 Kwang-Dong Choi
Kwang-Dong Choi Jeong-Yeon Kim1
Jeong-Yeon Kim1 Seo-Young Choi
Seo-Young Choi Eun Hye Oh
Eun Hye Oh Jae-Hwan Choi
Jae-Hwan Choi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol. , 18 June 2021
Sec. Neuro-Otology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.690078
This article is part of the Research Topic Epidemiology and Genetics of Vestibular Disorders View all 18 articles
The 22q11.2 deletion syndrome (22q11.2DS), caused by a microdeletion on the long arm of chromosome 22, is characterized by congenital heart disease, hypoparathyroidism, immunodeficiency, developmental delay, and velopharyngeal insufficiency. Anatomic malformations of the middle and inner ears are frequently present, leading to high prevalence of hearing impairment. We present a first case of 22q11.2DS showing fluctuating hearing loss with recurrent vertigo attacks, resembling Ménière's disease. A 38-year-old male known to have 22q11.2DS developed recurrent vertigo, tinnitus, and fluctuating hearing loss in the left ear during a 10-year follow-up period. During vertigo attack, he had spontaneous left-beating nystagmus with downbeat components, but bithermal caloric and video head impulse tests showed normal vestibulo-ocular reflex functions. Sequential pure tone audiograms demonstrated fluctuating sensorineural hearing loss (SNHL) in both ears, which finally progressed to permanent hearing loss in the left ear. Computed tomography imaging of the temporal bone exhibited bilaterally malformed lateral semicircular canals, and delayed 3D-FLAIR sequences revealed cochlear endolymphatic hydrops with dilation of the scala media in the left ear. This case shows that acute vertigo with SNHL can be one of the audiovestibular presentations in 22q11.2DS caused by disturbance of endolymphatic flow.
The 22q11.2 deletion syndrome (22q11.2DS), also known as DiGeorge syndrome, is caused by a microdeletion on the long arm of chromosome 22 (1). Patients with 22q11.2DS exhibit highly variable phenotypes that include congenital heart disease, hypoparathyroidism, immunodeficiency, developmental delay, and velopharyngeal insufficiency. Anatomic malformations of the middle and inner ears are frequently present, leading to high prevalence of hearing impairment (2, 3). While abnormal morphology of the vestibule and the lateral semicircular canal (LSCC) has been also reported, most patients do not present with vestibular symptoms (4). In this report, we present a first case of 22q11.2DS showing fluctuating hearing loss with recurrent vertigo attacks, resembling Ménière's disease.
A 38-year-old male known to have 22q11.2DS presented with recurrent vertigo lasting for hours and fluctuating tinnitus in the left ear. He had a history of neonatal seizure, hypocalcemia due to hypoparathyroidism, and intellectual disability, and underwent cardiac surgery due to congenital heart disease (tetralogy of Fallot) at 15 years of age. On examination, the patient showed characteristic facial features of 22q11.2DS such as hypertelorism, a short and broad nose, and a deeply grooved philtrum. There was no spontaneous nystagmus between vertigo attacks, and bedside head impulse and bithermal caloric tests were normal. The pure tone audiogram (PTA) demonstrated sensorineural hearing loss (SNHL) in the left ear (Figure 1). Computed tomography (CT) imaging of the temporal bone revealed bilateral widening of the vestibules, soft tissue densities in the middle ear, and decreased mastoid air cells (Figure 2A). The patient was diagnosed with Ménière's disease, and treated with a low-salt diet and diuretics. The vertigo attacks resolved, and follow-up PTA revealed mild improvement of SNHL in the left ear (Figure 1).
