 Hongyan Bi1*
Hongyan Bi1* Hui Guo2Qianfei Wang3Xiao Zhang1Yaming Zhao1Jimei Li1Weiqin Zhao1Houzhen Tuo1Yongbo Zhang1
Hui Guo2Qianfei Wang3Xiao Zhang1Yaming Zhao1Jimei Li1Weiqin Zhao1Houzhen Tuo1Yongbo Zhang1- 1Department of Neurology, Beijing Friendship Hospital, Capital Medical University, Beijing, China
- 2Center for Medical Genetics and Hunan Key Laboratory of Medical Genetics, School of Life Sciences, Central South University, Changsha, China
- 3CAS Key Laboratory of Genomic and Precision Medicine, Collaborative Innovation Center of Genetics and Development, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China
Background: Bilateral striatal necrosis (BSN) is characterized by symmetrical degeneration, predominantly of the caudate and putamen nucleus, in the basal ganglia. It is associated with numerous acquired and hereditary neuro-developmental and motor dysfunction-related pathological conditions. BSN results in high morbidity and mortality among infants and children, and its diagnosis is clinically challenging due to several overlapping disease phenotypes. Therefore, a precise genetic diagnosis is urgently needed for accurate genetic counseling and improved prognostic outcomes as well.
Objective: To identify novel missense mutations in the NDUFAF5 gene as a cause of childhood BSN in members of a Chinese family and summarize the clinical characteristics of patients with the NDUFAF5 gene mutations.
Methods: This study included a large family living in a remote northwestern area of China. Three siblings developed a neurological disorder characterized by generalized dystonia within the first decade of their lives. Cerebral computed tomography (CT) and magnetic resonance imaging (MRI) showed bilateral lesions of the putamen. Biochemical and genetic approaches were used to identify the cause of BSN.
Results: Sequence analysis showed no pathogenic variation in PANK2, SLC25A19, SLC19A3, and NUP62 genes and in the entire mitochondrial genome as well. Whole-exome sequencing revealed compound heterozygous mutations consisting of NDUFAF5:c.425A > C(p.E142A) and c.836T > G (p.M279R). The father, a healthy sister, and a healthy brother of the affected siblings carried the c.836T > G mutation, and the mother carried the c.425A > C mutation. These variants were absent in 100 ethnically matched non-BSN controls. In silico analysis demonstrated that the E142A and M279R mutations in NDUFAF5 protein significantly perturbed the normal conformation of the protein due to alterations in the hydrogen bonding patterns around the evolutionarily conserved catalytic domains, leading to its loss of function in the early stage of mitochondrial complex I assembly.
Conclusions: We identified a novel compound heterozygous mutation (c.425A > C and c.836T > G) in the NDUFAF5 gene as the potential cause of autosomal recessive childhood BSN, which extended the pathogenic variation spectrum of the NDUFAF5 gene. This study provides substantial evidence for further improvement of genetic counseling and better clinical management of BSN affected individuals.
Introduction
Bilateral striatal necrosis (BSN) represents a rare form of neurological disorders involving neostriata and is characterized by initial swelling of the dorsal striatum and neostriatum of the basal ganglia, especially the putamen and caudate nucleus, followed by progressive degeneration and necrosis of neuronal cells (1). It can be familial or sporadic in origin. BSN pathology may also involve globus pallidus, tegmental nucleus, and substantia nigra brain regions under various diseased conditions (2). The most commonly associated pyramidal symptoms include spasticity, hyperreflexia, and weakness primarily related to upper motor neuron degeneration, leading to developmental regression or cognitive as well as motor deficits (3).
Diverse pathogenic mutations in genes related to neuronal maturation, nuclear transport and mitochondrial respiratory system as well as severe biochemical defects have been implicated in various familial BSN pathogenesis, e.g., infantile autosomal recessive BSN-associated missense mutation (Q391P) in nucleoporin-62 (NUP62) (4); homozygous missense mutation c.373G > A in the SLC25A19 gene encoding mitochondrial thiamine pyrophosphate transporter (5); SLC25A19 gene mutation in pediatric recessive biotin-responsive basal ganglia disorder (6); homoplasmic mitochondrial 3697G > A mutation and DNA deletions (7); mitochondrial adenosine triphosphatase 6 (ATP6) gene mutations at 9176T > C and 8851T > C with symptoms resembling Leigh syndrome (8); childhood organic acidurias with parkinsonian rigidity (9); and Wilson's disease-like pathology visible in MRI as striatal necrosis in basal ganglia and atrophy (10), etc. Inherited forms of BSN manifest as dystonia, parkinsonian rigidity, eye movement abnormalities, chorea and athetosis overlap syndrome, myoclonus, seizures, and psychomotor retardation (11).
Biochemical and metabolic abnormalities in mitochondrial complex I have been frequently implicated in Leigh syndrome and BSN (12). Several pathogenic gene mutations have been identified in the components of complex I that are involved in ATP production and mitochondrial electrochemical gradient maintenance (13). Many of the NADH: Ubiquinone Oxidoreductase complex assembly factor (NDUFAF) family proteins act as the putative assembly factor for complex I, and their genetic mutations have been correlated with neurological disorders, including Leigh syndrome (14). Human NDUFAF5 (C20orf7) (MIM No.*612360), located in 20p12.1, encodes an important mitochondrial complex I assembly factor, arginine hydroxylase, which is imported from the nucleus into the mitochondrial inner membrane matrix. NDUFAF5 has been shown to hydroxylate NDUFS7 at Arg-73 at an early stage of mitochondrial complex I assembly (15). Biallelic pathogenic variants in NDUFAF5 cause a rare type of autosomal recessive BSN, namely mitochondrial complex I deficiency, nuclear type 16 (MC1DN16, MIM No. #618238) (https://www.omim.org/entry/612360) and neonatal mitochondrial disease (16).
Here, we demonstrate a previously unknown compound mutation in NDUFAF5 identified by whole-exome sequencing (WES) associates with a rare form of isolated BSN in childhood. Furthermore, with the help of protein structure prediction tools, we have investigated the implications of these missense mutations in functional modulations of NDUFAF5. Therefore, our findings highlight the importance of using an integrated approach that combines clinical evaluation with exome data analysis to reveal NDUFAF5 pathogenic mutation-associated disease mechanisms in lethal neuro-developmental diseases like BSN.
