
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 25 June 2021
Sec. Movement Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.677551
This article is part of the Research TopicNetworks in Movement DisordersView all 12 articles
 Andreas Traschütz1,2
Andreas Traschütz1,2 Selina Reich1,2
Selina Reich1,2 Astrid D. Adarmes3,4
Astrid D. Adarmes3,4 Mathieu Anheim5,6,7
Mathieu Anheim5,6,7 Mahmoud Reza Ashrafi8Jonathan Baets9,10,11
Mahmoud Reza Ashrafi8Jonathan Baets9,10,11 A. Nazli Basak12
A. Nazli Basak12 Enrico Bertini13
Enrico Bertini13 Bernard Brais14Cynthia Gagnon15Janina Gburek-Augustat16
Bernard Brais14Cynthia Gagnon15Janina Gburek-Augustat16 Hasmet A. Hanagasi17Anna Heinzmann18
Hasmet A. Hanagasi17Anna Heinzmann18 Rita Horvath19Peter de Jonghe9,10,11
Rita Horvath19Peter de Jonghe9,10,11 Christoph Kamm20
Christoph Kamm20 Peter Klivenyi21
Peter Klivenyi21 Thomas Klopstock22,23,24
Thomas Klopstock22,23,24 Martina Minnerop25,26,27
Martina Minnerop25,26,27 Alexander Münchau28Mathilde Renaud29,30
Alexander Münchau28Mathilde Renaud29,30 Richard H. Roxburgh31,32
Richard H. Roxburgh31,32 Filippo M. Santorelli33
Filippo M. Santorelli33 Tommaso Schirinzi34,35
Tommaso Schirinzi34,35 Deborah A. Sival36
Deborah A. Sival36 Dagmar Timmann37Stefan Vielhaber38,39,40Michael Wallner41
Dagmar Timmann37Stefan Vielhaber38,39,40Michael Wallner41 Bart P. van de Warrenburg42
Bart P. van de Warrenburg42 Ginevra Zanni13
Ginevra Zanni13 Stephan Zuchner43Thomas Klockgether44,45
Stephan Zuchner43Thomas Klockgether44,45 Rebecca Schüle1,2
Rebecca Schüle1,2 Ludger Schöls1,2 PREPARE Consortium
Ludger Schöls1,2 PREPARE Consortium Matthis Synofzik1,2*
Matthis Synofzik1,2*Autosomal recessive cerebellar ataxias (ARCAs) form an ultrarare yet expanding group of neurodegenerative multisystemic diseases affecting the cerebellum and other neurological or non-neurological systems. With the advent of targeted therapies for ARCAs, disease registries have become a precious source of real-world quantitative and qualitative data complementing knowledge from preclinical studies and clinical trials. Here, we review the ARCA Registry, a global collaborative multicenter platform (>15 countries, >30 sites) with the overarching goal to advance trial readiness in ARCAs. It presents a good clinical practice (GCP)- and general data protection regulation (GDPR)-compliant professional-reported registry for multicenter web-based capture of cross-center standardized longitudinal data. Modular electronic case report forms (eCRFs) with core, extended, and optional datasets allow data capture tailored to the participating site's variable interests and resources. The eCRFs cover all key data elements required by regulatory authorities [European Medicines Agency (EMA)] and the European Rare Disease (ERD) platform. They capture genotype, phenotype, and progression and include demographic data, biomarkers, comorbidity, medication, magnetic resonance imaging (MRI), and longitudinal clinician- or patient-reported ratings of ataxia severity, non-ataxia features, disease stage, activities of daily living, and (mental) health status. Moreover, they are aligned to major autosomal-dominant spinocerebellar ataxia (SCA) and sporadic ataxia (SPORTAX) registries in the field, thus allowing for joint and comparative analyses not only across ARCAs but also with SCAs and sporadic ataxias. The registry is at the core of a systematic multi-component ARCA database cluster with a linked biobank and an evolving study database for digital outcome measures. Currently, the registry contains more than 800 patients with almost 1,500 visits representing all ages and disease stages; 65% of patients with established genetic diagnoses capture all the main ARCA genes, and 35% with unsolved diagnoses are targets for advanced next-generation sequencing. The ARCA Registry serves as the backbone of many major European and transatlantic consortia, such as PREPARE, PROSPAX, and the Ataxia Global Initiative, with additional data input from SPORTAX. It has thus become the largest global trial-readiness registry in the ARCA field.
Autosomal recessive cerebellar ataxias (ARCAs) are a heterogeneous group of ultrarare multisystemic neurodegenerative diseases affecting the cerebellum and/or its afferent tracts, often accompanied by damage to other neurological (e.g., corticospinal tract, basal ganglia, vestibular system, and peripheral nerves) or non-neurological systems (e.g., muscle, heart, and pancreas) (1, 2). The number of ARCA genes is continuously expanding, extending far above >100 genes, and the first ARCAs now come into reach of targeted treatment options (2).
Disease registries have been important for identification, characterization, and aggregation of rare neurological diseases. However, the real-world quantitative and qualitative evidence in registries and registry-based natural history and outcome measure studies have now also become a precious source for planning of treatment trials and modeling trial designs and endpoints, thereby complementing the knowledge available from preclinical studies and clinical trials (3). The ARCA Registry was launched in 2013 in order to apply this concept to the field of ARCAs, and it remains the only multicenter registry fully dedicated to ARCAs and early-onset ataxias (EOAs), which are known to be enriched but not exclusive for ARCAs (1, 2). The overarching goal of the ARCA Registry is to become a key facilitator enabling trial readiness by
• providing an easily accessible, web-based, good clinical practice (GCP)-conforming, and general data protection regulation (GDPR)-compliant multicenter multi-trial registry infrastructure platform as a backbone for global trial-readiness efforts in ARCAs;
• building cohorts of sufficient size for trial-readiness studies and upcoming treatment trials through aggregating ARCA patients in an accessible, standardized, multicenter fashion around the world;
• characterizing the phenotypic spectra for ARCAs, which will inform treatment trial design and especially outcome selection for future treatment trials;
• collecting real-world natural history data for ARCAs acquired during daily clinical life across a large range of centers across the world, thereby informing design, planning, and modeling of treatment trials; and
• providing a continuous database backbone for trial-readiness ataxia consortia around the world, e.g., the German DZNE ARCA-EOA network (4), the PREPARE consortium (5), PROSPAX (6), and ARCA GLOBAL (7).
