 Tetsuro Abe
Tetsuro Abe Tomoki Kuwahara*
Tomoki Kuwahara*- Department of Neuropathology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
Previous genetic studies on hereditary Parkinson's disease (PD) have identified a set of pathogenic gene mutations that have strong impacts on the pathogenicity of PD. In addition, genome-wide association studies (GWAS) targeted to sporadic PD have nominated an increasing number of genetic variants that influence PD susceptibility. Although the clinical and pathological characteristics in hereditary PD are not identical to those in sporadic PD, α-synuclein, and LRRK2 are definitely associated with both types of PD, with LRRK2 mutations being the most frequent cause of autosomal-dominant PD. On the other hand, a significant portion of risk genes identified from GWAS have been associated with lysosomal functions, pointing to a critical role of lysosomes in PD pathogenesis. Experimental studies have suggested that the maintenance or upregulation of lysosomal activity may protect against neuronal dysfunction or degeneration. Here we focus on the roles of representative PD gene products that are implicated in lysosomal pathway, namely LRRK2, VPS35, ATP13A2, and glucocerebrosidase, and provide an overview of their disease-associated functions as well as their cooperative actions in the pathogenesis of PD, based on the evidence from cellular and animal models. We also discuss future perspectives of targeting lysosomal activation as a possible strategy to treat neurodegeneration.
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease after Alzheimer's disease, affecting about 10 million people worldwide. PD is clinically characterized by bradykinesia, tremor, rigidity, and postural instability as well as olfactory abnormalities and sleep disturbances. The motor symptoms of PD are mainly attributable to the selective loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc), causing dopamine deficiency (1). An important pathological hallmark in PD lesions is the intraneuronal inclusions called Lewy bodies that consist of aggregated α-synuclein phosphorylated at Ser129 residue (2–4). It is widely accepted that α-synuclein aggregates or oligomeric species spread to interconnected brain regions in a prion-like manner, although the processes are not fully understood (5).
Although the majority of PD cases (~90%) are sporadic, some forms of PD are hereditary and the responsible genes have been identified. SNCA encoding α-synuclein was the first gene identified, and the mutations in other genes such as leucine-rich repeat kinase 2 (LRRK2) and vacuolar protein sorting-associated protein 35 (VPS35) are also established as the cause for autosomal-dominant PD. On the other hand, genes associated with autosomal-recessive PD include PRKN, PINK1, and ATP13A2 (6). Importantly, accumulating evidence has pointed to a greater contribution of genetic determinants in sporadic PD (7, 8). Especially, past meta-analyses of genome-wide association studies (GWAS) targeting sporadic PD have repeatedly identified two of the above familial PD genes—LRRK2 and SNCA—as major risk factors, indicating that the impact of these two genes is more common in the general population (9–11). These GWAS for sporadic PD have succeeded in nominating a number of additional genes that were not identified from linkage analyses of familial PD cases, and GBA1 in particular is the most representative of such genes.
Importantly, a significant proportion of PD-associated genes (e.g., LRRK2, GBA1, ATP13A2, VPS35, and TMEM175) have been functionally implicated in the endolysosomal system in cells (12–16). Especially, GBA1 is well-known as a responsible gene for Gaucher disease, the most common lysosomal storage disorder. Moreover, the recent expansion of genetic, transcriptomic, and epigenetic studies in sporadic PD has nominated an increasing number of lysosomal pathway genes as a risk factor for PD (17–19). Endolysosomal dysfunctions are also frequently described in other neurodegenerative diseases such as Alzheimer's disease (AD), Huntington's disease (HD), frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS), all of which accompany neuronal accumulation of misfolded proteins (20, 21).
In addition to the evidence from genetics, the involvement of lysosomal dysfunction in PD has been implicated from pathological and biochemical studies using postmortem disease samples. The reduction in the immunoreactivity of lysosomal markers, such as LAMP1 and cathepsin D, was detected in PD and Lewy body disease (22, 23), and lysosomal breakdown, autophagosomal accumulation and the colocalization of autophagosomal markers with Lewy bodies were also detected in PD brains (24). Cathepsin D immunoreactivity has been shown to colocalize with α-synuclein pre-aggregates in nigral neurons in PD (25). The levels of lysosomal enzymes have been reported to be altered in cerebrospinal fluid and blood samples from PD patients (26–28). Thus, the role of lysosomes in PD pathogenesis is receiving increasing attention.
However, the detailed mechanisms on how lysosomal dysfunction leads to the neurodegeneration in PD remain largely elusive. There is a wide range of functions of PD-causative genes that are related to lysosomes, and much research has been focused on the elucidation of disease-related functions as well as the relationship among these genes. A common mechanism assumed by many researchers is that lysosomal dysfunction ultimately leads to α-synuclein accumulation and propagation in neurons. In fact, the role of lysosomes in α-synuclein degradation has long been attracted attention, and many studies on PD genes have also examined their effects on α-synuclein intracellular dynamics (i.e., metabolism, aggregation, secretion, and internalization).
In this article, we first summarize the current knowledge about the mechanisms of α-synuclein degradation in lysosomes, and then focus on the roles of other well-analyzed PD gene products, namely LRRK2, VPS35, ATP13A2 and GBA, in terms of their individual and co-operative regulations of endolysosomes and α-synuclein dynamics. Finally, we will discuss the potential of targeting endolysosomal system, especially the strategies to enhance lysosomal activity, in the future treatment of PD.
α-Synuclein: The Central Effector Degraded in Lysosomes
Missense mutation in SNCA gene encoding α-synuclein was first identified in 1997 as a cause of autosomal-dominant PD (29). Later on, more mutations in SNCA gene have been identified to date, including A53T, A30P, E46K, H50Q, G51D, and A53E (29–34). Furthermore, gene triplication and duplication of SNCA locus without missense mutations have also been reported as a cause of familial PD (35–37). This means that the increase of α-synuclein level by itself is sufficient to develop PD, and therefore proper clearance of α-synuclein is required for the prevention of disease onset. Multiple lines of evidence have suggested that α-synuclein is degraded in two major proteolytic pathways: the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal pathway (ALP) (38, 39). The metabolism in ALP has been the focus of much attention, especially in relation to the clearance of aggregated α-synuclein species.
Previous studies have shown that both extracellular and intracellular α-synuclein species are transported into lysosomes via the endosomal system or autophagy (40). It has been reported that α-synuclein is mainly degraded by cathepsins, especially cathepsin D, in lysosomes (41, 42). Cathepsin D level is shown to influence α-synuclein aggregation and toxicity in vivo (43). Treatment of cells with a lysosomal inhibitor bafilomycin A1 has been reported to not only affect α-synuclein metabolism but also to promote its propagation (44, 45).
Conversely, it has also been shown that the aggregated α-synuclein itself inhibits the function of lysosomes as well as other organelles. For example, α-synuclein pre-formed fibrils (PFFs) act on lysosomal membranes and cause its rupture (46–48). Another study has reported that α-synuclein impedes the lysosomal stress response mediated by the SNARE protein ykt6 (49). ykt6 is known as a regulator of ER-Golgi trafficking that is also reported to be disrupted by accumulated α-synuclein (50, 51), suggesting the possibility that the effect of α-synuclein on lysosomes is not necessarily direct. Collectively, it is assumed that lysosome inhibition exacerbates α-synuclein toxicity and α-synuclein accumulation in turn inhibits lysosomes, forming a vicious cycle that leads to the development of the disease.
Autophagy has also been established as a key mechanism regulating α-synuclein metabolism and toxicity. Macroautophagy is a major autophagy machinery that processes the degradation of a large portion of the cytoplasmic components through the formation of double-membrane structures called autophagosomes. The autophagosomes fuse with primary lysosomes to form autolysosomes where their contents are degraded and then either disposed or recycled back to the cell (52, 53). Inhibition of autophagosome-lysosome fusion by treatment with bafilomycin A1 or chloroquine enhanced α-synuclein release and transfer in human neuroglioma cells and rat primary cortical neurons (54, 55). In a mouse model of PD expressing human α-synuclein, impairment of macroautophagy under DA neuron-specifc knockout of Atg7 gene caused the aggravation of neuropathology, although the behavior of mice was paradoxically improved (56). In humans, it has been reported that the majority of Lewy bodies (~80%) composed of α-syuclein in the SNpc of PD patients were strongly immonoreactive for LC3 (24), and similar observation for LC3 immunoreactivity was observed in Lewy bodies of dementia with Lewy bodies (DLB) patients (57). These reports collectively implicate the impaired macroautophagy in the pathogenetic processes involving α-synuclein, although we should note that there is little direct evidence of α-synuclein degradation by macroautophagy.
On the other hand, another type of autophagy called chaperone-mediated autophagy (CMA) has been considered as a possible mechanism of PD (58). CMA mediates the lysosomal degradation of a specific subset of soluble cytosolic proteins containing a KFERQ-like motif, which can be recognized by the cytosolic chaperone heat shock cognate protein 70 (Hsc70). Proteins targeted by Hsc70 are directly transported into the lysosomes for degradation through association with lysosome-associated membrane protein 2A (LAMP2A). It has been shown that wild-type α-synuclein can be degraded in CMA whereas mutant α-synuclein interferes with the lysosomal transport process in CMA, suggesting a possible link between defective CMA activity and PD (59).
Accumulating evidence has suggested that these ALP machineries may be modified by several PD-associated gene products, including LRRK2, VPS35, ATP13A2, and GBA. In the following sections, we will discuss the possible roles of these proteins in ALP and α-synuclein metabolism, focusing on the pathological relevance in PD (Figure 1).
