
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 13 April 2021
Sec. Epilepsy
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.665334
This article is part of the Research Topic Glial Dysfunction in Epileptogenesis View all 16 articles
 Mani Ratnesh S. Sandhu1†
Mani Ratnesh S. Sandhu1† Benjamin F. Gruenbaum2†Shaun E. Gruenbaum2
Benjamin F. Gruenbaum2†Shaun E. Gruenbaum2 Roni Dhaher3Ketaki Deshpande1
Roni Dhaher3Ketaki Deshpande1 Melissa C. Funaro4Tih-Shih W. Lee5
Melissa C. Funaro4Tih-Shih W. Lee5 Hitten P. Zaveri6
Hitten P. Zaveri6 Tore Eid1*
Tore Eid1*The enzyme glutamine synthetase (GS), also referred to as glutamate ammonia ligase, is abundant in astrocytes and catalyzes the conversion of ammonia and glutamate to glutamine. Deficiency or dysfunction of astrocytic GS in discrete brain regions have been associated with several types of epilepsy, including medically-intractable mesial temporal lobe epilepsy (MTLE), neocortical epilepsies, and glioblastoma-associated epilepsy. Moreover, experimental inhibition or deletion of GS in the entorhinal-hippocampal territory of laboratory animals causes an MTLE-like syndrome characterized by spontaneous, recurrent hippocampal-onset seizures, loss of hippocampal neurons, and in some cases comorbid depressive-like features. The goal of this review is to summarize and discuss the possible roles of astroglial GS in the pathogenesis of epilepsy.
Astrocytes have historically been thought to serve a primarily structural role by supporting surrounding neurons (1). Over the last 40 years, however, a growing body of evidence has suggested that astrocytes serve important roles in normal brain function, and are critical for axonal growth, energy metabolism, neurotransmitter homeostasis and water/electrolyte balance (2–11). Moreover, abnormal astrocyte function has been postulated to contribute to the pathogenesis of a wide range of neurological and psychiatric disorders (12–17).
Following an acute central nervous system (CNS) injury, astrocytes undergo several morphological and functional changes. These “reactive astrocytes” are present in several pathological conditions While reactive astrocytes were originally thought to reflect scar tissue in response to neuronal injury and loss, recent studies have suggested that reactive astrocytes may in fact play important roles in the causation of many disorders, including epilepsy (18–20).
Glutamine synthetase (GS, also known as glutamate-ammonia-ligase, EC 6.3.1.2), an enzyme that is highly abundant in astrocytes, is of particular interest due to its roles in health and disease (21). Systemic mutations of the GS gene have been associated with brain malformations, seizures, multiorgan failure, and early death (22, 23). Studies have further suggested that acquired GS deficiencies in discrete areas of the brain might play a causative role in various neurological disorders and psychiatric conditions including Alzheimer's disease, hepatic encephalopathy, suicide/depression schizophrenia, and epilepsy (24–32). The goal of this review is to discuss the significance of GS in the pathogenesis of focal epilepsies, particularly mesial temporal lobe epilepsy (MTLE), which is one of the most common types of medication-refractory epilepsies in humans (26, 32–35).
GS serves important roles in nitrogen metabolism, acid-base homeostasis, and cell signaling in many species of prokaryotes and eukaryotes (36–38). GS is thought to eliminate or reduce the toxic effects of glutamate and ammonia in the mammalian CNS by metabolizing these compounds (39–41). Moreover, multiple physiological processes, including synthesis of glutamate and GABA, synthesis of proteins, and osmoregulation, rely on a steady supply of glutamine (42). Because GS is the only enzyme capable of synthesizing large amounts of glutamine in the human body, changes in its expression and activity are expected to have significant consequences for normal physiology.
After its release from the presynaptic neuron, neurotransmitter glutamate interacts with receptors in the postsynaptic membrane, followed by its removal from the synapse into astrocytes. After uptake into astrocytes, glutamate may be converted to glutamine via glutamine synthetase (Figure 1). Glutamine can subsequently be transferred from astrocytes to glutamatergic neurons (44, 45). Once in the glutamatergic neuron, the mitochondrial enzyme phosphate-activated glutaminase (PAG) converts glutamine to glutamate (46, 47), which is concentrated in synaptic vesicles and subsequently released into the extracellular space in response to an action potential. The glutamate is either taken up by astrocytes and converted back to glutamine via GS or taken up by inhibitory neurons and metabolized to GABA, which is used during inhibitory neurotransmission (Figure 1). Astrocytic glutamine can be taken up by GABAergic neuron as precursor for the inhibitory neurotransmitter γ-aminobutyric acid (GABA) via glutamate (48, 49). While most of the released GABA is taken up by the pre-synaptic neuron, some enters astrocytes where it is converted back to glutamine (50). Thus, astroglial GS is crucial for both excitatory (glutamatergic) and inhibitory (GABAergic) neurotransmission (51).