Materials and Methods
Subjects
This case study included 7 members of a family living in the northwest of China. The proband and two of her sisters were affected by BSN symptoms. This study was approved by the Ethics Committee of Beijing Friendship Hospital, Capital Medical University.
Brain MRI and CT Scans
Brain MRI examination was conducted using a Discovery MR750 3.0T scanner. Conventional T1-weighted spin echo (T1WI) sequences were obtained with the following parameters: repetition time (TR) = 250 ms, echo time (TE) = 2.5 ms, FoV = 306 × 220 mm; parameters for conventional T2-weighted spin echo (T2WI) sequences: TR = 4,000 ms, TE = 157 ms, FoV = 306 × 220 mm. The slice thickness and the slice gap were 5 mm and 6.5 mm, respectively.
Genetic Analysis
DNA Extraction
Blood samples were collected from all family members of the proband. Total DNA was extracted from the peripheral blood leukocytes using Blood and Tissue DNA isolation kit (Qiagen, Germany). The left biceps brachii muscle of the proband was biopsied, and the frozen sample was used for diagnostic studies.
Polymerase Chain Reaction (PCR) and Direct DNA Sequencing
Exonic and flanking intronic regions of nuclear genes NUP62, SLC19A3, SLC25A19, and PANK2 and all mitochondrial genes including MT-ATP6, MT-ND1, MT-ND3, MT-ND4, and MT-ND6 were amplified using specific primers (sequences available on request). Afterward, the coding regions and exon-intron junctions of these genes were analyzed by Sanger sequencing.
Whole-Exome Sequencing (WES) and Data Analysis
Exon-enrichment was carried out using SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA, USA). The exon-enriched DNA libraries were subjected to paired-end sequencing with the HiSeq 2500 system (Illumina, San Diego, CA, USA). Raw data were filtered and processed using CASAVA v1.8 (Illumina). Calls with variant quality <20 were filtered out to obtain clean reads, and at least 95% of the targeted nucleotides were covered sufficiently to pass the thresholds for calling single-nucleotide mutations (SNPs) and small insertion-deletion (InDel) sequences. The sequencing reads were aligned to the human reference genome (Vhg19) using the Burrows-Wheeler Aligner (BWA), and PCR duplicates were removed using the Picard 1.27–1 software (http://picard.sourceforge.net/). A pipeline incorporating MuTect (Broad Institute) to identify the single-nucleotide variants and ANNOVAR (17) was employed for variant calling. FASTQ, BAM, and variant call format (VCF) files were then used for sequence analysis. The minor allele frequency (MAF) was analyzed using the 1000 Genomes Project database, SNP database (dbSNP), NHLBI exome sequencing project (ESP) (http://evs.gs.washington.edu/EVS/), and Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org). Non-synonymous, loss-of-function, indel, duplication, and splicing variants were used to identify candidate variants. Protein biological function was predicted using the ConSurf (http://consurf.tau.ac.il) (18), Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi), SIFT (http://sift.bii.a-star.edu.sg) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) servers.
Variant Confirmation by Sanger Sequencing
To confirm that the identified variants were responsible for BSN, we screened the 7 members in this family by Sanger sequencing using ABI 3500 system (Thermo Fisher, USA). Additionally, we analyzed 100 control specimens from our in-house sample library (obtained from individuals without BSN) to exclude polymorphisms.
Molecular Modeling
Protein structure modeling was conducted using SWISS-MODEL with default parameters based on the available homologous models indexed in PDB database (https://swissmodel.expasy.org/). NDUFAF5 mutations were predicted using SWISS-MODEL (PDB: 2p35.1.A; Modeling GMQE score of 0.28, Modeling QMEAN score of 0.28–5.41; Seq Identity of 17.19%).
Results
Characteristics of the Participants
Figure 1A exhibits the pedigree diagram of the participating subjects in this study as the proband (II-2), her father (I-1), mother (I-2), three sisters (II-1, II-3, and II-4), and one brother (II-5). BSN was diagnosed in the proband (II-2) at her early childhood and 2 of her sibling sisters (II-1 and II-3). The disease phenotypes included varying degrees of dystonic posture and movement disorders, dysarthric speech, dysphagia, and vision loss. The auditory and cognitive functions of the subjects were tested normal.
