 Giulia Giannini
Giulia Giannini Federica Provini
Federica Provini Pietro Cortelli
Pietro Cortelli Giovanna Calandra-Buonaura
Giovanna Calandra-Buonaura
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 14 June 2021
Sec. Movement Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.677213
This article is part of the Research TopicAutonomic Dysfunction in Multiple System AtrophyView all 5 articles
A higher frequency of motor and breathing sleep-related disorders in multiple system atrophy (MSA) populations is reported. REM sleep behaviour disorder (RBD) is one of the most robust markers of an underlying alpha-synucleinopathy. Although a large corpus of literature documented the higher prevalence of RBD in MSA, few studies have systematically investigated the prevalence of RBD as mode of disease onset and its role in disease progression. Moreover, there has been increasing interest in phenoconversion into synucleinopathies of cohorts of patients with isolated RBD (iRBD). Finally, some studies investigated RBD as predictive factor of conversion in isolated autonomic failure, a synucleinopathy presenting with autonomic failure as the sole clinical manifestation that could convert to a manifest central nervous system synucleinopathy. As the field of neurodegenerative disorders moves increasingly towards developing disease-modifying therapies, detecting individuals in the prodromal stage of these synucleinopathies becomes crucial. The aims of this review are to summarise (1) the prevalence of RBD during the course of MSA and as presenting feature of MSA (iRBD), (2) the RBD features in MSA, (3) MSA progression and prognosis in the subgroup of patients with RBD predating disease onset, and (4) the prevalence of MSA conversion in iRBD cohorts. Moreover, we summarise previous results on the role of RBD in the context of isolated autonomic failure as marker of phenoconversion to other synucleinopathies and, in particular, to MSA.
Multiple system atrophy (MSA) is a progressive neurodegenerative disorder characterised by a heterogeneous combination of autonomic failure, cerebellar syndrome, parkinsonian features poorly responsive to levodopa, and pyramidal signs (1).
The diagnostic criteria define three degrees of certainty for diagnosis (possible, probable, and definite) and two phenotypes: parkinsonian (MSA-P) or cerebellar (MSA-C), according to the predominant feature at the time of evaluation (1). Disease onset is currently defined as the initial presentation of any motor sign whether parkinsonian or cerebellar, or autonomic features with the exception of male erectile dysfunction (1).
Sleep disorders are frequently seen in MSA populations. Some of these overlap across the other parkinsonisms, while others are either unique to or far more prevalent in this disease. Sleep disorders in MSA are multifactorial and include nocturnal and diurnal manifestations. Nocturnal sleep is characterised by reduced sleep efficiency, fragmented sleep, and frequent awakenings, resulting in poor sleep quality. These disturbances are due to the neurodegeneration itself, linked to motor (akinesia and rigidity) and autonomic symptoms (nocturia) related to the disease, but also to the presence of concomitant sleep motor or behaviour disorders such as REM sleep behaviour disorder (RBD), restless legs syndrome (RLS), periodic limb movements (PLMs), or sleep-related breathing disorders (2–5). Few studies reported diurnal manifestations including excessive daytime sleepiness, evaluated through questionnaires, and spontaneous or levodopa-induced sleep attacks, but these results were not confirmed in other studies (5–9).
RBD is a parasomnia characterised by loss of the physiological muscle atonia during REM sleep associated with complex, sometimes violent, and dangerous motor behaviours during which patients act out the content of their dreams (10). Although RBD is readily clinically suspected, video-polysomnography (V-PSG) is required to demonstrate REM sleep without atonia (RSWA) and, therefore, to confirm the diagnosis. The presence of RBD could be over- or underestimated when based only on clinical history. On one side, different sleep disorders may mimic RBD (obstructive sleep apnoea, somnambulism, nocturnal epilepsy, hallucinations, confusional arousals, and RLS with PLMs), and on the other side, a proportion of patients are unaware of their abnormal behaviours during the night (11).
RBD is one of the most robust markers of an underlying alpha-synucleinopathy, but its presence does not distinguish MSA from other alpha-synucleinopathies like Parkinson's disease (PD) and dementia with Lewy bodies (DLB) (12, 13). Moreover, RBD could predate the motor/autonomic onset of these diseases (12, 13). Although a large corpus of literature documented the higher prevalence of RBD in MSA, few studies have systematically investigated the prevalence of RBD as mode of disease onset and its role in disease progression.
In recent years, there has been increasing interest in cohorts of patients with isolated RBD (iRBD) aiming to calculate the rate and latency of phenoconversion into synucleinopathies and to detect clinical and instrumental features acting as reliable markers of these neurodegenerative subtypes (MSA, PD, DLB) (12, 13). Some studies investigated RBD as predictive factor of conversion into isolated autonomic failure (IAF), a synucleinopathy presenting with autonomic failure as the sole clinical manifestation that could convert to a manifest central nervous system synucleinopathy (14–16). As the field of neurodegenerative disorders moves increasingly towards developing disease-modifying therapies, detecting individuals in the prodromal stage of these synucleinopathies becomes crucial. Identifying specific features predicting a particular subtype of these neurodegenerative diseases in cohorts of iRBD patients remains, to date, a challenge.
The aims of this narrative review are to summarise the available literature on (1) the prevalence of RBD during the course of MSA and as presenting features of MSA, (2) the RBD features in MSA, (3) MSA progression and prognosis in the subgroup of patients with RBD predating disease onset, and (4) the prevalence of MSA conversion in iRBD cohorts. Finally, we summarise previous results on the role of RBD in the context of IAF as marker of phenoconversion to other synucleinopathies and, in particular, to MSA.
Publications in English language issued before July 2020 were initially searched in PubMed database by using the following search criteria: [(“Shy-Drager”) OR (“multiple system atrophy”) OR (“striatonigral degeneration”) OR (“olivopontocerebellar”) OR (“MSA”) OR (“autonomic failure”) OR (“synucleinopathies”)] AND [(Sleep) OR (REM sleep behaviour disorder) OR (RBD) OR (Isolated RBD) OR (Idiopathic RBD) OR (REM sleep without atonia)]. In addition, a manual search of bibliographies of included studies and related reviews was carried out to find additional references. No unpublished data or data from abstracts were encountered or used. A subsequent review process with update of the literature search was performed between August 2020 and January 2021.