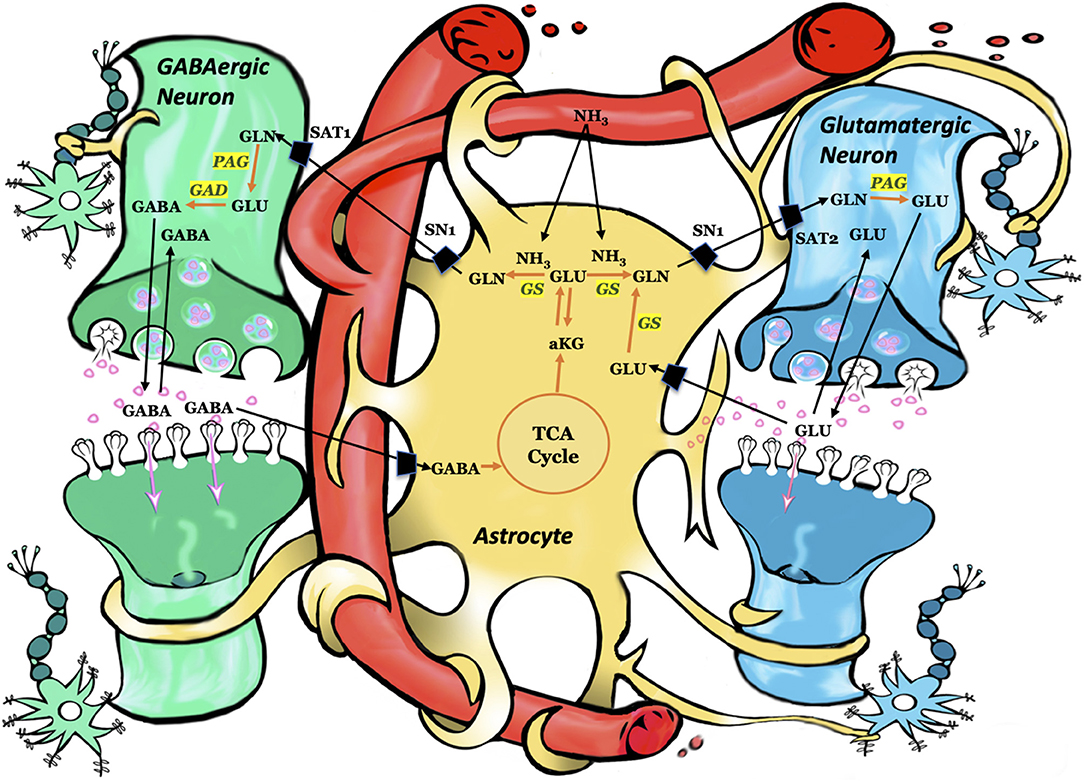
Figure 1. Schematic representation of key pathways involving astrocytes, glutamatergic neurons, and GABAergic neurons. Each arrow signifies several reactions. Using ammonia, glutamine synthetase converts glutamate to glutamine. Subsequently, glutamine is taken up by the adjacent neurons and converted to glutamate or GABA. Astrocytes take up the synaptic glutamate (glutamine–glutamate cycle) or GABA (glutamine–glutamate–GABA cycle) and converts these neurotransmitters to glutamine via glutamine synthetase. GLN, glutamine; GLU, glutamate; GABA, gamma-aminobutyric acid; aKG, alpha ketoglutarate; TCA, tricarboxylic acid cycle; GS, glutamine synthetase; PAG, phosphate-activated glutaminase; GAD, glutamic acid decarboxylase; SN1, system N transporter 1; SAT1, system A transporter and SAT2, system A transporter 2. Figure adapted with permission from (43).
The human body contains large amounts of ammonia, produced mainly by the action of bacterial enzymes on colonic content and from the hydrolysis of glutamine in the small and large intestinal cells. While the gut-derived ammonia is mostly metabolized by the liver, via the urea cycle and the GS reaction, a small amount of ammonia (10–30 μmol/L) remains in the plasma under normal conditions. Much higher plasma ammonia concentrations are detected in pathological conditions such as liver disease and urea cycle disorders. Because ammonia is neurotoxic and easily crosses the blood-brain-barrier, an efficient mechanism for clearing brain ammonia is essential (52). Astroglial GS is critical for such clearance, because the CNS lacks a functional urea cycle (52). GS utilizes ammonia, that is taken up by astrocytes, to convert glutamate to glutamine (Figure 1). The important role of astrocytes in this process is underscored by the presence of astrocytic end-feet surrounding the brain endothelial cells, which serve as a metabolic buffer between the blood and the brain, thereby reducing the toxic load of ammonia on neurons (53–55).