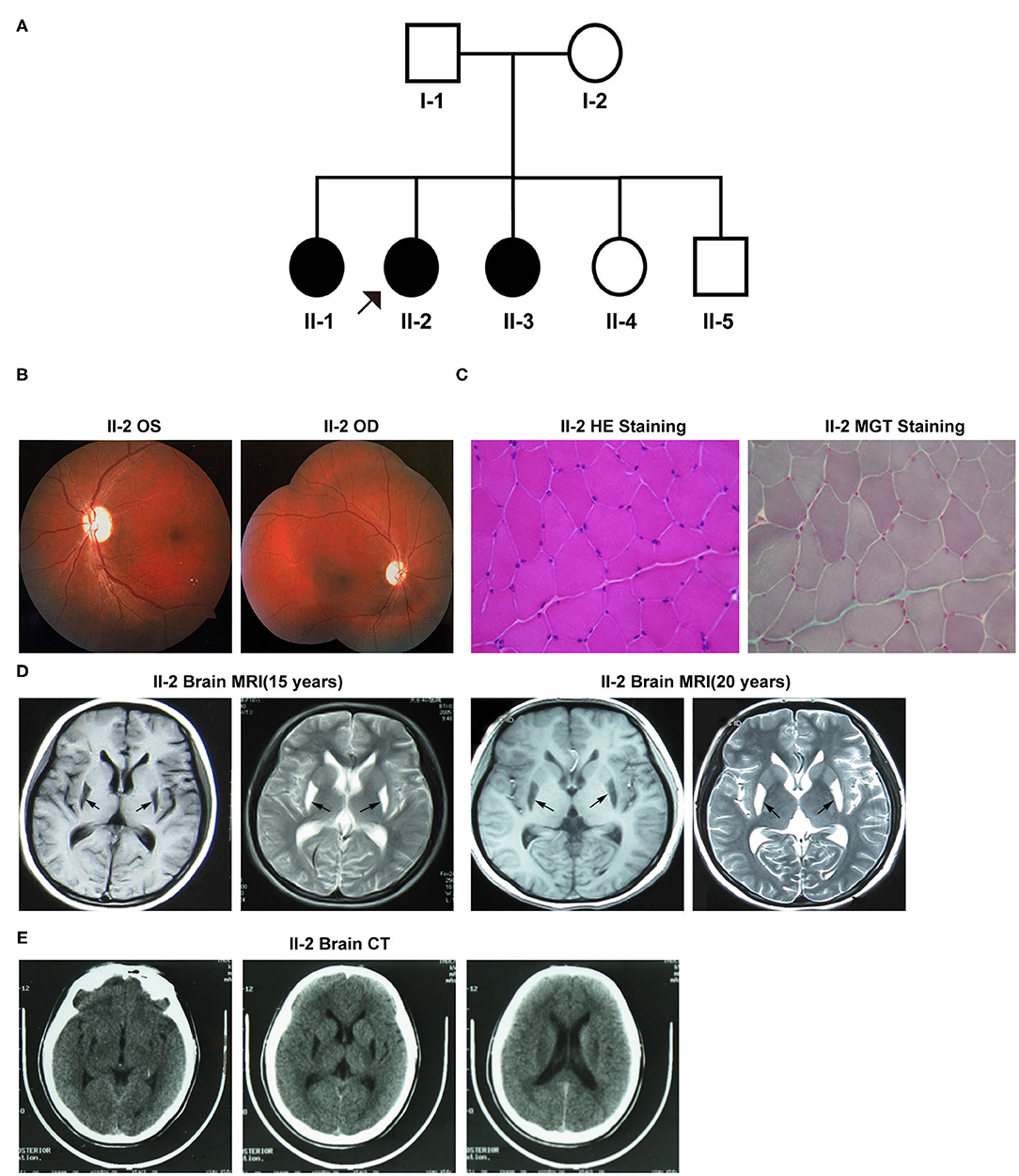
Figure 1. (A) The pedigree diagram of the family included in this study. (B–E) Clinical manifestations of the proband (II-2). (B) Fundus fluorescein angiography. (C) Muscle biopsy with specific staining. (D) Brain MRI at 15 and 20 years old. (E) Brain CT. OS, oculus sinister; OD, oculus dextrus; HE, hematoxylin-eosin; MGT, modified Gömöri trichrome.
Patient II-2
Patient II-2 (the proband), the second child of her parents, was 20 years old at the first evaluation at our hospital. At the age of six, she started to exhibit the first onset of symptoms like gait difficulties, spasticity, and dysarthria aggravated by emotional stress at times, and the symptoms were progressively exacerbated with age. At her 13-years, she developed visual impairment due to optic nerve atrophy that lasted for more than 10 days. During her late adolescence (18 years of age), she exhibited the flexed dystonic postures that included inward rotation of legs and bilateral inversion of the foot, followed by dysphagia, dystonic movements, and decreased fine motor abilities. Neurological examinations revealed severe dystonic movements of the facial and trunk muscles, dysarthric speech, and other common pathological signs of BSN. A visual assessment revealed vision loss in both eyes-the visual acuity was 0.25D in the left eye and “counting fingers” in the right eye. Fundus fluorescein angiography showed bilateral pallor of the papilla in the retina (Figure 1B). Moreover, her muscle biopsy examination result was unremarkable for any pathological alterations in the structure of muscle cells (Figure 1C). However, her mitochondrial complex I NADH dehydrogenase activity was detected 86.8 nmol/min/mg mitochondrial protein (ref-range, ≥76.6 nmol/min/mg), which was much lower than that of her mother (129.5 nmol/l/min/mg mitochondrial protein). Brain MRI results at 15 and 20 years of age revealed abnormal signals with hypointense lesions on T1-weighted images and hyperintense lesions on T2-weighted images in the putamen of basal ganglia in the posterior region (Figure 1D). Both MRI and CT scans did not detect any signs of apparent calcifications in and around the affected brain regions (Figure 1E). Visual evoked potentials (VEPs) showed prolongation of the bilateral P100 latency, especially in the right eye, indicative of the demyelination of the optic nerve.
Patient II-1
The proband's elder sister was 23 years old at the time of her first diagnosis at the patient's home. She was first presented with walking impairment and dysarthric speech expression at 6 years of age. Progressive worsening of these pathological conditions led to the complete loss of autonomic gait and speech performances. Notably, she exhibited the most severe clinical symptoms of BSN amongst her family members. Neurological examinations revealed diffuse muscle atrophy leading to severe spastic quadriplegia and marked dystonic postures involving both the upper and lower limbs (Figure 2A).
