 Kathlyn Laval
Kathlyn Laval Lynn W. Enquist
Lynn W. Enquist- Department of Molecular Biology, Princeton University, Princeton, NJ, United States
Alzheimer's disease (AD) is a neurodegenerative disease affecting ~50 million people worldwide. To date, there is no cure and current therapies have not been effective in delaying disease progression. Therefore, there is an urgent need for better understanding of the pathogenesis of AD and to rethink possible therapies. Herpes simplex virus type 1 (HSV1) has recently received growing attention for its potential role in sporadic AD. The virus is a ubiquitous human pathogen that infects mucosal epithelia and invades the peripheral nervous system (PNS) of its host to establish a reactivable, latent infection. Upon reactivation, HSV1 spreads back to the epithelium and initiates a new infection, causing epithelial lesions. Occasionally, the virus spreads from the PNS to the brain after reactivation. In this review, we discuss current work on the pathogenesis of AD and summarize research results that support a potential role for HSV1 in the infectious hypothesis of AD. We also highlight recent findings on the neuroinflammatory response, which has been proposed to be the main driving force of AD, starting early in the course of the disease. Relevant rodent models to study neuroinflammation in AD and novel therapeutic approaches are also discussed. Throughout this review, we focus on several aspects of HSV1 pathogenesis, including its primary role as an invader of the PNS, that should be considered in the etiology of AD. We also point out some of the contradictory data and remaining knowledge gaps that require further research to finally fully understand the cause of AD in humans.
Introduction
Alzheimer's disease (AD) is one of the most common neurodegenerative diseases of humans with 50 million people affected worldwide (1). The disease was first described by a German psychiatrist and neuropathologist, Alois Alzheimer, in 1907. He reported a peculiar severe disease process of the cerebral cortex in a 50-year-old woman, named Auguste D, admitted to a psychiatric hospital for paranoia, progressive sleep and memory disturbance, confusion, and aggression (2). Clinically, AD begins with mild symptoms of memory loss and slowly leads to global cognitive impairment. The cognitive deficits are frequently accompanied by severe neurological and psychiatric symptoms in late-stage disease (3, 4). The most relevant AD risk factor is age, with the prevalence of AD rising exponentially after 65 years of age, with one in ten individuals affected (5). Early-onset AD (EOAD) are usually diagnosed before age 65 while late-onset AD (LOAD) develops after age 65. Approximately 95% of all AD cases are LOAD while a few percentages are attributed to EOAD. The LOAD is mainly sporadic with a complex multifactorial etiology (e.g., age, environmental and genetic factors, pathogens). In contrast, the EOAD is almost entirely genetic disease with 92–100% heritability (6–10). AD represents a growing public health burden as populations live longer. Indeed, epidemiological studies suggest that the overall prevalence of AD is expected to double within the next 20 years (11). On average, a person can live 4–8 years after diagnosis and in rare cases, up to 20 years. However, patients require total care in the later stages of the disease. Therefore, AD has also a significant financial impact on global healthcare systems. AD care costs have been predicted to be around $2 trillion in 2030 with ~75 million people affected worldwide if effective interventions are not found (12). So far, there is no cure and current therapies have not been effective in delaying progression. Donepezil, a cholinesterase inhibitor and FDA-approved drug, is currently the most common treatment for mild to severe forms of AD. This drug enhances acetylcholine availability and increases neuronal transmission in the brain by inhibiting its breakdown in the synapse. This treatment shows modest benefit in cognitive function and daily activities of some AD patients compared to placebo treatment (13). However, Donepezil presents severe adverse side-effects and patients who discontinue this drug experience accelerated cognitive decline (14). Consequently, there is an urgent need to gain a better understanding of the pathophysiology of AD in order to develop effective therapies.
The Pathogenesis of AD
The two neuropathological hallmarks of AD are the extracellular deposits of amyloid-β (Aβ) peptide and the intracellular accumulation of hyperphosphorylated tau protein in the brain. These pathologic features are commonly accompanied by the loss of neurons and synapses and reactive gliosis. Aβ peptide is produced by the sequential, proteolytic processing of the amyloid precursor protein (APP). APP is produced primarily by neurons and is transported anterograde in axons to synapse terminals (15). While its function remains largely unclear, APP has been suggested to play a role in synapse formation, synaptic transmission and neurogenesis (16, 17). APP has demonstrated neuroprotective properties following traumatic brain injury (18). However, its processing and cleavage represent the foundation of the “amyloid cascade hypothesis” in which, the accumulation of neurotoxic Aβ by-products in neurons causes AD (19, 20). APP processing can occur through 2 distinct pathways (21). In the non-amyloidogenic pathway, APP cleavage is mediated by the α-secretase and γ-secretase and results in the production of 3 fragments: a secreted C-terminal fragment (sAPP-α), p3, and the APP intracellular domain. In the amyloidogenic pathway, APP is cleaved by the β-secretase, which generates a large soluble extracellular secreted domain (sAPP-β). The remaining APP bound fragment is cleaved multiple times by the γ-secretase to produce neurotoxic Aβ peptides, variable in length, including Aβ-40 and Aβ-42 fragments. Genetic mutations in APP and the γ-secretase complex [presenilin-1 (PSEN1) and PSEN2] have been reported to induce aberrant APP processing and overproduction of Aβ in the brain and cause EOAD (9, 22, 23). Several lines of evidence suggest that the impaired clearance of Aβ rather than its overproduction leads to the accumulation of Aβ and amyloid deposition in the brain (24). The clearance of Aβ in the brain can be accomplished by several mechanisms, including non-enzymatic and enzymatic pathways. The non-enzymatic pathway consists of: (1) the interstitial fluid drainage of Aβ; (2) the uptake of Aβ by microglial phagocytosis and transport into the blood circulation; (3) the autophagy in microglia and further degradation of Aβ fibrils via the lysosomal system; (4) transcytosis of Aβ across the blood-brain barrier via the low-density lipoprotein receptor family (25–29). The enzymatic pathway comprises the activity of Aβ-degrading enzymes such as neprilysin, insulin-degrading enzyme, matrix metalloproteinase-9, and glutamate carboxypeptidase II (30). These mechanisms are not covered in this review as several excellent reviews have addressed these topics in more detail (31–33).