Eighteen prospective and retrospective studies (7, 17–33), most of which on a small sample, investigated the prevalence of clinically suspected RBD in MSA during the disease course (Table 1). Sixteen articles included the prevalence of RBD confirmed by PSG, 13 of them monitored with synchronised audiovisual recording (V-PSG) (Table 1). A meta-analysis performed in 2015 on 13 studies reported a summary prevalence of clinically suspected RBD in MSA ranging from 25 to 100%, with a summary prevalence of 73% (95% confidence interval [CI] = 62–84%) in a pooled sample of 324 patients. The prevalence of PSG-confirmed RBD in MSA ranged from 68.8 to 100%, with a summary prevalence of 88% (95% CI = 79–94%) in the pooled sample (30). Other five studies (27, 28, 31–33), not included in the meta-analysis, reported a prevalence of clinically suspected RBD ranging from 44.4 to 76.6% and of V-PSG-confirmed RBD ranging from 32.7 to 76.0% (Table 1).

Table 1. Studies focused on the prevalence of clinically suspected or V-PSG-confirmed RBD in MSA patients.
To note, several studies reported RBD diagnosis as video-polysomnographic finding also in MSA patients with a negative symptomatic history for this sleep disorder (17, 18, 26, 30, 34).
In patients with synucleinopathies, RBD frequently predates motor and cognitive deficits (11, 12). Despite its higher prevalence in MSA, the prevalence of RBD as initial manifestation of disease has been poorly investigated.
Few studies primarily investigated RBD as symptom of MSA onset. However, these data were reported also in other studies focused on different aims. To date, 12 studies have reported RBD preceding the clinical MSA onset (17, 19, 23, 25, 26, 28–32, 34, 35) (Table 2). Prevalence of RBD as first symptom of disease onset ranged from 10 to 60.0% when calculated in overall MSA samples and from 21.4 to 63.6% when calculated only in MSA patients with RBD (Table 2). This variability in results may be a consequence of the differences in study design, sample size, population characteristics, follow-up duration, and diagnostic criteria used to determine RBD. The first cause of heterogeneity derived from study design and population characteristics because the majority of the studies included all MSA patients, while other focused on RBD characteristics and, for this reason, included only MSA patients with clinically suspected RBD. Another cause of this variability is related to the sample size: the majority of studies included <50 patients (17, 19, 23, 25, 26, 28, 29, 34, 35), one study included between 50 and 100 patients (30), and two studies included >100 patients (31, 32). Moreover, in eight studies, RBD was diagnosed with V-PSG (17, 19, 23, 26, 28, 29, 32, 34), while in the other four studies (25, 30, 31, 35) the prevalence of iRBD was calculated on clinically suspected RBD because RBD diagnosis was based on history taking (31), V-PSG was performed only in a subgroup of the iRBD sample (35), or the rate of V-PSG-confirmed RBD in patients presenting with RBD was not specified in the results section (25, 30).
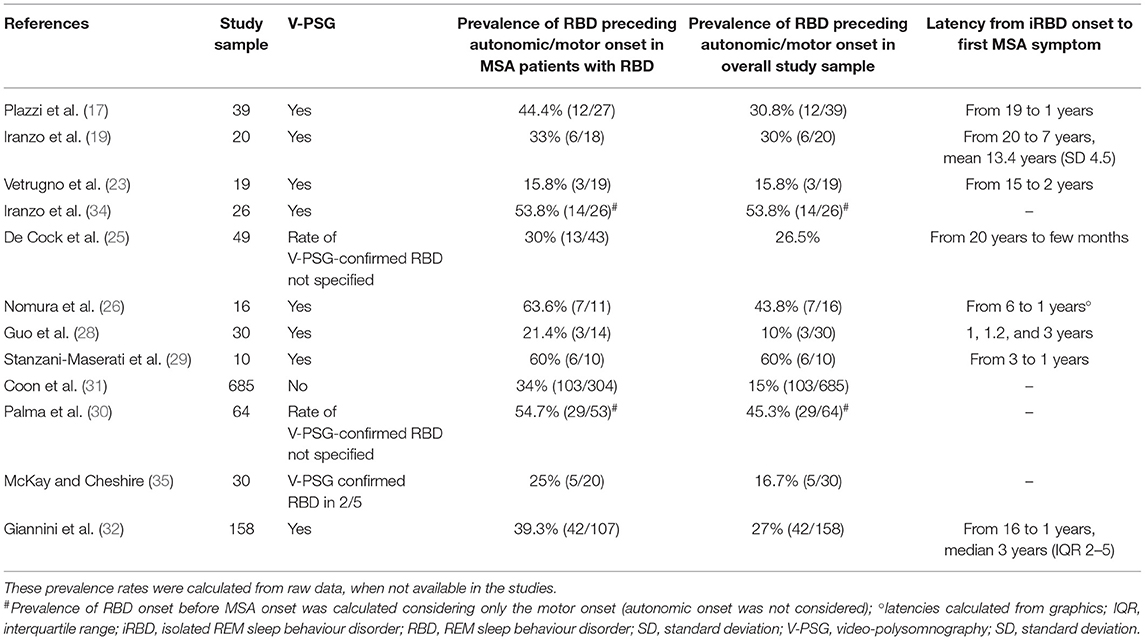
Table 2. Studies reporting isolated RBD as initial manifestation of MSA.
Among all studies investigating RBD predating MSA, two studies calculated prevalence and latency from RBD onset to disease onset taking into consideration only the motor onset and not the autonomic one (30, 34). Finally, some studies calculated prevalence on the overall sample to investigate the rate of iRBD as presenting feature of MSA, while others calculated this prevalence in MSA patients with RBD. Both values of prevalence and latency between RBD onset and MSA symptoms were reported in Table 2.
A retrospective study on 30 MSA patients sought to identify the very earliest symptoms in the natural history of disease and identified RBD as the third most frequent initial symptom of MSA (n = 5/30, 16.7%) after complete erectile failure and postural lightheadedness (35). Recently, a large monocentric study on 158 MSA patients compared disease onset according to the current international criteria (1) and considering RBD as a symptom of MSA onset revealed that this sleep disorder represents the most frequent first symptom of MSA. In this large cohort, patients with clinically suspected RBD had undergone V-PSG to confirm the diagnosis and, although the RBD onset was retrospectively reported by patients and caregivers, the event recording from V-PSG was shown to them to ensure it was the same reported in patient recall. Moreover, some MSA patients derived from the autonomic and sleep centres of the same Institute and were evaluated with closer follow-up before the onset of motor symptoms (32).