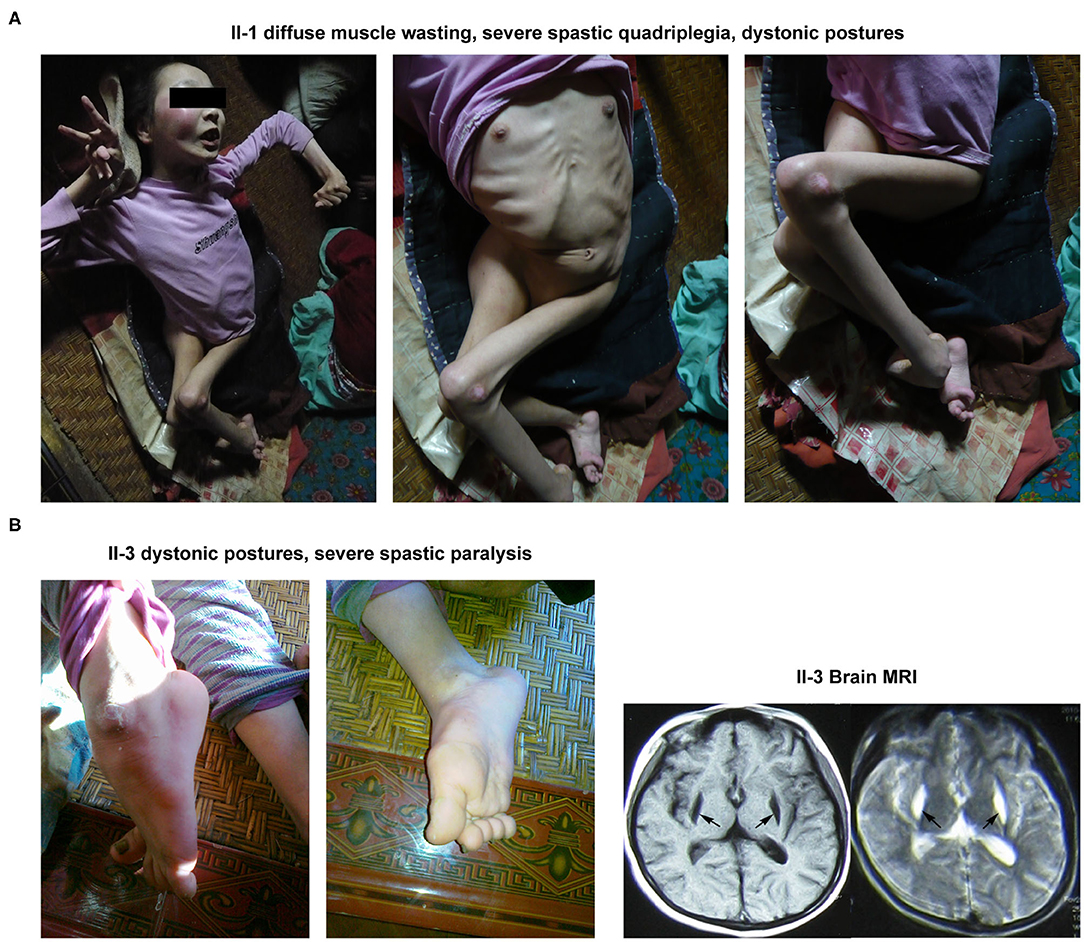
Figure 2. Clinical manifestations of the other 2 patients (II-1 and II-3) in this family. (A) The abnormal appearance of II-1. (B) The dystonic posture in lower limbs and brain MRI result of II-3.
Patient II-3
The younger sister of the proband was 18 years old during her first clinical examinations at her home. The patient had experienced 3 occasions of febrile convulsions between the age of 1 and 3. She began to display walking impairment and dysarthric speech expressions at the age of 5. At the time of clinical examinations, the patient was mostly bedridden and needed complete physical assistance with most of the daily motor performances, including walking. Neuromuscular examinations demonstrated dystonic postures involving both the upper and lower limbs as well as severe spastic paralysis of the right upper limb. Notably, her brain MRI examination revealed abnormal signals in the putamen (Figure 2B).
Genetic Analysis
Our WES data provided a coverage depth of more than 99.60 × and an average mapping rate of 99.91%, which were sufficient to support our analysis, according to the previously published reports (19). Mutational analysis of these samples revealed a novel compound heterozygous variant in the NDUFAF5 gene consisting of c. 425A > C (p.E142A) and c.836T > G (p.M279R) in 3 patients, suggesting the pathogenic role of NDUFAF5 missense mutations in the childhood onset of BSN pathology. Furthermore, subjects I-1, II-4, and II-5 exhibited the presence of heterozygous NDUFAF5 c.836T > G mutation, while subject I-2 carried the heterozygous c.425A > C mutation (Figure 3A). Furthermore, we matched this genomic sequence with 100 other unrelated healthy controls and concluded that these mutations were precisely related to BSN. Taken together, this study demonstrated that synergistic effects of two independent non-pathogenic paternal and maternal single-nucleotide mutations in the NDUFAF5 gene could lead to an aggressive novel compound mutation initiating BSN pathogenesis at early childhood.
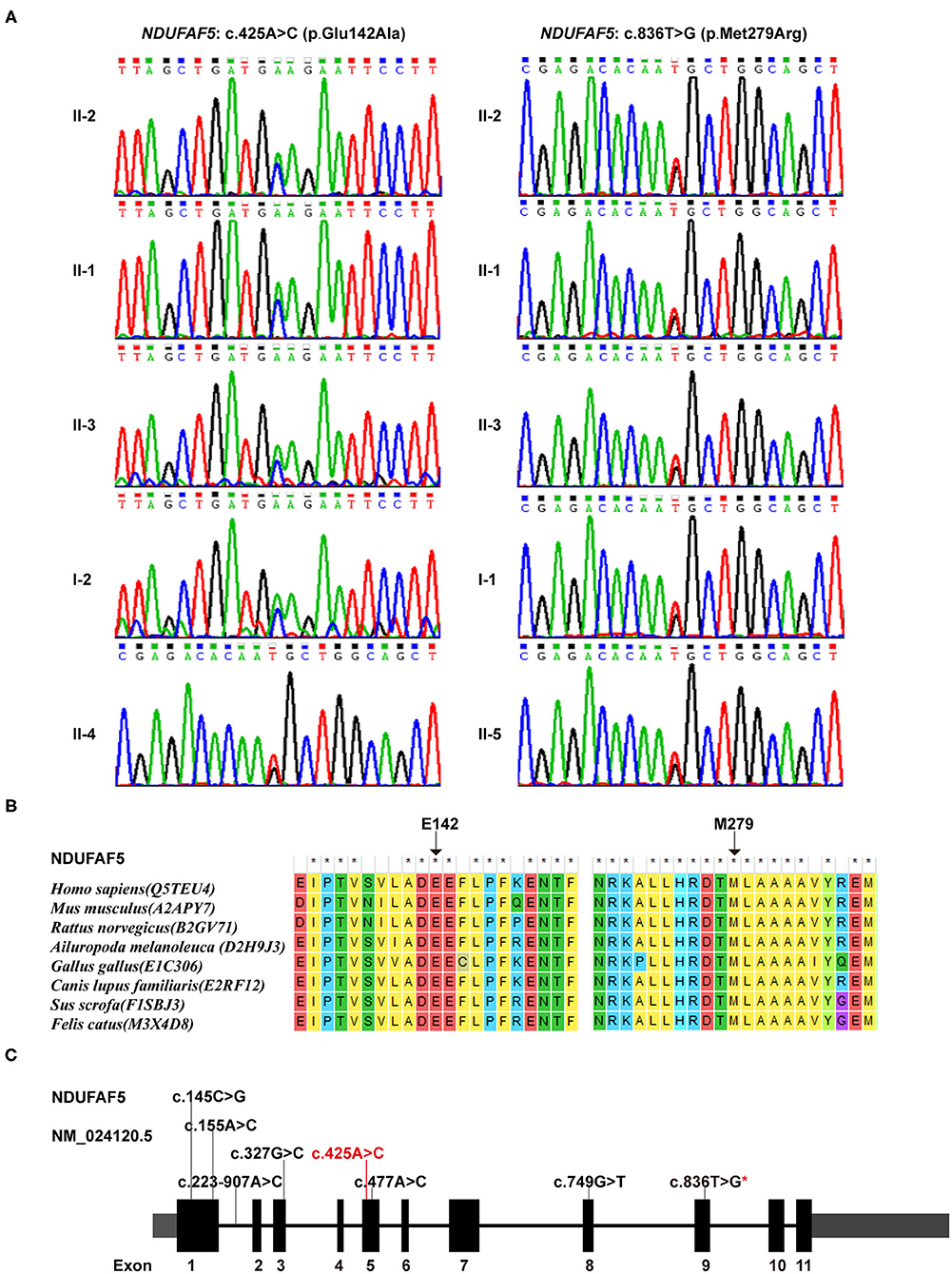
Figure 3. Genetic findings in this study. (A) Sequence variants and carrying status of each member in this family. (B) Conservatism of the 2 corresponding amino acids of the 2 missense variants among species. (C) The identified variants in NDUFAF5 gene so far (Red characters represented the variant identified in this study; red star means the one identified both in this study and in other studies).