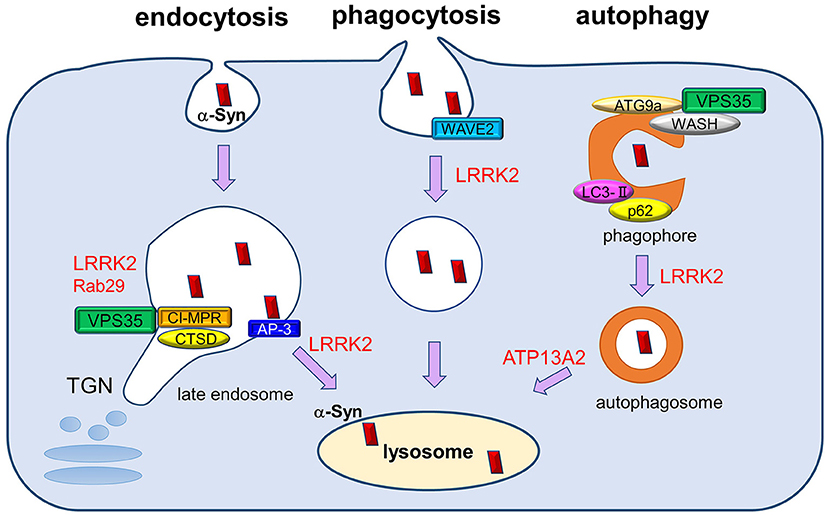
Figure 1. The roles of PD-associated proteins in endolysosomal pathways responsible for α-synuclein degradation. Extracellular and intracellular α-synuclein species (both soluble and aggregated) are transported into lysosomes for degradation through several pathways, including endocytosis, phagocytosis, and autophagy. PD-associated proteins VPS35, LRRK2, and Rab29 influence multiple steps of these degradation pathways, both individually and cooperatively. The retromer complex component VPS35 regulates the recycling of cathepsin D (CATD), the main lysosomal hydrolase responsible for α-synuclein degradation, by retrieving the lysosomal hydrolase receptor CI-MPR from endosome to the TGN. VPS35 pathogenic mutation may affect the recycling of CATD and thus impair α-synuclein degradation. LRRK2 and Rab29 further interact with VPS35 and regulate its function cooperatively. In endocytosis pathway, LRRK2 regulates AP-3-mediated intracellular recycling of lysosomal membrane proteins, whereas LRRK2 modulates the phagocytic activity by interacting with actin-cytoskeletal regulator WAVE2. VPS35 has been shown to function in macroautophagy pathway together with its interactors WASH complex and ATG9a, thereby regulating the transport of LC3-positive compartments. LRRK2 also regulates the autophagic flux, and ATP13A2 influences the clearance of autophagosomes. The perturbation of macroautophagy pathway is thought to contribute to the impaired degradation of α-synuclein, especially those of aggregated species.
LRRK2: A Multifaceted Kinase in the Endolysosomal System
Mutations in LRRK2 gene have been identified as the most common cause of autosomal-dominant PD (60, 61). LRRK2 is a large ~280 kDa protein and is widely expressed in human tissues including brains, although the expression is higher in the kidney, lung and immune cells (61–64). LRRK2 protein consists of multiple enzymatic and protein interaction domains including armadillo repeats (ARM), ankyrin repeats (ANK), leucine-rich repeats (LRR), Ras of complex (Roc), C-terminal of Roc (COR), kinase, and WD40 domains (61, 65, 66), suggesting that LRRK2 has diverse binding partners in distinct cellular pathways. LRRK2 has an ability to bind GTP through its ROC domain, and PD-associated mutations in LRRK2 have been shown to cause alterations in GTP binding and/or GTPase activity (64, 67, 68). A number of mutations in LRRK2 gene have been reported so far (69), and the following mutations are well-validated: N1437H, R1441C/G/H, Y1699C, G2019S, and I2020T. These mutations are located either in the ROC domain (N1437H, R1441C/G/H), COR domain (Y1699C) or kinase domain (G2019S, I2020T). Among these, G2019S is the most prevalent, followed by R1441C/G/H (60, 61, 69–72). It has been shown that G2019S mutation in LRRK2 increases its intrinsic kinase activity (73), whereas ROC/COR domain mutants affect GTPase activity or GTP binding (64, 68). These findings implicate the important roles of both GTPase/GTP binding and kinase activities of LRRK2 in PD pathomechanisms. Recent structural analyses of LRRK2 on microtubules using cryo-electron tomography/microscopy have shown that the kinase and GTPase domains are in close proximity (74, 75), suggesting that the activities of both domains are not independent but influence each other.
Recent studies have accumulated evidence that LRRK2 phosphorylates a subset of Rab family GTPases, including Rab3, Rab8, Rab10, Rab29, and Rab35, in their switch-II regions (76–80). Rab GTPases are the major regulators of intracellular membrane trafficking (81). It has been shown that LRRK2 and its substrate Rab GTPases, especially Rab8 and Rab10, are readily recruited onto lysosomes that are stressed by lysosomal overload (82, 83) or by membrane damage (84, 85). Under lysosomal overload stress, LRRK2 and Rab8 act against lysosomal hypertrophy, whereas LRRK2 and Rab10 facilitate the release of lysosomal contents. Under lysosomal membrane-damaging stress, LRRK2 recruits the ESCRT-III component CHMP4B via Rab8a (85) or the motor adaptor protein JIP4 via Rab10/Rab35 (84) to damaged lysosomes for membrane repair. The lysosomal recruitment of LRRK2 is further regulated by Rab29 (also known as Rab7L1), another interactor and substrate of LRRK2 (82, 83). Studies in C. elegans neurons have suggested that the orthologs of LRRK2 and Rab29 both regulate axon termination, and double mutant analysis has revealed their functions in a same genetic pathway that involves the clathrin adaptor protein complex 3 (AP-3), an important regulator of Golgi-lysosome transport of lysosomal membrane proteins (86).
A variety of studies have also reported the relationship between LRRK2 and autophagy. Studies of Lrrk2 KO mice have demonstrated the altered autophagic markers such as the autophagosome marker LC3-II and the autophagy substrate p62 (87, 88). The levels of these autophagic markers were changed in age-dependent and bi-phasic manners; LC3-II level was increased at 7 months of age but decreased at 20 months in Lrrk2 KO mice, whereas p62 was decreased at 7 months and increased at 20 months (87). In vitro studies have shown that the knockdown of LRRK2 in neuroblastoma SH-SY5Y cells caused a marked increase in LC3-II and p62 levels (89). In contrast, another study has shown that the knockdown of endogenous LRRK2 in macrophage or microglial cells decreased LC3-II levels and autophagy flux (90). Thus, although these changes in the levels of autophagic markers indicate the important role of LRRK2 in the proper regulation of autophagic flux, the effects of LRRK2 on autophagy depend on the conditions such as cell type and experimental methodology, and the mechanism of how LRRK2 regulates autophagy still remains unclear.
As for the relationship between LRRK2 and CMA, it has been reported that LRRK2-G2019S inhibits CMA by affecting LAMP2A-mediated internalization of the substrate proteins like α-synuclein into lysosomes, which results in α-synuclein accumulation in neurons (91). Consistently, a significant reduction in CMA or lysosomal markers such as LAMP1, LAMP2A, Hsc70, and cathepsin D has been described in whole brains or SNpc of PD patients (22, 24, 92, 93). LRRK2 may additionally regulate the phagocytic activity in myeloid cells, where LRRK2 binds and phosphorylates the actin remodeling protein Wiskott-Aldrich syndrome protein family verprolin-homologous protein 2 (WAVE2), which is important for the efficient promotion of phagocytosis (94).
In neurons, LRRK2 physically and functionally interacts with the retromer complex component VPS35, which is also known as a causative gene product for hereditary PD. Retromer complex functions on endosomes to selectively transport cargo proteins to the trans-Golgi network (TGN) or plasma membranes (95), and intirectly regulates lysosomal functions, as described later. The LRRK2-VPS35 functional interaction in various experimental context was further modulated by a LRRK2-binding protein Rab29 (96). Another report has demonstrated that a pathogenic VPS35 mutation (D620N) influences LRRK2 kinase activity with unknown mechanism; that is, LRRK2 activity to phosphorylate its substrate Rab GTPases was significantly enhanced in VPS35[D620N] knock-in cells compared to those without VPS35 mutation (97). Collectively, there is considerable evidence that LRRK2 acts on endolysosomal system, although further analysis is needed to determine which of these functions is particularly important in PD pathogenesis.
VPS35: An Indirect Regulator of Lysosomes
Mutations in vacuolar protein sorting-associated protein 35 (VPS35) gene are the genetic cause in PARK17, a locus for autosomal-dominant familial PD. Two independent groups have investigated Austrian and Swiss kindreds that develop PD and identified D620N mutation in VPS35 as the cause of the disease (98, 99). Patients with VPS35 D620N mutation have a mean age of onset in the 50s, and their clinical manifestations are similar to those of sporadic PD, such as resting tremor, bradycardia and L-DOPA reactivity (100, 101). Thus, although the presence of Lewy bodies in patient brains has not been confirmed, PD with VPS35 mutation and sporadic PD are thought to share some common pathogenetic mechanisms.
The VPS35 gene encodes a 796 amino acid protein that acts as a crucial component of the retromer complex, a mediator of the retrograde transport of endosomal proteins to TGN or plasma membranes (102–104). Retromer contains two subprotein complexes: a cargo recognition complex composed of VPS26–VPS29–VPS35 heterotrimer and a membrane-targeting dimer of sorting nexins (SNX1, SNX2, SNX5, SNX6, and SNX32) (105–108). VPS35 is located at the center of the complex and is important for the recognition and binding of the cytoplasmic domain of cargoes for retrograde transport (109). Particularly, retromer is responsible for the retrograde transport of cation-independent mannose 6-phosphate receptor (CI-MPR), a sorting receptor of lysosomal hydrolases including cathepsin D (110). Therefore, the dysfunction of VPS35 or retromer is thought to affect lysosomal activity through impaired delivery of lysosomal hydrolases, and this may also affect α-synuclein clearance, as cathepsin D is one of major enzymes responsible for the degaradation of α-synuclein (41, 43). In relation to PD, it has been reported that PD-associated D620N mutation in VPS35 causes defects in sorting of CI-MPR (111). Also, D620N mutation in VPS35 has been shown to affect retromer binding to the actin-nucleating Wiskott-Aldrich syndrome and SCAR homolog (WASH) complex, an important functional partner of retromer (16, 112).