Only one study investigated the disease progression and prognosis of MSA patients presenting with RBD (32). This study on 158 patients, 107 with V-PSG-confirmed RBD, showed that 42 patients presented RBD as first symptoms of MSA. Among patients with RBD predating MSA onset, 29 presented with history of autonomic failure (25 urinary symptoms, 6 symptomatic orthostatic hypotension), 17 with cerebellar syndrome, and 6 with parkinsonism (isolated or combined). The subgroups of patients with RBD preceding the disease onset showed a more rapid progression of disease. Comparing patients with RDB predating disease onset and those developing RBD after the disease onset, the former showed more frequently an autonomic onset and less frequently parkinsonism both at disease onset and during the disease course. Moreover, during the disease course, patients with RBD as presenting symptom revealed earlier onset of stridor, pyramidal signs, symptomatic orthostatic hypotension, and urinary symptoms and showed shorter latency of several milestones of disease progression (urinary catheterization, severe dysphagia, and wheelchair dependency). The risk of death estimated by Kaplan–Meier analysis was higher in patients with RBD before disease onset with a weak statistical significance (log-rank test, p = 0.05) (32).
Concerning the clinical features of RBD in MSA, no differences were found between MSA-C and MSA-P subgroups (25, 27). As parkinsonism disappears during RBD movements in PD patients (36), the same research group investigated whether cerebellar symptoms and parkinsonism disappear during RBD in MSA patients. In this study, the authors interviewed 49 MSA and 49 PD patients along with their 98 bed partners, by means of a structured questionnaire, comparing movements, speech, and facial expressions during RBD and wakefulness. Clinical RBD was observed in 43/49 MSA patients. Reports from bed partners who were able to evaluate movements during sleep indicate that 81% of MSA patients reported some form of improvement during RBD compared to wakefulness. Movements were improved in 73% of patients with MSA, including increased speed (67%), strength (52%), or smoothness (26%). Speech was improved in 59% of patients and was more intelligible (17%), better articulated (36%), or louder in volume (55%). Facial expressions were normal (with disappearance of amimia and expressions of smiling, frowning, or fear) in 50% of patients during RBD. The rate of improvement was higher in PD than in MSA, but no further difference was observed either between MSA-P and MSA-C or in MSA patients with a disease duration <5 years vs. ≥5 years. Moreover, the analysis of movements was also monitored by V-PSG in 22/49 MSA patients and revealed more expressive faces and movements that were faster and more ample when compared with facial expression and movements during wakefulness. These movements were still somewhat jerky but lacked any visible parkinsonism, while cerebellar signs were not assessable. These findings suggest that there are still functional pathways in MSA patients (25).
Two studies investigated the longitudinal progression of V-PSG recordings in MSA patients with RBD. Decreased behaviours/movements during the disease course were confirmed on V-PSG recording in one patient. Sleep talking became less frequent and intense. At a more advanced stage, the ratio of RSWA to the whole of REM sleep increased, while the sleep architecture as well as the percentage of REM sleep were maintained (37). A V-PSG report on two MSA patients described a reduced frequency of RBD episodes during the disease course and the appearance of a disrupted sleep pattern characterised by no longer identifiable sleep stages, ambiguous and rapid oscillation of state-determining polysomnographic variables, associated with a nearly continuous motor and verbal abnormal behaviours. This disrupted sleep pattern was recorded after 4 and 6 years from RBD onset, and after 3 and 2 years from autonomic/motor onset, respectively. Features of abnormal Stage 1 NREM and REM sleep, together with unstable chin muscle tone, recurred rapidly and irregularly, in a sort of undifferentiated sleep state. These features were consistent with status dissociatus and were interpreted as progression of RBD (38).
Although almost all patients eventually diagnosed with MSA experience RBD at some point in their disease progression, studies investigating the rate of phenoconversion to synucleinopathies in cohorts of patients with iRBD showed that a low percentage of those who converted actually developed MSA. iRBD is a rare condition, and population-based studies showed a prevalence of about 1% over the age of 60. Few studies investigated the prevalence of RBD, using PSG, in population-based cohorts. One study on 348 individuals from South Korea aged 60 years and over quantified REM tone with PSG. Participants with abnormal tone were contacted by phone and asked about dream-enactment behaviour, revealing an RBD prevalence estimate of 1.15% (39). Another study estimated an RBD prevalence of 0.74% (95% CI = 0.29–1.89) in a sample of 539 individuals aged 60 years or older from a Spanish community, using a validated single question for the screening of RBD followed, in those who screened positive, by clinical assessment and V-PSG (40). Analysing data from 1997 participants belonging to the population-based HypnoLaus study (mean age = 59 ± 11.1 years, 53.6% women) who completed the Munich Parasomnia Screening questionnaire and had a complete polysomnography at home, authors estimated a prevalence of RBD of 1.06% (95% CI = 0.61–1.50) (41).
In the last years, several international groups have performed follow-up studies on iRBD cohorts focusing on the rate of phenoconversion to an overt neurodegenerative disease and on the predictive role of clinical and instrumental markers for phenoconversion.
In 1996, Schenck et al. first reported the phenoconversion of a series of 29 males older than 50 years of age, and in 2013, they provided a 16 year update of their previous report (42, 43). Other two important research groups closely followed up iRBD cohorts throughout the disease course. Since 2014, Postuma et al. recruited up to 154 patients with iRBD in Montreal (Canada), publishing subsequent articles on the longitudinal evolution of this cohort (44–48). Similarly, Iranzo et al., from Barcelona (Spain), first followed iRBD patients diagnosed from 1991 to 2003 and, subsequently, followed up disease-free patients from 2005 to date in a systematic manner, publishing articles on an increasing number of patients including up to 203 participants in this cohort (49–53). Moreover, other international groups focused on this topic and recently the International RBD Study Group combined prospective follow-up data, making up the largest iRBD cohort counting 1,280 participants recruited from 24 centres (54, 55). It is important to specify that this multicentre international group included overlapping patients with previous monocentric studies of smaller samples.
Taken together, these observational studies have suggested that most iRBD patients who eventually develop a defined neurodegenerative disease were almost always diagnosed with a synucleinopathy (43, 48, 53–63) (Table 3). These studies, generally performed in single centres and on small samples, reported a frequency of phenoconversion ranging from 12.5 to 80.8%. This variability in results could be related to the heterogeneity of methods and follow-up duration (43, 48, 53, 56–63). More recently, a larger study analysed prospective follow-up data on 1,280 iRBD patients from 24 centres of the International RBD Study Group, showing that 352 (28%) converted to an overt neurodegenerative syndrome (median time to phenoconversion = 8.0 years), 16 (4.5%) of which converted into MSA (55).