The aggregation of Aβ proteins into plaques enhances the formation of neuro-fibrillary tangles (NFT), which consist of the hyperphosphorylation, misfolding, and aggregation of tau protein, a microtubule associated protein normally located in axons. There are multiple ways by which NFT may damage neurons and glial cells in the brain (34). For instance, by acting as physical barriers in the cytoplasm, compromising normal cellular functions, and homeostasis through inhibition of proteasomal activity (35). NFT may also cause neuronal toxicity by reducing normal tau functions such as tau-mediated regulation of microtubule dynamics (36). Ultimately, NFT are able to self-propagate, spread to synapses to promote synaptic and cognitive dysfunction in AD. While the toxic gain of function and the loss of normal tau functions are thought to play a role in inducing neuronal death and synaptic loss, their underlying mechanisms are still largely unclear.
Aberrant APP processing and overproduction of Aβ can promote EOAD, but it is not clear whether these events can also cause LOAD. Most of the previous AD research has supported the amyloid cascade hypothesis and focused on identifying the mechanisms underlying Aβ plaque deposition in LOAD. However, this hypothesis has been the source of increased controversy over the years (37, 38). For instance, the presence of Aβ deposition has been detected in brains of elderly normal individuals, suggesting that Aβ plaques might be non-toxic and related to aging (39, 40). In fact, treatments aimed at targeting and reducing Aβ protein levels have largely failed in clinical trials (41, 42). Several alternative approaches for understanding AD pathogenesis are now being considered. For example, if the accumulation of Aβ in the brain occurs 10–20 years before the onset of cognitive impairment, early interventions could be more successful than those given to people with late-stage disease (43). An alternative view is the tau hypothesis, in which tau abnormalities are as important if not more than Aβ for disease progression (44). Because the role of Aβ accumulation and aggregation as a primary cause of LOAD remains debatable, there is an urgent need to investigate the pathogenesis of AD from a new angle and to rethink possible therapeutic strategies.
Herpes Simplex Virus Type 1 (HSV1): A Potential Player in the Infectious Hypothesis of AD
In the last years, the inflammatory-infectious hypothesis of AD has gained support as an alternative to the amyloid hypothesis that has dominated the AD field for decades. Several infectious agents, including bacteria and viruses have been implicated in the pathogenesis of AD. Human herpes simplex virus type 1 (HSV1) stands out as a possible significant player (45–50). Numerous publications have linked evidence for HSV1 infection with the development of sporadic AD (see section The Involvement of HSV1 in AD). It was proposed that reactivation of latent HSV1 infections may cause local neuronal damage and inflammation, which over time may lead to the deposition of Aβ and abnormal phosphorylation of tau in the brain. However, these data are correlative and do not demonstrate a direct causal relationship between HSV1 and AD. In this section, we aim to provide an understanding of the pathogenesis of HSV1 with a focus on HSV1 reactivation and latency in humans and in vivo models. This knowledge is essential as we discuss the most relevant studies looking into the role of HSV1 in the pathogenesis of AD.
The Pathogenesis of HSV1
HSV1 is a neuroinvasive pathogen, which commonly causes mucosal lesions, infectious blindness (herpes keratitis), as well as rare cases of encephalitis in humans. HSV1 infection is widespread around the world, with seropositivity among older adults (>65 years old) estimated to be 60–90% (51, 52). After primary replication in the mucosal epithelia, HSV1 reaches neurons of peripheral nervous system (PNS) that innervate the infected tissue. In the PNS, active replication does not occur and a lifelong latent infection is established in peripheral sensory and autonomic neurons [e.g., trigeminal ganglia (TG) and ciliary ganglia]. These quiescent infections may reactivate periodically (53, 54). Upon reactivation, newly replicated virus particles travel back to the mucosal epithelium. Secondary replication in the mucosal epithelium can cause local lesions in a subset of individuals while asymptomatic shedding can occur in others (55, 56). HSV1 can also infect the central nervous system (CNS) from peripheral infections and cause HSV encephalitis (HSE). While HSE is the most common fatal sporadic encephalitis in humans, its incidence is relatively rare with ~2–4 cases/1,000,000 people worldwide (57, 58).
Several studies demonstrated the presence of HSV1 DNA in postmortem brain tissues from asymptomatic patients by in situ hybridization and polymerase chain reaction (PCR) (59–63). The exact mechanism by which HSV1 gains access to the CNS in humans is unclear. The most likely route includes HSV1 retrograde transport to the brainstem via the trigeminal nerve that innervates the orofacial or corneal epithelium to the brainstem (64). An alternative route may involve viral spread via the olfactory tract (65). Direct CNS infection from the periphery as well as the possibility of reactivation of latent HSV1 within the brain are also considered but these possibilities are still an area of research and debate (66). Some studies with mice are informative. For instance, in the in vivo study of Kastrukoff et al., 10 mice were inoculated via the oral mucosa using a scarification method. The presence of HSV1 DNA and antigen positive cells in the brain were determined by in situ hybridization and immunohistochemistry (IHC) techniques, respectively (67). The authors demonstrated that HSV1 DNA and antigen positive cells were detected sequentially in the TG and throughout the brain as the infection progressed (first, the brainstem followed by the cerebellum and then the cerebral hemispheres). These areas are widely separated from each other and imply spread via neuronal circuitry. These findings are in accordance with previous reports on the spread and localization of HSV1 from the PNS to the CNS (68, 69). Interestingly, the authors also found “focal” areas of HSV1 antigen positive neuronal and glial cells. The authors showed that despite the clearance of viral antigen positive cells over time, HSV1 DNA was detected in the mouse brain 25 days after inoculation. This finding implies that a latent HSV1 infection was established in the brain. Indeed, the subsequent reactivation of HSV1 from these localized sites of CNS latent infection may cause sub-clinical or mild encephalitis in mice. Such processes, which may occur sporadically, would be difficult or impossible to detect or diagnose in asymptomatic patients.