VPS35 has also been associated with other cellular processes such as autophagy (102). It has been shown that VPS35 regulates macroautophagy by controlling the endosomal localization of WASH complex as well as ATG9a, a multipass transmembrane protein that is considered to regulate the early steps of autophagosome formation (16). Specifically, the transport of ATG9a is affected by D620N mutant VPS35, which then causes the impairment of autophagosome formation. Another study has suggested the role of VPS35 in CMA, where VPS35 mediates endosome-to-Golgi retrieval of LAMP2A receptor (113). Mice with reduced Vps35 level or D620N mutation showed alterations in lysosomal morphology with a decrease in the level of LAMP2A. This may be due to impaired recovery of LAMP2A from the endosome to the Golgi, which then leads to the enhanced degradation at the lysosomes. This reduction in LAMP2A level is expected to cause a decrease in CMA-mediated α-synuclein degradation. Actually, Vps35-deficient mice showed multiple PD-like phenotypes such as the accumulation of α-synuclein in DA neurons, reduced level of the catecholamine-synthesizing enzyme tyrosine hydroxylase (TH) and DA transmitters, dystrophic TH-positive neurites/axons, and impaired motor behaviors (113). Another group has reported that lentivirus-mediated overexpression of human wild-type VPS35, but not PD-linked P316S mutant, rescues α-synuclein accumulation as well as α-synuclein-mediated neuronal loss and astrogliosis in α-synuclein transgenic mice (114). In humans, the alterations in the protein levels of CMA markers (LAMP2A and Hsc70) are documented in SNpc and amygdala of PD patients (115). These findings collectively suggest the role of VPS35 as an indirect controller of lysosomes through the regulation of intracellular trafficking of lysosomal enzyme adaptors or multiple autophagic regulators.
ATP13A2: A Unique Cation Transporter on Lysosomes
Recessive mutations in ATP13A2 (polyamine-transporting ATPase 13A2), a gene located in a PD-associated locus PARK9, have been identified as the genetic cause for Kufor-rakeb syndrome (KRS), which is a type of Parkinsonian syndromes. KRS is clinically characterized by L-DOPA-responsive juvenile parkinsonism as well as cognitive impairment and myoclonus (116), and pathologically characterized by diffuse cerebral and cerebellar atrophy (117). Loss-of-function mutations in ATP13A2 have additionally been reported to cause neuronal ceroid lipofuscinosis (118, 119). ATP13A2 is a lysosomal P5-type transport ATPase that is involved in the transport of divalent metal cations as well as polyamines on lysosomal membranes (120). Loss of ATP13A2 causes lysosomal accumulation of polyamines (e.g., spermine) and lysosomal rupture, leading to cell toxicity (121). ATP13A2 has also been suggested to regulate multiple cellular functions related to lysosomes, including heavy metal homeostasis and mitochondrial homeostasis (15, 122). For example, a recent study using SH-SY5Y cells, patient-derived fibroblasts and the nematode C. elegans has identified a conserved cell protective pathway that counters mitochondrial oxidative stress via ATP13A2-mediated lysosomal spermine export (123).
A number of previous studies have pointed to the essential role of ATP13A2 in the homeostasis of lysosomal function (124). Studies with PD patient-derived mutant ATP13A2 fibroblasts and ATP13A2-knockdown DA neurons have shown that PD-linked mutations in ATP13A2 lead to several lysosomal alterations, including impaired lysosomal acidification, decreased activity of lysosomal enzymes, reduced degradation of lysosomal substrates and defective clearance of autophagosomes (125). Conversely, overexpression of wild-type ATP13A2 in ATP13A2-deficient cells restores lysosomal function and prevents cell death (125). Other studies have demonstrated that ATP13A2 regulates endolysosomal cargo sorting through its cytosolic N terminal domain, independent of its catalytic activity (126), and ATP13A2 regulates mTORC1-TFEB pathway together with another PD-associated gene product synaptotagmin 11 (SYT11) to induce autophagy as well as α-synuclein clearance (127). ATP13A2 deficiency and mutation have also been shown to cause the reduction in the level of cathepsin D, a main α-synuclein-degrading enzyme in lysosomes, in human neuroblastoma SH-SY5Y cells and in medaka fish (128).
The relevance of ATP13A2 defects to α-synuclein accumulation has been more directly demonstrated from other studies. Depletion of ATP13A2 in primary cortical neurons using a short hairpin RNA promoted the aggregation of α-synuclein by reducing lysosomal activity, which ultimately led to cell death (15, 129). On the other hand, overexpression of ATP13A2 in α-synuclein-stable SH-SY5Y cells lowered intracellular α-synuclein levels and instead promoted extracellular secretion of α-synuclein (130). Another study has reported that overexpression of ATP13A2 rescued DA neuron degeneration caused by overexpressed α-synuclein in rat primary midbrain cultures and in C. elegans (131).
In vivo, Atp13a2 knockout mice exhibit a neuronal ceroid lipofuscinosis-like phenotype, accumulation of mitochondrial ATP synthase subunit C (132), α-synuclein accumulation, dopaminergic pathology and late-onset sensorimotor deficits (133, 134). More specifically, ATP13A2 deficiency causes dysfunctions in the fusion of autophagic vacuoles with lysosomes as well as the impairment of lysosome-mediated degradation of proteins including α-synuclein (135). Analyses of postmortem PD patient brains have shown the presence of ATP13A2 in the Lewy bodies and a decrease in the levels of lysosomal components including ATP13A2 in DA neurons (125, 136). Although the mutations in ATP13A2 are rare in humans, these studies have collectively pointed to the important roles of ATP13A2 in ALP that may be involved in the neurodegenerative processes.
Glucocerebrosidase: The Lysosomal Enzyme Linked to Sporadic PD
Homozygous or compound heterozygous mutations in GBA1 gene are well-known to cause Gaucher disease (GD), a lysosomal storage disorder, whereas heterozygous mutations that in the homozygous state lead to GD have been reported to increase the risk for developing PD (137–139). Also, a higher incidence of Parkinsonism in patients with GD harboring GBA1 homozygous mutations has been reported (140, 141). Moreover, a number of genome-wide association studies (GWAS) have identified GBA1 as a most common genetic risk factor for idiopathic PD (9, 11, 142). Compared to non-GBA1-associated PD, GBA1-associated PD shows an earlier onset of the disease and a higher prevalence of non-motor symptoms, such as cognitive decline. They also tend to have a family history of dementia, and non-motor symptoms often manifest before the onset of motor symptoms (143, 144). GBA1 mutations are also a risk factor for dementia with Lewy bodies (DLB) (145, 146), and PD patients with GBA1 mutation have about a 3-fold higher risk of progressing to dementia than those without mutation (147). They also exhibit a faster progression of visual hallucinations, dysautonomia and motor symptoms, with a resultant decrease in survival rate (143, 145, 148).
GBA1 gene encodes the lysosomal enzyme glucocerebrosidase (GCase) that hydrolyzes glucosylceramide (GlcCer) to ceramide and glucose. GBA1 mutations have been shown to cause the reduction in the enzymatic activity of GCase (149, 150) and prevent GCase from reaching the lysosome, causing the accumulation of GlcCer in neurons (151–153). The significant correlation between the severity of the specific GBA1 mutation and that of clinical phenotypes (e.g., odds ratios for PD, age at onset, risk for dementia) has been reported (145, 147, 154), suggesting major impact of GCase activity in the pathogenetic processes. Importantly, idiopathic PD patients without GBA1 mutations also showed lower enzymatic activity and levels of GCase in brain tissue samples (155–157) and in dried blood spots (149). The reduction in GCase activity was further demonstrated in PD patient-derived DA neurons without GBA1 mutations (158, 159). These observations suggest that GCase dysfunction is a common pathogenic mechanism in idiopathic PD.
The reduced function of GCase are expected to contribute to the accumulation of α-synuclein in PD lesions (160). Indeed, treatment with a GCase inhibitor Conduritol B epoxide (CBE) caused a large increase in the levels of α-synuclein, without increasing α-synuclein mRNA, in human neuroblastoma SH-SY5Y cells and in mice (161). The association between reduced GCase and increased α-synuclein is further implicated in human PD postmortem brains (157). The accumulation of GlcCer in neurons as a result of GCase deficiency is thought to promote the formation of toxic α-synuclein aggregates (162), as lipids like GlcCer may strongly interact with α-synuclein and accelerate its fibril formation (163, 164). Another study has suggested a model where α-synuclein deposition and reduced GCase activity may influence each other and form a positive feedback loop that leads to a vicious cycle of disease progression (156).
On the other hand, the activity and function of GCase in microglia or related phagocytic cells have also been focused, as GCase is highly expressed in monocyte lineage cells. In mice, genetic depletion or pharmacological inhibition of GCase caused microglial activation (165, 166). Lower GCase activity was detected in monocytes, but not lymphocytes, from PD patients, when compared with those from healthy subjects (167). Importantly, such reduction in GCase activity was detected in those from patients without GBA1 mutations. As monocyte lineage cells contain a large number of well-developed lysosomes, it is possible to assume that the dysfunction of lysosomal GCase in these cells greatly influences α-synuclein metabolism.
Recently, much attention has been paid to the relationship between GBA1 and LRRK2. An increasing number of patients harboring both GBA1 and LRRK2 mutations have been reported, and these patients tend to develop PD at an earlier age than carriers of LRRK2 or GBA1 mutation alone (168–170). These reports suggest the cooperative effect of GBA1 and LRRK2 mutations for the development of PD. In experiments using DA neurons derived from PD patients, reduced GCase activity was observed in cells with LRRK2 mutations, and the inhibition of LRRK2 kinase activity restored GCase activity (171). Furthermore, treatment of GBA1 mutant knock-in astrocytes with LRRK2 kinase inhibitor rescued the lysosomal abnormalities such as pH increase and the reduction in cathepsin B activity (172). These observations collectively suggest that the functions of LRRK2 and GCase in terms of lysosomal regulation are closely interrelated.
