 Chiara Scoppola1,2*
Chiara Scoppola1,2* Giorgio Magli1,2Marta Conti1,2Maria Fadda1,2
Giorgio Magli1,2Marta Conti1,2Maria Fadda1,2 Giovanni M. Luzzu1,2Delia M. Simula1,2
Giovanni M. Luzzu1,2Delia M. Simula1,2 Alessandra Carta2Stefano Sotgiu2Susanna Casellato1,2
Alessandra Carta2Stefano Sotgiu2Susanna Casellato1,2- 1Center for Diagnosis and Care of Pediatric Epilepsy, University Hospital of Sassari, Sassari, Italy
- 2Department of Medical, Surgical and Experimental Sciences, Section of Child Neuropsychiatry, University of Sassari, Sassari, Italy
Background: Glucose-transporter-1 deficiency syndrome (GLUT1-DS), due to SLC2A1 gene mutation, is characterized by early-onset seizures, which are often drug-resistant, developmental delay, and hypotonia. Hemiplegic migraine (HM) is a rare form of migraine, defined by headache associated with transient hemiplegia, and can be caused by mutations in either CACNA1A, ATP1A2, or SCN1A. Paroxysmal movements, other transient neurological disorders, or hemiplegic events can occur in GLUT1-DS patients with a mild phenotype.
Case: We report on a girl with GLUT1-DS, due to SLC2A1 mutation, with a mild phenotype. In early childhood, she developed epilepsy and mild cognitive impairment, balance disorders, and clumsiness. At the age of 9, the patient reported a first hemiplegic episode, which regressed spontaneously. Over the next 3 years, two similar episodes occurred, accompanied by headache. Therefore, in the hypothesis of HM, genetic testing was performed and CACNA1A mutation was identified. The treatment with Lamotrigine avoided the recurrence of HM episodes.
Discussion: To our knowledge, among the several cases of GLUT1-DS with HM symptoms described in the literature, genetic testing was only performed in two of them, which eventually proved to be negative. In all other cases, no other genes except for SLC2A1 were examined. Consequently, our patient would be the first description of GLUT1-DS with HM due to CACNA1A mutation. We would emphasize the importance of performing specific genetic testing in patients with GLUT1-DS with symptoms evocative of HM, which may allow clinicians to use specific pharmacotherapy.
Introduction
Glucose-transporter-1 deficiency syndrome (GLUT1-DS), due to SLC2A1 gene mutation, is characterized by seizures, developmental delay, and hypotonia, although the clinical picture can vary between mild to severe phenotypes (1, 2).
Additional symptoms in GLUT1-DS might consist of paroxysmal movements or other transient neurological disorders. Notably, among them, patients with a GLUT1-DS mild phenotype can also experience transient hemiplegic events (3).
Hemiplegic migraine (HM) is a rare form of migraine, which can be caused by mutations in either CACNA1A, ATP1A2, or SCN1A. HM attacks are characterized by headache associated with transient neurological deficits such as hemiparesis, impairment in speech, and sensory and visual disturbances (4–6).
We report on a girl presenting with SLC2A1 and CACNA1A mutation.
Case Description
A 19-year-old girl who had experienced the onset of frequent sudden falls from the age of 34 months was admitted to the Unit of Child Neuropsychiatry, University Hospital of Sassari (clinical history summarized in Figure 1). On neurological examination, psychomotor development featured a slight delay, together with motor dyspraxia and a wide-based gait. A follow-up evaluation performed at 3 years and 5 months pointed out mild speech delay and dyslalia.