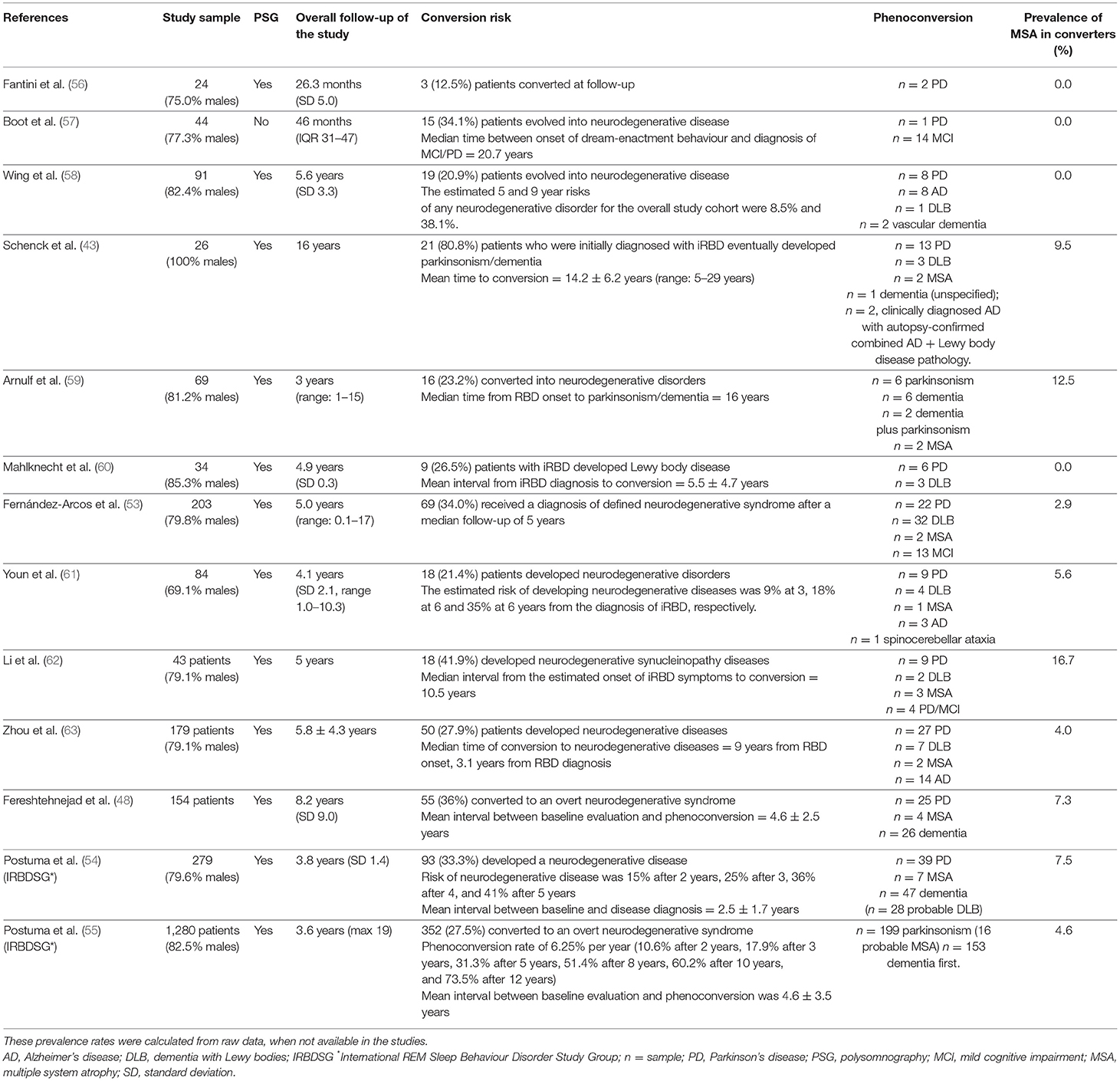
Table 3. Studies investigating phenoconversion in cohorts of patients with iRBD.
Considering only the sample who phenoconverted from iRBD, the rate of those who developed MSA ranged from 0% (56–58, 60) to 16.7% (62), with the remaining patients developing PD, DLB, or dementia (Table 3).
One retrospective study (64) focused on a subgroup of patients who experienced iRBD for at least 15 years before evolving into PD, PD dementia (PDD), DLB, or MSA. In 27 patients (88.9% males) with clinical history of RBD, 14 of whom with V-PSG-confirmed RBD, initial parkinsonian symptomatology was observed in 13 patients, initial cognitive impairment was reported by 13 patients, and primary autonomic symptomatology was the first symptom in one patient. At the last evaluation, the following diagnoses were established: MSA in 1 (3.7%) patient, PD in 4 (14.8%) patients, PDD in 6 (22.2%) patients, PD-MCI in 3 (11.1%) patients, MCI in 2 (7.4%) patients, and DLB in 11 (40.8%) patients. The median latency from RBD to neurodegenerative syndrome symptoms onset was 25 years (range 15–50 years) (64).
Few studies investigated predictors of phenoconversion in patients with iRBD. One recent study on the largest cohort of iRBD patients (n = 1,280) investigated potential predictors of phenoconversion identifying motor symptoms, objective motor examination, olfactory deficit, mild cognitive impairment, erectile dysfunction, abnormal DAT scan, colour vision abnormalities, constipation, RSWA, and age as markers (55). However, this study considered factors predicting the overall conversion rate from iRBD to an overt neurodegenerative disease, but not the ones specific for MSA conversion. A recent study analysed 154 polysomnography-proven patients with iRBD, of whom 55 phenoconverted to defined parkinsonism (n = 25 PD, n = 4 MSA) or dementia (n = 26 DLB) (48). Compared to the other synucleinopathies (DLB/PD), the four patients who eventually developed MSA were significantly younger (54.2 ± 7.8 vs. 70.4 ± 7.5, p = 0.001) and had preserved cognition (MoCA = 26.8 ± 2.5 vs. 21.3 ± 5.5, p = 0.044) and colour vision (FM100 = 87.0 ± 60.1 vs. 227.0 ± 122.9, p = 0.005) at the time of phenoconversion. MSA-converters may also have had less impaired olfaction (UPSIT % normal = 77.3 ± 44.4 vs. 57.6 ± 26.6) and more severe urinary symptoms (1.2 ± 0.4 vs. 0.7 ± 0.9) at phenoconversion (with marginal statistical significance levels) (48).