In this overview, we will describe the main methodological features and assets of the ARCA Registry, with examples on how it is already being utilized to improve trial readiness in the field of ARCAs, including its current use by multiple research networks. It will also illustrate the registry's potential for expansion to other partners worldwide to promote trial readiness for ARCAs.
The ARCA Registry is built on WebSpirit (2mt Software, Ulm, Germany), a web-based electronic data-capture system currently used in a variety of national and international medical research consortia (Figure 1). The web-based implementation allows direct access by registered clinicians and study teams from any computer worldwide, as required for easy access in a global multicenter setting. The fact that it uses the same technical registry platform (WebSpirit) as one of the largest autosomal dominant ataxia registries, namely, the spinocerebellar ataxia (SCA)/ESMI registry (8, 9) and SCA Global (10), as well as the large sporadic ataxia registry (SPORTAX) (11, 12) and the Hereditary Spastic Paraplegia (HSP) Registry, allows for cross talk and joint analysis not only across the manifold ARCAs captured in the ARCA Registry itself but also with SCAs, sporadic ataxias, and even HSPs. This is further facilitated by aligning all key electronic case report forms (eCRFs) between these major ataxia registries. The registry platform is GCP-compliant: an audit trail is maintained to track changes to recorded data, a detailed rights and role management system limits access to entered data for each individual system user, and quality assurance is supported by an integrated online-monitoring system. Moreover, it is fully compliant with the European Union GDPR based on the following features: use of unique pseudonyms generated with a secure one-way hash function to restrict the use of personally identifiable data to local sites; separation of processing activities through assignment of user roles (e.g., data entry, monitoring, and data management) and restriction of access to data; record and transfer only of pseudonymized data; all access to data through encrypted connections; and servers located within the EU. Participating sites maintain access to their data entered in the ARCA Registry, with the possibility to easily export and systematically analyze locally aggregated datasets. Access to full multisite datasets is provided for specific projects upon request by a standardized project template and provided to the project submitter after evaluation of the request.
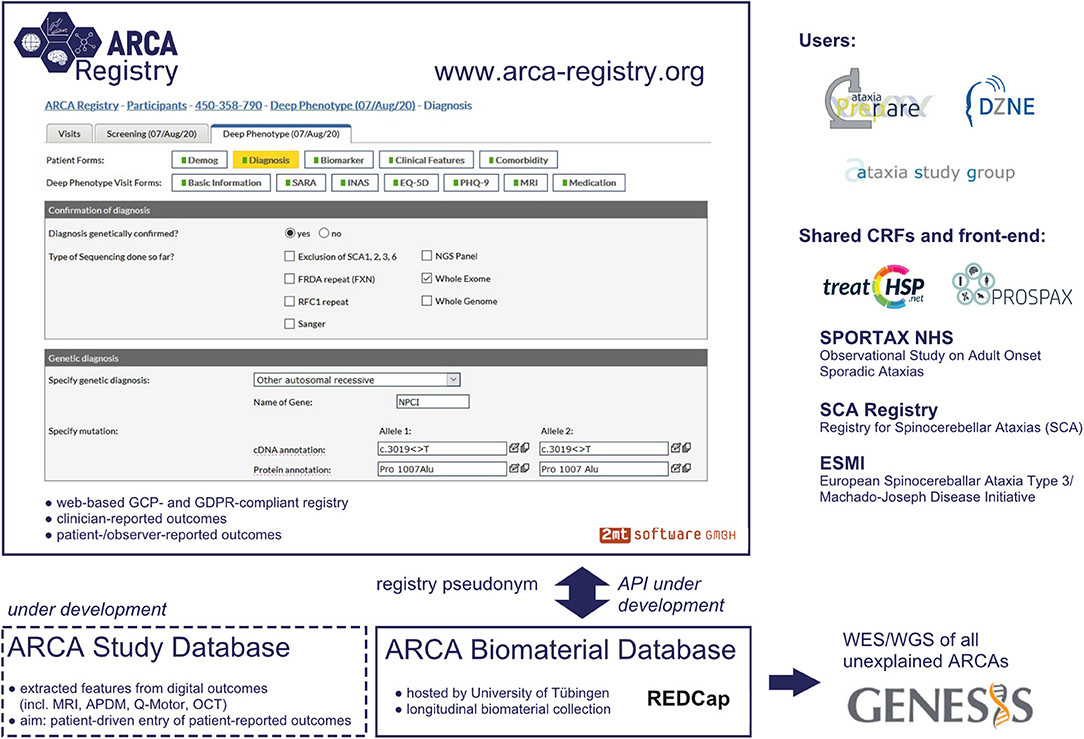
Figure 1. Platform and database infrastructure of the ARCA Registry. Graphical user interface of the web-based ARCA Registry, with display of a representative electronic case report form (eCRF) and embedding in the larger database infrastructure. The front-end of the software and the main core of eCRFs are shared with other major ataxia and rare disease registries (e.g., the HSP registry used by the TreatHSP network), making the ARCA Registry user friendly and convenient for joint analysis of data across genetic ataxias, sporadic ataxias, and hereditary spastic paraplegia (HSP) registries.
The physician-reported multidomain datasets in the ARCA Registry are the core of a larger systematic multi-component ARCA database cluster (Figure 1). For longitudinal collection of biomaterials, the ARCA Registry is linked to an ARCA biomaterial database built on REDCap. To facilitate whole-exome and whole-genome sequencing in all patients with unsolved ARCA, the ARCA Registry is moreover linked to next-generation sequencing (NGS) data on the genomics research platform GENESIS (13, 14). GENESIS is a user-friendly collaborative cloud-based analysis and matchmaking platform that encompasses the largest ataxia NGS dataset collection worldwide (>2,000 ataxia NGS datasets), aggregated via the PREPARE consortium (PREPARE-GENESIS) (see below). While the ARCA Registry and the GENESIS platform are two distinct databases, subjects from the registry are linked to the GENESIS platform via an ID generated by the ARCA biomaterial database. Ongoing developments of this multi-component ARCA database cluster will include an ARCA multi-study database as a repository for features of digital outcomes such as magnetic resonance imaging (MRI), digital-motor sensors (APDM, Q-Motor), and optical coherence tomography and for patient-driven entry of patient-reported outcome measures (PROMs).