Impact Prediction of the Variants
Individual impact prediction analysis of the heterozygous variants exhibited that the E142A variant had a disease risk score of 0.965 (sensitivity: 0.78, specificity: 0.95), and that of M279R variant was 0.930 (sensitivity: 0.81, specificity: 0.94). Additionally, analyses using ConSurf server and BLAST software demonstrated that the E142 and M279 residues were evolutionarily conserved among several mammalian species, including human, mouse, and rat (Figure 3B). Thus, the two mutational variants in the NDUFAF5 gene led to two different amino acid substitutions in NDUFAF5 protein affecting the arginine hydroxylase enzymatic activities, leading to childhood BSN pathogenesis.
Prediction of the Effects of the Mutations on Protein Conformation
Since NDUFAF5 forms homodimer, we performed simulated mutations on chain A (wildtype) and B (mutant), and then summed up the effects (Figure 4). Interestingly, E142A mutation significantly perturbed the normal hydrogen bond formation between E142 and G97 residues, leading to a conformational change in the mutant NDUFAF5. In chain A, the distance of the hydrogen bond between E142 and G97 was 3.15 Å, whereas it was absent in the B chain as A142 could not form hydrogen bonds with G97 (top left and top right; Figure 4A). Instead, a new hydrogen bond was formed in the B chain between A142 and D141 of 3.00 Å (lower left and lower right; Figure 4A). In variant M279R, a neutral methionine residue was replaced by a basic arginine moiety. Notably, M279R mutation significantly decreased the number of hydrogen bonds from 3 to 2 in the mutant NDUFAF5, resulting in decreased protein stability (M279, top left and top right, Figure 4B; Arg279: lower left and lower right, Figure 4B). Taken together, these results suggest that the novel missense mutation (E142A) could play a synergistic role in combination with a previously identified missense mutation (M279R) by negatively modulating the crucial enzymatic activities of NDUFAF5 under pathological conditions.
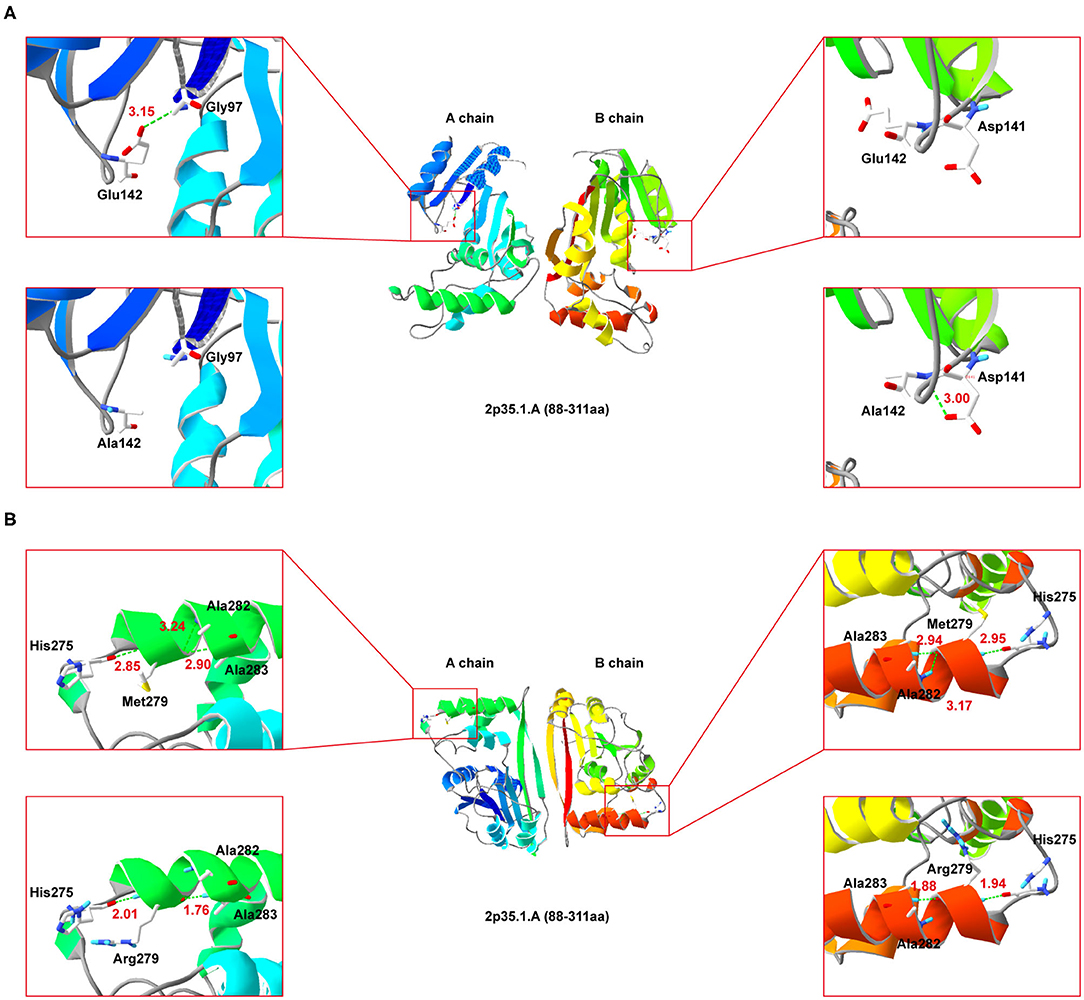
Figure 4. Secondary-structure of NDUFAF5 (residues 88–311) is shown. The intermediate region of (A,B) present a full picture of the structure of the protein dimer. Top left and top right of the (A,B) show magnified view of the outlined region containing the conserved residues. Lower left and lower right of (A,B) show magnified view of the outlined region containing the mutant residues.