Two ex vivo studies demonstrated that infectious HSV1 could be recovered from dissociated mouse TG as well as brain explants (70, 71). The authors found that following eye inoculation with HSV1 KOS and the more virulent McKrae strain, 71% and 80% of brainstem samples reactivated latent HSV1 at 30 days post-infection, respectively. A higher percentage of TG samples (100 and 90%) reactivated latent HSV1. Using the hyperthermia reactivation model, Yao et al. detected higher levels of HSV1 genomes in the brainstem of mice latently infected with HSV1 294.1 and McKrae strains than those in the TG (72). Moreover, HSV1 294.1 and McKrae reactivations occurred earlier in the mouse brainstem and TG (13 and 16 h after hypothermic stimulation). The authors suggested that the poor viability of brain explant cultures might explain the discrepancy in reactivation sites observed between ex vivo and in vivo results. A recent in vivo study by Doll et al. reported contradictory results. These authors found more infectious HSV1 particles and a higher frequency of viral reactivation in the TG compared to the brainstem of latently infected mice (73). In addition, HSV1 antigen was detected in the TG, but not the brainstem of these animals, 24 h after post-reactivation. Overall, these studies confirm that HSV1 can establish latency in the PNS and CNS after peripheral infection. They also suggest that HSV1 spreads to the CNS after reactivation from latency in the PNS. These studies point out the importance of initial infection of the PNS and possible effects in the brain that might have implications for AD research.
The Involvement of HSV1 in AD
In the early 1990's, two studies from Jamieson et al. first demonstrated that HSV1 DNA is present in a higher proportion of brains of elderly people with or without dementia compared to young people (60, 61). Additional studies showed that HSV1 DNA is detected in more AD brain samples than non-AD ones (74, 75). PCR results specifically correlated HSV1 DNA with β-amyloid deposits in the cerebral cortex. HSV1 antigens were also detected in the cytoplasm of cortical neurons of AD brain samples by IHC. However, these and other studies do not prove that HSV1 DNA is directly associated with Aβ plaques in the brain. On the one hand, 72% of HSV1 DNA was associated with β-amyloid plaques in AD brains compared to 24% in aged normal brains (76, 77). On the other hand, another study showed that only 55% of HSV1 DNA is plaque-associated in AD brains (63). Differences in the methodology used between the two studies (in situ PCR and immunohistochemistry vs. TaqMan PCR and serology) and sample sizes might explain the difference between results.
In the late 2000's, a population-based cohort study showed that the risk of AD increased in elderly subjects correlated with positive titers of anti-HSV1 IgM antibodies. These antibodies are indicators of primary infection or recent reactivation and replication of the virus (78). The authors suggested that the presence of anti-HSV1 IgM antibodies is highly correlated with incidence of AD. These findings were consistent with those of Féart et al., who correlated anti-HSV1 antibodies and plasma levels of Aβ1−40 and Aβ1−42 (two biomarkers of the disease) in a large study sample (79). This work demonstrated that high IgM levels, but not IgG levels, are significantly associated with low plasma levels of Aβ1−40 and Aβ1−42. The authors proposed that HSV1 reactivation leads to the accumulation of Aβ-deposits in the brain and subsequent decrease of Aβ plasma levels. Another HSV1 antibody study, by Lovheim et al. involved a larger number of participants (N = 3,432) with a longer follow-up (11 years). This study confirmed the presence of anti-HSV1 IgM antibodies at baseline in serum samples and almost doubled the risk for AD (80). A recent systematic review and meta-analysis further compiles up-to-date evidence that HSV1 is a major risk factor for AD (81).
In the last decade, several studies and animal models have been developed to determine the specific role of HSV1 reactivation in the progression of AD (82). Using a mouse model of encephalitis, Martin et al. showed that HSV1 immediate early protein ICP4 is expressed in TG by 7 days after intranasal infection, and also in the cerebral cortex by 60-days post-infection (dpi) (83). The expression of the HSV1 ICP4 protein was accompanied by the increased expression in the brain of neuroinflammatory (toll-like receptor 4; TLR-4) and early neurodegenerative markers (phospho-tau and TauC3) in the brain. The authors suggested that the presence of this HSV1 protein in the brain at 60 dpi correlated with HSV1 reactivation from CNS latency sites and the induction of neuroinflammation and neurodegeneration. These data must be interpreted with caution because the correlation of HSV1 ICP4 expression in both TG and cerebral cortex at 60 dpi does not prove that HSV1 reactivated from CNS latency sites only. Perhaps, HSV1 reactivated from PNS or both PNS and CNS latency sites. Recently, De Chiara et al. developed a model of recurrent HSV1 in mice experiencing repeated cycles of viral reactivation (84). This study was the first to report a connection between HSV1 and AD in a long-term in vivo study. After labial HSV1 inoculation, infectious virus particles were detected in the TG and several brain regions of infected mice after 4 dpi. They showed that HSV1 replicated in the brain of infected mice in response to a reactivation stimulus. Moreover, the repeated HSV1 reactivations induced the accumulation of Aβ and deposition in amyloid plaques as well as increased levels of phospho-tau in the brain. In addition, repeated reactivations resulted in impairment of cognitive functions in infected mice. Note that Aβ and phosphor-tau levels were not evaluated in the TG following viral reactivation. It also was unclear if all of the infected, unstressed mice actually had HSV1 in the CNS, as approximately of 21% of mice were negative by PCR for the virus in the brain. These results should be validated in future studies. Overall, the consensus hypothesis to date is that repeated asymptomatic HSV1 reactivations can occur in the brain during episodes of stress or immunosuppression, which induces cumulative neuronal damage and AD pathology. It was also suggested that periodic HSV1 reactivations occur more frequently in elderly brains as the immune system declines with age (45). However, these studies focused primarily on HSV1 CNS reactivation and latency in the pathogenesis of AD. A simple question is how does HSV1 get to the CNS? It is well-known that HSV1 invades the PNS where it establishes latency and that the PNS is connected to the CNS. Perhaps the virus enters the CNS from the periphery by axonal transport. Future studies should clarify the role of reactivation events in the PNS and the subsequent contributions of these peripheral processes to the initiation and development of AD in the brain.