Glutamate is the predominant excitatory neurotransmitter in the adult brain, and perturbed extracellular brain glutamate levels have been implicated in the pathogenesis of epilepsy, particularly MTLE. Notably, extracellular glutamate is chronically elevated in the epileptogenic hippocampus (the seizure onset area) in human patients with MTLE, as ascertained by simultaneous depth electrode EEG and in vivo brain microdialysis (56–59). In addition, during seizure activity, a six-fold increase in the extracellular hippocampal glutamate above the basal (chronic) level was observed, followed by a slow decline to basal levels over a period of several minutes (60). Furthermore, many animal studies have suggested a causational relationship between increased brain glutamate signaling and epilepsy (39, 61, 62). Therefore, it is possible that an excessive amount of extracellular glutamate in the seizure onset area of the brain acts as a central metabolic cause of the neuronal loss and spontaneous seizures associated with MTLE (39, 61, 62).
Because GS is thought to be critical for glutamate metabolism following uptake into astrocytes, deficiency in this enzyme has been postulated as a possible basis for the increased glutamate observed in the extracellular fluid of the epileptogenic areas of the brain (26, 62). Moreover, isotopic tracer (C13) studies suggest that a slowing of the glutamate-glutamine cycle metabolism in the epileptogenic hippocampus is responsible for the accumulation and reduced clearance of glutamate in MTLE (63). Given the essentiality of GS in the glutamate-glutamine cycle, we and others sought to quantify activity of astroglial GS in the epileptogenic hippocampus in human patients with MTLE (26, 32). Intriguingly, the protein content and functional activity of GS was reduced by ~40% in subfields of the epileptogenic hippocampal formation in patients with MTLE and concomitant mesial temporal sclerosis (26, 32). Similarly, GS deficiency in the amygdala occurs in some patients with neocortical epilepsies (64) and in the tumor tissue of patients with malignant gliomas and secondary epilepsy (65).
Furthermore, several other studies support the idea that GS deficiency is a causative or contributing factor in some types of epilepsies besides MTLE. In the first three known cases of congenital, homozygous mutations in the GS gene, the patients had severe brain malformations and epileptic seizures (22, 66). Two of the patients died shortly after birth (22). Similarly, the high morbidity and mortality rates that accompany genetic GS deficiencies in humans are also observed in transgenic mouse models. For example, animals with prenatal excisions of the GS gene in all cell types do not survive past early embryonic development (67), and mice with selective gene deletions in GFAP-positive astrocytes survive until postnatal day 3 (68). However, mice with selective deletions in the neocortex and hippocampus are born without any apparent malformations but develop neurodegeneration and spontaneous seizures that seem to increase in frequency with age (34).
A common approach to assess the effect of GS deficiencies in specific brain regions involves the use of methionine sulfoximine (MSO). MSO impedes the ability of GS to catalyze the conversion of glutamate and ammonia to glutamine by irreversibly binding to the catalytic sites of GS (69). In one study, a continuous infusion of MSO into the right hippocampal formation of normal adult Sprague-Dawley rats was compared to a continuous infusion of normal saline into the same region of a separate group of rats. The animals were monitored by continuous video-intracranial electroencephalogram (EEG) recordings for several weeks and the brains were analyzed for GS activity. MSO resulted in a reduction of GS activity in the infused hippocampal formation of ~80% (28), and the animals displayed repetitive seizures that began several hours after the onset of infusion. The initial repetitive seizures, which lasted between 24 and 48 h, were followed by a quiet period of variable length before spontaneous, recurrent seizures commenced (70). The MSO-treated animals sometimes exhibited glial proliferation and patterned neuron loss in the infused hippocampal formation, similar to that of human MTLE (28, 33).
Another study analyzed whether the neuroanatomical site of GS inhibition is an important determinant for the epileptogenic process and for the epileptic phenotype. MSO was infused unilaterally into different limbic regions of adult rats, including the angular bundle, the deep entorhinal cortex, area CA1, the molecular layer of the subiculum, the hilus of the dentate gyrus, the lateral ventricle, and the central nucleus of the amygdala (71, 72). Recurrent seizures were observed in all animals infused with MSO into the brain tissue, and the seizures increased in severity (Racine grade) over a period of several weeks with variations in the seizure frequency and severity patterns between brain regions. Moreover, animals infused with MSO into the central nucleus of the amygdala displayed recurrent seizures with depressive-like behaviors, as observed by a reduction of sucrose consumption in the sucrose preference test (72). Collectively, these studies suggest that the neuroanatomical site of GS inhibition affects the epileptogenic process as well as the overall phenotype of the disease.
