. Du W, Bautista JF, Yang H, Diez-Sampedro A, You S-A, Wang L, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. (2005) 37:733–8. doi: 10.1038/ng1585
13. Dale RC, Melchers A, Fung VSC, Grattan-Smith P, Houlden H, Earl J. Familial paroxysmal exercise-induced dystonia: atypical presentation of autosomal dominant GTP-cyclohydrolase 1 deficiency. Dev Med Child Neurol. (2010) 52:583–6. doi: 10.1111/j.1469-8749.2010.03619.x
14. Erro R, Stamelou M, Ganos C, Skorvanek M, Han V, Batla A, et al. The clinical syndrome of paroxysmal exercise-induced dystonia: diagnostic outcomes and an algorithm. Mov Disord Clin Pract. (2014) 1:57–61. doi: 10.1002/mdc3.12007
15. Olgiati S, Skorvanek M, Quadri M, Minneboo M, Graafland J, Breedveld GJ, et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov Disord. (2016) 31:1041–8. doi: 10.1002/mds.26610
16. Mahajan A, Constantinou J, Sidiropoulos C. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: case presentation and initial benefit of intervention. J Neurol. (2017) 264:185–7. doi: 10.1007/s00415-016-8381-z
17. Garone G, Capuano A, Travaglini L, Graziola F, Stregapede F, Zanni G, et al. Clinical and genetic overview of paroxysmal movement disorders and episodic ataxias. Int J Mol Sci. (2020) 21:3603. doi: 10.3390/ijms21103603
18. Ebrahimi-Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain. (2015) 138:3476–95. doi: 10.1093/brain/awv317
19. Blakeley J, Jankovic J. Secondary paroxysmal dyskinesias. Mov Disord. (2002) 17:726–34. doi: 10.1002/mds.10178
20. On behalf of the ACMG Laboratory Quality Assurance Committee, Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–23. doi: 10.1038/gim.2015.30
21. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1530/ey.17.14.3
22. Kuang Q, Purhonen P, Hebert H. Structure of potassium channels. Cell Mol Life Sci. (2015) 72:3677–93. doi: 10.1007/s00018-015-1948-5
23. Paulhus K, Ammerman L, Glasscock E. Clinical spectrum of KCNA1 mutations: new insights into episodic ataxia and epilepsy comorbidity. Int J Mol Sci. (2020) 21:2802. doi: 10.3390/ijms21082802
24. Masnada S, Hedrich UBS, Gardella E, Schubert J, Kaiwar C, Klee EW, et al. Clinical spectrum and genotype–phenotype associations of KCNA2-related encephalopathies. Brain. (2017) 140:2337–54. doi: 10.1093/brain/awx184
25. Yifrach O, MacKinnon R. Energetics of pore opening in a voltage-gated K+ channel. Cell. (2002) 111:231–9. doi: 10.1016/S0092-8674(02)01013-9
26. Soler-Llavina GJ, Chang T-H, Swartz KJ. Functional interactions at the interface between voltage-sensing and pore domains in the shaker Kv channel. Neuron. (2006) 52:623–34. doi: 10.1016/j.neuron.2006.10.005
27. Leen WG, Klepper J, Verbeek MM, Leferink M, Hofste T, van Engelen BG, et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain. (2010) 133:655–70. doi: 10.1093/brain/awp336
28. Zaman SM, Mullen SA, Petrovski S, Maljevic S, Gazina EV, Phillips AM, et al. Development of a rapid functional assay that predicts GLUT1 disease severity. Neurol Genet. (2018) 4:e297. doi: 10.1212/NXG.0000000000000297. Available online at: https://ng.neurology.org/content/4/6/e297
29. Castellotti B, Ragona F, Freri E, Solazzi R, Ciardullo S, Tricomi G, et al. Screening of SLC2A1 in a large cohort of patients suspected for Glut1 deficiency syndrome: identification of novel variants and associated phenotypes. J Neurol. (2019) 266:1439–48. doi: 10.1007/s00415-019-09280-6
30. Rajakulendran S, Schorge S, Kullmann DM, Hanna MG. Episodic ataxia type 1: a neuronal potassium channelopathy. Neurotherapeutics. (2007) 4:258–66. doi: 10.1016/j.nurt.2007.01.010
31. D'Adamo MC, Hasan S, Guglielmi L, Servettini I, Cenciarini M, Catacuzzeno L, et al. New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front Cell Neurosci. (2015) 9:317. doi: 10.3389/fncel.2015.00317
32. Manville RW, Abbott GW. Isoform-selective KCNA1 potassium channel openers built from glycine. J Pharmacol Exp Ther. (2020) 373:391–401. doi: 10.1124/jpet.119.264507
33. Lauxmann S, Sonnenberg L, Koch NA, Boßelmann CM, Winter N, Schwarz N, et al. Therapeutic potential of sodium channel blockers as targeted therapy approach in KCNA1-associated episodic ataxia (EA1) and a comprehensive review of the literature. Front Neurol. (2021) (in review).