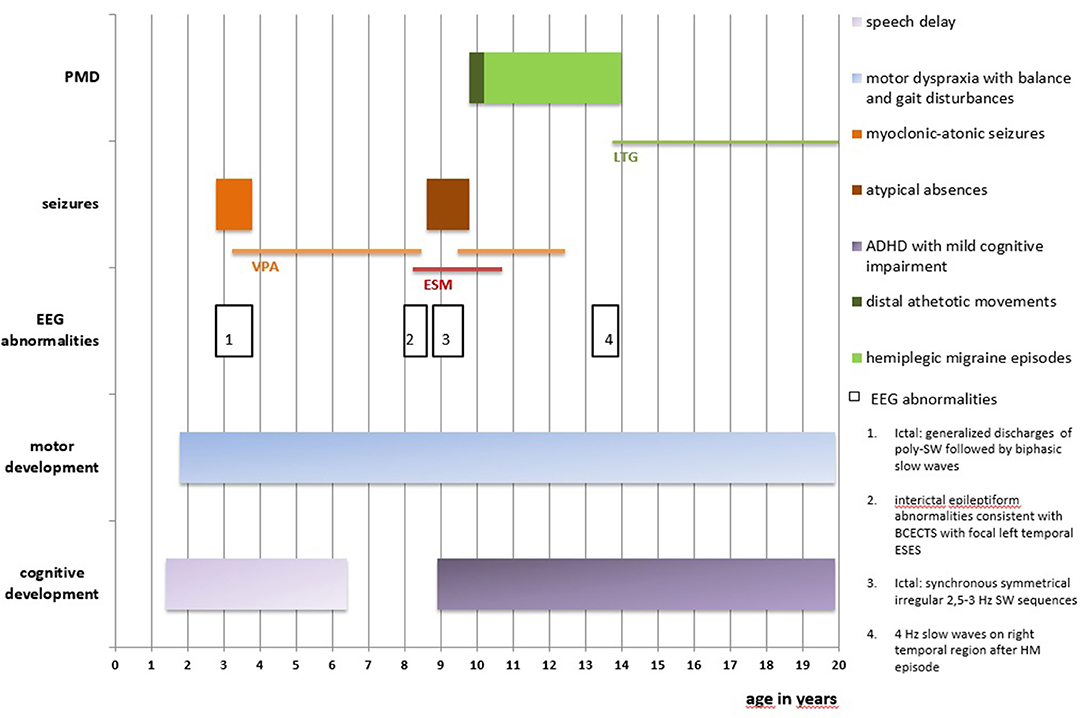
Figure 1. Clinical history is summarized in this figure. PMD, paroxysmal movement disorders; EEG, electroencephalography; BCECTS, Benign Childhood Epilepsy with Centro-Temporal Spikes; ESES, Electrical Status-Epilepticus during slow-waves Sleep; SW, spike-and-wave; poly-SW, polyspike-and-wave; HM, hemiplegic migraine; ADHFD, Attention Deficit and Hyperactivity Disorder; VPA, Sodium Valproate; ESM, Ethosuximide; LTG, Lamotrigine.
At age nine, Wechsler Intelligence Scale for Children-III (WISC-III) showed total I.Q. of 55 (Verbal I.Q. 59, Performance I.Q. 60) and a diagnosis of Attention Deficit and Hyperactivity Disorder, combined type (ADHD-C), with mild cognitive impairment, was formulated.
Seizures started at the age of 2 years and 10 months, characterized by sudden falls to the ground or sudden head drops, associated with loss of awareness, often anticipated by mild distal myoclonias involving both upper and lower limbs. These episodes were classified as myoclonic-atonic seizures. At seizure onset, electroencephalography (EEG) showed normal awake and sleep background activity; ictal EEG showed diffuse discharges of polyspike-and-wave 300–400 microV, followed by biphasic slow waves, synchronous with clinical myoclonic-atonic episodes. Monotherapy with Sodium Valproate (VPA) was effective on seizures' control and normalization of the EEG records.
Brain and spinal cord MRI, metabolic screening, electroneurography, electrocardiogram, and echocardiography were all reported to be normal.
At the age of eight, follow-up EEGs showed interictal epileptiform abnormalities consistent with Benign Childhood Epilepsy with Centro-Temporal Spikes (BCECTS) with focal left temporal Electrical Status-Epilepticus during slow-waves Sleep (ESES). Therefore, Ethosuximide (ESM) was prescribed, while VPA was gradually discontinued.
Six months later, concurrently with the complete withdrawal of VPA, episodes of staring and loss of awareness lasting 3 s were reported. EEG showed synchronous symmetrical irregular 2.5–3 Hz spike-and-wave sequences lasting 2 s, facilitated by hyperventilation, consistent with atypical absence seizures (Figure 2).