The eCRFs of the ARCA Registry are designed to characterize the clinical heterogeneity of phenotypic spectra and natural history phenotypic evolution of ARCAs, thus helping in the selection of outcomes and planning of upcoming treatment trials (sample size calculation, trial duration, etc.) as well-modeling of trial endpoints and treatment effects. Different degrees of eCRFs details—characterized as “core,” extended,” and “optional” datasets—allow data capture tailored to the participating site's variable interests and resources (Table 1). In brief, the eCRFs include clinical scales and composite measures, clinician-reported outcome measure and PROMs, biomarker outcomes, and quantitative performance measures:
• The core dataset in the ARCA Registry comprises demographic data (with ethnic background), genetic diagnosis (with types of sequencing performed), different scores to measure disease severity like the Friedreich Ataxia Rating Scale (FARS) Functional Stage (15), the Scale for the Assessment and Rating of Ataxia (SARA) (16), systematic phenotyping using the Inventory of Non-Ataxia Signs (INAS) (17) with customized amendments (e.g., bradykinesia, ptosis, or the head impulse test), the presence and onset of typical clinical ARCA features (e.g., ataxia, epilepsy, cognitive impairment, and diabetes), ARCA biomarkers (serum and neurophysiology), and relevant comorbidities (including alcohol intake).
• The extended dataset adds questionnaires on health status and depression (EQ-5D and PHQ-9) (18, 19), disease-relevant medication and treatment effects, and a summary of MRI findings.
• Optional datasets include the possibility to report pediatric features (e.g., pregnancy and birth or developmental milestones), and the ARSACS Disease Severity Index as a disease-specific outcome measure (20).
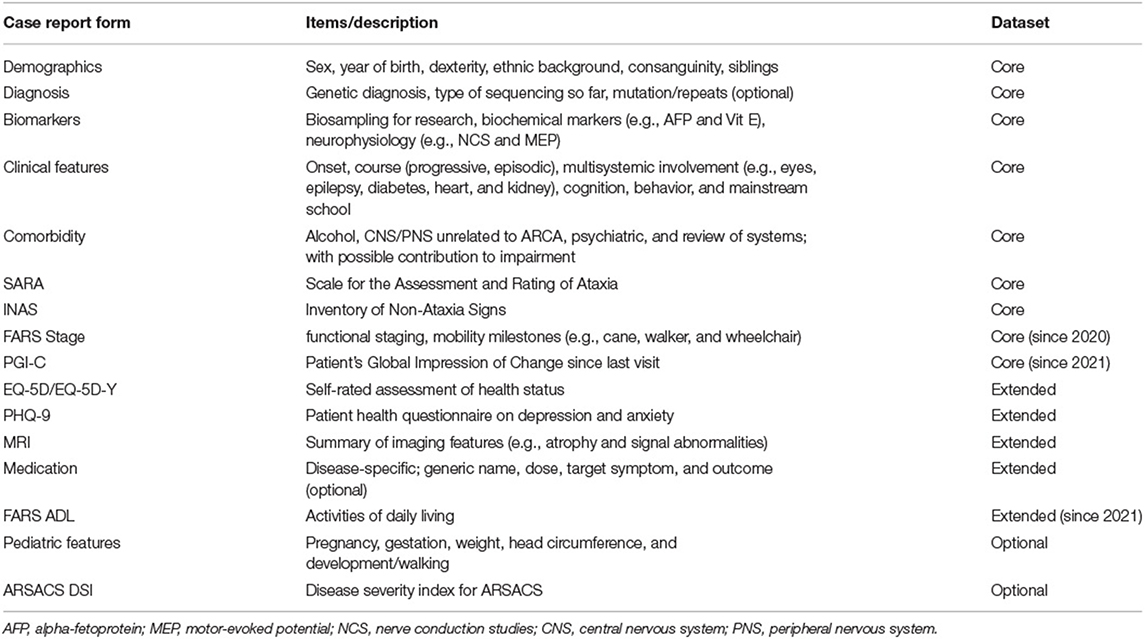
Table 1. Case report forms in the ARCA Registry.
The Patient's Global Impression of Change (PGI-C; core dataset) (21, 22) and the FARS Activity of Daily Living (ADL; extended datasets) (15) have recently been implemented as “anchor measures,” i.e., measures reflecting the patient's subjective experience of disease progression and functional impairment, which serve as reference measures helping to evaluate the significance of changes and effect sizes observed for the longitudinal clinical and biomarker data in the registry.
The ARCA Registry captures ARCAs from centers around the world (Figure 2). While initially mainly capturing centers from countries across Europe, the scope of the ARCA Registry has continuously grown in the last 5 years currently to now more than 30 sites from 15 countries. The registry has an active strategy to recruit centers from underrepresented countries to strengthen its global representation of ARCAs, regarding both disease prevalence and variable genetic/ethnic backgrounds. Participation is possible upon request. Minimum requirements are the commitment to contribute CRFs of at least the basic phenotype (see above) and to aim for longitudinal follow-ups.
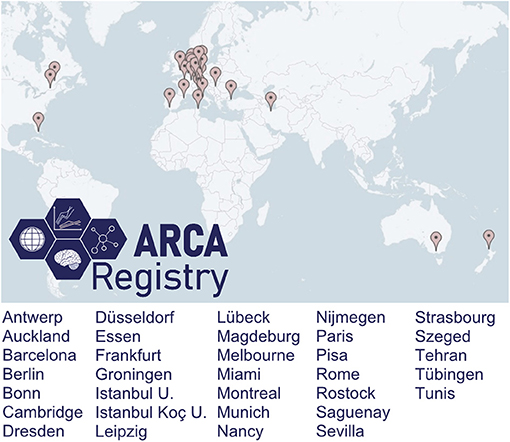
Figure 2. Contributing centers. The ARCA Registry is a growing, cross-continental multicenter registry, with more than 30 contributing sites in 15 countries by the end of 2020.