34. Hedrich UBS, Lauxmann S, Wolff M, Synofzik M, Bast T, Binelli A, et al. Effective precision therapy in KCNA2-related developmental and epileptic encephalopathy with 4-aminopyridine. Sci Trans Med. (in press).
35. Hebeisen S, Pires N, Loureiro AI, Bonifácio MJ, Palma N, Whyment A, et al. Eslicarbazepine and the enhancement of slow inactivation of voltage-gated sodium channels: A comparison with carbamazepine, oxcarbazepine and lacosamide. Neuropharmacology. (2015) 89:122–35. doi: 10.1016/j.neuropharm.2014.09.008
36. Bayraktar E, Liu Y, Hedrich UBS, Sara Y, Lerche H, Wuttke TV, et al. In vitro effects of S-Licarbazepine as a potential precision therapy on SCN8A variants causing neuropsychiatric disorders. bioRxiv [preprint]. (2021). doi: 10.1101/2021.04.24.441205
37. Zona C, Siniscalchi A, Mercuri NB. Bernardi G. Riluzole interacts with voltage-activated sodium and potassium currents in cultured rat cortical neurons. Neuroscience. (1998) 85:931–8. doi: 10.1016/S0306-4522(97)00604-0
38. Wamil AW, Schmutz M, Portet C, Feldmann KF, McLean MJ. Effects of oxcarbazepine and 10-hydroxycarbamazepine on action potential firing and generalized seizures. Eur J Pharmacol. (1994) 271:301–8. doi: 10.1016/0014-2999(94)90787-0
39. Suls A, Dedeken P, Goffin K, Van Esch H, Dupont P, Cassiman D, et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. (2008) 131:1831–44. doi: 10.1093/brain/awn113
40. Weber YG, Storch A, Wuttke TV, Brockmann K, Kempfle J, Maljevic S, et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest. (2008) 118:2157–68. doi: 10.1172/JCI34438
Keywords: paroxysmal dyskinesia, paroxysmal kinesiogenic dyskinesia, paroxysmal exercise induced dyskinesia, KCNA1, SLC2A1
Citation: Kegele J, Krüger J, Koko M, Lange L, Marco Hernandez AV, Martinez F, Münchau A, Lerche H and Lauxmann S (2021) Genetics of Paroxysmal Dyskinesia: Novel Variants Corroborate the Role of KCNA1 in Paroxysmal Dyskinesia and Highlight the Diverse Phenotypic Spectrum of KCNA1- and SLC2A1-Related Disorders. Front. Neurol. 12:701351. doi: 10.3389/fneur.2021.701351
Received: 27 April 2021; Accepted: 28 May 2021;
Published: 08 July 2021.
Edited by:
Anna De Rosa, Università di Napoli Federico II, ItalyReviewed by:
Florence Riant, Assistance Publique Hopitaux De Paris, FranceCopyright © 2021 Kegele, Krüger, Koko, Lange, Marco Hernandez, Martinez, Münchau, Lerche and Lauxmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Josua Kegele, am9zdWEua2VnZWxlQG1lZC51bmktdHVlYmluZ2VuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.