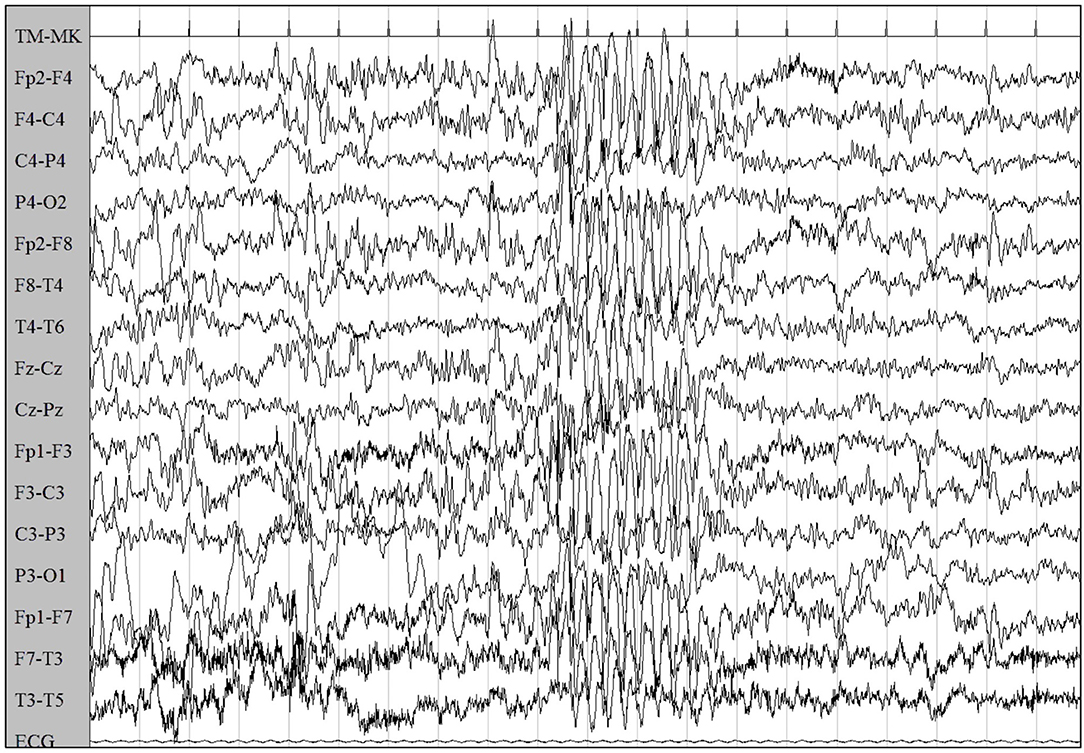
Figure 2. EEG showed synchronous symmetrical irregular 2.5–3 Hz spike-and-wave sequences, facilitated by hyperventilation, consistent with atypical absence seizure.
Treatment with VPA was then restored, while ESM was gradually discontinued, given the age-related disappearance of ESES and abnormalities consistent with BCECTS and its ineffectiveness on absences seizures. VPA treatment was successfully maintained for the following 3 years and discontinued after being clinically and EEG seizure-free for 2 years.
At age 9, the patient experienced episodes of right hemiparesthesia and dysarthria lasting up to 25 min, with spontaneous regression, together with episodes of sudden distal athetotic movements of hands and feet, without EEG abnormalities (Supplementary Video 1). Neurologic examination confirmed the persistence of the balance and gait disturbances (i.e., wide-based gait).
The clinical complexity led to the hypothesis of GLUT1-DS. Thus, the genetic investigation was performed and confirmed the presence of a heterozygous c.667C>T de novo mutation of exon 5 in the SLC2A1 gene, given that the same mutation was not detected in the parents' blood sample.
A ketogenic diet was initiated and discontinued, after two attempts, owing to poor compliance.
Further non-epileptic paroxysmal episodes presented at age 13, the first of which was characterized by right hemiparesthesia and dysarthria, combined with cephalalgia and emesis, followed by hemiparesis lasting 90 min. On another occasion, the girl experienced the same symptoms on the left hemisoma, without dysarthria. The EEG performed 2 days after this last episode showed 4 Hz slow waves, 40–60 microV, on the right temporal region, which disappeared on subsequent EEGs.
Due to the suspicion of HM, specific genetic testing was carried out, leading to the identification of a heterozygous c.3043G>A, p.(Glu1015Lys) mutation in the CACNA1A gene. We have not performed genetic testing for this mutation on the parents as the anamnesis was negative for episodes of migraine and HM.
Treatment with Lamotrigine was initiated with complete remission of HM episodes. Follow-up EEGs have been normal since then.
Discussion
Glucose-transporter type 1 (GLUT1), encoded by SLC2A1 gene on chromosome 1, is a transmembrane protein, which allows glucose to pass across the blood-brain barrier (BBB). SLC2A1 mutations cause GLUT1-DS, resulting in impaired glucose transport across the BBB and consequent hypoglycorrhachia (1).