Discussion
Here, we investigated a family with three siblings harboring compound heterozygous mutations in the NDUFAF5 gene. The proband and her immediate elder and younger sisters were presented with the symptoms of childhood-onset generalized dystonia caused by BSN. The proband had vision loss due to optic nerve atrophy at her mid-childhood. Brain MRI examinations exhibited bilateral lesions in the putamen that were mainly confined to the posterior region that did not enlarge or regress over a 5-year period. The clinical course and radiological changes observed in these patients were inconsistent with those reported as the most common causes of BSN and related pathogenesis, such as infantile BSN (20), BSN and progressive polyneuropathy (21), biotin-responsive basal ganglia disease (22), Leber's hereditary optic neuropathy and dystonia (23), MT-ATP6-related neuropathy, ataxia and retinitis pigmentosa syndrome (24), Hallervorden–Spatz syndrome (25), and Huntington's disease (10). Using WES, we identified a previously unknown compound heterozygous mutation in NDUFAF5 gene. Furthermore, protein conformational analysis of NDUFAF5 carrying missense mutations E142A and M279R revealed that considerable alterations in its conserved catalytic domains could be the possible reason for its inability to contribute to the normal physiological assembly of mitochondrial complex I subunits, leading to a severe loss of striatal neurons and motor impairments in BSN pathology.
NDUFAF5 protein possesses methyltransferase and arginine hydroxylase activity, which are critical for posttranslational modifications of other mitochondrial complex I assembly factors like NDUFS7 (26). This protein dysfunction has been implicated in mitochondrial complex I functional deficiency-induced fatal infantile neurodevelopmental diseases (27). Moreover, previous studies have shown the pathological relationship of various combinations of missense mutations in NDUFAF5 with familial BSN and related disease onset (28). These studies' main findings are summarized in Table 1, while the novel point-mutation c.425A > C (highlighted in red) is shown in Figure 3C. Notably, eleven of the thirteen patients described in these studies were not alive at the time of publications, with death occurring by the age of 7 years mostly due to cardiac arrest in 10 of the 11 patients, suggesting the aggressive influences of pathogenic NDUFAF5 missense mutations on BSN pathogenesis and subsequent disease progression. RNAi-mediated knockdown of NDUFAF5 in control fibroblasts has exhibited decreased complex I activity, suggesting that the pathological complex I deficiency could have a direct mechanistic relationship with NDUFAF5 mutation-associated dysfunctions. Besides, alternative splicing of exon5 of the NDUFAF5 gene results in the production of shorter isoform under pathological conditions (29).
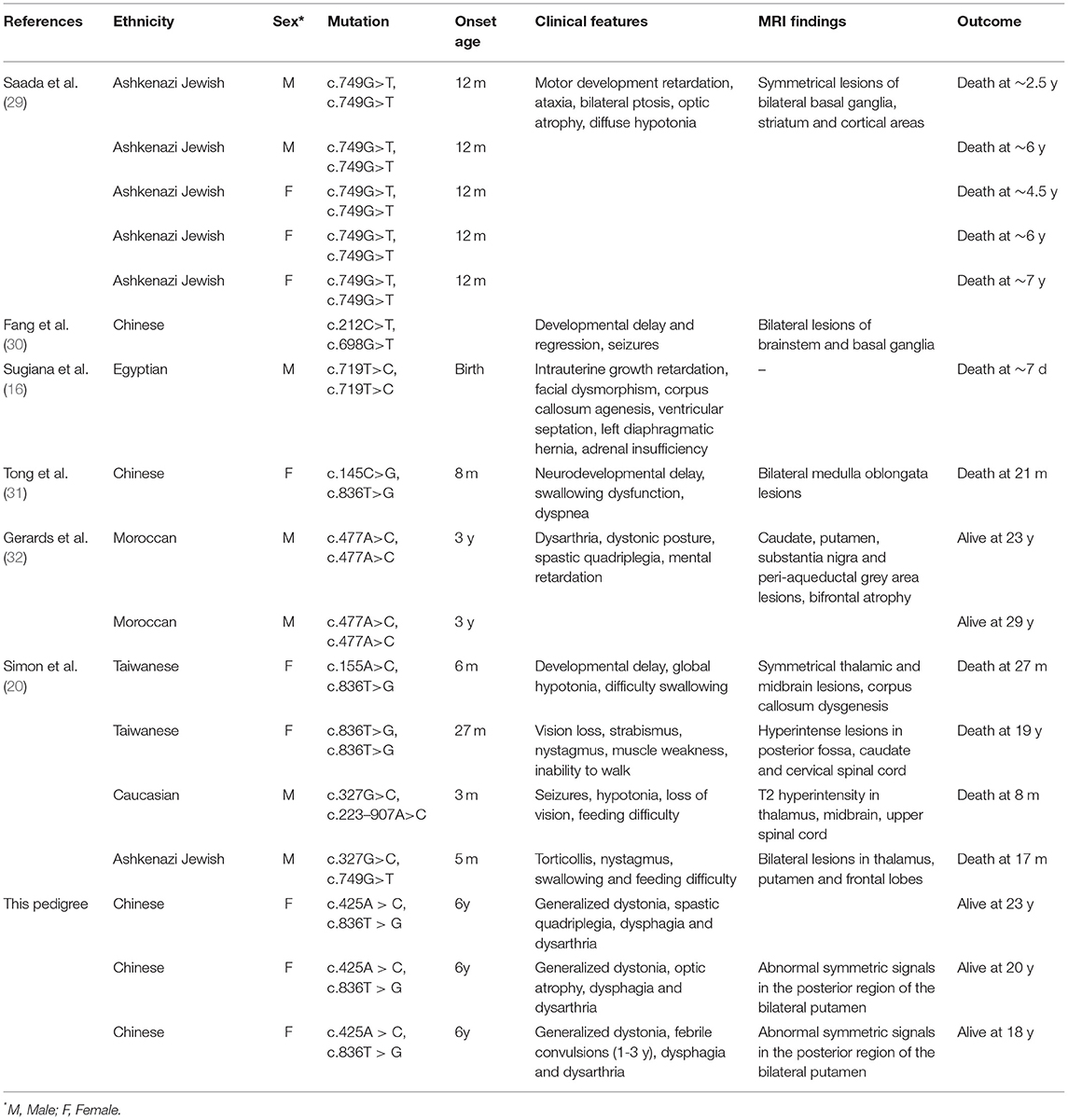
Table 1. Clinical features of patients with NDUFAF5 variations reported in literature.
Therefore, our results expand the genetic and phenotypic spectrum of BSN genetics to better understand the mechanistic implication of these missense mutations in different extents of disease aggressiveness. And the NDUFAF5-related BSN pathology should be included in the differential diagnosis of patients presenting with insidious dystonia in early childhood that progresses from gait disturbances to generalized dystonia with dysphagia and dysarthria.
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: SRA SRR14293161-SRR14293164 and BioSample SAMN18820150-SAMN18820153.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Beijing Friendship Hospital, Capital Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HB designed and supervised the study and wrote the manuscript. HG and QW performed genetic testing and related data analyses. XZ, YZhao, JL, and WZ conducted the subject recruitment, initial medical examination, and iconography testing. HT and YZhan performed detailed examinations. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81300976).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer QW declared a shared affiliation, with no collaboration, with the authors HB, XZ, YZ, JL, WZ, HT, and YZ at the time of the review.