Recently, three major studies demonstrated a direct role of HSV1 in the pathogenesis of AD. Tzeng et al. reported that HSV1 infection in humans led to a significant risk of later development of AD and that treatment with specific herpes antiviral drugs at the time of infection markedly reduced that risk by a factor of 10 (85, 86). This study was performed over a 10-year period with starting participants older than 50 years who had a newly diagnosed HSV1 infection. These findings are in accordance with a previous in vitro study showing that anti-HSV1 agents, such as acyclovir, penciclovir and foscarnet reduced Aβ and tau accumulation in HSV1-infected Vero cells (87). Another study by Eimer et al. demonstrated that in 3D human neural immortalized cells, Aβ oligomers inhibited HSV1 infection and significantly protected transgenic mice from acute HSV1 encephalitis following intracranial inoculations (88). The authors proposed that Aβ is an intrinsic antiviral response of neurons. Recently, Cairns et al. developed a 3D-brain tissue model using human-induced neural stem cells (hiNSCs). This study was the first to demonstrate that HSV1 infection directly causes a remarkable AD-like phenotype within 1 week after infection (89). This 3D brain tissue model demonstrated multiple aspects of AD physiology, including Aβ fibrillar plaque-like formations upon HSV1 infection, neuronal loss, neuroinflammation, and reactive gliosis. These findings corroborate previous in vitro studies showing that HSV1 infection induces APP and the accumulation of Aβ and other neurotoxic APP fragments in human and rat CNS neuronal cells (90, 91). Most importantly, the study from Cairns et al. showed that treatment with valacyclovir, a common herpesvirus antiviral drug, significantly reduced the AD-like phenotype in infected hiNSCs.
Alpha herpesviruses like HSV1 have evolved a well-defined relationship with neurons. They have evolved to survive in their host with limited damage to neurons and to disseminate to other hosts by establishing a latent, reactivable infection in neurons. It has been proposed that neurons produce certain synaptic proteins (e.g., Aβ) as an ancient protective mechanism against viral infections. This phenomenon could justify the use of HSV1 antiviral treatments and HSV1 vaccines as clinical treatments of AD. Antiviral drug treatments have already shown promising results to reduce AD pathology in vivo and in vitro. The development of a HSV1 vaccine may represent another effective treatment. Other more general interventions to block or reduce viral infections may be useful. For example, a pilot study demonstrated that treatment of 42 AD patients with IFN-β slightly, but not significantly, reduced disease progression compared to control group (92). IFN-β was well-tolerated with limited adverse effects, which may warrant further investigation in larger studies.
Neuroinflammation: At the Center of the Infectious Hypothesis of AD
Two critical elements of the infectious hypothesis are that the infectious agent must persist for years or periodically infect the host and that the immune response to this agent involves the nervous system. HSV1 infections meet these two requirements. It is a ubiquitous human infection that infects the nervous system and established a reactivable latent infection. Despite a strong immune response, the infection is not cleared and lasts for the life of the host.
Neuroinflammation is an important component of the infectious hypothesis of AD. It is defined as an inflammatory response in the nervous system to infection or injury. Neuroinflammation is considered a major driving factor in neurodegeneration and AD pathology, which starts early in the course of the disease, prior to the formation of Aβ plaques in the brain (93, 94). It may be triggered by infectious agents including viruses (95). Neuroinflammation may represent a promising drug target to prevent neurodegeneration and AD. In this section, we summarize key findings of the role of the neuroinflammation in the viral etiology of AD.
The Neuroinflammation in the Pathogenesis of AD
Microglia, astrocytes and oligodendrocyte are the main glia cells in the brain (96). These cells control the microenvironment of CNS neurons and are crucial in synaptic remodeling, tissue repair, and neuronal survival following CNS injury (97–100). Neuroinflammation stimulates the local activation of glia cells surrounding damaged or infected neurons. Upon activation, microglia and astrocytes cells undergo a series of morphological and functional changes and acquire a “reactive” phenotype (101). For instance, activated microglia migrate to the site of infection/lesion and phagocytose cellular debris. In addition, activated glia cells release a wide range of proinflammatory mediators, including IL-1β, IL-6, TNF-α, aimed at preventing further damage to the CNS (102). Astrocytes can also be activated by the release of inflammatory mediators from microglia (103). The interaction between activated microglia and astrocytes plays a key role in the neuroinflammatory response (104, 105). While the activation of glia cells is part of a protective immune response to tissue injury or infections, its long-lasting and uncontrolled activation can be deleterious, causing chronic inflammation, and neurodegeneration.
In the early pathogenesis of AD, it has been suggested that microglia activation reduces the accumulation of Aβ in the brain by increasing its phagocytosis, clearance, and degradation (106, 107). Microglia bind soluble Aβ oligomers and fibrils via receptors, including class A scavenger receptor A1, toll-like receptors (e.g., TLR2, TLR4) and coreceptor CD14, and subsequently phagocytose and clear Aβ. However, the persistent microglia activation stimulated by the binding of microglia to Aβ can increase the production of inflammatory mediators and reactive oxygen species (ROS), which amplifies the neuroinflammatory response in the brain (108). Using transgenic PS1-APP mice, an established model of AD, Hickman et al. demonstrated that the continuous production of proinflammatory cytokines reduces the expression of the phagocytosis receptors expressed in microglia and alters their functions (109). The sustained activation of microglia cells has been shown to exacerbate both amyloid and tau pathology and may serve as a link in the pathogenesis of AD (110, 111). Indeed, the prolonged activation results in an impaired clearance of Aβ and increased accumulation in the CNS (112). Moreover, the increase of inflammatory cytokines expression contributes to tau hyperphosphorylation and neuronal loss. In particular, IL-6 has been shown to induce tau hyperphosphorylation in rat hippocampal neurons (113). Ultimately, the prolonged priming of microglia to an inflammatory state causes neuronal damage and neurodegeneration and contributes to the progression of AD.
Moreover, the activation of pattern recognition receptors (PRRs) expressed by microglia can influence the neuroinflammation and AD pathology. PRRs are either membrane-bound [e.g., Toll-like receptors (TLRs)] or intracellular, such as the nucleotide-binding domain and leucine-rich repeat-containing receptors (NLRs), AIM2-like receptors (ALRs), and the tripartite motif-containing (TRIM) family member pyrin (114). The sensing of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) by cytosolic PRRs results in the assembly of the inflammasome (115). This multiprotein complex is involved in the initiation of inflammatory responses (e.g., production of interleukins IL-1β and IL-18) and in the induction of pyroptosis, a highly-pyrogenic inflammatory form of cell death (116, 117). Several inflammasomes, including the pyrin domain-containing 3 (NLRP3) inflammasome, have been shown to play a crucial role in the development and progression of Aβ plaque formation and tau-induced pathology (118–120). Interestingly, Ising et al. demonstrated that the accumulation and deposition of Aβ and NFT formation are detected by cytosolic PRRs, triggering NLRP3 inflammasome activation in microglia (118). This mechanism ultimately exacerbates the neuroinflammation and AD pathology.