The prevalent idea that the neuroanatomical and electrophysiological substrates of focal epilepsies occur only in a circumscribed brain region, the seizure focus, has been questioned more recently (73–77). Many clinical and EEG studies have suggested that large-scale, aberrant “brain networks” play key roles in seizure initiation and propagation, and that aberrant network activity may be present even during the time between seizures (76–85).
Albright et al. analyzed changes in neuronal networks during epileptogenesis in the intrahippocampal MSO-infusion model of MLTE (86). Intracranial EEG recordings and c-Fos immunohistochemistry were used to record seizure-associated neuronal activation at different stages during epileptogenesis. It was found that low-grade seizures during the earliest stages of epileptogenesis activated neurons in the entorhinal-hippocampal territory, the basolateral amygdala, the piriform cortex, the midline thalamus, and the anterior olfactory area. However, during later stages of epileptogenesis, when the seizures were more severe, neuronal activation was evident in extensive areas of the brain, such as the neocortex, the bed nucleus of the stria terminalis, the mediodorsal thalamus, and the central nucleus of the amygdala. These areas were activated in addition to the areas activated during early epileptogenesis.
In another study, whole brain diffusion tensor imaging (DTI) was used to assess any structural changes during early and late epileptogenesis in the hippocampal MSO-infusion model of MTLE (33). There were significant changes in fractional anisotropy (FA) in multiple brain regions in MSO-infused vs. phosphate buffered saline (PBS)-infused control animals. The changes in FA were markedly different in early epileptogenesis compared to late epileptogenesis, suggesting that inhibition of GS in one hippocampal formation results in structural changes that affect multiple brain regions and change over a period of several weeks (33, 71, 86).
An increasing number of studies in humans and animal models have linked astroglial GS dysfunction to the pathogenesis of focal epilepsies, particularly MTLE. Even very small and circumscribed deficiencies in GS affecting a subfield of the hippocampal formation or a nucleus of the amygdala can lead to epileptic seizures and comorbid, depressive-like features in laboratory animals. Moreover, such small deficiencies result in widespread and progressive changes in neuronal network activation and in the structure of the brain. While the mechanism by which GS dysfunction causes epilepsy remains unclear, several scenarios are possible such as reduced clearance of extracellular brain glutamate and ammonia, glutamine deficiency, and perturbed glutamatergic and GABAergic neurotransmission. Further studies are needed to determine the causes of astroglial GS deficiency so that more effective treatments can be developed to prevent such deficiencies and combat the development and progression of GS-associated epilepsies.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by grants from the Swebilius Family Trust, the National Institutes of Health (NIH, NS058674, NS070824, NS109062, and NS109734), the National Center for Advancing Translational Sciences (NCATS, a component of the NIH; RR024139), and Citizens United for Research in Epilepsy (CURE). The contents of the publication are solely the responsibility of the authors and do not necessarily represent the official view of NCATS or NIH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge Garrett Sendlewski for his outstanding illustrative work for Figure 1.
1. Vasile F, Dossi E, Rouach N. Human astrocytes: structure and functions in the healthy brain. Brain Struct Funct. (2017) 222:2017–29. doi: 10.1007/s00429-017-1383-5
2. Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. (1966) 29:788–806. doi: 10.1152/jn.1966.29.4.788
3. Katz MJ, Lasek RJ, Silver J. Ontophyletics of the nervous system: development of the corpus callosum and evolution of axon tracts. Proc Natl Acad Sci USA. (1983) 80:5936–40. doi: 10.1073/pnas.80.19.5936
4. Magistretti PJ, Pellerin L. Astrocytes couple synaptic activity to glucose utilization in the brain. Physiology. (1999) 14:177–82. doi: 10.1152/physiologyonline.1999.14.5.177
5. Verkhratsky A, Steinhäuser C. Ion channels in glial cells. Brain Res Brain Res Rev. (2000) 32:380–412. doi: 10.1016/S0165-0173(99)00093-4
6. Danbolt NC. Glutamate uptake. Prog Neurobiol. (2001) 65:1–105. doi: 10.1016/S0301-0082(00)00067-8
7. Lemke G. Glial Control of Neuronal Development. Ann Rev Neurosci. (2001) 24:87–105. doi: 10.1146/annurev.neuro.24.1.87
8. Pellerin L, Bonvento G, Chatton JY, Pierre K, Magistretti PJ. Role of neuron-glia interaction in the regulation of brain glucose utilization. Diabetes Nutr Metab. (2002) 15:268–73; discussion 273.