From its foundation in 2013 until now, more than 800 patients with almost 1,500 visits have been recruited to the ARCA Registry. In the past 5 years, there has been a considerable increase in longitudinal data, currently reaching up to eight annual follow-up visits in the first patients (Figure 3A). Follow-up core datasets including SARA or INAS from at least two visits and 13 sites are available in >300 patients. Follow-up extended datasets such as MRI summary data or the self-rated assessment of health status by EQ-5D from at least two visits are available from >200 patients. In addition to its longitudinal coverage, the ARCA Registry also captures patients with a broad range of ages and disease stages (Figure 3B). While—in keeping with the early onset of ARCAs (1, 2) −60% of patients have symptom onset before 40 years of age, 40% of patients have later onset, up to 80 years of age. Ataxia severity at baseline visits have been recorded as mild (SARA: ≤8), moderate (8–16), and severe (>16) in 16, 41, and 43% of patients, respectively. Sixty-five percent of patients have an established genetic diagnosis. The most frequent diagnoses in the ARCA Registry are ARSACS (~120 patients, 14%), Friedreich ataxia (~90 patients, 11%), and SPG7 (~40 patients, 4%; see Figure 3C for the 10 most frequent ARCAs). Except for the enrichment of ARSACS—which is an overrepresentation due to the major contribution of participating sites in Quebec—the ARCA Registry provides prevalence data that are generally consistent with expectations from the literature (1, 23). Patients in the ARCA Registry who do not have a genetic diagnosis yet (currently 35%) are included in a coordinated NGS effort on a continuous basis to make a diagnosis or to identify novel genes, via the PREPARE-GENESIS platform (see above).
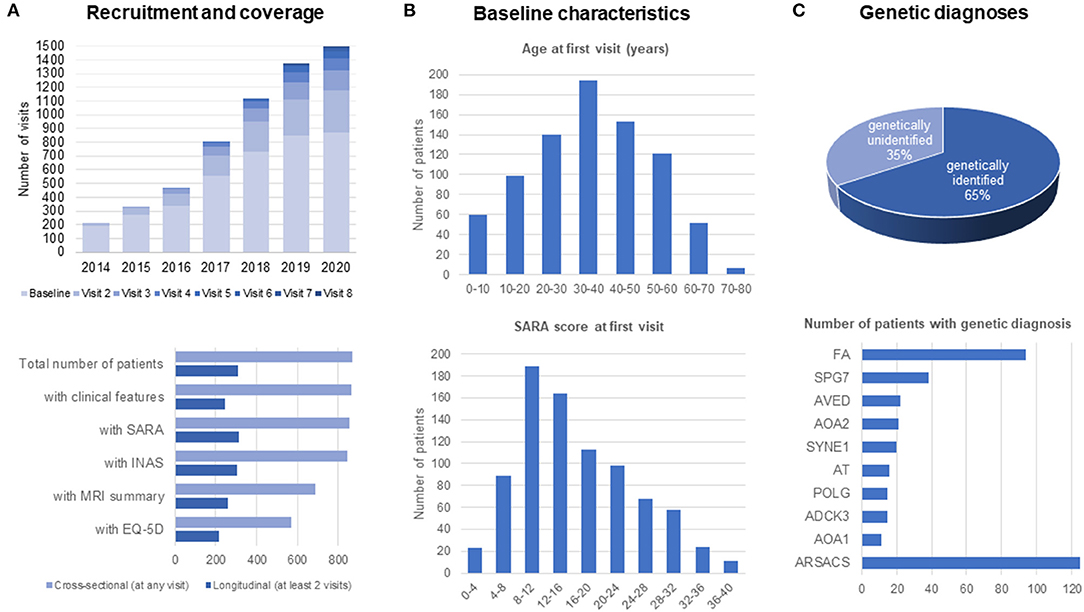
Figure 3. Recruitment and patient characteristics in the ARCA Registry. (A) Recruitment and coverage. Recruitment of patients is continuous since 2013, reaching more than 800 patients with up to eight longitudinal assessments by the end of 2020. Longitudinal data with at least two assessments are available in about 200–300 patients. (B) Baseline characteristics of patients. Patients in the ARCA Registry span the full range of ages and disease stages. (C) Genetic diagnoses in the ARCA Registry, with a list of the 10 most frequent genetic causes.
The ARCA Registry with its phenotypic and longitudinal data has enabled large clinico-genetic cohort studies to delineate the phenotypic spectrum and longitudinal disease progression of major and novel ARCAs. It thus fulfills the requirements of primary datasets that can be used to describe natural history progression models and plan treatment trials in almost all of the most frequent ARCAs, including pharmacometric modeling of outcome measures and treatment effects. For example, for RFC1-ataxia, it has helped to reveal multisystemic phenotypes mimicking cerebellar type multiple system atrophy and progressive supranuclear palsy, and hyperkinetic movement disorders such as chorea and dystonia, and provided first sample size calculations based on longitudinal SARA assessments (24). For COQ8A/ADCK3-ataxia, the ARCA Registry facilitated the delineation of clinico-genetic associations, and the longitudinal analysis of SARA scores has provided the first systematic, group-based evidence for a possible treatment effect of coenzyme Q10 (25). Similarly, the ARCA Registry has enabled the natural history of POLG-related ataxia to be documented through longitudinal SARA and INAS assessments (26). The systematic assessment of patients with as-yet-unknown genetic molecular diagnoses means that—when the underlying gene is discovered—there are already established longitudinal progression data, as exemplified for patients found to carry pathogenic variants in the novel ARCA gene PRDX3 (27).
European authorities including the European Medicines Agency (EMA) have highlighted the potential of disease registries to provide real-world evidence that can complement preclinical, clinical, and even post-marketing data especially for rare diseases (3). At the same time, however, they have also put forward clear standards for data collection and quality criteria for disease registries that aim to meet this goal.