GLUT1-DS occurs with variable clinical phenotypes, which are usually classified into two groups: “classical” or severe and “non-classical” or mild phenotype. The severe form is characterized by microcephaly, infantile-onset epilepsy, which is often drug-resistant, intellectual disability, and disabling movement disorders; conversely, the mild phenotype involves paroxysmal movement disorders, childhood epilepsy with atypical absences, and/or myoclonic-atonic epilepsy (1–3).
Hence, GLUT1-DS is categorized as developmental and epileptic encephalopathy (DEE) (7).
Symptom severity depends on glucose intake, with symptoms worsening during fasting and improving with meals. Moreover, clinical signs can often vary in the single patient's history, which makes the diagnosis even more challenging. Generally, epileptic seizures precede the onset of movement disorders and, in some cases, a progressive remission of epilepsy is described; it is likely that modifications of glucose metabolism in the cerebral cortex, related to growth, could explain symptoms' heterogeneity and variability over time (2).
Three different types of paroxysmal movement disorders (PMD) are described in GLUT1-DS, namely exercise-induced paroxysmal dyskinesia, which is the most frequent, paroxysmal kinesigenic dyskinesia, and paroxysmal non-kinesigenic dyskinesia (PNKD). In 10% of GLUT1-DS identified as mild phenotype, the main clinical manifestation is PMD (3).
The ketogenic diet represents a specific therapy for GLUT1-DS, as it improves epilepsy management, PMD, and cognitive development, especially if it is started in the early stage of the disease (1).
Hemiplegic Migraine (HM) is a rare type of migraine, featuring transient neurological deficits such as hemiparesis, speech impairment, and sensory and visual disturbances, often accompanied by headache (4–6). Patients are mostly females, with the onset occurring mostly in their first or second decade and an age-related decrease of attack frequency (4, 5).
In the International Classification of Headache Disorders-3, two different HM subtypes are included: familial and sporadic hemiplegic migraine (6).
Mutations in the ion transportation genes CACNA1A, ATP1A2, and SCN1A are well-known to cause HM; despite this, mutations in these genes are found in <20% of HM cases (4, 8). Other genes with roles in synaptic function and neurotransmission have also been implicated in the pathogenesis of HM and HM-like disorders such as PRRT2 mutations (4, 8, 9). The management of HM is not specific; among the most commonly used prophylactic and effective treatments, Verapamil, Lamotrigine, Flunarizine, Naloxone, and Acetazolamide are described (4, 5).
Transient hemiplegic episodes, resembling HM, can be experienced in patients with a GLUT1-DS, particularly in those with mild phenotype, although only nine cases of GLUT1-DS with HM attacks are described in the literature (3, 9–14). To our knowledge genetic testing for HM was only performed in two of them, which eventually resulted to be negative (3, 12). In all other cases, only the SLC2A1 gene was examined (9–11, 13, 14).
Moreover, in a large population of HM patients with no mutation on the main genes (CACNA1A, ATP1A2, or SCN1A), 1.3% of cases presented with a mutation on SLC2A1 and having HM episodes as the main clinical feature (8).
Conversely, other studies postulated that SLC2A1 mutations alone might not be sufficient for the development of isolated migraine symptoms and they were not described in patients with HM as a unique clinical manifestation (9).
Our patient presented with mild psychomotor delay, as well as language development delay, which characterized the clinical picture for many years, together with ADHD-C. Epileptic history evolved from early-onset myoclonic-atonic seizures to atypical absence seizures. Thus, we could classify our patient as a mild or “non-classic” phenotype of GLUT1-DS, due to a de novo mutation in the SLC2A1 gene. VPA in monotherapy was effective, with good clinical and EEG response.
During follow-up, interictal centro-temporal EEG abnormalities were interpreted as independent from the genetic mutations described and disappeared with age.
Later, she presented with non-epileptic paroxysmal events, characterized by involuntary distal athetotic movements, which are also a GLUT1-DS clinical manifestation, especially in mild phenotype (3).
In addition, the girl experienced other paroxysmal episodes, marked by transitory hemiplegia, often associated with headache. Such symptoms suggested the hypothesis of HM, leading to specific genetic testing, which revealed the pathogenetic mutation on the CACNA1A gene. Lamotrigine monotherapy was effective in preventing HM relapses over time.
Interestingly, our case presented with GLUT1-DS together with HM, due to CACNA1A mutation. To the best of our knowledge, the co-existence of such mutations in the same patient has not been described yet.