Acknowledgments
We thank the participants for agreeing to take part in this study.
References
1. Tonduti D, Chiapparini L, Moroni I, Ardissone A, Zorzi G, Zibordi F, et al. Neurological disorders associated with striatal lesions: classification and diagnostic approach. Curr Neurol Neurosci Rep. (2016) 16:54. doi: 10.1007/s11910-016-0656-3
2. Zhong S, Wen S, Qiu Y, Yu Y, Xin L, He Y, et al. Bilateral striatal necrosis due to homoplasmic mitochondrial 3697g>a mutation presents with incomplete penetrance and sex bias. Mol Genet Genomic Med. (2019) 7:e541. doi: 10.1002/mgg3.541
3. Basel-Vanagaite L, Muncher L, Straussberg R, Pasmanik-Chor M, Yahav M, Rainshtein L, et al. Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann Neurol. (2006) 60:214–22. doi: 10.1002/ana.20902
4. Kohrogi K, Imagawa E, Muto Y, Hirai K, Migita M, Mitsubuchi H, et al. Biotin-responsive basal ganglia disease: a case diagnosed by whole exome sequencing. J Hum Genet. (2015) 60:381–5. doi: 10.1038/jhg.2015.35
5. Cirillo G, Cirillo M, Panetsos F, Virtuoso APapa M. Selective vulnerability of basal ganglia: insights into the mechanisms of bilateral striatal necrosis. J Neuropathol Exp Neurol. (2019) 78:123–9. doi: 10.1093/jnen/nly123
6. De Meirleir L, Seneca S, Lissens W, Schoentjes E, Desprechins B. Bilateral striatal necrosis with a novel point mutation in the mitochondrial atpase 6 gene. Pediatr Neurol. (1995) 13:242–6. doi: 10.1016/0887-8994(95)00184-H
7. Thyagarajan D, Shanske S, Vazquez-Memije M, De Vivo D, DiMauro S. A novel mitochondrial atpase 6 point mutation in familial bilateral striatal necrosis. Ann Neurol. (1995) 38:468–72. doi: 10.1002/ana.410380321
8. Gascon GG, Ozand PT, Brismar J. Movement disorders in childhood organic acidurias. Clinical, neuroimaging, and biochemical correlations. Brain Dev. (1994) 16(Suppl):94–103. doi: 10.1016/0387-7604(94)90102-3
9. Aniello MS, Martino D, Petruzzella V, Eleopra R, Mancuso M, Dell'Aglio R, et al. Bilateral striatal necrosis, dystonia and multiple mitochondrial DNA deletions: case study and effect of deep brain stimulation. Mov Disord. (2008) 23:114–8. doi: 10.1002/mds.21760
10. Spiegel R, Shaag A, Edvardson S, Mandel H, Stepensky P, Shalev SA, et al. Slc25a19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann Neurol. (2009) 66:419–24. doi: 10.1002/ana.21752
11. Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, et al. The assembly pathway of mitochondrial respiratory chain complex i. Cell Metab. (2017) 25:128–39. doi: 10.1016/j.cmet.2016.09.002
12. Herzer M, Koch J, Prokisch H, Rodenburg R, Rauscher C, Radauer W, et al. Leigh disease with brainstem involvement in complex i deficiency due to assembly factor ndufaf2 defect. Neuropediatrics. (2010) 41:30–4. doi: 10.1055/s-0030-1255062
13. Baertling F, Sánchez-Caballero L, Timal S, van den Brand MA, Ngu LH, Distelmaier F, et al. Mutations in mitochondrial complex i assembly factor ndufaf3 cause leigh syndrome. Mol Genet Metab. (2017) 120:243–6. doi: 10.1016/j.ymgme.2016.12.005
14. Floyd BJ, Wilkerson EM, Veling MT, Minogue CE, Xia C, Beebe ET, et al. Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol Cell. (2016) 63:621–32. doi: 10.1016/j.molcel.2016.06.033
15. Rhein VF, Carroll J, Ding S, Fearnley IM, Walker JE. Ndufaf5 hydroxylates ndufs7 at an early stage in the assembly of human complex i. J Biol Chem. (2016) 291:14851–60. doi: 10.1074/jbc.M116.734970
16. Sugiana C, Pagliarini DJ, McKenzie M, Kirby DM, Salemi R, Abu-Amero KK, et al. Mutation of c20orf7 disrupts complex i assembly and causes lethal neonatal mitochondrial disease. Am J Hum Genet. (2008) 83:468–78. doi: 10.1016/j.ajhg.2008.09.009