9. Benfenati V, Amiry-Moghaddam M, Caprini M, Mylonakou MN, Rapisarda C, Ottersen OP, et al. Expression and functional characterization of transient receptor potential vanilloid-related channel 4 (TRPV4) in rat cortical astrocytes. Neuroscience. (2007) 148:876–92. doi: 10.1016/j.neuroscience.2007.06.039
10. Hertz L. Astrocytic energy metabolism and glutamate formation–relevance for 13C-NMR spectroscopy and importance of cytosolic/mitochondrial trafficking. Magn Reson Imaging. (2011) 29:1319–29. doi: 10.1016/j.mri.2011.04.013
11. Schousboe A, Bak LK, Madsen K, Waagepetersen H. Amino acid neurotransmitter synthesis and removal. In: Kettenmann H, Ransom BR, editors, Neuroglia, Third Edition, Oxford: Oxford University Press (2012). p. 443–56. doi: 10.1093/med/9780199794591.003.0035
12. Robinson SR. Neuronal expression of glutamine synthetase in Alzheimer's disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem Int. (2000) 36:471–82. doi: 10.1016/S0197-0186(99)00150-3
13. Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia. (2005) 50:281–6. doi: 10.1002/glia.20205
14. Binder DK, Steinhäuser C. Functional changes in astroglial cells in epilepsy. Glia. (2006) 54:358–68. doi: 10.1002/glia.20394
15. Sharma R, Fischer MT, Bauer J, Felts PA, Smith KJ, Misu T, et al. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol. (2010) 120:223–36. doi: 10.1007/s00401-010-0704-z
16. Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, et al. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: a PET study with [11C]DAA1106. Int J Neuropsych. (2010) 13:943–50. doi: 10.1017/S1461145710000313
17. Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. (2012) 62:63–77. doi: 10.1016/j.neuropharm.2011.07.036
18. Gloor P. Mesial temporal sclerosis: historical background and an overview from a modern perspective. Epilepsy surgery. (1991) 689–703.
19. Cohen-Gadol AA, Pan JW, Kim JH, Spencer DD, Hetherington HH. Mesial temporal lobe epilepsy: a proton magnetic resonance spectroscopy study and a histopathological analysis. J Neurosurg. (2004) 101:613–20. doi: 10.3171/jns.2004.101.4.0613
20. Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, et al. Role of microglia in central nervous system infections. Clin Microb Rev. (2004) 17:942–64. doi: 10.1128/CMR.17.4.942-964.2004
21. Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science. (1977) 195:1356–8. doi: 10.1126/science.14400
22. Haberle J, Gorg B, Rutsch F, Schmidt E, Toutain A, Benoist JF, et al. Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med. (2005) 353:1926–33. doi: 10.1056/NEJMoa050456
23. Häberle J, Görg B, Toutain A, Rutsch F, Benoist JF, Gelot A, et al. Inborn error of amino acid synthesis: human glutamine synthetase deficiency. J Inherit Metab Dis. (2006) 29:352–8. doi: 10.1007/s10545-006-0256-5
24. Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, et al. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci USA. (1991) 88:10540–3. doi: 10.1073/pnas.88.23.10540
25. Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem. (1995) 65:2146–56. doi: 10.1046/j.1471-4159.1995.65052146.x
26. Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. (2004) 363:28–37. doi: 10.1016/S0140-6736(03)15166-5
27. Bruneau EG, Mccullumsmith RE, Haroutunian V, Davis KL, Meador-Woodruff JH. Increased expression of glutaminase and glutamine synthetase mRNA in the thalamus in schizophrenia. Schizophr Res. (2005) 75:27–34. doi: 10.1016/j.schres.2004.12.012
28. Eid T, Ghosh A, Wang Y, Beckström H, Zaveri HP, Lee TS, et al. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain. (2008) 131:2061–70. doi: 10.1093/brain/awn133
29. Klempan TA, Sequeira A, Canetti L, Lalovic A, Ernst C, Ffrench-Mullen J, et al. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol Psychiatry. (2009) 14:175–89. doi: 10.1038/sj.mp.4002110