A study from Wyss-Coray et al. demonstrated that reactive mouse astrocytes internalize and degrade Aβ, suggesting a neuroprotective role of these cells in AD (121). However, other studies showed that reactive astrocytes have neurotoxic properties and their persistent inflammatory phenotype contribute to the loss of functions (122, 123). Astrocyte dysfunction has been shown to result in an increase of pro-inflammatory mediators release, decrease of glutamate uptake, loss of synapses, and ultimately cognitive deficits in AD (124). A number of recent reviews further examine particular aspects of the role of microglia and astrocytes in the neuroinflammatory response in AD, including phenotypical and functional changes, genetic variants of the innate microglial immune receptor TREM2, which has been associated with an increased risk of LOAD (105, 125–129).
Several rodent models of neuroinflammation have been developed to study the inflammatory hypothesis of AD and to test potential new therapeutics. Ideally, these models should demonstrate early chronic neuroinflammation prior to the formation of Aβ plaques and NFT. An interesting model is polyI:C-induced neuroinflammation, which consists of the systemic injection of polyI:C, a synthetic double-stranded RNA that induces an innate immune analogous to an acute viral infection. Systemic injection of polyI:C on gestational day 17 mice increased IL-1 and IL-6 levels in the plasma and brains of treated animals compared to controls, starting from 3 weeks post-injection and was sustained throughout aging (130). This prenatal immune challenge also demonstrated a significant, age-dependent increase of tau hyperphosphorylation starting from 3 months of age and of APP, starting as late as 12 months of age. Cognitive impairment was detected in mice 20 months post-injection. Importantly, a second systemic immune challenge performed when mice were fully mature, resulted in exacerbated AD neuropathologies, such as APP deposition, tau aggregation, microglia activation and astrogliosis in brain tissues. The authors concluded that chronic inflammation induces AD-like pathologies in mice in an age-dependent manner. The streptozotocin-induced neuroinflammation model relies on peripheral injection of the glucosamine-nitrosourea compound streptozotocin (STZ). STZ induces oxidative stress and impairs brain glucose metabolism associated with insulin signal transduction failure. This drug produces pancreatic insulitis and causes diabetes mellitus in mice (131). In addition, the intracerebroventricular injection of STZ in rats caused chronic inflammation (astrocytosis and microgliosis) accompanied by neuronal loss and neurodegenerative lesions in the brain (132, 133). In this model, neuroinflammation was detected 1-week post-injection and tau hyperphosphorylation and amyloid deposition were detected as early as 3 and 12 weeks, respectively (134). Finally, p25 and 3xTg transgenic mouse models are two additional and relevant neuroinflammatory models. First, p25 transgenic mice overexpress human p25, an activator of the cyclin-dependent kinase 5 (cdk5) that regulates cell cycle and plays a key role in brain development (135). In this model, astrogliosis and the increased expression of proinflammatory cytokines and chemokines (IL-1, TNF-α, TGF-β, and MIP-1α) were detected at 1 week of induction of p25 expression (136). Microgliosis, tau hyperphosphorylation and amyloid pathology were detected only at 4 and 8 weeks, and cognitive deficits were observed within 6 weeks after p25 induction. The authors suggested that in p25 transgenic mice, neuroinflammation is an early event in the pathogenesis of AD and is independent of β-amyloid and tau phosphorylation. Second, the triple transgenic mouse of AD (3xTg) is the only transgenic model to express three major genes (PS1M146V, APPSwe, and tauP301L) associated with EOAD and behavioral and neurological changes that are observed in the human form (137, 138). This model exhibits both extracellular Aβ deposits and hyperphosphorylated tau tangles. In addition, it shows CNS inflammation such as astrogliosis, activated microglia and neurodegeneration including synapse loss, already at pre-symptomatic stage (5–20 post-natal weeks) (139). Systemic injection of polyI:C exacerbated neuroinflammation and AD pathology in 3Txg mice (130). Overall, these two neuroinflammatory mouse models provide similar chronological progression of AD pathologies as seen in humans. Therefore, they are considered suitable models to study the inflammatory hypothesis of AD and to discover effective therapeutic agents.
So far in mouse AD models, therapeutic approaches targeting different components of the neuroinflammatory response have shown promising results. As microglia activation is a key step in the neuroinflammatory pathogenesis of AD, depletion of these cells in adult mice has been shown to significantly reduce tau progression (140). Recently, Spangenberg et al. demonstrated the elimination of more than 95% of microglia in an AD mouse model using the drug PLX3397. This molecule inhibits colony-stimulating factor 1 receptor (CSF1R), which microglia need to survive. Feeding animals with this orally bioavailable and brain-penetrant inhibitor resulted in the reduced accumulation of Aβ inside neurons and prevented the formation of neuritic plaques in the brain (141). Moreover, cromolyn sodium, an FDA-approved drug for the treatment of asthma, has been shown to inhibit Aβ aggregation both in vitro and in vivo (142). This drug also has proved successful in shifting microglia from a proinflammatory/neurotoxic to a pro-phagocytic/neuroprotective activation state in transgenic mice, thus enhancing efficient uptake of Aβ (143). Finally, specific targeting of TLR2 has been proposed to be an important step for the attenuation of microglia activation. Indeed, neutralizing antibodies against TLR2 blocked Aβ-induced expression of proinflammatory cytokines in mouse primary microglia (144). TLR2 involvement in microglia activation was further confirmed using TLR2 deficient mouse microglia. In this study, the authors suggested that Aβ peptide binds to the TLR2 receptor expressed on microglia and primed these cells to a proinflammatory state via TLR2 signaling pathway activation. The α-melanocyte-stimulating hormone (MSH), a neuropeptide and member of the melanocortin family, has demonstrated anti-inflammatory properties in the treatment of CNS inflammatory disease such as experimental autoimmune encephalomyelitis (EAE) (145). Treatment of primary rat microglia cultures with an analog of MSH, the synthetic peptide NDP-MSH, resulted in the inhibition of TLR2- and TLR4-mediated TNF-α release (146). In addition, NSD-MSH treatment reduced the levels of Aβ and tau phosphorylation, inflammation, neuronal apoptosis, and improved cognitive functions in 3Txg transgenic mice compared to control groups (147). All together, these findings suggested that NDP-MSH promotes the development of microglia into an anti-inflammatory M2-like phenotype. Potentially, this molecule could be beneficial in preventing the neuroinflammation associated with AD. While anti-inflammatory therapy, such as NSAIDs, has been shown to reduce AD pathology in animal models, it has not been proven to be effective in human clinical trials (148–150). However, the inhibition of specific proinflammatory cytokines represents a more promising therapeutic strategy for AD. Blocking antibodies that bind to IL-1β have been shown to attenuate tau pathology and microglia activation in the brain of 3Txg AD mice (151). In addition, the administration of a monoclonal antibody against TNF-α (Infliximab) has been demonstrated to reduce amyloid plaques and tau phosphorylation in APP/PS1 transgenic mice (152).