Another pre-motor cohort is represented by patients with IAF, a synucleinopathy presenting with autonomic failure as the sole clinical manifestation that could convert to a manifest central nervous system synucleinopathy. As there is an overlap of autonomic dysfunction in patients with MSA and those with pure autonomic failure (PAF) (5 year history of IAF with normal neurologic examination), it may be challenging to differentiate these two disorders in early stages, when only IAF is present.
The presence of RBD in the context of autonomic failure was suggested to be confined to those evolving to MSA and to be absent in those with PAF in a previous case series, making RBD a clinical marker of MSA (65). However, subsequent case series have reported RBD in patients with PAF (66–68), with symptoms occurring after the onset of autonomic failure (68).
Disagreement in results may be a consequence of the differences in design, sample size, population characteristics, follow-up duration, and diagnostic criteria used in the studies.
Few studies on cohorts of patients with IAF have investigated the role of RBD in predicting phenoconversion.
The largest follow-up studies reported to date on the natural history of a cohort of patients with a 5 year history of IAF, who fulfil current criteria for PAF (n = 50, mean disease duration 13.8 ± 7.1), documented a global percentage of 32% of phenoconversion to a manifest synucleinopathy (converters n = 16, 10 with MSA). This study found that the latency of RBD onset, not its presence, allows the discrimination of the converters group. In fact, V-PSG-confirmed RBD, which was more frequent in the converters group without reaching statistical significance, occurred significantly earlier in this group during the disease course (2 [−2; 5] vs. 10 [4; 13] years, p = 0.0281). These data entail a higher risk of phenoconversion to other synucleinopathies in patients with IAF with early-onset RBD (hazard ratio = 8.05, p = 0.016) (16).
A multicentric prospective study on the natural history of 100 patients with IAF, 74 of those followed up longitudinally, reported a cumulative incidence of phenoconversion to central synucleinopathy of 34% (converters n = 25, 6 with MSA) during a limited 4 year follow-up period (≈14%/year). This study did not report RBD in the small group of patients retaining the PAF phenotype at the last follow-up (n = 12) and showed that the presence of RBD was strongly associated with phenoconversion to other synucleinopathies (14). In a monocentric retrospective cohort study on 318 patients (79 patients followed up for at least 3 years) with IAF, in which the estimated conversion rate ranged between 12 and 48% (38 of 318 and 38 of 79, respectively), 22 patients developed MSA. Conversely, compared to previous findings, RBD was not found to be associated with a higher risk of phenoconversion and was seen in about half of patients with stable PAF, but its presence or absence was not consistently documented (15).
However, the latter two studies included all patients with IAF, irrespective of the disease duration, with subtle non-specific neurologic deficits at the time of enrolment and followed patients up for a short period (4 year follow-up). Our previous study (16) included only patients with IAF with a disease duration of >5 years without subtle non-specific neurologic deficits at the time of enrolment who were followed up for a longer period, allowing better estimation of the phenoconversion rate in patients currently defined as having PAF.
RBD is a key feature of MSA, playing a role in the presentation, prognosis, and progression of disease. Almost all MSA patients experience RBD at some point of the disease course, without differences in prevalence values between MSA-C and MSA-P groups. Although clinically suspected MSA is frequently reported in the majority of MSA patients, previous results demonstrated that V-PSG is needed to diagnose RBD, especially in those who do not report symptoms or do not have a sleep partner. Despite the high prevalence of RBD during the disease course, the rate of RBD as initial manifestation of MSA has been poorly investigated because only few studies primarily focused on this aspect. Prevalence of RBD as first symptom of disease onset ranged from 10% to 60.0% when calculated in overall MSA samples. One recent study (32), comparing the mode of MSA presentation according to the current international criteria (1) and to iRBD onset, reported that this sleep disorder represents the most frequent first symptom of MSA with a prevalence of 27%. Overall, data on the prevalence of RBD in MSA and of RBD as first symptom of disease argue for the inclusion of RBD as supportive feature of MSA diagnosis. The latest version of MSA diagnostic criteria in 2008 included additional features that allowed the diagnosis at an earlier stage in individuals who do not yet fulfil the requirements for “probable MSA” (1). Although the introduction of additional features led to improved diagnostic accuracy at the early disease stage (1), the overall sensitivity of these criteria remains suboptimal (69). This is related to the difficulty in distinguishing MSA from other more common synucleinopathies, especially at the early stages, but also from other mimics. Indeed, a recent study, performed on 203 cases, revealed that 160 (78.8%) patients were correctly diagnosed in life and had pathologically confirmed MSA, while the remaining 43 (21.2%) had alternative pathological diagnoses (70). For this reason, the introduction of RBD as supportive feature of MSA in the early disease stage could improve diagnostic accuracy.
Moreover, these studies suggest that RBD could be introduced as mode of MSA onset in forthcoming consensus criteria to evaluate the disease duration and also as a clinical feature of pre-motor and pre-autonomic presentation of MSA. Prodromal diagnostic criteria are required for MSA. Diagnostic criteria for prodromal MSA should optimise sensitivity in capturing an underlying alpha-synucleinopathy in patients presenting at a disease stage prior to that of “possible MSA” and should retain enough specificity to distinguish it from PD and DLB (13, 71). Prodromal diagnostic criteria for MSA could guarantee homogenous patient selection for clinical trials with therapies able to slow or prevent the neurodegenerative process.
However, although RBD is a robust marker of underlying alpha-synucleinopathy, its presence alone does not stratify among these different neurodegenerative disorders and other clinical and/or instrumental features in addition to RBD should be validated to increase diagnostic accuracy (e.g., neuroimaging, olfactory dysfunction, and central or peripheral autonomic failure).
Finally, patients who presented with RBD at disease onset showed a more rapid and severe disease progression, mainly due to a rapid involvement of autonomic features and to an early achievement of milestones of disease progression. These data entail the highly topographic and functional interconnection of brainstem neuronal networks, whose degeneration in MSA has been widely documented (72, 73), and suggest that a close follow-up is required to early recognise and treat stridor and autonomic disorders in this subgroup of patients.
GG: analysis of literature and drafting the manuscript. FP and PC: critical revision of the manuscript. GC-B: conception of the study and critical revision of the manuscript. All authors read and approved the final manuscript.