17. Wang K, Li M, Hakonarson H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
18. Iyer A, Lapointe NE, Zielke K, Berdynski M, Guzman E, Barczak A, et al. A novel mapt mutation, g55r, in a frontotemporal dementia patient leads to altered tau function. PLoS ONE. (2013) 8:e76409. doi: 10.1371/journal.pone.0076409
19. Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. (2009) 461:272–6. doi: 10.1038/nature08250
20. Simon MT, Eftekharian SS, Stover AE, Osborne AF, Braffman BH, Chang RC, et al. Novel mutations in the mitochondrial complex i assembly gene ndufaf5 reveal heterogeneous phenotypes. Mol Genet Metab. (2019) 126:53–63. doi: 10.1016/j.ymgme.2018.11.001
21. Straussberg R, Shorer Z, Weitz R, Basel L, Kornreich L, Corie CI, et al. Familial infantile bilateral striatal necrosis: clinical features and response to biotin treatment. Neurology. (2002) 59:983–9. doi: 10.1212/WNL.59.7.983
22. Zeng WQ, Al-Yamani E, Acierno JS Jr., Slaugenhaupt S, Gillis T, MacDonald ME, et al. Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in slc19a3. Am J Hum Genet. (2005) 77:16–26. doi: 10.1086/431216
23. De Vries DD, Went LN, Bruyn GW, Scholte HR, Hofstra RM, Bolhuis PA, et al. Genetic and biochemical impairment of mitochondrial complex i activity in a family with leber hereditary optic neuropathy and hereditary spastic dystonia. Am J Hum Genet. (1996) 58:703–11.
24. Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucleotide pair 14459 of the nadh dehydrogenase subunit 6 gene associated with maternally inherited leber hereditary optic neuropathy and dystonia. Proc Natl Acad Sci USA. (1994) 91:6206–10. doi: 10.1073/pnas.91.13.6206
25. Spruijt L, Smeets HJ, Hendrickx A, Bettink-Remeijer MW, Maat-Kievit A, Schoonderwoerd KC, et al. A melas-associated nd1 mutation causing leber hereditary optic neuropathy and spastic dystonia. Arch Neurol. (2007) 64:890–3. doi: 10.1001/archneur.64.6.890
26. Wang K, Takahashi Y, Gao ZL, Wang GX, Chen XW, Goto J, et al. Mitochondrial nd3 as the novel causative gene for leber hereditary optic neuropathy and dystonia. Neurogenetics. (2009) 10:337–45. doi: 10.1007/s10048-009-0194-0
27. Watanabe M, Mita S, Takita T, Goto Y, Uchino M, Imamura S. Leber's hereditary optic neuropathy with dystonia in a japanese family. J Neurol Sci. (2006) 243:31–4. doi: 10.1016/j.jns.2005.11.003
28. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (pank2) is defective in hallervorden-spatz syndrome. Nat Genet. (2001) 28:345–9. doi: 10.1038/ng572
29. Saada A, Edvardson S, Shaag A, Chung WK, Segel R, Miller C, et al. Combined oxphos complex i and iv defect, due to mutated complex i assembly factor c20orf7. J Inherit Metab Dis. (2012) 35:125–31. doi: 10.1007/s10545-011-9348-y
30. Fang F, Shen Y, Shen DM, Liu ZM, Ding CH, Zhang WC, et al. [Clinical and genetic characteristics of children with Leigh syndrome]. Zhonghua er ke za zhi = Chinese J Pediatr. (2017) 55:205–9. doi: 10.3760/cma.j.issn.0578-1310.2017.03.008
31. Tong W, Wang Y, Lu Y, Ye T, Song C, Xu Y, et al. Whole-exome sequencing helps the diagnosis and treatment in children with neurodevelopmental delay accompanied unexplained dyspnea. Sci Rep. (2018) 8:5214.
Keywords: bilateral striatal necrosis, NDUFAF5, mitochondrial complex I deficiency, whole-exome sequencing, novel variation
Citation: Bi H, Guo H, Wang Q, Zhang X, Zhao Y, Li J, Zhao W, Tuo H and Zhang Y (2021) A Novel Variation in the Mitochondrial Complex I Assembly Factor NDUFAF5 Causes Isolated Bilateral Striatal Necrosis in Childhood. Front. Neurol. 12:675616. doi: 10.3389/fneur.2021.675616
Received: 03 March 2021; Accepted: 13 May 2021;
Published: 10 June 2021.
Edited by:
Mathias Toft, University of Oslo, NorwayReviewed by:
Manabu Funayama, Juntendo University, JapanQun Wang, Capital Medical University, China
Copyright © 2021 Bi, Guo, Wang, Zhang, Zhao, Li, Zhao, Tuo and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyan Bi, aHliaTE5OTZAc2luYS5jb20=