Perspectives on the Therapeutic Strategies Targeting Lysosomes
As described above, ALP can be regulated by PD-associated genes LRRK2, VPS35, ATP13A2, and GBA1 not only individually but also cooperatively. Especially, cooperative maintenance of lysosomes by these genes is considered as one of key mechanisms related to PD (Figure 2). For example, lysosomal morphology under lysosomal overload stress is maintained by LRRK2 kinase activity (82) that is enhanced in cells harboring VPS35 pathogenic mutation, although the mechanism of enhancement is unclear (97). As lysosomes apparently play important roles in the accumulation and toxicity of α-synuclein, a number of studies have focused on enhancing ALP as a possible therapeutic strategy for α-synucleinopathies (173).
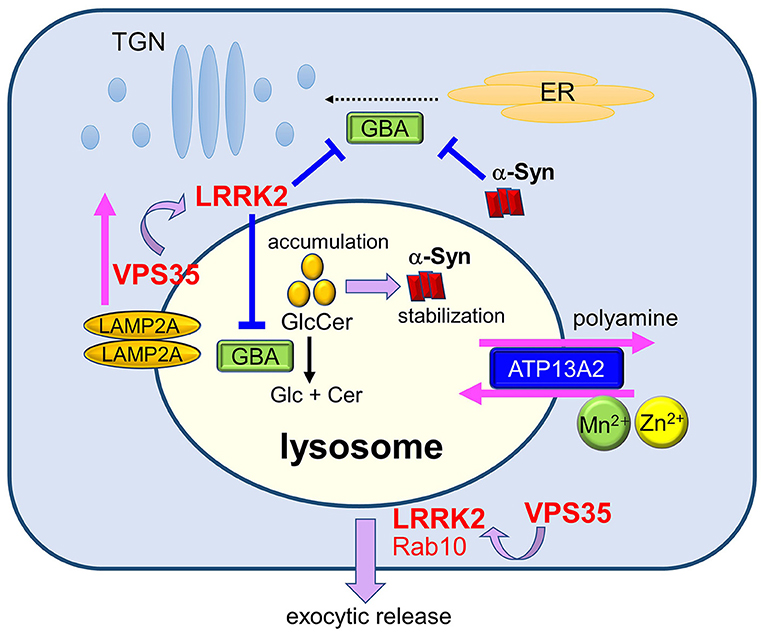
Figure 2. Maintenance of lysosomes by PD gene products and its relevance to PD. Lysosomal homeostasis is regulated by several PD genes such as ATP13A2, LRRK2, GBA, and VPS35. Deficiency in ATP13A2, a P5-type ATPase localized to the lysosomal membranes, is expected to affect lysosomal functions via dysregulated transport of several divalent metal cations and polyamines. LRRK2 functions in the maintenance of stressed lysosomes by facilitating the exocytic release of lysosomal contents together with its substrate Rab10 under lysosomal overload stress. LRRK2 may also negatively regulate the activity of lysosomal hydrolase glucocerebrosidase (GCase). Decreased activity of GCase causes the accumulation of its substrate glucosylceramide (GlcCer), which stabilizes toxic α-synuclein species. Accumulation of α-synuclein has been shown to block ER-to-Golgi trafficking of GCase, causing a further decrease in lysosomal GCase. The retromer component VPS35 mediates the retrieval of a CMA receptor LAMP2A on endolysosomal membranes, and mutation in VPS35 leads to the enhanced degradation of LAMP2A at the lysosomes, causing CMA defects and α-synuclein accumulation. VPS35 mutation also causes the enhancement of LRRK2 kinase activity, which may then affect the lysosomal maintenance and GCase activity.
Enhancement of lysosomal activity is one of plausible approaches to facilitate α-synuclein degradation. Among the PD-associated gene products introduced above, GCase has been the most well-studied as a target that contributes to lysosomal activation and α-synuclein metabolism. It has been shown that lysosomal GCase activity can be enhanced by treatment with ambroxol hydrochloride, a clinically used expectorant drug and an effective pharmacological chaperone for GCase (174–176). Oral administration of ambroxol to wild-type and α-synuclein transgenic mice caused the increase in brain GCase activity as well as the reduction in the levels of total and phosphorylated α-synuclein (177). Amboxol administration in rats also resulted in the restoration of decreased GCase activity and the decrease of α-synuclein pathology that were induced by 6-hydroxydopamine (6-OHDA) treatment (178). Additionally, oral administration of another molecular chaperone for GCase, AT2101 (afegostat-tartrate, isofagomine), to α-synuclein transgenic mice improved motor and non-motor function, abolished microglial response in the substantia nigra, and reduced the number of small α-synuclein aggregates (179). Adeno-associated virus (AAV)-mediated overexpression of GCase in hippocampus ameliorated α-synuclein accumulation as well as cognitive impairment in transgenic mice expressing mutant GCase (D409V/D409V) or A53T α-synucein (180, 181). Using the same mice models, the researchers have also shown that the administration of a brain-penetrant inhibitor of GlcCer synthase (GCS), GZ667161, ameliorated α-synuclein accumulation and cognitive deficits (182). These reports indicated that proper GlcCer metabolism is important to control α-synuclein accumulation.
Farnesyltransferease inhibitors (FTIs) are recently attracting significant attention as a promising lysosomal activator. It has been reported that FTI treatment in α-synuclein transgenic mice enhanced GCase activity and rescued pathological α-synuclein aggregation (49). FTI treatment has also been reported to reduce tau pathology in tauopathy model mice by activating lysosomes (183). Importantly, one of FTIs, lonafarnib, has been approved by FDA very recently for the treatment of Hutchinson-Gilford progeria syndrome, a rare and fatal premature aging disease (184). Thus, it will be of particular interest to see if such therapeutic strategies are actually effective in the treatment of PD or related neurodegenerative disorders.
Another plausible approach to activate lysosomes is the expression of transcription factor EB (TFEB), a master transcriptional regulator of ALP. Overexpression of TFEB has been shown to rescue midbrain DA neurons from α-synuclein-induced toxicity in transgenic rat models (185). In addition to α-synuclein, overexpression of constitutively active TFEB has been shown to reduce protein aggregates in old quiescent neural stem cells (qNSCs) (186) and in p53-induced senescent fibroblast cells (187). Nuclear translocation of TFEB is induced by inhibition of mammalian target of rapamycin (mTOR) (188), a well-known negative regulator of macroautophagy and ALP, and therefore mTOR inhibition has also been focused as a promising strategy. Intra-cerebral infusion of an mTOR inhibitor rapamycin for 2 weeks in α-synuclein transgenic mice resulted in reduced accumulation of α-synuclein (189), and long-term feeding a rapamycin diet (~24 weeks) improved motor performance in A53T α-synuclein transgenic mice (190). However, due to the side effects of rapamycin that have been noted to be problematic with long-term use (191), the use of rapamycin in the treatment of PD is expected to be challenging.
On the other hand, an mTOR-independent activator of autophagy, trehalose, has been shown to activate macroautophagy and enhance the clearance of wild-type or mutant forms of α-synuclein (192–195). Mechanistically, trehalose has been shown to activate macroautophagy by inhibiting the glucose transporter SLC2A, which ultimately leads to the activation of an energy-sensing kinase AMPK that stimulates autophagy (196). Oral administration of trehalose to A53T α-synuclein transgenic mice for 1 week induced macroautophagy and reduced the level of insoluble α-synuclein in the brain (197). Similarly, oral administration of trahalose to AAV-based rat model expressing A53T α-synuclein for 6 weeks caused a significant attenuation in α-synuclein-mediated motor deficits and DA neurodegeneration as well as α-synuclein accumulation (198). In addition to trehalose, a tyrosine kinase inhibitor nilotinib is another drug that stimulates autophagy by activating AMPK (199); chronic administration of nilotinib for 3–6 weeks in human A53T α-synuclein transgenic mice resulted in the decrease of α-synuclein levels, suppression of DA neuronal loss and improvement of motor behavior (200).
Activation of the CMA pathway is considered as an alternative strategy to increase the clearance of α-synuclein. Overexpression of LAMP2A has been shown to upregulate CMA, decrease α-synuclein accumulation and protect against α-synuclein toxicity in human neuroblastoma SH-SY5Y cells, rat primary cortical neurons, and nigral dopaminergic neurons in vivo (201). Inhibition of signaling through retinoic acid receptor α (RARα), a negative regulator of CMA, has also been focused; treatment with the RARα inhibitor all-trans-retinoic acid and its synthetic derivatives has been shown to activate CMA and protect against oxidative stress and proteotoxicity in cells (202). A specific subset of miRNAs that downregulate CMA has also been identified (203), and treatment with Geniposide, a bioactive iridoid glycoside that acts as a down-regulator of miRNAs especially for miR-21, increased LAMP2A expression and reduced α-synuclein levels in SH-SY5Y cells and MPTP-treated PD model mice (204).
In conclusion, a variety of strategies that aim to activate ALP have been developed and shown to modulate α-synuclein accumulation as well as PD-related phenotypes. The strategies that were tested for in vivo phenotypic modulation are summarized in Table 1. Several of the compounds used in these strategies are now being examined in clinical trials for PD and related disorders [e.g., ambroxol (205) and nilotinib (206, 207), see ClinicalTrials.gov]. These compounds or related products with similar mechanisms is expected to be available in the future as disease-modifying therapies. Moreover, as overviewed above, ALP is regulated in various ways by PD gene products—including LRRK2, VPS35, ATP13A2, GCase, and other risk factors not mentioned in this review—and among these, not only GCase (activator, ambroxol) but also LRRK2 (inhibitor) are being targeted in clinical trials (208). Further clarification of the functional relationships among PD-causing genes and their regulation to ALP may lead to the proposal of new therapeutic targets. It is hoped that further basic analysis of cellular and animal models, such as those described in this review, will accelerate the development of fundamental therapeutic agents.