The study was supported by the strategic research program Bando ricerca finalizzata 2016 of the Italian Ministry of Health (reference number RF-2016-02361597). This review is part of the Ph.D. thesis of GG.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. (2008) 71:670–6. doi: 10.1212/01.wnl.0000324625.00404.15
2. Abbott SM, Videnovic A. Sleep disorders in atypical parkinsonism. Mov Disord Clin Pract. (2014) 1:89–96. doi: 10.1002/mdc3.12025
3. Jecmenica-Lukic M, Poewe W, Tolosa E, Wenning GK. Premotor signs and symptoms of multiple system atrophy. Lancet Neurol. (2012) 11:361–8. doi: 10.1016/S1474-4422(12)70022-4
4. Cortelli P, Calandra-Buonaura G, Benarroch EE, Giannini G, Iranzo A, Low PA, et al. Stridor in multiple system atrophy: consensus statement on diagnosis, prognosis, and treatment. Neurology. (2019) 93:630–9. doi: 10.1212/WNL.0000000000008208
5. Moreno-López C, Santamaría J, Salamero M, Del Sorbo F, Albanese A, Pellecchia MT, et al. Excessive daytime sleepiness in multiple system atrophy (SLEEMSA study). Arch Neurol. (2011) 68:223e30. doi: 10.1001/archneurol.2010.359
6. Högl B, Seppi K, Brandauer E, Wenning G, Poewe W. Irresistible onset of sleep during acute levodopa challenge in a patient with multiple system atrophy (MSA): placebo-controlled, polysomnographic case report. Mov Disord. (2001) 16:1177–9. doi: 10.1002/mds.1207
7. Ghorayeb I, Yekhlef F, Chrysostome V, Balestre E, Bioulac B, Tison F. Sleep disorders and their determinants in multiple system atrophy. J Neurol Neurosurg Psychiatry. (2002) 72:798–800. doi: 10.1136/jnnp.72.6.798
8. Martinez-Rodriguez JE, Seppi K, Cardozo A, Iranzo A, Stampfer-Kountchev M, Wenning G, et al. Cerebrospinal fluid hypocretin-1 levels in multiple system atrophy. Mov Disord. (2007) 22:1822–4. doi: 10.1002/mds.21668
9. Seppi K, Högl B, Diem A, Peralta C, Wenning GK, Poewe W. Levodopa-induced sleepiness in the Parkinson variant of multiple system atrophy. Mov Disord. (2006) 21:1281–3. doi: 10.1002/mds.20898
10. American Academy of Sleep Medicine. International Classification of Sleep Disorders - Third Edition (ICSD-3). Darien, IL: American Academy of Sleep Medicine (2014).
11. Iranzo A, Santamaria J, Tolosa E. The clinical and pathophysiological relevance of REM sleep behavior disorder in neurodegenerative diseases. Sleep Med Rev. (2009) 13:385–401. doi: 10.1016/j.smrv.2008.11.003
12. Högl B, Stefani A, Videnovic A. Idiopathic REM sleep behaviour disorder and neurodegeneration - an update. Nat Rev Neurol. (2018) 14:40–55. doi: 10.1038/nrneurol.2017.157
13. Xia C, Postuma RB. Diagnosing multiple system atrophy at the prodromal stage. Clin Auton Res. (2020) 30:197–205. doi: 10.1007/s10286-020-00682-5
14. Kaufmann H, Norcliffe-Kaufmann L, Palma JA, Biaggioni I, Low PA, Singer W, et al. Natural history of pure autonomic failure: a United States prospective cohort. Ann Neurol. (2017) 81:287–97. doi: 10.1002/ana.24877
15. Singer W, Berini SE, Sandroni P, Fealey RD, Coon EA, Suarez MD, et al. Pure autonomic failure: predictors of conversion to clinical CNS involvement. Neurology. (2017) 88:1129–36. doi: 10.1212/WNL.0000000000003737
16. Giannini G, Calandra-Buonaura G, Asioli GM, Cecere A, Barletta G, Mignani F, et al. The natural history of idiopathic autonomic failure: the IAF-BO cohort study. Neurology. (2018) 91:e1245–54. doi: 10.1212/WNL.0000000000006243
17. Plazzi G, Corsini R, Provini F, Pierangeli G, Martinelli P, Montagna P, et al. REM sleep behavior disorders in multiple system atrophy. Neurology. (1997) 48:1094–7. doi: 10.1212/WNL.48.4.1094
18. Tachibana N, Kimura K, Kitajima K, Shinde A, Kimura J, Shibasaki H. REM sleep motor dysfunction in multiple system atrophy: with special emphasis on sleep talk as its early clinical manifestation. J Neurol Neurosurg Psychiatry. (1997) 63:678–81. doi: 10.1136/jnnp.63.5.678
19. Iranzo A, Santamaria J, Tolosa E. Continuous positive air pressure eliminates nocturnal stridor in multiple system atrophy. Barcelona Multiple System Atrophy Study Group. Lancet. (2000) 356:1329–30. doi: 10.1016/S0140-6736(00)02824-5
20. Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord. (2000) 15:699–704. doi: 10.1002/1531-8257(200007)15:4<699::AID-MDS1015>3.0.CO;2-L
21. Wetter TC, Collado-Seidel V, Pollmacher T, Yassouridis A, Trenkwalder C. Sleep and periodic leg movement patterns in drug-free patients with Parkinson's disease and multiple system atrophy. Sleep. (2000) 23:361–7. doi: 10.1093/sleep/23.3.1c
22. Boeve BF, Silber MH, Ferman TJ, Lucas JA, Parisi JE. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. (2001) 16:622–30. doi: 10.1002/mds.1120
23. Vetrugno R, Provini F, Cortelli P, Plazzi G, Lotti EM, Pierangeli G, et al. Sleep disorders in multiple system atrophy: a correlative video-polysomnographic study. Sleep Med. (2004) 5:21–30. doi: 10.1016/j.sleep.2003.07.002
24. Schmeichel AM, Buchhalter LC, Low PA, Parisi JE, Boeve BW, Sandroni P, et al. Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy. Neurology. (2008) 70:368–73. doi: 10.1212/01.wnl.0000298691.71637.96
25. De Cock VC, Debs R, Oudiette D, Leu S, Radji F, Tiberge M, et al. The improvement of movement and speech during rapid eye movement sleep behaviour disorder in multiple system atrophy. Brain. (2011) 134:856–62. doi: 10.1093/brain/awq379
26. Nomura T, Inoue Y, Hogl B, Uemura Y, Yasui K, Sasai T, et al. Comparison of the clinical features of rapid eye movement sleep behavior disorder in patients with Parkinson's disease and multiple system atrophy. Psychiatry Clin Neurosci. (2011) 65:264–71. doi: 10.1111/j.1440-1819.2011.02201.x
27. Muntean ML, Sixel-Döring F, Trenkwalder C. No difference in sleep and RBD between different types of patients with multiple system atrophy: a pilot video- Polysomnographical study. Sleep Disord. (2013) 2013:258390. doi: 10.1155/2013/258390
28. Guo XY, Cao B, Lei F, Huang L, Chen K, Song W, et al. Clinical and polysomnographic features of patients with multiple system atrophy in Southwest China. Sleep Breath. (2013) 17:1301–7. doi: 10.1007/s11325-013-0839-y