Our report highlights the importance of distinguishing between different PMDs classically experienced by patients with GLUT1-DS mild phenotype. Therefore, genetic testing, with the aim to achieve better etiological definition, is encouraged when transient hemiplegic events occur, to start specific treatment at an early stage.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CS: study design, execution, acquisition of data, drafting, writing, and revising. GM and SS: drafting, writing, and revising. MC, DS, and AC: drafting and revising. MF and GL: acquisition of data. SC: conceptual contribution, drafting, writing, and revising. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.679354/full#supplementary-material
References
1. Koch H, Weber YG. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav. (2018) 91:90–3. doi: 10.1016/j.yebeh.2018.06.010
2. Kim H, Lee JS, Lee Y, Kim SY, Lim BC, Kim KJ, et al. Diagnostic challenges associated with GLUT1 deficiency: phenotypic variabilities and evolving clinical features. Yonsei Med J. (2019) 60:1209–15. doi: 10.3349/ymj.2019.60.12.1209
3. Gburek-Augustat J, Heinze A, Abou Jamra R, Merkenschlager A. Hemiplegic migraine in glut1 deficiency syndrome and paroxysmal dyskinesia at ketogenic diet induction: case report and literature review. Mov Disord Clin Pract. (2020) 7:965–70. doi: 10.1002/mdc3.1308
4. Kumar A, Samanta D, Emmady PD, Arora R. Hemiplegic Migraine. Treasure Island, FL: StatPearls Publishing (2020).
5. Russell MB, Ducros A. Sporadic and familial hemiplegic migraine: pathophysiological mechanisms, clinical characteristics, diagnosis, and management. Lancet Neurol. (2011) 10:457–70. doi: 10.1016/S1474-4422(11)70048-5
6. Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition (beta version). Cephalalgia. (2013) 33:629–808. doi: 10.1177/0333102413485658
7. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
8. Sutherland HG, Maksemous N, Albury CL, Ibrahim O, Smith RA, Lea RA, et al. Comprehensive exonic sequencing of hemiplegic migraine-related genes in a cohort of suspected probands identifies known and potential pathogenic variants. Cells. (2020) 9:2368. doi: 10.3390/cells9112368
9. Weller CM, Leen WG, Neville BGR, Duncan JS, de Vries B, Geilenkirchen MA, et al. A novel SLC2A1 mutation linking hemiplegic migraine with alternating hemiplegia of childhood. Cephalalgia. (2015) 35:10–15. doi: 10.1177/0333102414532379
10. Almuqbil M, Rivkin MJ, Takeoka M, Yang E, Rodan LH. Transient regional cerebral hypoperfusion during a paroxysmal hemiplegic event in GLUT1 deficiency syndrome. Eur J Paediatr Neurol. (2018) 22:544–7. doi: 10.1016/j.ejpn.2018.02.005
11. Pellegrin S, Cantalupo G, Opri R, Dalla Bernardina B, Darra F. EEG findings during “paroxysmal hemiplegia” in a patient with GLUT1-deficiency. Eur J Paediatr Neurol. (2017) 21:580–2. doi: 10.1016/j.ejpn.2017.01.002
12. Mohammad SS, Coman D, Calvert S. Glucose transporter 1 deficiency syndrome and hemiplegic migraine as a dominant presenting clinical feature. J Paediatr Child Health. (2014) 50:1025–6. doi: 10.1111/jpc.12613
13. Pons R, Collins A, Rotstein M, Engelstad K, De Vivo DC. The spectrum of movement disorders in Glut-1 deficiency. Mov Disord. (2010) 25:275–81. doi: 10.1002/mds.22808
Keywords: GLUT1 deficiency syndrome, hemiplegic migraine, CACNA1A, epilepsy, headache, SLC2A1
Citation: Scoppola C, Magli G, Conti M, Fadda M, Luzzu GM, Simula DM, Carta A, Sotgiu S and Casellato S (2021) CACNA1A-Linked Hemiplegic Migraine in GLUT 1 Deficiency Syndrome: A Case Report. Front. Neurol. 12:679354. doi: 10.3389/fneur.2021.679354
Received: 11 March 2021; Accepted: 19 April 2021;
Published: 31 May 2021.
Edited by:
Maurizio Elia, Oasi Research Institute (IRCCS), ItalyReviewed by:
Vincenzo Belcastro, Lodi Hospital, ItalyAntonietta Coppola, University of Naples Federico II, Italy
Copyright © 2021 Scoppola, Magli, Conti, Fadda, Luzzu, Simula, Carta, Sotgiu and Casellato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Scoppola, Y2hpYXJhLnNjb3Bwb2xhQGdtYWlsLmNvbQ==