Table 1. Strategies to enhance lysosomal activity for the modulation of PD-related phenotypes in vivo.
Author Contributions
TA and TK conceived and wrote the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by JSPS KAKENHI Grant No. 19K07816.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our lab members for their helpful suggestions and discussions.
References
1. Braak H, Braak E. Pathoanatomy of Parkinson's disease. J Neurol. (2000) 247(Suppl 2):II3–10. doi: 10.1007/PL00007758
2. Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, et al. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. (1998) 152:879–84.
3. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. (2002) 4:160–4. doi: 10.1038/ncb748
4. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. (1997) 388:839–40. doi: 10.1038/42166
5. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
6. Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harb Perspect Med. (2012) 2:a008888. doi: 10.1101/cshperspect.a008888
7. Lubbe S, Morris HR. Recent advances in Parkinson's disease genetics. J Neurol. (2014) 261:259–66. doi: 10.1007/s00415-013-7003-2
8. Zhang PL, Chen Y, Zhang CH, Wang YX, Fernandez-Funez P. Genetics of Parkinson's disease and related disorders. J Med Genet. (2018) 55:73–80. doi: 10.1136/jmedgenet-2017-105047
9. Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. (2017) 49:1511–6. doi: 10.1038/ng.3955
10. Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. (2019) 18:1091–102. doi: 10.1016/S1474-4422(19)30320-5
11. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. (2014) 46:989–93. doi: 10.1038/ng.3043
12. Jinn S, Drolet RE, Cramer PE, Wong AH, Toolan DM, Gretzula CA, et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α -synuclein aggregation. Proc Natl Acad Sci USA. (2017) 114:2389–94. doi: 10.1073/pnas.1616332114
13. Schapansky J, Khasnavis S, DeAndrade MP, Nardozzi JD, Falkson SR, Boyd JD, et al. Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble α-synuclein in neurons. Neurobiol Dis. (2018) 111:26–35. doi: 10.1016/j.nbd.2017.12.005
14. Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, et al. Glucosylsphingosine promotes α-Synuclein pathology in mutant GBA-associated Parkinson's Disease. J Neurosci. (2017) 37:9617–31. doi: 10.1523/JNEUROSCI.1525-17.2017
15. Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci. (2012) 32:4240–6. doi: 10.1523/JNEUROSCI.5575-11.2012
16. Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, et al. Mutation in VPS35 associated with Parkinson's disease impairs WASH complex association and inhibits autophagy. Nat Commun. (2014) 5:3828. doi: 10.1038/ncomms4828
17. Bandres-Ciga S, Saez-Atienzar S, Bonet-Ponce L, Billingsley K, Vitale D, Blauwendraat C, et al. The endocytic membrane trafficking pathway plays a major role in the risk of Parkinson's disease. Mov Disord. (2019) 34:460–8. doi: 10.1002/mds.27614
18. Hopfner F, Mueller SH, Szymczak S, Junge O, Tittmann L, May S, et al. Rare variants in specific lysosomal genes are associated with Parkinson's disease. Mov Disord. (2020) 35:1245–8. doi: 10.1002/mds.28037
19. Kia DA, Zhang D, Guelfi S, Manzoni C, Hubbard L, Reynolds RH, et al. Identification of candidate Parkinson disease genes by integrating genome-wide association study, expression, and epigenetic data sets. JAMA Neurol. (2021) 78:464–72. doi: 10.1001/jamaneurol.2020.5257
20. Bourdenx M, Koulakiotis NS, Sanoudou D, Bezard E, Dehay B, Tsarbopoulos A. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: examples of amyloidopathies, tauopathies and synucleinopathies. Prog Neurobiol. (2017) 155:171–93. doi: 10.1016/j.pneurobio.2015.07.003
21. Soria FN, Pampliega O, Bourdenx M, Meissner WG, Bezard E, Dehay B. Exosomes, an unmasked culprit in neurodegenerative diseases. Front Neurosci. (2017) 11:26. doi: 10.3389/fnins.2017.00026
22. Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to α-synuclein inclusions. Neurobiol Dis. (2009) 35:385–98. doi: 10.1016/j.nbd.2009.05.023
23. Gurney R, Davidson YS, Robinson AC, Richardson A, Jones M, Snowden JS, et al. Lysosomes, autophagosomes and Alzheimer pathology in dementia with Lewy body disease. Neuropathology. (2018) 38:347–60. doi: 10.1111/neup.12472
24. Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, et al. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci. (2010) 30:12535–44. doi: 10.1523/JNEUROSCI.1920-10.2010
25. Puska G, Lutz MI, Molnar K, Regelsberger G, Ricken G, Pirker W, et al. Lysosomal response in relation to α-synuclein pathology differs between Parkinson's disease and multiple system atrophy. Neurobiol Dis. (2018) 114:140–52. doi: 10.1016/j.nbd.2018.02.019
26. Niimi Y, Ito S, Mizutani Y, Murate K, Shima S, Ueda A, et al. Altered regulation of serum lysosomal acid hydrolase activities in Parkinson's disease: a potential peripheral biomarker? Parkinsonism Relat Disord. (2019) 61:132–7. doi: 10.1016/j.parkreldis.2018.10.032
27. Parnetti L, Chiasserini D, Persichetti E, Eusebi P, Varghese S, Qureshi MM, et al. Cerebrospinal fluid lysosomal enzymes and α-synuclein in Parkinson's disease. Mov Disord. (2014) 29:1019–27. doi: 10.1002/mds.25772
28. van Dijk KD, Persichetti E, Chiasserini D, Eusebi P, Beccari T, Calabresi P, et al. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson's disease. Mov Disord. (2013) 28:747–54. doi: 10.1002/mds.25495
29. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
30. Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, et al. α-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. (2013) 28:811–3. doi: 10.1002/mds.25421
31. Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat Genet. (1998) 18:106–8. doi: 10.1038/ng0298-106
32. Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. (2013) 73:459–71. doi: 10.1002/ana.23894
33. Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, et al. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology. Neurobiol Aging. (2014) 35:2180.e2181–5. doi: 10.1016/j.neurobiolaging.2014.03.024
34. Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. (2004) 55:164–73. doi: 10.1002/ana.10795
35. Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. α-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. (2004) 364:1167–9. doi: 10.1016/S0140-6736(04)17103-1
36. Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between α-synuclein gene duplication and familial Parkinson's disease. Lancet. (2004) 364:1169–71. doi: 10.1016/S0140-6736(04)17104-3
37. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. α-Synuclein locus triplication causes Parkinson's disease. Science. (2003) 302:841. doi: 10.1126/science.1090278
38. Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK. How is α-synuclein cleared from the cell? J Neurochem. (2019) 150:577–90. doi: 10.1111/jnc.14704
39. Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. α-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. (2003) 278:25009–13. doi: 10.1074/jbc.M300227200
40. Xilouri M Brekk OR Stefanis L. α-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. (2013) 47:537–51. doi: 10.1007/s12035-012-8341-2
41. McGlinchey RP, Lee JC. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc Natl Acad Sci USA. (2015) 112:9322–7. doi: 10.1073/pnas.1500937112
42. Sevlever D, Jiang P, Yen SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of α-synuclein and generation of its carboxy-terminally truncated species. Biochemistry. (2008) 47:9678–87. doi: 10.1021/bi800699v
43. Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, et al. Cathepsin D expression level affects α-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. (2009) 2:5. doi: 10.1186/1756-6606-2-5
44. Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci USA. (2009) 106:13010–5. doi: 10.1073/pnas.0903691106
45. Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. (2010) 113:1263–74. doi: 10.1111/j.1471-4159.2010.06695.x
46. Flavin WP, Bousset L, Green ZC, Chu Y, Skarpathiotis S, Chaney MJ, et al. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. (2017) 134:629–53. doi: 10.1007/s00401-017-1722-x
47. Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, et al. α-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE. (2013) 8:e62143. doi: 10.1371/journal.pone.0062143
48. Samuel F, Flavin WP, Iqbal S, Pacelli C, Sri Renganathan SD, Trudeau LE, et al. Effects of Serine 129 Phosphorylation on α-synuclein aggregation, membrane association, and internalization. J Biol Chem. (2016) 291:4374–85. doi: 10.1074/jbc.M115.705095
49. Cuddy LK, Wani WY, Morella ML, Pitcairn C, Tsutsumi K, Fredriksen K, et al. Stress-induced cellular clearance is mediated by the SNARE protein ykt6 and disrupted by α-Synuclein. Neuron. (2019) 104:869–84.e811. doi: 10.1016/j.neuron.2019.09.001
50. Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, et al. α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. (2006) 313:324–8. doi: 10.1126/science.1129462
51. Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC. α-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell. (2010) 21:1850–63. doi: 10.1091/mbc.e09-09-0801
52. Klionsky DJ, Ohsumi Y. Vacuolar import of proteins and organelles from the cytoplasm. Annu Rev Cell Dev Biol. (1999) 15:1–32. doi: 10.1146/annurev.cellbio.15.1.1
53. Xilouri M Brekk OR Stefanis L. Autophagy and α-synuclein: relevance to Parkinson's disease and related synucleopathies. Mov Disord. (2016) 31:178–92. doi: 10.1002/mds.26477