29. Stanzani-Maserati M, Gallassi R, Calandra-Buonaura G, Alessandria M, Oppi F, Poda R, et al. Cognitive and sleep features of multiple system atrophy: review and prospective study. Eur Neurol. (2014) 72:349–59. doi: 10.1159/000364903
30. Palma JA, Fernandez-Cordon C, Coon EA, Low PA, Miglis MG, Jaradeh S, et al. Prevalence of REM sleep behavior disorder in multiple system atrophy: a multicenter study and meta-analysis. Clin Auton Res. (2015) 25:69–75. doi: 10.1007/s10286-015-0279-9
31. Coon EA, Sletten DM, Suarez MD, Mandrekar JN, Ahlskog JE, Bower JH, et al. Clinical features and autonomic testing predict survival in multiple system atrophy. Brain. (2015) 138:3623–31. doi: 10.1093/brain/awv274
32. Giannini G, Mastrangelo V, Provini F, Droghini A, Cecere A, Barletta G, et al. Progression and prognosis in multiple system atrophy presenting with REM behavior disorder. Neurology. (2020) 94:e1828–34. doi: 10.1212/WNL.0000000000009372
33. Wang H, An R, Chen Y, Mu X, Yang B, Zhao Q, et al. Clinical features of multiple system atrophy with or without rapid eye movement behavior disorder: a cross-sectional study in southwest China. Clin Auton Res. (2020) 30:239–45. doi: 10.1007/s10286-019-00651-7
34. Iranzo A, Santamaría J, Rye DB, Valldeoriola F, Martí MJ, Muñoz E, et al. Characteristics of idiopathic REM sleep behaviour disorder and that associated with MSA and PD. Neurology. (2005) 65:247–52. doi: 10.1212/01.wnl.0000168864.97813.e0
35. McKay JH, Cheshire WP. First symptoms in multiple system atrophy. Clin Auton Res. (2018) 28:215–21. doi: 10.1007/s10286-017-0500-0
36. De Cock VC, Vidailhet M, Leu S, Texeira A, Apartis E, Elbaz A, et al. Restoration of normal motor control in Parkinson's disease during REM sleep. Brain. (2007) 130:450–6. doi: 10.1093/brain/awl363
37. Tachibana N, Oka Y. Longitudinal change in REM sleep components in a patient with multiple system atrophy associated with REM sleep behavior disorder: paradoxical improvement of nocturnal behaviors in a progressive neurodegenerative disease. Sleep Med. (2004) 5:155–8. doi: 10.1016/j.sleep.2003.09.007
38. Vetrugno R, Alessandria M, D'Angelo R, Plazzi G, Provini F, Cortelli P, et al. Status dissociatus evolving from REM sleep behaviour disorder in multiple system atrophy. Sleep Med. (2009) 10:247–52. doi: 10.1016/j.sleep.2008.01.009
39. Kang SH, Yoon IY, Lee SD, Han JW, Kim TH, Kim KW. REM sleep behavior disorder in the Korean elderly population: prevalence and clinical characteristics. Sleep. (2013) 36:1147–52. doi: 10.5665/sleep.2874
40. Pujol M, Pujol J, Alonso T, Fuentes A, Pallerola M, Freixenet J, et al. Idiopathic REM sleep behavior disorder in the elderly Spanish community: a primary care center study with a two stage design using video-polysomnography. Sleep Med. (2017) 40:116–21. doi: 10.1016/j.sleep.2017.07.021
41. Haba-Rubio J, Frauscher B, Marques-Vidal P, Toriel J, Tobback N, Andries D, et al. Prevalence and determinants of REM sleep behavior disorder in the general population. Sleep. (2018) 41:zsx197. doi: 10.1093/sleep/zsx197
42. Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology. (1996) 46:388–93. doi: 10.1212/WNL.46.2.388
43. Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med. (2013) 14:744–8. doi: 10.1016/j.sleep.2012.10.009
44. Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. (2009) 72:1296–300. doi: 10.1212/01.wnl.0000340980.19702.6e
45. Postuma RB, Gagnon JF, Vendette M, Desjardins C, Montplaisir JY. Olfaction and color vision identify impending neurodegeneration in rapid eye movement sleep behavior disorder. Ann Neurol. (2011) 69:811–8. doi: 10.1002/ana.22282
46. Postuma RB, Lang AE, Gagnon JF, Pelletier A, Montplaisir JY. How does parkinsonism start? Prodromal parkinsonism motor changes in idiopathic REM sleep behaviour disorder. Brain. (2012) 135:1860–70. doi: 10.1093/brain/aws093
47. Postuma RB, Gagnon JF, Bertrand JA, Genier Marchand D, Montplaisir JY. Parkinson risk in idiopathic REM sleep behavior disorder: preparing for neuroprotective trials. Neurology. (2015) 84:1104–13. doi: 10.1212/WNL.0000000000001364
48. Fereshtehnejad SM, Yao C, Pelletier A, Montplaisir JY, Gagnon JF, Postuma RB. Evolution of prodromal Parkinson's disease and dementia with Lewy bodies: a prospective study. Brain. (2019) 142:2051–67. doi: 10.1093/brain/awz111
49. Iranzo A, Molinuevo JL, Santamaría J, Serradell M, Martí MJ, Valldeoriola F, et al. Rapid-eye-movement sleep behaviour disorder as an early marker for a neurodegenerative disorder: a descriptive study. Lancet Neurol. (2006) 5:572–7. doi: 10.1016/S1474-4422(06)70476-8
50. Iranzo A, Lomeña F, Stockner H, Valldeoriola F, Vilaseca I, Salamero M, et al. Decreased striatal dopamine transporter uptake and substantia nigra hyperechogenicity as risk markers of synucleinopathy in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol. (2010) 9:1070–7. doi: 10.1016/S1474-4422(10)70216-7
51. Iranzo A, Tolosa E, Gelpi E, Molinuevo JL, Valldeoriola F, Serradell M, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol. (2013) 12:443–53. doi: 10.1016/S1474-4422(13)70056-5
52. Iranzo A, Fernandez-Arcos A, Tolosa E, Serradell M, Molinuevo JL, Valldeoriola F, et al. Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PLoS ONE. (2014) 9:e89741. doi: 10.1371/journal.pone.0089741
53. Fernandez-Arcos A, Iranzo A, Serradell M, Gaig C, Santamaria J. The clinical phenotype of idiopathic rapid eye movement sleep behavior disorder at presentation: a study in 203 consecutive patients. Sleep. (2016) 39:121–32. doi: 10.5665/sleep.5332
54. Postuma RB, Iranzo A, Hogl B, Arnulf I, Ferini-Strambi L, Manni R, et al. Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: a multicenter study. Ann Neurol. (2015) 77:830–9. doi: 10.1002/ana.24385
55. Postuma RB, Iranzo A, Hu M, Högl B, Boeve BF, Manni R, et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain. (2019) 142:744–59. doi: 10.1093/brain/awz030
56. Fantini ML, Farini E, Ortelli P, Zucconi M, Manconi M, Cappa S, et al. Longitudinal study of cognitive function in idiopathic REM sleep behavior disorder. Sleep. (2011) 34:619–25.