The ARCA Registry captures the key EMA data elements (3), including administrative information (e.g., site, contact dates, registry entry and exit dates, and reason for registry exit), patient data (e.g., age, sex, and alcohol as lifestyle factor), disease features (including diagnosis, disease duration, severity/staging, genetic information, and biochemical tests if appropriate), relevant comorbidities, and disease-related or relevant concomitant medical treatment (Table 2). The ARCA Registry is also enrolled in the European Directory of Registries of the European Rare Disease Registry Infrastructure (ERDRI.dor). As a constituent registry of the evolving Rare Neurological Disease Registry of the European Research Network on Rare Neurological Diseases (ERN-RND) (28), the ARCA Registry will provide the common data elements defined by the ERDRI (29), which adds more systematic coding of diagnosis (Orpha code), genetic diagnosis (HGVS) or phenotype (HPO), resources for research (e.g., biosampling and link to biobank), and a classification of disability (Table 3). As a cross-disease database, the ERN-RND registry collects general information on demographics, genetics, and phenotype, which allows the identification of centers that look after specific (and often genetically defined) patient groups. By contrast, as a disease-group specific database, the ARCA Registry collects complementary in-depth data to enable trial readiness in recessive ataxias. Both registries are well-interconnected, as the common ERN dataset can be easily extracted from the ARCA Registry and imported into the ERN-RND registry. This link with the ERN-RND registry also adopts the FAIR principles in the ARCA Registry, ensuring that its data are f indable, accessible, interoperable, and reusable between countries (30).
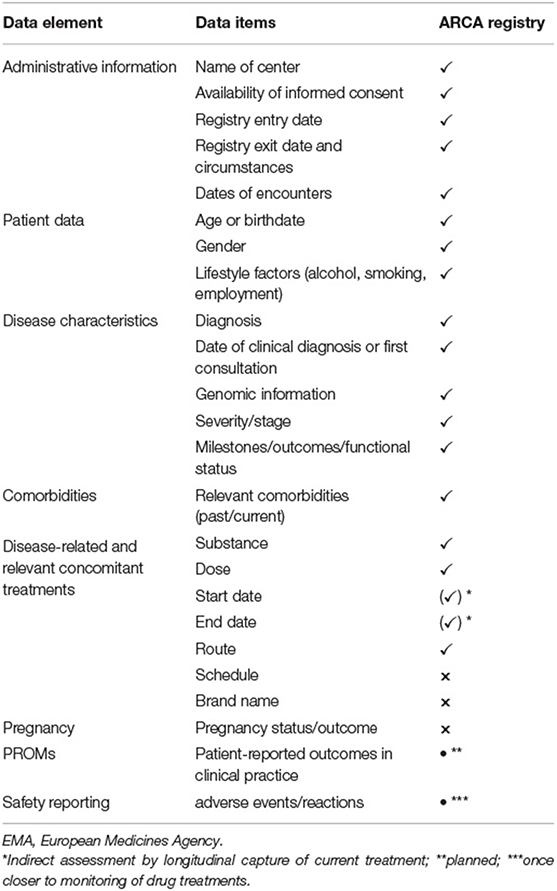
Table 2. Implementation of EMA guidelines on patient registries.
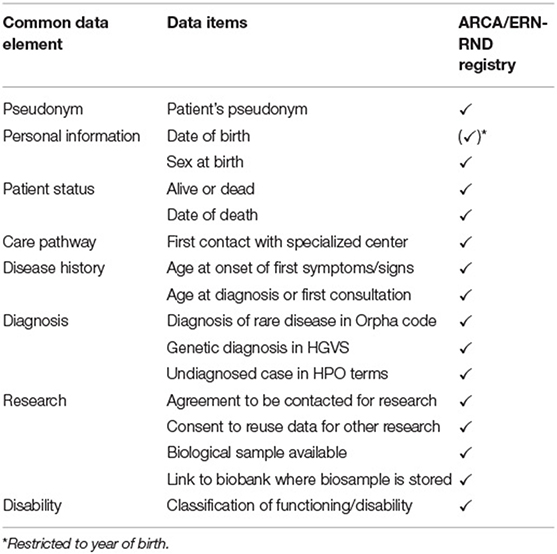
Table 3. Implementation of common data elements of the EU Rare Disease platform.
In line with the EMA standards of data quality (3), the ARCA Registry aims for consistency, completeness, accuracy, and timeliness. Consistency of data is facilitated by common standardized eCRFs, by clearly defined variables and selection of questions with binary outcomes, and by the implementation of clinical scales with high interrater reliability, especially SARA, INAS, and FARS stage or ADL (15, 16, 31). Completeness of data in core datasets is automatically checked online as the first step of a continuous database-embedded monitoring process; this resulted in 95% (e.g., for clinical features of ARCAs) to 99% (e.g., for SARA or INAS) CRF completion rate. Following automated online control of data plausibility and consistency within and between different CRFs at the time of data entry, accuracy of data is afterwards controlled offline in the second monitoring step. Recruitment numbers including availability and completeness are regularly disseminated in systematic, standardized reports of networks that use the ARCA Registry.
The ARCA Registry is being used not only by more than 30 single sites but also by several leading ARCA networks in Europe and worldwide.
The German network on ARCAs and EOAs, launched in 2013 by the German Center for Neurodegenerative Diseases (DZNE), comprises five major German ataxia sites (Tübingen, Bonn, Munich, Magdeburg, and Rostock). The network has established the first version of the ARCA Registry, which was co-hosted by the Ataxia Study Group. Every effort was made to ensure that the Registry is fully aligned in its data fields and database system with other major SCA and sporadic ataxia registries (32), likewise hosted by the Ataxia Study Group. Since then, the German ARCA/EOA network has contributed >400 subjects to the ARCA Registry and provides monthly reports on its recruitments to the ARCA Registry.
PREPARE (Preparing for therapies in autosomal recessive ataxias) was launched in 2016 as an EU-funded (E-RARE JTC 2015) rare disease network; it was one of the first dedicated ARCA trial-readiness networks. Utilizing the complementary expertise from many ARCA centers and international ARCA networks, it was established to facilitate all crucial translational steps from genetic profiling (including discovery of new genes) to standardized preclinical trials, developing FDA-compliant outcome measures, and registry-inventoried transnational trial-ready cohorts, hereby fully building on the ARCA Registry as its backbone. The network began with seven centers from across Europe and Canada and has now expanded to >13 centers including centers in Turkey, Iran, and New Zealand. Using the extended site network and longitudinal dataset provided by the ARCA Registry, PREPARE has run phenotype and natural history studies on several ARCAs, e.g., RFC1 (24), COQ8A/ADCK3 (25), RFC1, POLG (26), and PRDX3 (27).