30. Sequeira A, Mamdani F, Ernst C, Vawter MP, Bunney WE, Lebel V, et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS ONE. (2009) 4:e6585. doi: 10.1371/journal.pone.0006585
31. Brusilow SW, Koehler RC, Traystman RJ, Cooper AJ. Astrocyte glutamine synthetase: importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics. (2010) 7:452–70. doi: 10.1016/j.nurt.2010.05.015
32. Van Der Hel WS, Notenboom RG, Bos IW, Van Rijen PC, Van Veelen CW, De Graan PN. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology. (2005) 64:326–33. doi: 10.1212/01.WNL.0000149636.44660.99
33. Wang H, Huang Y, Coman D, Munbodh R, Dhaher R, Zaveri HP, et al. Network evolution in mesial temporal lobe epilepsy revealed by diffusion tensor imaging. Epilepsia. (2017) 58:824–34. doi: 10.1111/epi.13731
34. Zhou Y, Dhaher R, Parent M, Hu QX, Hassel B, Yee SP, et al. Selective deletion of glutamine synthetase in the mouse cerebral cortex induces glial dysfunction and vascular impairment that precede epilepsy and neurodegeneration. Neurochem Int. (2019) 123:22–33. doi: 10.1016/j.neuint.2018.07.009
35. Sandhu MRS, Dhaher R, Gruenbaum SE, Raaisa R, Spencer DD, Pavlova MK, et al. Circadian-like rhythmicity of extracellular brain glutamate in epilepsy. Front Neurol. (2020) 11:398. doi: 10.3389/fneur.2020.00398
36. Elliott WH. Adenosinetriphosphate in glutamine synthesis. Nature. (1948) 161:128–9. doi: 10.1038/161128a0
37. Speck JF. The enzymatic synthesis of glutamine, a reaction utilizing adenosine triphosphate. J Biol Chem. (1949) 179:1405–26. doi: 10.1016/S0021-9258(18)56802-2
38. Wu C. Glutamine synthetase. I a comparative study of its distribution in animals and its inhibition by dl-allo-delta-hydroxylysine. Comp Biochem Physiol. (1963) 9:335–51. doi: 10.1016/0010-406X(63)90169-5
39. Olney JW, Sharpe LG, Feigin RD. Glutamate-induced brain damage in infant primates. J Neuropathol Exp Neurol. (1972) 31:464–88. doi: 10.1097/00005072-197207000-00006
40. Albrecht J, Jones EA. Hepatic encephalopathy: molecular mechanisms underlying the clinical syndrome. J Neurol Sci. (1999) 170:138–46. doi: 10.1016/S0022-510X(99)00169-0
41. Butterworth RF. Pathophysiology of hepatic encephalopathy: a new look at ammonia. Metab Brain Dis. (2002) 17:221–7. doi: 10.1023/A:1021989230535
42. Norenberg MD, Jayakumar AR, Rama Rao KV, Panickar KS. New concepts in the mechanism of ammonia-induced astrocyte swelling. Metab Brain Dis. (2007) 22:219–34. doi: 10.1007/s11011-007-9062-5
43. Gruenbaum S, Chen E, Sandhu MR, Deshpande K, Dhaher R, Hersey D, et al. Branched-chain amino acids and seizures: a systematic review of the literature. CNS Drugs. (2019) 33:755–70. doi: 10.1007/s40263-019-00650-2
44. Chaudhry FA, Reimer RJ, Edwards RH. The glutamine commute: take the N line and transfer to the A. J Cell Biol. (2002) 157:349–55. doi: 10.1083/jcb.200201070
45. Jenstad M, Quazi AZ, Zilberter M, Haglerød C, Berghuis P, Saddique N, et al. System A transporter SAT2 mediates replenishment of dendritic glutamate pools controlling retrograde signaling by glutamate. Cereb Cortex. (2009) 19:1092–106. doi: 10.1093/cercor/bhn151
46. Svenneby G. Pig brain glutaminase: purification and identification of different enzyme forms. J Neurochem. (1970) 17:1591–9. doi: 10.1111/j.1471-4159.1970.tb03729.x
47. Kvamme E, Torgner IA, Roberg B. Kinetics and localization of brain phosphate activated glutaminase. J Neurosci Res. (2001) 66:951–8. doi: 10.1002/jnr.10041
48. Reubi J-C, Van Der Berg C, Cuenod M. Glutamine as precursor for the GABA and glutamate trasmitter pools. Neurosci Lett. (1978) 10:171–4. doi: 10.1016/0304-3940(78)90030-7
49. Sonnewald U, Westergaard N, Schousboe A, Svendsen J, Unsgård G, Petersen S. Direct demonstration by [13C] NMR spectroscopy that glutamine from astrocytes is a precursor for GABA synthesis in neurons. Neurochem Int. (1993) 22:19–29. doi: 10.1016/0197-0186(93)90064-C