HSV1-Induced Neuroinflammation
So far, most studies to investigate neuroinflammation in AD pathogenesis have focused on understanding the mechanisms underlying glia cell activation and downstream inflammatory events in the late stages of AD (e.g., Aβ accumulation). Very little attention has been given to how glia cells initially become activated and whether in the early stages of AD, an infectious agent may trigger their activation.
The role of HSV1 infection in the activation of glia cells has been well-studied in the context of HSE. HSV1-infected primary microglia and astrocytes produce high levels of proinflammatory cytokines, including IL-6 (153, 154). Moreover, the anti-inflammatory cytokine IL-10 inhibits the production of HSV1-induced inflammation in human microglia (155). These results suggest that microglia are key mediators in the neuroinflammatory response to HSV1 infection. Several in vivo studies reported that HSV1-induced inflammation in mouse microglia and astrocytes is mediated by TLR2 signaling pathway (156–158). Moreover, Marques et al. showed that microglia remained activated in the brain of HSV1-infected mice at 30 dpi, a time when neither infectious virus nor viral replication could be detected (159). The authors suggested that persistent microglia activation may contribute to neuronal damage and long-term neurological sequelae observed in HSE patients. Recurrent HSV1 infections of mice have been proposed to be one of the main causes of prolonged glia cell activation and neuroinflammation in the CNS, leading to long-term CNS damage (83, 84). Multiple HSV1 reactivation events in the brain induced gliosis and increased brain levels of IL-1β and IL-6. These events were also accompanied by the increased expression of neurodegenerative markers (e.g., Aβ and tau phosphorylation levels) and cognitive deficits. It was recently proposed that CNS asymptomatic HSV1 reactivation events can occur in recurrent HSV1 infections and lead to a mild and chronic inflammation of the brain with a non-fatal outcome. While microglia and astrocytes not only have pro-inflammatory properties, they also orchestrate the antiviral response to HSV1, such as the production of type I IFN (160, 161). The production of type I IFN in microglia and astrocytes is mediated by TLR3-dependent mechanisms (162). Defects in the TLR3-type I IFN signaling pathway were correlated with severe HSE in some patients (163). While a large number of humans are infected with HSV1, only a small minority ever experience HSE. This fact suggests that while glia cells efficiently contribute to antiviral defense in HSV1 infection, they cause limited CNS symptoms (from asymptomatic reactivations to mild HSE). However, even though most people experience recurrent HSV1 infections with no to mild symptoms, recurrent infections may actually might predispose them more to develop persistent inflammation in the brain and subsequent neurodegenerative diseases.
Herpesvirus-Induced Neuroinflammation in the PNS: Where It All Starts?
A second aspect of the infectious hypothesis for AD is that the immune response to the infection must be centered in the brain. One possible complication is the fact that herpesviruses, such as HSV1, are primary invaders of the PNS, not the CNS. However, because the PNS and CNS are intimately connected, it may be that PNS responses to infection will have direct and indirect effects in the CNS. This possibility should be further examined in regards to AD pathogenesis. Several animal models of peripheral HSV1 infection already exist such as the rabbit and mouse eye models and the guinea pig models (164, 165). These models have been widely used to study multiple aspects of HSV1 biology, including viral reactivation in the PNS (166–170). However, they have rarely been used to investigate the direct role of herpesvirus-induced neuroinflammatory responses in the PNS in initiating AD in the brain.
Recently, Laval et al. dissected the mechanisms of herpesvirus-induced neuroinflammatory responses in the PNS in vivo and its direct impact on the CNS (171, 172). This work was done using pseudorabies virus (PRV), an alpha herpesvirus distantly related to HSV1. PRV has been used extensively to study the mechanisms of neuronal spread from the site of primary infection to the PNS and CNS, neuro-circuitry, and immune responses to herpesvirus infections (173). PRV is a swine alpha herpesvirus, which can infect many other animal species, including mice and rats (174). PRV infection has much in common with HSV1 infection. Using a mouse footpad inoculation model, the authors demonstrated that a virulent strain of PRV induced a specific neuroinflammatory response (171). There was a strong correlation between the amount of infectious PRV detected in the PNS neurons and the production of two specific pro-inflammatory cytokines, IL-6, and granulocyte colony-stimulating factor (G-CSF) in a wide array of other tissues. Recently, Laval et al. further characterized the early events of the neuroinflammatory response to PRV infection in mice. They demonstrated that peripheral PRV infection primes PNS neurons to a state of inflammation very early after infection (within hours) (172). This priming resulted in an increase of proinflammatory cytokines (IL-6 and G-CSF) levels in the PNS and CNS simultaneously, and without active viral replication in the brain. They also showed that TLR2 and IFN type I play crucial roles in modulating this early neuroinflammatory response after peripheral infection. This long-distance immune signaling from PNS to CNS during herpesvirus infection was further supported by a study with vesicular stomatitis virus demonstrating that virus infection could activate long-distance interferon signaling in uninfected regions of the brain (175). The footpad inoculation model was useful for characterizing herpesvirus-induced neuroinflammatory responses in vivo because it was possible to separate local events at the site of infection and events in the PBS from those occurring in the CNS in both space and time. Infection in this model travels a greater distance from the periphery to the PNS and CNS neurons via the sciatic nerve and to the CNS via the spinal cord. As a result, it provides a clear assessment of viral kinetics and associated immunopathological processes initiated in the PNS and its potential role in long-distance immune-signaling to the CNS (176). These results provided a solid foundation for the role of peripheral herpesvirus infection in the global inflammatory priming of the brain. Future work should focus specifically on the role of HSV1-induced neuroinflammation in the PNS and its long-term neurological effects on the CNS.