The network PROSPAX (An integrated multimodal PROgression chart in SPastic atAXias), launched in 2020 and funded by the European Joint Program on Rare Diseases (EJP RD), will establish a paradigmatic integrated trial-ready model of disease progression and mechanistic evolution in spastic ataxias. It hereby builds on a rigorous trial-like multicenter natural history center study on the two flagship recessive ataxias ARSACS and SPG7, combining longitudinal clinician- and patient-reported digital and molecular outcomes for these spastic ARCAs. It unites all major European ARCA and HSP networks and includes Canadian ARCA centers (>7 centers) to run this transatlantic natural history study. PROSPAX hereby utilizes a “spin-off” study registry version, which directly builds on the ARCA Registry, with fully compatible pseudonymization procedure and eCRFs, and where datasets will be integrated into the ARCA Registry (and equally the HSP registry) at the end of the study. PROSPAX also draws on all the other components of the multi-component ARCA database cluster described above (ARCA biomaterial database, GENESIS, and ARCA multi-study database). By sharing the same core eCRFs and front-end with the HSP Registry used by the TreatHSP network (33), this spin-off version of the ARCA Registry enables direct cross talk with the HSP registry and joint analysis with HSPs, which is of high importance given the large genetic, molecular, and clinical overlap between ataxias and HSPs (34).
ARCA GLOBAL and its sister platform SCA GLOBAL together comprise the Ataxia Global Initiative (AGI). The AGI presents a worldwide multi-stakeholder (academia, industry, and patient organizations) platform coordinating and preparing all necessary steps for trial readiness in autosomal-dominant (SCA GLOBAL) and autosomal recessive (ARCA GLOBAL) ataxias (7, 10). With establishing trial-ready cohorts and cross-center harmonized clinician-reported outcome measures and PROMs as one of its key tasks, the AGI uses the ARCA Registry as one of its key registries. This reflects the fact that the ARCA Registry already captures all outcome measures that were stipulated by the AGI as the common core set of clinical outcome measures to be used by ataxia centers worldwide. Moreover, the AGI builds on the ARCA Registry as one of its major trial-readiness resources, as this registry readily allows data-download and dataset preparation for further workup, e.g., by the Critical Path to Therapeutics for the Ataxias (CPTA) consortium of the Critical Path Institute (C-Path), which aims to prepare regulatory approval by the FDA and the EMA for clinical endpoints in genetic ataxias.
In addition, the SPATAX network, which includes all types (i.e., not only autosomal recessive) of ataxias and HSPs, has contributed subsets of data to the ARCA Registry.
The ARCA Registry faces several limitations and open challenges that remain be to be addressed:
• The sustainability of the ARCA Registry depends on strong commitment by the contributing centers as well as project-based funding, with fluctuations in patient recruitment, participating sites, and monitoring performance. Technical improvements and new software implementations often come along with variable latencies. Even the “core dataset” may exceed the possibilities of a clinical appointment in many ataxia centers, but further minimization of the core dataset would need to be carefully weighed against the minimal required data necessary to really help in preparing trial readiness as well as against registry standards put forward by public authorities. The timeliness of monitoring is governed by batch monitoring of each site by site, which provides the opportunity for focused local revision of data and files but also leads to periodic delays for those sites that had just been monitored.
• Patients with the same genetically defined ARCA are still dispersed in different ataxia registries, e.g., because of identification of novel autosomal recessive genes in sporadic late-onset ataxia patients (e.g., RFC1) who have so far been collected in a sporadic ataxia registry (e.g., the SPORTAX registry).
• The ARCA Registry does not cover all aspects of each ARCA disease, or may cover it with measures that are too broad for clinical trial design in a specific ARCA disease. While the global phenotype or the progression of ataxia as measured by the SARA score is assessed, more fine-grained motor (e.g., walking speed) and especially a larger array of non-motor features (e.g., the cerebellar cognitive-affective syndrome) are not captured. Moreover, selected eCRFs and non-ataxia scales like the INAS were primarily implemented to systematically capture disease phenotypes but might show less responsiveness to change. Thus, for capturing the natural history of certain ARCAs, the ARCA Registry might need to be complemented by additional eCRFs. Registry spin-offs such as PROSPAX registry, however, exemplify that the registry infrastructure can indeed be readily adapted to meet the needs of such natural history studies.
• Finally, recruitment into the ARCA Registry is still biased toward patients from Europe or of European descent, which leads to an underrepresentation of ARCAs with other ethnic/genetic as well as sociocultural backgrounds. Structural disadvantages (especially availability of local person and funding resources for data-entry) and language barriers may hamper a more global dissemination of the ARCA Registry. The eCRFs, the registry software, and templates for an application to a local institutional review board are all available in English language, but additional translations and country-specific adaptations may help to increase the scope.
The ARCA Registry has (i) enabled the harmonization of clinical outcomes across ataxia centers around the world; (ii) has demonstrated its capacity to act as a centralized database for genotype–phenotype and natural history studies in the >100 ARCAs, already exemplified for COQ8A-, RFC1-, and POLG-related ataxias; and (iii) aggregates the necessary large-scale longitudinal progression datasets for calculating sample sizes, modeling trial designs and randomization procedures, and running pharmacometric models simulating treatment effect sizes for anticipated clinical trials. Given its adoption by many international ARCA sites and networks; its GCP, GDPR, and EMA compliance; its web-based data capture; and its connections to a constantly growing multi-component ARCA database cluster, the ARCA Registry is well-placed to become a global trial-readiness registry for ARCAs.