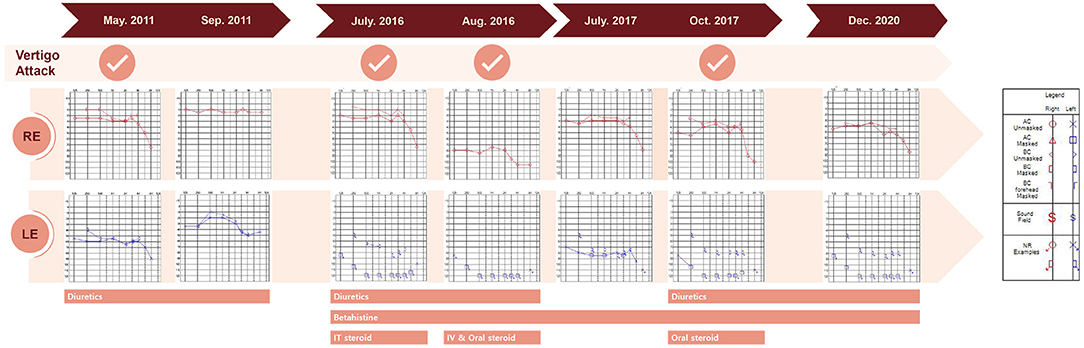
Figure 1. Time course of vertigo attacks, pure tone audiogram results, and treatments. AC, air conduction; BC, bone conduction; IT, intratympanic; IV, intravenous, LE, left ear, NR, no response; RE, right ear.
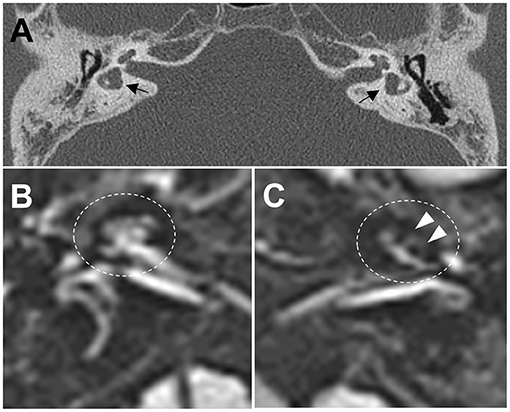
Figure 2. (A) Computed tomography images of the temporal bone demonstrate bilateral widening of the vestibules with an abnormally small bony island of lateral semicircular canals (black arrows) and soft tissue densities in the middle ear, and decreased mastoid air cells. (B,C) Axial three-dimensional fluid-attenuated inversion recovery images obtained 4 h after intravenous administration of gadolinium reveal cochlear endolymphatic hydrops (EH) with dilation of the scala media in the left ear (C, white arrows), but no EH in the right ear (B).
Five years after his initial presentation, the patient developed recurrent vertigo, hearing loss, and tinnitus in the left ear again. A physical examination demonstrated spontaneous left-beating nystagmus with downbeat components that increased during left eccentric gaze. Bithermal caloric tests were normal, and video head impulse tests showed normal vestibulo-ocular reflex (VOR) gains for all six semicircular canals (SCCs). PTA revealed profound SNHL in the left ear (Figure 1). Treatment with diuretics, betahistine, and intratympanic steroid injection to the left ear failed to attenuate the vertigo attacks. A 1-month follow-up PTA revealed additional hearing loss in the right ear without improvement of the left SNHL (Figure 1). The patient received an intravenous steroid injection followed by a tapering dose of oral steroids, resulting in a gradual reduction of the frequency of vertigo attacks. At the 1-year follow-up PTA, there was improvement of hearing loss in both ears, particularly in the right ear (Figure 1). Since the vertigo attacks recurred after stopping diuretics, the patient was maintained on combination therapy with betahistine and diuretics. During a 3-year follow-up period, he remained symptom-free despite the persistence of left SNHL (Figure 1). At the latest follow-up, the patient underwent delayed 3T magnetic resonance imaging (MRI; MAGNETOM Vida, Siemens, Erlangen, Germany) using a 64-channel array head coil, 4 h after intravenous gadolinium (12 ml of gadoterate meglumine, Dotarem; Guerbet, Aulnay-sous-Bois, France) to evaluate the presence of endolymphatic hydrops (EH) (5). We performed the 3D fluid-attenuated inversion recovery (3D-FLAIR) images with the following parameters; field of view (FOV): 160 × 160 mm, repetition time (RT): 7,000 ms, echo time (ET): 303 ms, inversion time (IT): 2,050 ms, matrix size: 256 × 230, flip angle: 120°, number of excitation (NES): 2, and scan time of 6 min 39 s. Axial 3D-FLAIR revealed cochlear EH with dilation of the scala media in the left ear (Figures 2B,C), but no cochlear enhancement or vestibular EH.