57. Boot BP, Boeve BF, Roberts RO, Ferman TJ, Geda YE, Pankratz VS, et al. Probable rapid eye movement sleep behavior disorder increases risk for mild cognitive impairment and Parkinson disease: a population-based study. Ann Neurol. (2012) 71:49–56. doi: 10.1002/ana.22655
58. Wing YK, Li SX, Mok V, Lam SP, Tsoh J, Chan A, et al. Prospective outcome of rapid eye movement sleep behaviour disorder: psychiatric disorders as a potential early marker of Parkinson's disease. J Neurol Neurosurg Psychiatry. (2012) 83:470–72. doi: 10.1136/jnnp-2011-301232
59. Arnulf I, Neutel D, Herlin B, Golmard JL, Leu-Semenescu S, Cochen de Cock V, et al. Sleepiness in idiopathic REM sleep behavior disorder and Parkinson disease. Sleep. (2015) 38:1529–35. doi: 10.5665/sleep.5040
60. Mahlknecht P, Iranzo A, Hogl B, Frauscher B, Muller C, Santamaria J, et al. Olfactory dysfunction predicts early transition to a Lewy body disease in idiopathic RBD. Neurology. (2015) 84:654–8. doi: 10.1212/WNL.0000000000001265
61. Youn S, Kim T, Yoon IY, Jeong J, Kim HY, Han JW, et al. Progression of cognitive impairments in idiopathic REM sleep behaviour disorder. J Neurol Neurosurg Psychiatry. (2016) 87:890–6. doi: 10.1136/jnnp-2015-311437
62. Li Y, Kang WK, Yang Q, Zhang L, Zhang L, Dong F, et al. Predictive markers for early conversion of IRBD to neurodegenerative synucleinopathy diseases. Neurology. (2017) 88:1493–500. doi: 10.1212/WNL.0000000000003838
63. Zhou J, Zhang J, Lam SP, Chan JW, Mok V, Chan A, et al. Excessive daytime sleepiness predicts neurodegeneration in idiopathic REM sleep behavior disorder. Sleep. (2017) 40:zsx041. doi: 10.1093/sleep/zsx041
64. Claassen DO, Josephs KA, Ahlskog JE, Silber MH, Tippmann-Peikert M, Boeve BF. REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology. (2010) 75:494–9. doi: 10.1212/WNL.0b013e3181ec7fac
65. Plazzi G, Cortelli P, Montagna P, De Monte A, Corsini R, Contin M, et al. REM sleep behaviour disorder differentiates pure autonomic failure from multiple system atrophy with autonomic failure. J Neurol Neurosurg Psychiatry. (1998) 64:683–5. doi: 10.1136/jnnp.64.5.683
66. Weyer A, Minnerop M, Abele M, Klockgether T. REM sleep behavioral disorder in pure autonomic failure (PAF). Neurology. (2006) 66:608–9. doi: 10.1212/01.WNL.0000198251.94422.F3
67. Kashihara K, Ohno M, Kawada S, Imamura T. Frequent nocturnal vocalization in pure autonomic failure. J Int Med Res. (2008) 36:489–95. doi: 10.1177/147323000803600313
68. Miglis MG, Muppidi S, During E, Jaradeh S. A case series of REM sleep behavior disorder in pure autonomic failure. Clin Auton Res. (2017) 27:41–4. doi: 10.1007/s10286-016-0386-2
69. Osaki Y, Ben-Shlomo Y, Lees AJ, Wenning GK, Quinn NP. A validation exercise on the new consensus criteria for multiple system atrophy. Mov Disord. (2009) 24:2272–6. doi: 10.1002/mds.22826
70. Miki Y, Foti SC, Asi YT, Tsushima E, Quinn N, Ling H, et al. Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study. Brain. (2019) 142:2813–27. doi: 10.1093/brain/awz189
71. Stankovic I, Quinn N, Vignatelli L, Antonini A, Berg D, Coon E, et al. Movement Disorder Society Multiple System Atrophy Study Group. A critique of the second consensus criteria for multiple system atrophy. Mov Disord. (2019) 34:975–84. doi: 10.1002/mds.27701
72. Benarroch EE. Brainstem integration of arousal, sleep, cardiovascular, and respiratory control. Neurology. (2018) 91:958–66. doi: 10.1212/WNL.0000000000006537
Keywords: disease progression, sleep disorders, phenoconversion, prodromic phase, review, autonomic failure, REM sleep behaviour disorder, multiple system atrophy
Citation: Giannini G, Provini F, Cortelli P and Calandra-Buonaura G (2021) REM Sleep Behaviour Disorder in Multiple System Atrophy: From Prodromal to Progression of Disease. Front. Neurol. 12:677213. doi: 10.3389/fneur.2021.677213
Received: 07 March 2021; Accepted: 04 May 2021;
Published: 14 June 2021.
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Phillip Low, Mayo Clinic, United StatesCopyright © 2021 Giannini, Provini, Cortelli and Calandra-Buonaura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanna Calandra-Buonaura, Z2lvdmFubmEuY2FsYW5kcmFAdW5pYm8uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.