Sarah Doss, Department of Neurology, Charité University Medicine, Berlin, Germany; Jun-Suk Kang, Department of Neurology, University Hospital, Frankfurt, Germany; Ivana Ricca, IRCCS Fondazione Stella Maris, Pisa, Italy; S. Ben Sassi, Neurology Department, National Institute of Neurology, Tunis, Tunisia; Laszlo Szpisjak, Interdisciplinary Excellence Centre, Department of Neurology, Faculty of Medicine, Albert Szent-Györgyi Clinical Center, University of Szeged, Szeged, Hungary; Andreas Thieme, Department of Neurology, Essen University Hospital, University of Duisburg-Essen, Essen, Germany
AT and MS have designed and conceptualized the review and analyzed the data. All authors have contributed data to the ARCA Registry and/or drafted or revised the manuscript for intellectual content, contributed to the article, and approved the submitted version.
This work was supported via the European Union's Horizon 2020 research and innovation program by the BMBF under the frame of the E-Rare-3 network PREPARE (01GM1607; to MS, MA, and BW), the DFG under the frame of EJP-RD network PROSPAX (No. 441409627; MS, RS, FS, BB, CG, RH, AB, DT, and BW), and grant 779257 “Solve-RD” (to MS, RS, and BW). The study was further funded by the Federal Ministry of Education and Research, Germany, through the TreatHSP network (01GM1905 to RS and LS), and the National Institute of Neurological Diseases and Stroke (R01NS072248 to SZ and RS). BW receives additional research support from ZonMW, Hersenstichting, Gossweiler Foundation, uniQure, and Radboud University Medical Centre. LS, TKlop, TKlock, EB, GZ, BW, RS, and MS are members of the European Reference Network for Rare Neurological Diseases—Project ID No 739510. AT receives funding from the University of Tübingen, Medical Faculty, for the Clinician Scientist Program Grant #439-0-0. PK receives financial support by the Hungarian Brain Research Program 2017-1.2.1-NKP-2017_VI/4. MRA received funding from NIMAD, proposal No 971846. JB is supported by a Senior Clinical Researcher mandate of the Research Fund—Flanders (FWO) under grant agreement number 1805021N and is a member of the μNEURO Research Centre of Excellence of the University of Antwerp. Several authors of this publication are members of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO-NMD) and of the European Reference Network for Rare Neurological Diseases (ERN-RND). RR and the University of Auckland CBR Neurogenetic Research Clinic receive funding from the Friedreich's Ataxia Clinical Outcome Measures study and the Duncan Foundation (NZ).
MW is the director of 2mt Software GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JT declared a shared affiliation, with no collaboration, with one of the authors AM to the handling Editor.
ADLs, activities of daily living; AGI, Ataxia Global Initiative; ARCA, autosomal recessive cerebellar ataxia; DZNE, German Center for Neurodegenerative Diseases; ESMI, European Spinocerebellar Ataxia Type 3/Machado-Joseph Disease Initiative; eCRF, electronic case report form; EMA, European Medicines Agency; EOA, early-onset ataxia; ERDRI, European Rare Disease Registry Infrastructure; ERN-RND, European Reference Network for Rare Neurological Diseases; FARS, Friedreich Ataxia Rating Scale; GCP, good clinical practice; GDPR, general data protection regulation; HSP, hereditary spastic paraplegia; INAS, Inventory of Non-Ataxia Signs; MRI, magnetic resonance imaging; NGS, next-generation sequencing; PGI-C, Patient's Global Impression of Change; PHQ-9, patient health questionnaire on depression and anxiety; PROM, patient-reported outcome measure; SARA, Scale for the Assessment and Rating of Ataxia; SPORTAX, Consortium “Sporadic Degenerative Ataxia with Adult Onset”; PREPARE, Consortium “Preparing for therapies in autosomal recessive ataxias”; PROSPAX, Consortium “An integrated multimodal PROgression chart in SPastic atAXias”; SCA, spinocerebellar ataxia.
1. Synofzik M, Nemeth AH. Recessive ataxias. Handb Clin Neurol. (2018) 155:73–89. doi: 10.1016/B978-0-444-64189-2.00005-6
2. Synofzik M, Puccio H, Mochel F, Schöls L. Autosomal recessive cerebellar ataxias: paving the way toward targeted molecular therapies. Neuron. (2019) 101:560–83. doi: 10.1016/j.neuron.2019.01.049
3. European Medical Agency. Guideline on Registry-Based Studies. Available online at: https://www.ema.europa.eu/en/guideline-registry-based-studies (accessed January 6, 2021).
4. DZNE Clinical Studies: Early Onset Ataxias. Available online at: https://www.dzne.de/en/research/studies/clinical-studies/eoa/ (accessed January 6, 2021).
5. Preparing for Therapies in Autosomal Recessive Ataxias (PREPARE) - A Global Consortium. Available online at: https://www.prepare-ataxia.com/ (accessed January 6, 2021).
6. PROSPAX: An Integrated Multimodal Progression Chart in Spastic Ataxias. Available online at: https://www.prospax.net/ (accessed January 6, 2021).
7. Ataxia Global Initiatives: ARCA Global. Available online at: http://ataxia-global-initiatives.net/arca-global/ (accessed January 6, 2021).
8. Jacobi H, du Montcel ST, Bauer P, Giunti P, Cook A, Labrum R, et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol. (2015) 14:1101–8. doi: 10.1016/S1474-4422(15)00202-1
9. The European Spinocerebellar Ataxia Type 3 Initiative (ESMI). Available online at: http://www.ataxia-study-group.net/html/studies/esmi (accessed January 6, 2021).
10. Ataxia Global Initiatives: SCA Global. Available online at: http://ataxia-global-initiatives.net/sca-global/ (accessed January 6, 2021).
11. Giordano I, Harmuth F, Jacobi H, Paap B, Vielhaber S, Machts J, et al. Clinical and genetic characteristics of sporadic adult-onset degenerative ataxia. Neurology. (2017) 89:1043–9. doi: 10.1212/WNL.0000000000004311
12. Sporadic Degenerative Ataxia With Adult Onset: Natural History Study (SPORTAX). Available online at: http://www.ataxia-study-group.net/html/studies/sportax (accessed January 6, 2021).