Among the affected genes in 22q11.2DS, deletion of TBX1 is responsible for the main features of the disease, such as heart problems, hypoplasia of the thymus and parathyroid glands, and velopharyngeal insufficiency (6). TBX1 is also expressed in the otic vesicle and the periotic mesenchyme during development, and was shown to be necessary for the development of the inner ear in mice (7). Thus, homozygous TBX1 mutant mice developed inner ear malformations that are characterized by an absent or hypoplastic vestibular apparatus with poorly developed SCCs, and a lack of coiled cochlear duct (6, 7). Likewise, patients with 22q11.2DS frequently present with anatomic malformations of the middle and inner ears in CT imaging of the temporal bone, primarily malformation of the LSCC with an abnormally small bony island, and incomplete partition type II of the cochlea. With regard to the highly prevalent malformations of the vestibule and LSCC in 22q11.2DS, a recent cross-sectional study found that, although vestibular dysfunctions such as caloric hypofunction and abnormal cervical vestibular-evoked myogenic potentials were common in patients with 22q11.2DS, none had experienced sudden vertigo (4). Postural imbalance has been described in patients with 22q11.2DS, but in most cases, it was associated with general muscle hypotonia or motor delay. Only one case had disequilibrium associated with severe malformations of bilateral LSCCs (8).
Remarkably, our patient presented with Ménière's disease-like symptoms such as whirling-type vertigo with nystagmus, fluctuating hearing loss, and tinnitus. He had bilaterally widened vestibules and LSCCs with an abnormally small bony island, but his VOR functions were spared. These anatomic malformations may have contributed to the development of Ménière's disease-like symptoms in our patient, presumably due to the formation of EH. This hypothesis is supported by the presence of cochlear EH on 3D-FLAIR images, which is concordant with PTA finding showing total deafness in the left ear. Because the patient was maintaining a vertigo-free state with normal VOR functions at the time of MRI examination, vestibular EH might not have been observed. Ménière's disease-like symptoms have been reported in patients with LSCC dysplasia, and histopathologic study revealed EH with hypoplastic endolymphatic sacs (9, 10). MRI evaluation of EH have found enlarged vestibular endolymph in LSCC dysplasia, and a strong negative correlation between the areas of the bony island and the endolymphatic space (11). Likewise, our patient may have had disturbance of endolymphatic flow due to inner ear malformations that caused Ménière's disease-like symptoms. Alternatively, the existence of genetic modifiers interacting with TBX1 may cause phenotypic variability of audiovestibular dysfunction in 22q11.2DS. A recent study found that non-coding variants in CRKL were significantly associated with risk for conotruncal heart defects in individuals with 22q11.2DS (12). Many genes have been related to hereditary non-syndromic hearing loss and Ménière's disease (13–16). It is therefore possible that rare variants in these genes could modify audiovestibular phenotype by interacting with TBX1 in 22q11.2DS.
Even though it is a rare occurrence, the case described herein shows that acute vertigo with SNHL can be one of the audiovestibular presentations in 22q11.2DS.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
All experiments followed the tenets of the Declaration of Helsinki, and informed consents were obtained after the nature and possible consequences of this study had been explained to the participants. This study was approved by the institutional review boards of Pusan National University Yangsan Hospital.