50. Schousboe A, Bak LK, Waagepetersen HS. Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front Endocrinol. (2013) 4:102. doi: 10.3389/fendo.2013.00102
51. Walls AB, Waagepetersen HS, Bak LK, Schousboe A, Sonnewald U. The glutamine–glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res. (2015) 40:402–9. doi: 10.1007/s11064-014-1473-1
52. Cooper A, Plum F. Biochemsitry and Physiology of brain ammonia. Physiol Rev. (1987) 67:440–519. doi: 10.1152/physrev.1987.67.2.440
53. Cooper A, Mcdonald J, Gelbard A, Gledhill R, Duffy T. The metabolic fate of 13N-labeled ammonia in rat brain. J Biol Chem. (1979) 254:4982–92. doi: 10.1016/S0021-9258(18)50550-0
54. Cooper ÅJ, Mora SN, Cruz NF, Gelbard AS. Cerebral ammonia metabolism in hyperammonemic rats. J Neurochem. (1985) 44:1716–23. doi: 10.1111/j.1471-4159.1985.tb07159.x
55. Braet K, Paemeleire K, D'herde K, Sanderson MJ, Leybaert L. Astrocyte–endothelial cell calcium signals conveyed by two signalling pathways. Europ J Neurosci. (2001) 13:79–91. doi: 10.1046/j.1460-9568.2001.01372.x
56. Petroff OA, Errante LD, Kim JH, Spencer DD. N-acetyl-aspartate, total creatine, and myo-inositol in the epileptogenic human hippocampus. Neurology. (2003) 60:1646–51. doi: 10.1212/01.WNL.0000068020.85450.8B
57. Kim JH, Je SJ, Petroff O, Spencer SS, Hwang JY, Spencer DD. Hippocampal Glial Density in Temporal Lobe Epilepsy. Epilepsia: Wiley (2004).
58. Petroff EA, Cavus I, Kim JH, Spencer DD. Interictal Extracellular Glutamate Concentrations Are Increased in Hippocampal Sclerosis. Wiley (2004).
59. Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. (2005) 57:226–35. doi: 10.1002/ana.20380
60. During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. (1993) 341:1607–10. doi: 10.1016/0140-6736(93)90754-5
61. Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanisms of epileptic brain damage. Adv Neurol. (1986) 44:857–77.
62. Eid T, Williamson A, Lee TS, Petroff OA, De Lanerolle NC. Glutamate and astrocytes–key players in human mesial temporal lobe epilepsy? Epilepsia. (2008) 49 Suppl 2:42–52. doi: 10.1111/j.1528-1167.2008.01492.x
63. Petroff O.a.C., Errante LD, Rothman DL, Kim JH, Spencer DD. Glutamate–glutamine cycling in the epileptic human hippocampus. Epilepsia. (2002) 43:703–10. doi: 10.1046/j.1528-1157.2002.38901.x
64. Steffens M, Huppertz HJ, Zentner J, Chauzit E, Feuerstein TJ. Unchanged glutamine synthetase activity and increased NMDA receptor density in epileptic human neocortex: implications for the pathophysiology of epilepsy. Neurochem Int. (2005) 47:379–84. doi: 10.1016/j.neuint.2005.06.001
65. Rosati A, Poliani PL, Todeschini A, Cominelli M, Medicina D, Cenzato M, et al. Glutamine synthetase expression as a valuable marker of epilepsy and longer survival in newly diagnosed glioblastoma multiforme. Neuro Oncol. (2013) 15:618–25. doi: 10.1093/neuonc/nos338
66. Häberle J, Shahbeck N, Ibrahim K, Hoffmann GF, Ben-Omran T. Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol Genet Metab. (2011) 103:89–91. doi: 10.1016/j.ymgme.2011.02.001
67. He Y, Hakvoort TB, Vermeulen JL, Lamers WH, Van Roon MA. Glutamine synthetase is essential in early mouse embryogenesis. Dev Dyn. (2007) 236:1865–75. doi: 10.1002/dvdy.21185
68. He Y, Hakvoort TB, Vermeulen JL, Labruyère WT, De Waart DR, Van Der Hel WS, et al. Glutamine synthetase deficiency in murine astrocytes results in neonatal death. Glia. (2010) 58:741–54. doi: 10.1002/glia.20960
69. Eisenberg D, Gill HS, Pfluegl GM, Rotstein SH. Structure-function relationships of glutamine synthetases. Biochim Biophys Acta. (2000) 1477:122–45. doi: 10.1016/S0167-4838(99)00270-8
70. Wang Y, Zaveri HP, Lee TS, Eid T. The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video-intracranial electroencephalographic study. Exp Neurol. (2009) 220:293–302. doi: 10.1016/j.expneurol.2009.08.034