54. Minakaki G, Menges S, Kittel A, Emmanouilidou E, Schaeffner I, Barkovits K, et al. Autophagy inhibition promotes SNCA/ α-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy. (2018) 14:98–119. doi: 10.1080/15548627.2017.1395992
55. Poehler AM, Xiang W, Spitzer P, May VE, Meixner H, Rockenstein E, et al. Autophagy modulates SNCA/ α-synuclein release, thereby generating a hostile microenvironment. Autophagy. (2014) 10:2171–92. doi: 10.4161/auto.36436
56. Hunn BHM, Vingill S, Threlfell S, Alegre-Abarrategui J, Magdelyns M, Deltheil T, et al. Impairment of macroautophagy in dopamine neurons has opposing effects on Parkinsonian pathology and behavior. Cell Rep. (2019) 29:920–31.e927. doi: 10.1016/j.celrep.2019.09.029
57. Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol Dis. (2011) 43:690–7. doi: 10.1016/j.nbd.2011.05.022
58. Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. (2014) 24:92–104. doi: 10.1038/cr.2013.153
59. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science. (2004) 305:1292–5. doi: 10.1126/science.1101738
60. Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. (2004) 44:595–600. doi: 10.1016/j.neuron.2004.10.023
61. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. (2004) 44:601–7. doi: 10.1016/j.neuron.2004.11.005
62. Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, et al. Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci. (2007) 8:102. doi: 10.1186/1471-2202-8-102
63. Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, et al. Parkinson's disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm. (2011) 118:795–808. doi: 10.1007/s00702-011-0653-2
64. Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. (2007) 103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x
65. Marin I. The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol. (2006) 23:2423–33. doi: 10.1093/molbev/msl114
66. Rideout HJ, Stefanis L. The neurobiology of LRRK2 and its role in the pathogenesis of Parkinson's disease. Neurochem Res. (2014) 39:576–92. doi: 10.1007/s11064-013-1073-5
67. Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, et al. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. (2007) 46:1380–8. doi: 10.1021/bi061960m
68. Lewis PA, Greggio E, Beilina A, Jain S, Baker A, Cookson MR. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. (2007) 357:668–71. doi: 10.1016/j.bbrc.2007.04.006
69. Di Fonzo A, Rohe CF, Ferreira J, Chien HF, Vacca L, Stocchi F, et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet. (2005) 365:412–5. doi: 10.1016/S0140-6736(05)17829-5
70. Aasly JO, Vilarino-Guell C, Dachsel JC, Webber PJ, West AB, Haugarvoll K, et al. Novel pathogenic LRRK2 p.Asn1437His substitution in familial Parkinson's disease. Mov Disord. (2010) 25:2156–63. doi: 10.1002/mds.23265
71. Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, et al. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol. (2005) 57:918–21. doi: 10.1002/ana.20484
72. Zabetian CP, Samii A, Mosley AD, Roberts JW, Leis BC, Yearout D, et al. A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. (2005) 65:741–4. doi: 10.1212/01.WNL.0000172630.22804.73
73. West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, et al. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. (2005) 102:16842–7. doi: 10.1073/pnas.0507360102
74. Deniston CK, Salogiannis J, Mathea S, Snead DM, Lahiri I, Matyszewski M, et al. Structure of LRRK2 in Parkinson's disease and model for microtubule interaction. Nature. (2020) 588:344–9. doi: 10.1038/s41586-020-2673-2
75. Watanabe R, Buschauer R, Bohning J, Audagnotto M, Lasker K, Lu TW, et al. The in situ structure of Parkinson's disease-linked LRRK2. Cell. (2020) 182:1508–18.e1516. doi: 10.1016/j.bpj.2019.11.2690
76. Fujimoto T, Kuwahara T, Eguchi T, Sakurai M, Komori T, Iwatsubo T. Parkinson's disease-associated mutant LRRK2 phosphorylates Rab7L1 and modifies trans-Golgi morphology. Biochem Biophys Res Commun. (2018) 495:1708–15. doi: 10.1016/j.bbrc.2017.12.024
77. Kuwahara T, Iwatsubo T. The emerging functions of LRRK2 and Rab GTPases in the endolysosomal system. Front Neurosci. (2020) 14:227. doi: 10.3389/fnins.2020.00227
78. Liu Z, Bryant N, Kumaran R, Beilina A, Abeliovich A, Cookson MR, et al. LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum Mol Genet. (2018) 27:385–95. doi: 10.1093/hmg/ddx410
79. Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife. (2017) 6:e31012. doi: 10.7554/eLife.31012.018
80. Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, et al. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. (2016) 5:e12813. doi: 10.7554/eLife.12813.023
81. Pfeffer SR. Rab GTPases: master regulators that establish the secretory and endocytic pathways. Mol Biol Cell. (2017) 28:712–5. doi: 10.1091/mbc.e16-10-0737
82. Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci USA. (2018) 115:E9115–24. doi: 10.1073/pnas.1812196115
83. Kuwahara T, Funakawa K, Komori T, Sakurai M, Yoshii G, Eguchi T, et al. Roles of lysosomotropic agents on LRRK2 activation and Rab10 phosphorylation. Neurobiol Dis. (2020) 145:105081. doi: 10.1016/j.nbd.2020.105081
84. Bonet-Ponce L, Beilina A, Williamson CD, Lindberg E, Kluss JH, Saez-Atienzar S, et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv. (2020) 6:eabb2454. doi: 10.1126/sciadv.abb2454
85. Herbst S, Campbell P, Harvey J, Bernard EM, Papayannopoulos V, Wood NW, et al. LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J. (2020) 39:e104494. doi: 10.15252/embj.2020104494
86. Kuwahara T, Inoue K, D'Agati VD, Fujimoto T, Eguchi T, Saha S, et al. LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci Rep. (2016) 6:29945. doi: 10.1038/srep29945
87. Tong Y, Giaime E, Yamaguchi H, Ichimura T, Liu Y, Si H, et al. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol Neurodegener. (2012) 7:2. doi: 10.1186/1750-1326-7-2
88. Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher, et al. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of α-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci USA. (2010) 107:9879–84. doi: 10.1073/pnas.1004676107
89. Saez-Atienzar S, Bonet-Ponce L, Blesa JR, Romero FJ, Murphy MP, Jordan J, et al. The LRRK2 inhibitor GSK2578215A induces protective autophagy in SH-SY5Y cells: involvement of Drp-1-mediated mitochondrial fission and mitochondrial-derived ROS signaling. Cell Death Dis. (2014) 5:e1368. doi: 10.1038/cddis.2014.320
90. Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet. (2014) 23:4201–14. doi: 10.1093/hmg/ddu138
91. Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. (2013) 16:394–406. doi: 10.1038/nn.3350
92. Mamais A, Manzoni C, Nazish I, Arber C, Sonustun B, Wray S, et al. Analysis of macroautophagy related proteins in G2019S LRRK2 Parkinson's disease brains with Lewy body pathology. Brain Res. (2018) 1701:75–84. doi: 10.1016/j.brainres.2018.07.023
93. Murphy KE, Gysbers AM, Abbott SK, Spiro AS, Furuta A, Cooper A, et al. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson's disease. Mov Disord. (2015) 30:1639–47. doi: 10.1002/mds.26141
94. Kim KS, Marcogliese PC, Yang J, Callaghan SM, Resende V, Abdel-Messih E, et al. Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson's disease. Proc Natl Acad Sci USA. (2018) 115:E5164–73. doi: 10.1073/pnas.1718946115
95. Seaman MN. The retromer complex - endosomal protein recycling and beyond. J Cell Sci. (2012) 125:4693–702. doi: 10.1242/jcs.103440
96. MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. (2013) 77:425–39. doi: 10.1016/j.neuron.2012.11.033
97. Mir R, Tonelli F, Lis P, Macartney T, Polinski NK, Martinez TN, et al. The Parkinson's disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem J. (2018) 475:1861–83. doi: 10.1042/BCJ20180248
98. Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. (2011) 89:162–7. doi: 10.1016/j.ajhg.2011.06.001
99. Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. (2011) 89:168–75. doi: 10.1016/j.ajhg.2011.06.008
100. Ando M, Funayama M, Li Y, Kashihara K, Murakami Y, Ishizu N, et al. VPS35 mutation in Japanese patients with typical Parkinson's disease. Mov Disord. (2012) 27:1413–7. doi: 10.1002/mds.25145
101. Kumar KR, Weissbach A, Heldmann M, Kasten M, Tunc S, Sue CM, et al. Frequency of the D620N mutation in VPS35 in Parkinson disease. Arch Neurol. (2012) 69:1360–4. doi: 10.1001/archneurol.2011.3367
102. Eleuteri S, Albanese A. VPS35-Based approach: a potential innovative treatment in Parkinson's disease. Front Neurol. (2019) 10:1272. doi: 10.3389/fneur.2019.01272
103. Seaman MN, McCaffery JM, Emr SD. A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J Cell Biol. (1998) 142:665–81. doi: 10.1083/jcb.142.3.665
104. Williams ET, Chen X, Moore DJ. VPS35, the retromer complex and Parkinson's disease. J Parkinsons Dis. (2017) 7:219–33. doi: 10.3233/JPD-161020
105. Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. (2008) 20:427–36. doi: 10.1016/j.ceb.2008.03.009
106. Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. (2004) 165:111–22. doi: 10.1083/jcb.200312034
107. van Weering JR, Sessions RB, Traer CJ, Kloer DP, Bhatia VK, Stamou D, et al. Molecular basis for SNX-BAR-mediated assembly of distinct endosomal sorting tubules. EMBO J. (2012) 31:4466–80. doi: 10.1038/emboj.2012.283
108. Wassmer T, Attar N, Bujny MV, Oakley J, Traer CJ, Cullen PJ. A loss-of-function screen reveals SNX5 and SNX6 as potential components of the mammalian retromer. J Cell Sci. (2007) 120:45–54. doi: 10.1242/jcs.03302