Conclusions and Future Perspectives
AD represents a major global health challenge with serious economical and personal impact on those affected and their families. Despite more than 3 decades of research on AD, there is no effective therapy to prevent or cure the disease. This fact makes it all the more necessary to better understand the pathogenesis of AD and design novel therapeutics. While the amyloid cascade hypothesis has been a central focus of attention, the idea that a common infectious agent could contribute to the development of AD is gaining support (the infectious hypothesis). There are two critical elements of the infectious hypothesis: (1) the infectious agent must be a common human infection and must persist in the host for years or must periodically infect the host and (2) the immune response to this agent involves the nervous system. HSV1 and its remarkable capacity to establish a reactivable infection in the nervous system is attracting more attention for a possible role in AD. It is noteworthy that treatment with specific anti HSV1 antiviral agents are reported to efficiently reduce AD pathology in both in vitro and in vivo systems. While HSV1 might a contributing factor in AD, additional factors (e.g., host and environmental factors) are likely to be involved in the sporadic manifestation of AD (177). Moreover, neuroinflammation has now emerged as an important component of AD pathogenesis. Several anti-inflammatory therapies have been tested and showed promising results in AD mouse models. Their efficacy in humans awaits clinical trials. The current infectious/inflammation hypothesis of AD suggests that recurrent HSV1 infections or periodic reinfections may cause persistent inflammation and long-term CNS damage leading to neurodegeneration. Reactivation of latent infections in the PNS may be sufficient to cause CNS inflammation without the presence of virus in the brain. Another idea is that latent infections can occur in the CNS and that reactivation of these latent infections may be important. While the neuroinflammatory response occurred before the appearance of Aβ plaques in the CNS, future work should also examine this response at earlier stages of the disease. Our lab demonstrated how herpesvirus infection of the PNS triggers specific inflammatory responses in both the PNS and CNS within hours post-infection. We therefore propose a model focused on HSV1 infection/reactivation in the PNS that, over time, provides the trigger for the initiation and development of AD (Figure 1). Targeting the PNS might represent a novel therapeutic approach to prevent AD. For instance, preventing HSV1 reactivation from PNS latency sites (e.g., TG) could efficiently stop the spread of new progeny virions from the TG to the brain. It might also reduce the priming of PNS neurons by infection and subsequent initiation of CNS neuroinflammation. Ultimately, this strategy might prevent the establishment of HSV1 latency and halt the development of AD pathology.
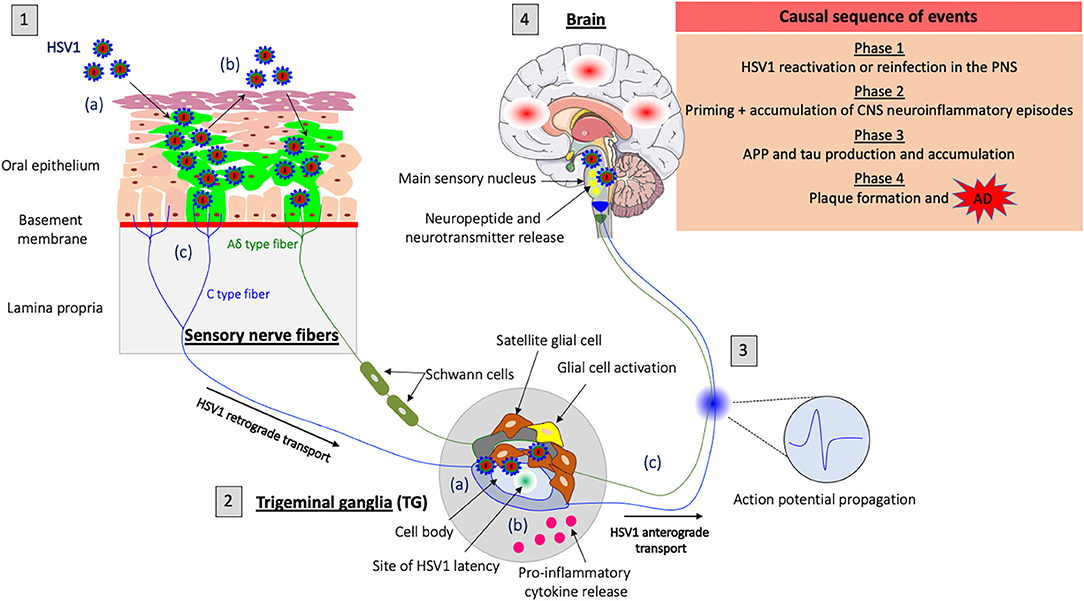
Figure 1. Model of HSV1-induced neuroinflammation in the PNS as a trigger for Alzheimer's disease (AD). (1) HSV1 first replicates in the oral mucosa epithelial cells; (a) HSV1 infection; (b) Viral spread within the mucosal epithelium and viral shedding; (c) HSV1 enters nerve endings of the PNS, including those coming from the trigeminal ganglia (TG) and spreads in the retrograde direction in axons to the ganglia. (2) HSV1 initiates a productive infection in TG neurons; (a) HSV1 replicates in cell bodies of TG neurons; (b) HSV1 induces a neuroinflammatory response (e.g., proinflammatory cytokine release and glial cell activation) in the PNS following primary infection or reactivation from latency; (c) New progeny virions can further spread in the anterograde direction and infect the CNS. (3) Synaptic transmission of the inflammatory response accompanied by the release of neuropeptides, neurotransmitters and the propagation of specific action potentials from the TG to the main sensory nucleus located in the brainstem. (4) Priming of the brain to a similar inflammatory state following transduction of inflammatory stimuli from the PNS. HSV1 reactivation or new primary infection of the PNS will thus enhance inflammatory responses in the brain. These events are likely to trigger the production of APP and tau proteins in the brain and repeated inflammation will lead to the accumulation of these proteins and the development of Aβ plaques that cause AD.
Author Contributions
KL suggested the idea and wrote the review. LWE critically reviewed, corrected, and guided the completion of the review. All authors read and approved the final manuscript.