13. Gonzalez M, Falk MJ, Gai X, Postrel R, Schüle R, Zuchner S. Innovative genomic collaboration using the GENESIS (GEM. app) platform. Hum Mutat. (2015) 36:950–6. doi: 10.1002/humu.22836
14. GENESIS Software Platform. Available online at: https://www.genesis-app.com/ (accessed January 6, 2021).
15. Subramony S, May W, Lynch D, Gomez C, Fischbeck K, Hallett M, et al. Measuring Friedreich ataxia: interrater reliability of a neurologic rating scale. Neurology. (2005) 64:1261–2. doi: 10.1212/01.WNL.0000156802.15466.79
16. Schmitz-Hübsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. (2006) 66:1717–20. doi: 10.1212/01.wnl.0000219042.60538.92
17. Schmitz-Hübsch T, Coudert M, Bauer P, Giunti P, Globas C, Baliko L, et al. Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology. (2008) 71:982–9. doi: 10.1212/01.wnl.0000325057.33666.72
18. Group TE. EuroQol-a new facility for the measurement of health-related quality of life. Health Policy. (1990) 16:199–208. doi: 10.1016/0168-8510(90)90421-9
19. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. (2001) 16:606–13. doi: 10.1046/j.1525-1497.2001.016009606.x
20. Gagnon C, Brais B, Lessard I, Lavoie C, Côté I, Mathieu J. Development and validation of a disease severity index for ataxia of Charlevoix-Saguenay. Neurology. (2019) 93:e1543-9. doi: 10.1212/WNL.0000000000008313
21. Schmitz-Hübsch T, Fimmers R, Rakowicz M, Rola R, Zdzienicka E, Fancellu R, et al. Responsiveness of different rating instruments in spinocerebellar ataxia patients. Neurology. (2010) 74:678–84. doi: 10.1212/WNL.0b013e3181d1a6c9
22. de Vet HC, Terwee CB, Ostelo RW, Beckerman H, Knol DL, Bouter LM. Minimal changes in health status questionnaires: distinction between minimally detectable change and minimally important change. Health Qual Life Outcom. (2006) 4:1–5. doi: 10.1186/1477-7525-4-54
23. Coutelier M, Hammer MB, Stevanin G, Monin ML, Davoine CS, Mochel F, et al. Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol. (2018) 75:591–9. doi: 10.1001/jamaneurol.2017.5121
24. Traschütz A, Cortese A, Reich S, Dominik N, Faber J, Jacobi H, et al. Natural history, phenotypic spectrum, and discriminative features of multisystemic RFC1-disease. Neurology. (2021) 96:e1369–82. doi: 10.1212/WNL.0000000000011528
25. Traschutz A, Schirinzi T, Laugwitz L, Murray NH, Bingman CA, Reich S, et al. Clinico-genetic, imaging and molecular delineation of COQ8A-ataxia: a multicenter study of 59 patients. Ann Neurol. (2020) 88:251–63. doi: 10.1002/ana.25751
26. Bender F, Timmann D, Van de Warrenburg B, Adarmes-Gomez AD, Bender B, Thieme A, et al. Natural history of polymerase gamma related ataxia (Submitted). (2021).
27. Rebelo A, Eidhof I, Cintra VP, Pereira CV, Timmann D, Traschütz A, et al. Biallelic loss-of-function variations in PRDX3 cause cerebellar ataxia. Brain. (2021) 2021:awab071. doi: 10.1093/brain/awab071
28. European Reference Network - Rare Neurological Diseases (ERN-RND) Registry. Available online at: http://www.ern-rnd.eu/ern-rnd-registry/ (accessed February 23, 2021).
29. EU RD Platform: The European Rare Disease Registry Infrastructure (ERDRI). Available online at: https://eu-rd-platform.jrc.ec.europa.eu/erdri-description_en (accessed January 6, 2021).
30. Lochmüller H, Badowska DM, Thompson R, Knoers NV, Aartsma-Rus A, Gut I, et al. RD-Connect, NeurOmics and EURenOmics: collaborative European initiative for rare diseases. Eur J Hum Genet. (2018) 26:778–85. doi: 10.1038/s41431-018-0115-5
31. Jacobi H, Rakowicz M, Rola R, Fancellu R, Mariotti C, Charles P, et al. Inventory of Non-Ataxia Signs (INAS): validation of a new clinical assessment instrument. Cerebellum. (2013) 12:418–28. doi: 10.1007/s12311-012-0421-3
32. Ataxia Study Group (ASG). Available online at: www.ataxia-study-group.net (accessed February 23, 2021).
33. The TreatHSP Consortium. Available online at: https://www.treathsp.net (accessed February 23, 2021).
Keywords: ataxia, registry, network, natural history, trial readiness
Citation: Traschütz A, Reich S, Adarmes AD, Anheim M, Ashrafi MR, Baets J, Basak AN, Bertini E, Brais B, Gagnon C, Gburek-Augustat J, Hanagasi HA, Heinzmann A, Horvath R, de Jonghe P, Kamm C, Klivenyi P, Klopstock T, Minnerop M, Münchau A, Renaud M, Roxburgh RH, Santorelli FM, Schirinzi T, Sival DA, Timmann D, Vielhaber S, Wallner M, van de Warrenburg BP, Zanni G, Zuchner S, Klockgether T, Schüle R, Schöls L, PREPARE Consortium and Synofzik M (2021) The ARCA Registry: A Collaborative Global Platform for Advancing Trial Readiness in Autosomal Recessive Cerebellar Ataxias. Front. Neurol. 12:677551. doi: 10.3389/fneur.2021.677551
Received: 07 March 2021; Accepted: 20 May 2021;
Published: 25 June 2021.
Edited by:
Antonio Pisani, University of Pavia, ItalyReviewed by:
Alessandro Filla, University of Naples Federico II, ItalyCopyright © 2021 Traschütz, Reich, Adarmes, Anheim, Ashrafi, Baets, Basak, Bertini, Brais, Gagnon, Gburek-Augustat, Hanagasi, Heinzmann, Horvath, de Jonghe, Kamm, Klivenyi, Klopstock, Minnerop, Münchau, Renaud, Roxburgh, Santorelli, Schirinzi, Sival, Timmann, Vielhaber, Wallner, van de Warrenburg, Zanni, Zuchner, Klockgether, Schüle, Schöls, PREPARE Consortium and Synofzik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthis Synofzik, bWF0dGhpcy5zeW5vZnppa0B1bmktdHVlYmluZ2VuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.