K-DC conducted the interpretation of the data and wrote the manuscript. J-YK, S-YC, EO, H-ML, and JR contributed to the interpretation and analysis of the data. J-HC conducted the design, conceptualized the study, interpreted the data, and revised the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2020R1I1A307161711).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. Verheij E, Elden L, Crowley TB, Pameijer FA, Zackai EH, McDonald-McGinn DM, et al. Anatomic malformations of the middle and inner ear in 22q11.2 deletion syndrome: case series and literature review. AJNR Am J Neuroradiol. (2018) 39:928–34. doi: 10.3174/ajnr.A5588
3. Jiramongkolchai P, Kumar MS, Chinnadurai S, Wootten CT, Goudy SL. Prevalence of hearing loss in children with 22q11.2 deletion syndrome. Int J Pediatr Otorhinolaryngol. (2016) 87:130–3. doi: 10.1016/j.ijporl.2016.06.005
4. Willaert A, Van Eynde C, Verhaert N, Desloovere C, Vander Poorten V, Devriendt K, et al. Vestibular dysfunction is a manifestation of 22q11.2 deletion syndrome. Am J Med Genet A. (2019) 179:448–54. doi: 10.1002/ajmg.a.7
5. van Steekelenburg JM, van Weijnen A, de Pont LMH, Vijlbrief OD, Bommeljé CC, Koopman JP, et al. Value of endolymphatic hydrops and perilymph signal intensity in suspected Ménière disease. AJNR Am J Neuroradiol. (2020) 41:529–34. doi: 10.3174/ajnr.A6410
6. Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. (2001) 27:286–91. doi: 10.1038/85845
7. Vitelli F, Viola A, Morishima M, Pramparo T, Baldini A, Lindsay E. TBX1 is required for inner ear morphogenesis. Hum Mol Genet. (2003) 12:2041–8. doi: 10.1093/hmg/ddg216
8. Moxham LMR, Mallinson AI. Vestibular function correlates with radiologic findings in a gymnast with 22q11.2DS. Am J Case Rep. (2020) 21:e922908. doi: 10.12659/AJCR.922908
9. Maekawa C, Kitahara T, Horii A, Miyabe J, Kubo T. Vestibular type of Mondini anomalies with BPPV and Meniere's disease-like symptoms. Auris Nasus Larynx. (2009) 36:218–20. doi: 10.1016/j.anl.2008.04.010
10. Hamed AA, Gadre AK, Linthicum FH Jr. Hypoplastic endolymphatic sac, hydrops, and Mondini deformity: a case report. Laryngoscope. (1992) 102:1043–8. doi: 10.1288/00005537-199209000-00015
11. Naganawa S, Kawai H, Sone M, Ikeda M. Ratio of vestibular endolymph in patients with isolated lateral semicircular canal dysplasia. Magn Reson Med Sci. (2015) 14:203–10. doi: 10.2463/mrms.2014-0112
12. Zhao Y, Diacou A, Johnston HR, Musfee FI, McDonald-McGinn DM, McGinn D, et al. Complete sequence of the 22q11.2 allele in 1,053 subjects with 22q11.2 deletion syndrome reveals modifiers of conotruncal heart defects. Am J Hum Genet. (2020) 106:26–40. doi: 10.1016/j.ajhg.2019.11.010
13. Mei C, Dong H, Nisenbaum E, Thielhelm T, Nourbakhsh A, Yan D, et al. Genetics and the Individualized Therapy of Vestibular Disorders. Front Neurol. (2021) 12:633207. doi: 10.3389/fneur.2021.633207
14. Oh EH, Shin JH, Kim HS, Cho JW, Choi SY, Choi KD, et al. Rare variants of putative candidate genes associated with sporadic meniere's disease in east Asian population. Front Neurol. (2020) 10:1424. doi: 10.3389/fneur.2019.01424
15. Escalera-Balsera A, Roman-Naranjo P, Lopez-Escamez JA. Systematic review of sequencing studies and gene expression profiling in familial meniere disease. Genes. (2020) 11:1414. doi: 10.3390/genes11121414
Keywords: 22q112 deletion syndrome, Ménière's disease, endolymphatic hydrops, case report, vertigo, sensorineural hearing loss
Citation: Choi K-D, Kim J-Y, Choi S-Y, Oh EH, Lee H-M, Roh J and Choi J-H (2021) Case Report: Ménière's Disease-Like Symptoms in 22q11.2 Deletion Syndrome. Front. Neurol. 12:690078. doi: 10.3389/fneur.2021.690078
Received: 02 April 2021; Accepted: 14 May 2021;
Published: 18 June 2021.
Edited by:
Jose Antonio Lopez-Escamez, Andalusian Autonomous Government of Genomics and Oncological Research (GENYO), SpainReviewed by:
Juan M. Espinosa-Sanchez, Virgen de las Nieves University Hospital, SpainCopyright © 2021 Choi, Kim, Choi, Oh, Lee, Roh and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jae-Hwan Choi, cmFjaGVsYm9sYW5AaGFubWFpbC5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.