71. Dhaher R, Wang H, Gruenbaum SE, Tu N, Lee TS, Zaveri HP, et al. Effects of site-specific infusions of methionine sulfoximine on the temporal progression of seizures in a rat model of mesial temporal lobe epilepsy. Epilepsy Res. (2015) 115:45–54. doi: 10.1016/j.eplepsyres.2015.05.005
72. Gruenbaum SE, Wang H, Zaveri HP, Tang AB, Lee TS, Eid T, et al. Inhibition of glutamine synthetase in the central nucleus of the amygdala induces anhedonic behavior and recurrent seizures in a rat model of mesial temporal lobe epilepsy. Epilepsy Behav. (2015) 51:96–103. doi: 10.1016/j.yebeh.2015.07.015
73. Spencer SS. Neural networks in human epilepsy: evidence of and implications for treatment. Epilepsia. (2002) 43:219–27. doi: 10.1046/j.1528-1157.2002.26901.x
74. Kramer MA, Cash SS. Epilepsy as a disorder of cortical network organization. Neuroscientist. (2012) 18:360–72. doi: 10.1177/1073858411422754
75. Dickten H, Porz S, Elger CE, Lehnertz K. Weighted and directed interactions in evolving large-scale epileptic brain networks. Scient Rep. (2016) 6:34824. doi: 10.1038/srep34824
76. Bartolomei F, Lagarde S, Wendling F, Mcgonigal A, Jirsa V, Guye M, et al. Defining epileptogenic networks: contribution of SEEG and signal analysis. Epilepsia. (2017) 58:1131–47. doi: 10.1111/epi.13791
77. Spencer DD, Gerrard JL, Zaveri HP. The roles of surgery and technology in understanding focal epilepsy and its comorbidities. Lancet Neurol. (2018) 17:373–82. doi: 10.1016/S1474-4422(18)30031-0
78. Zaveri HP, Pincus SM, Goncharova Ii, Duckrow RB, Spencer DD, Spencer SS. Localization-related epilepsy exhibits significant connectivity away from the seizure-onset area. Neuroreport. (2009) 20:891–5. doi: 10.1097/WNR.0b013e32832c78e0
79. Varotto G, Tassi L, Franceschetti S, Spreafico R, Panzica F. Epileptogenic networks of type II focal cortical dysplasia: a stereo-EEG study. Neuroimage. (2012) 61:591–8. doi: 10.1016/j.neuroimage.2012.03.090
80. Constable RT, Scheinost D, Finn ES, Shen X, Hampson M, Winstanley FS, et al. Potential use and challenges of functional connectivity mapping in intractable epilepsy. Front Neurol. (2013) 4:39. doi: 10.3389/fneur.2013.00039
81. Englot DJ, Konrad PE, Morgan VL. Regional and global connectivity disturbances in focal epilepsy, related neurocognitive sequelae, and potential mechanistic underpinnings. Epilepsia. (2016) 57:1546–57. doi: 10.1111/epi.13510
82. Smith EH, Schevon CA. Toward a mechanistic understanding of epileptic networks. Curr Neurol Neurosci Rep. (2016) 16:97. doi: 10.1007/s11910-016-0701-2
83. Nissen IA, Stam CJ, Reijneveld JC, Van Straaten IE, Hendriks EJ, Baayen JC, et al. Identifying the epileptogenic zone in interictal resting-state MEG source-space networks. Epilepsia. (2017) 58:137–48. doi: 10.1111/epi.13622
84. Sinha N, Dauwels J, Kaiser M, Cash SS, Brandon Westover M, Wang Y, et al. Predicting neurosurgical outcomes in focal epilepsy patients using computational modelling. Brain. (2017) 140:319–32. doi: 10.1093/brain/aww299
85. Tomlinson SB, Porter BE, Marsh ED. Interictal network synchrony and local heterogeneity predict epilepsy surgery outcome among pediatric patients. Epilepsia. (2017) 58:402–11. doi: 10.1111/epi.13657
Keywords: epilepsy, epileptogenesis, glutamine synthetase, astrocyte, epilepsy network, mesial temporal lobe epilepsy
Citation: Sandhu MRS, Gruenbaum BF, Gruenbaum SE, Dhaher R, Deshpande K, Funaro MC, Lee T-SW, Zaveri HP and Eid T (2021) Astroglial Glutamine Synthetase and the Pathogenesis of Mesial Temporal Lobe Epilepsy. Front. Neurol. 12:665334. doi: 10.3389/fneur.2021.665334
Received: 07 February 2021; Accepted: 19 March 2021;
Published: 13 April 2021.
Edited by:
Kjell Heuser, Oslo University Hospital, NorwayReviewed by:
Peter Bedner, University Hospital Bonn, GermanyCopyright © 2021 Sandhu, Gruenbaum, Gruenbaum, Dhaher, Deshpande, Funaro, Lee, Zaveri and Eid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tore Eid, dG9yZS5laWRAeWFsZS5lZHU=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.