109. Nothwehr SF, Ha SA, Bruinsma P. Sorting of yeast membrane proteins into an endosome-to-Golgi pathway involves direct interaction of their cytosolic domains with Vps35p. J Cell Biol. (2000) 151:297–310. doi: 10.1083/jcb.151.2.297
110. Arighi CN, Hartnell LM, Aguilar RC, Haft CR, Bonifacino JS. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J Cell Biol. (2004) 165:123–33. doi: 10.1083/jcb.200312055
111. Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S, et al. The Vps35 D620N mutation linked to Parkinson's disease disrupts the cargo sorting function of retromer. Traffic. (2014) 15:230–44. doi: 10.1111/tra.12136
112. McGough IJ, Steinberg F, Jia D, Barbuti PA, McMillan KJ, Heesom KJ, et al. Retromer binding to FAM21 and the WASH complex is perturbed by the Parkinson disease-linked VPS35(D620N) mutation. Curr Biol. (2014) 24:1670–6. doi: 10.1016/j.cub.2014.06.024
113. Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, et al. VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for α-synuclein degradation and prevention of pathogenesis of Parkinson's disease. J Neurosci. (2015) 35:10613–28. doi: 10.1523/JNEUROSCI.0042-15.2015
114. Dhungel N, Eleuteri S, Li LB, Kramer NJ, Chartron JW, Spencer B, et al. Parkinson's disease genes VPS35 and EIF4G1 interact genetically and converge on α-synuclein. Neuron. (2015) 85:76–87. doi: 10.1016/j.neuron.2014.11.027
115. Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. (2010) 67:1464–72. doi: 10.1001/archneurol.2010.198
116. Hampshire DJ, Roberts E, Crow Y, Bond J, Mubaidin A, Wriekat AL, et al. Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J Med Genet. (2001) 38:680–2. doi: 10.1136/jmg.38.10.680
117. Estiar MA, Leveille E, Spiegelman D, Dupre N, Trempe JF, Rouleau GA, et al. Clinical and genetic analysis of ATP13A2 in hereditary spastic paraplegia expands the phenotype. Mol Genet Genomic Med. (2020) 8:e1052. doi: 10.1002/mgg3.1052
118. Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet. (2012) 21:2646–50. doi: 10.1093/hmg/dds089
119. Farias FH, Zeng R, Johnson GS, Wininger FA, Taylor JF, Schnabel RD, et al. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis. (2011) 42:468–74. doi: 10.1016/j.nbd.2011.02.009
120. van Veen S, Sorensen DM, Holemans T, Holen HW, Palmgren MG, Vangheluwe P. Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson's disease and other neurological disorders. Front Mol Neurosci. (2014) 7:48. doi: 10.3389/fnmol.2014.00048
121. van Veen S, Martin S, Van den Haute C, Benoy V, Lyons J, Vanhoutte R, et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature. (2020) 578:419–24. doi: 10.1038/s41586-020-1968-7
122. Schmidt K, Wolfe DM, Stiller B, Pearce DA. Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem Biophys Res Commun. (2009) 383:198–202. doi: 10.1016/j.bbrc.2009.03.151
123. Vrijsen S, Besora-Casals L, van Veen S, Zielich J, Van den Haute C, Hamouda NN, et al. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc Natl Acad Sci USA. (2020) 117:31198–207. doi: 10.1073/pnas.1922342117
124. Dehay B, Martinez-Vicente M, Ramirez A, Perier C, Klein C, Vila M, et al. Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy. (2012) 8:1389–91. doi: 10.4161/auto.21011
125. Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci USA. (2012) 109:9611–6. doi: 10.1073/pnas.1112368109
126. Demirsoy S, Martin S, Motamedi S, van Veen S, Holemans T, Van den Haute C, et al. ATP13A2/PARK9 regulates endo-/lysosomal cargo sorting and proteostasis through a novel PI(3, 5)P2-mediated scaffolding function. Hum Mol Genet. (2017) 26:1656–69. doi: 10.1093/hmg/ddx070
127. Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC. The Parkinson's disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun. (2016) 7:11803. doi: 10.1038/ncomms11803
128. Matsui H, Sato F, Sato S, Koike M, Taruno Y, Saiki S, et al. ATP13A2 deficiency induces a decrease in cathepsin D activity, fingerprint-like inclusion body formation, and selective degeneration of dopaminergic neurons. FEBS Lett. (2013) 587:1316–25. doi: 10.1016/j.febslet.2013.02.046
129. Lopes da Fonseca T, Pinho R, Outeiro TF. A familial ATP13A2 mutation enhances α-synuclein aggregation and promotes cell death. Hum Mol Genet. (2016) 25:2959–71. doi: 10.1093/hmg/ddw147
130. Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, et al. Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum Mol Genet. (2014) 23:2816–33. doi: 10.1093/hmg/ddu099
131. Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, et al. α-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. (2009) 41:308–15. doi: 10.1038/ng.300
132. Sato S, Koike M, Funayama M, Ezaki J, Fukuda T, Ueno T, et al. Lysosomal storage of subunit c of mitochondrial ATP synthase in brain-specific Atp13a2-deficient mice. Am J Pathol. (2016) 186:3074–82. doi: 10.1016/j.ajpath.2016.08.006
133. Dirr ER Ekhator OR Blackwood R Holden JG Masliah E Schultheis PJ . Exacerbation of sensorimotor dysfunction in mice deficient in Atp13a2 and overexpressing human wildtype α-synuclein. Behav Brain Res. (2018) 343:41–9. doi: 10.1016/j.bbr.2018.01.029
134. Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. (2013) 22:2067–82. doi: 10.1093/hmg/ddt057
135. Wang R, Tan J, Chen T, Han H, Tian R, Tan Y, et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J Cell Biol. (2019) 218:267–84. doi: 10.1083/jcb.201804165
136. Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun. (2013) 1:11. doi: 10.1186/2051-5960-1-11
137. Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab. (2004) 81:70–3. doi: 10.1016/j.ymgme.2003.11.004
138. O'Regan G, deSouza RM, Balestrino R, Schapira AH. Glucocerebrosidase mutations in Parkinson disease. J Parkinsons Dis. (2017) 7:411–22. doi: 10.3233/JPD-171092
139. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. (2012) 11:986–98. doi: 10.1016/S1474-4422(12)70190-4
140. Machaczka M, Rucinska M, Skotnicki AB, Jurczak W. Parkinson's syndrome preceding clinical manifestation of Gaucher's disease. Am J Hematol. (1999) 61:216–7. doi: 10.1002/(SICI)1096-8652(199907)61:3andlt;216::AID-AJH12andgt;3.0.CO;2-B
141. Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, et al. Occurrence of Parkinson's syndrome in type I Gaucher disease. QJM. (1996) 89:691–64. doi: 10.1093/qjmed/89.9.691
142. Iwaki H, Blauwendraat C, Leonard HL, Liu G, Maple-Grodem J, Corvol JC, et al. Genetic risk of Parkinson disease and progression: AN analysis of 13 longitudinal cohorts. Neurol Genet. (2019) 5:e348. doi: 10.1212/NXG.0000000000000348
143. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. (2009) 361:1651–61. doi: 10.1056/NEJMoa0901281
144. Zhang Y, Sun QY, Zhao YW, Shu L, Guo JF, Xu Q, et al. Effect of GBA mutations on phenotype of Parkinson's disease: a study on Chinese population and a meta-analysis. Parkinsons Dis. (2015) 2015:916971. doi: 10.1155/2015/916971
145. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson's disease: the mutation matters. Ann Neurol. (2016) 80:662–73. doi: 10.1002/ana.24777
146. Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. (2013) 70:727–35. doi: 10.1001/jamaneurol.2013.1925
147. Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, et al. Specifically neuropathic Gaucher's mutations accelerate cognitive decline in Parkinson's. Ann Neurol. (2016) 80:674–85. doi: 10.1002/ana.24781
148. Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, et al. Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community-based incident cohort. Brain. (2013) 136:392–9. doi: 10.1093/brain/aws318
149. Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain. (2015) 138:2648–58. doi: 10.1093/brain/awv179
150. Huh YE, Chiang MSR, Locascio JJ, Liao Z, Liu G, Choudhury K, et al. beta-Glucocerebrosidase activity in GBA-linked Parkinson disease: the type of mutation matters. Neurology. (2020) 95:e685–96. doi: 10.1212/WNL.0000000000009989
151. Butters TD. Gaucher disease. Curr Opin Chem Biol. (2007) 11:412–8. doi: 10.1016/j.cbpa.2007.05.035
152. Ron I, Horowitz M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet. (2005) 14:2387–98. doi: 10.1093/hmg/ddi240
153. Schmitz M, Alfalah M, Aerts JM, Naim HY, Zimmer KP. Impaired trafficking of mutants of lysosomal glucocerebrosidase in Gaucher's disease. Int J Biochem Cell Biol. (2005) 37:2310–20. doi: 10.1016/j.biocel.2005.05.008
154. Gan-Or Z Amshalom I Kilarski LL Bar-Shira A Gana-Weisz M Mirelman A . Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. (2015) 84:880–7. doi: 10.1212/WNL.0000000000001315
155. Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. (2012) 72:455–63. doi: 10.1002/ana.23614
156. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. (2011) 146:37–52. doi: 10.1016/j.cell.2011.06.001
157. Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, et al. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson's disease. Brain. (2014) 137:834–48. doi: 10.1093/brain/awt367
158. Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science. (2017) 357:1255–61. doi: 10.1126/science.aam9080
159. Nguyen M, Krainc D. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson's disease. Proc Natl Acad Sci USA. (2018) 115:5576–81. doi: 10.1073/pnas.1717590115
160. Balestrino R, Schapira AHV. Glucocerebrosidase and Parkinson disease: molecular, clinical, therapeutic implications. Neuroscientist. (2018) 24:540–59. doi: 10.1177/1073858417748875
161. Manning-Bog AB, Schule B, Langston JW. α-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology. (2009) 30:1127–32. doi: 10.1016/j.neuro.2009.06.009
162. Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible conformational conversion of α-Synuclein into toxic assemblies by glucosylceramide. Neuron. (2018) 97:92–107.e110. doi: 10.1016/j.neuron.2017.12.012
163. Fecchio C, Palazzi L, de Laureto PP. α-Synuclein and Polyunsaturated fatty acids: molecular basis of the interaction and implication in neurodegeneration. Molecules. (2018) 23:1531. doi: 10.3390/molecules23071531
164. Morabito G, Giannelli SG, Ordazzo G, Bido S, Castoldi V, Indrigo M, et al. AAV-PHP.B-Mediated global-scale expression in the mouse nervous system enables GBA1 gene therapy for wide protection from synucleinopathy. Mol Ther. (2017) 25:2727–42. doi: 10.1016/j.ymthe.2017.08.004
165. Mus L, Siani F, Giuliano C, Ghezzi C, Cerri S, Blandini F. Development and biochemical characterization of a mouse model of Parkinson's disease bearing defective glucocerebrosidase activity. Neurobiol Dis. (2019) 124:289–96. doi: 10.1016/j.nbd.2018.12.001
166. Soria FN, Engeln M, Martinez-Vicente M, Glangetas C, Lopez-Gonzalez MJ, Dovero S, et al. Glucocerebrosidase deficiency in dopaminergic neurons induces microglial activation without neurodegeneration. Hum Mol Genet. (2017) 26:2603–15. doi: 10.1093/hmg/ddx120
167. Atashrazm F, Hammond D, Perera G, Dobson-Stone C, Mueller N, Pickford R, et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson's disease. Sci Rep. (2018) 8:15446. doi: 10.1038/s41598-018-33921-x
168. Duran R, Mencacci NE, Angeli AV, Shoai M, Deas E, Houlden H, et al. The glucocerobrosidase E326K variant predisposes to Parkinson's disease, but does not cause Gaucher's disease. Mov Disord. (2013) 28:232–6. doi: 10.1002/mds.25248
169. Spitz M, Pereira JS, Nicareta DH, Abreu Gde M, Bastos EF, Seixas TL, et al. Association of LRRK2 and GBA mutations in a Brazilian family with Parkinson's disease. Parkinsonism Relat Disord. (2015) 21:825–6. doi: 10.1016/j.parkreldis.2015.03.029
170. Yahalom G, Greenbaum L, Israeli-Korn S, Fay-Karmon T, Livneh V, Ruskey JA, et al. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson's disease: Risk estimates and genotype-phenotype correlations. Parkinsonism Relat Disord. (2019) 62:179–84. doi: 10.1016/j.parkreldis.2018.12.014
171. Ysselstein D, Nguyen M, Young TJ, Severino A, Schwake M, Merchant K, et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson's disease patients. Nat Commun. (2019) 10:5570. doi: 10.1038/s41467-019-13413-w
172. Sanyal A, Novis HS, Gasser E, Lin S, LaVoie MJ. LRRK2 kinase inhibition rescues deficits in lysosome function due to heterozygous GBA1 expression in human iPSC-derived neurons. Front Neurosci. (2020) 14:442. doi: 10.3389/fnins.2020.00442
173. Lie PPY, Nixon RA. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis. (2019) 122:94–105. doi: 10.1016/j.nbd.2018.05.015
174. Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson's disease cells. Neurobiol Dis. (2015) 82:235–42. doi: 10.1016/j.nbd.2015.06.008
175. Bendikov-Bar I, Maor G, Filocamo M, Horowitz M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol Dis. (2013) 50:141–5. doi: 10.1016/j.bcmd.2012.10.007
176. McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. (2014) 137:1481–95. doi: 10.1093/brain/awu020
177. Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann Neurol. (2016) 80:766–75. doi: 10.1002/ana.24790
178. Mishra A, Krishnamurthy S. Neurorestorative effects of sub-chronic administration of ambroxol in rodent model of Parkinson's disease. Naunyn Schmiedebergs Arch Pharmacol. (2020) 393:429–44. doi: 10.1007/s00210-019-01737-9
179. Richter F, Fleming SM, Watson M, Lemesre V, Pellegrino L, Ranes B, et al. A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics. (2014) 11:840–56. doi: 10.1007/s13311-014-0294-x
180. Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, et al. CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci USA. (2011) 108:12101–6. doi: 10.1073/pnas.1108197108
181. Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci USA. (2013) 110:3537–42. doi: 10.1073/pnas.1220464110
182. Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci USA. (2017) 114:2699–704. doi: 10.1073/pnas.1616152114
183. Hernandez I, Luna G, Rauch JN, Reis SA, Giroux M, Karch CM, et al. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci Transl Med. (2019) 11: eaat3005. doi: 10.1126/scitranslmed.aat3005
184. Mullard A. FDA outlines steps for individualized drug development. Nat Rev Drug Discov. (2021) 20:91. doi: 10.1038/d41573-021-00009-7
185. Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci USA. (2013) 110:E1817–26. doi: 10.1073/pnas.1305623110
186. Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, Pollina EA, et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science. (2018) 359:1277–83. doi: 10.1126/science.aag3048
187. Johmura Y, Yamanaka T, Omori S, Wang TW, Sugiura Y, Matsumoto M, et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science. (2021) 371:265–70. doi: 10.1126/science.abb5916
188. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. (2012) 8:903–14. doi: 10.4161/auto.19653
189. Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of α-synucleinopathy. PLoS ONE. (2010) 5:e9313. doi: 10.1371/journal.pone.0009313
190. Bai X, Wey MC, Fernandez E, Hart MJ, Gelfond J, Bokov AF, et al. Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol Aging Age Relat Dis. (2015) 5:28743. doi: 10.3402/pba.v5.28743
191. Blagosklonny MV. Rapamycin for longevity: opinion article. Aging. (2019) 11:8048–67. doi: 10.18632/aging.102355
192. Hoffmann AC, Minakaki G, Menges S, Salvi R, Savitskiy S, Kazman A, et al. Extracellular aggregated α-synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci Rep. (2019) 9:544. doi: 10.1038/s41598-018-35811-8
193. Lan DM, Liu FT, Zhao J, Chen Y, Wu JJ, Ding ZT, et al. Effect of trehalose on PC12 cells overexpressing wild-type or A53T mutant α-synuclein. Neurochem Res. (2012) 37:2025–32. doi: 10.1007/s11064-012-0823-0
194. Rodríguez-Navarro JA, Rodríguez L, Casarejos MJ, Solano RM, Gómez A, Perucho J, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. (2010) 39:423–38. doi: 10.1016/j.nbd.2010.05.014
195. Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J Biol Chem. (2007) 282:5641–52. doi: 10.1074/jbc.M609532200
196. DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE, et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal. (2016) 9:ra21. doi: 10.1126/scisignal.aac5472
197. Tanji K, Miki Y, Maruyama A, Mimura J, Matsumiya T, Mori F, et al. Trehalose intake induces chaperone molecules along with autophagy in a mouse model of Lewy body disease. Biochem Biophys Res Commun. (2015) 465:746–52. doi: 10.1016/j.bbrc.2015.08.076
198. He Q, Koprich JB, Wang Y, Yu WB, Xiao BG, Brotchie JM, et al. Treatment with trehalose prevents behavioral and neurochemical deficits produced in an AAV α-Synuclein rat model of Parkinson's disease. Mol Neurobiol. (2016) 53:2258–68. doi: 10.1007/s12035-015-9173-7
199. Yu HC, Lin CS, Tai WT, Liu CY, Shiau CW, Chen KF. Nilotinib induces autophagy in hepatocellular carcinoma through AMPK activation. J Biol Chem. (2013) 288:18249–59. doi: 10.1074/jbc.M112.446385
200. Hebron ML, Lonskaya I, Moussa CE. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson's disease models. Hum Mol Genet. (2013) 22:3315–28. doi: 10.1093/hmg/ddt192
201. Xilouri M Brekk OR Landeck N Pitychoutis PM Papasilekas T Papadopoulou-Daifoti Z . Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain. (2013) 136:2130–46. doi: 10.1093/brain/awt131
202. Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol. (2013) 9:374–82. doi: 10.1038/nchembio.1230
203. Jegga AG, Schneider L, Ouyang X, Zhang J. Systems biology of the autophagy-lysosomal pathway. Autophagy. (2011) 7:477–89. doi: 10.4161/auto.7.5.14811
204. Su C, Yang X, Lou J. Geniposide reduces α-synuclein by blocking microRNA-21/lysosome-associated membrane protein 2A interaction in Parkinson disease models. Brain Res. (2016) 1644:98–106. doi: 10.1016/j.brainres.2016.05.011
205. Mullin S, Smith L, Lee K, D'Souza G, Woodgate P, Elflein J, et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. (2020) 77:427–34. doi: 10.1001/jamaneurol.2019.4611
206. Pagan FL, Wilmarth B, Torres-Yaghi Y, Hebron ML, Mulki S, Ferrante D, et al. Long-term safety and clinical effects of nilotinib in Parkinson's disease. Mov Disord. (2021) 36:740–9. doi: 10.1002/mds.28389
207. Simuni T, Fiske B, Merchant K, Coffey CS, Klingner E, Caspell-Garcia C, et al. Efficacy of nilotinib in patients with moderately advanced Parkinson disease: a randomized clinical trial. JAMA Neurol. (2021) 78:312–20. doi: 10.1001/jamaneurol.2020.4725
Keywords: lysosome, α-synuclein, LRRK2, VPS35, ATP13A2, GBA
Citation: Abe T and Kuwahara T (2021) Targeting of Lysosomal Pathway Genes for Parkinson's Disease Modification: Insights From Cellular and Animal Models. Front. Neurol. 12:681369. doi: 10.3389/fneur.2021.681369
Received: 16 March 2021; Accepted: 20 May 2021;
Published: 14 June 2021.
Edited by:
Luca Marsili, University of Cincinnati, United StatesReviewed by:
Lena F. Burbulla, Northwestern University, United StatesTito Calì, University of Padua, Italy
Jolanta Dorszewska, Poznan University of Medical Sciences, Poland
Copyright © 2021 Abe and Kuwahara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoki Kuwahara, a3V3YWhhcmFAbS51LXRva3lvLmFjLmpw