Funding
KL and LWE were thankful to the National Institute of Neurological Disorders and Stroke (NINDS) grants R01 NS33506 and NS060699 that funded this research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Patterson C. World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers. London: Alzheimer's Disease International (2018).
2. Hippius H, Neundörfer G. The discovery of Alzheimer's disease. Dialogues Clin Neurosci. (2003) 5:101–8. doi: 10.31887/DCNS.2003.5.1/hhippius
3. Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. (2006) 368:387–403. doi: 10.1016/s0140-6736(06)69113-7
4. Lyketsos CG, Carrillo MC, Ryan JM, Khachaturian AS, Trzepacz P, Amatniek J, et al. Neuropsychiatric symptoms in Alzheimer's disease. Alzheimers Dement. (2011) 7:532–9. doi: 10.1016/j.jalz.2011.05.2410
5. Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. (2019) 15:565–81. doi: 10.1038/s41582-019-0244-7
6. Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Genl Psychiatry. (2006) 63:168–74. doi: 10.1001/archpsyc.63.2.168
7. Wingo TS, Lah JJ, Levey AI, Cutler DJ. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch Neurol. (2012) 69:59–64. doi: 10.1001/archneurol.2011.221
8. Piaceri I, Nacmias B, Sorbi S. Genetics of familial and sporadic Alzheimer's disease. Front Biosci. (2013) 5:167–77. doi: 10.2741/e605
9. Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early-onset Alzheimer's disease revisited. Alzheimers Dement. (2016) 12:733–48. doi: 10.1016/j.jalz.2016.01.012
10. Dorszewska J, Prendecki M, Oczkowska A, Dezor M, Kozubski W. Molecular basis of familial and sporadic Alzheimer's disease. Curr Alzheimer Res. (2016) 13:952–63. doi: 10.2174/1567205013666160314150501
11. Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. (2013) 9:63–75.e62. doi: 10.1016/j.jalz.2012.11.007
12. Prince M, Wimo A, Guerchet M, Ali G, Wu Y, Prina M. World Alzheimer's Report 2015, The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends. Alzheimer's Disease International (2015).
13. Birks JS, Harvey RJ. Donepezil for dementia due to Alzheimer's disease. Cochrane Database Syst Rev. (2018) 6:CD001190. doi: 10.1002/14651858.CD001190.pub3
14. Adlimoghaddam A, Neuendorff M, Roy B, Albensi BC. A review of clinical treatment considerations of donepezil in severe Alzheimer's disease. CNS Neurosci Ther. (2018) 24:876–88. doi: 10.1111/cns.13035
15. O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. (2011) 34:185–204. doi: 10.1146/annurev-neuro-061010-113613
16. Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. (2006) 26:7212. doi: 10.1523/JNEUROSCI.1450-06.2006
17. Zhou Z-D, Chan CH-S, Ma Q-H, Xu X-H, Xiao Z-C, Tan E-K. The roles of amyloid precursor protein (APP) in neurogenesis: implications to pathogenesis and therapy of Alzheimer disease. Cell Adh Migr. (2011) 5:280–92. doi: 10.4161/cam.5.4.16986
18. Plummer S, Van den Heuvel C, Thornton E, Corrigan F, Cappai R. The neuroprotective properties of the amyloid precursor protein following traumatic brain injury. Aging Dis. (2016) 7:163–79. doi: 10.14336/AD.2015.0907
19. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. (1992) 256:184–5. doi: 10.1126/science.1566067
20. Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. (2006) 26:6011–8. doi: 10.1523/jneurosci.1189-06.2006
21. Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer's disease. Mol Brain. (2011) 4:3. doi: 10.1186/1756-6606-4-3
22. Dimitrov M, Alattia JR, Lemmin T, Lehal R, Fligier A, Houacine J, et al. Alzheimer's disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat Commun. (2013) 4:2246. doi: 10.1038/ncomms3246
23. Dai MH, Zheng H, Zeng LD, Zhang Y. The genes associated with early-onset Alzheimer's disease. Oncotarget. (2018) 9:15132–43. doi: 10.18632/oncotarget.23738
24. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. (2010) 330:1774. doi: 10.1126/science.1197623
25. Lee CYD, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neuro Trans. (2010) 117:949–60. doi: 10.1007/s00702-010-0433-4
26. Pflanzner T, Janko MC, André-Dohmen B, Reuss S, Weggen S, Roebroek AJ, et al. LRP1 mediates bidirectional transcytosis of amyloid-β across the blood-brain barrier. Neurobiol Aging. (2011) 32:2323.e1–e11. doi: 10.1016/j.neurobiolaging.2010.05.025
27. Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ, et al. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. (2014) 10:1761–75. doi: 10.4161/auto.29647
28. Arbel-Ornath M, Hudry E, Eikermann-Haerter K, Hou S, Gregory JL, Zhao L, et al. Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer's disease mouse models. Acta Neuropathol. (2013) 126:353–64. doi: 10.1007/s00401-013-1145-2
29. Jiang T, Zhang YD, Gao Q, Zhou JS, Zhu XC, Lu H, et al. TREM1 facilitates microglial phagocytosis of amyloid beta. Acta Neuropathol. (2016) 132:667–83. doi: 10.1007/s00401-016-1622-5
30. Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer's disease. Brain Pathol. (2008) 18:240–52. doi: 10.1111/j.1750-3639.2008.00132.x
31. Yoon S-S, Jo SA. Mechanisms of amyloid-β peptide clearance: potential therapeutic targets for Alzheimer's disease. Biomol Ther. (2012) 20:245–55. doi: 10.4062/biomolther.2012.20.3.245
32. Yu Y, Ye RD. Microglial Aβ receptors in Alzheimer's disease. Cell Mol Neurobiol. (2015) 35:71–83. doi: 10.1007/s10571-014-0101-6
33. Bakker EN, Bacskai BJ, Arbel-Ornath M, Aldea R, Bedussi B, Morris AW, et al. Lymphatic clearance of the brain: perivascular, paravascular and significance for neurodegenerative diseases. Cell Mol Neurobiol. (2016) 36:181–94. doi: 10.1007/s10571-015-0273-8
34. Guerrero-Muñoz MJ, Gerson J, Castillo-Carranza DL. Tau oligomers: the toxic player at synapses in Alzheimer's disease. Front Cell Neurosci. (2015) 9:464. doi: 10.3389/fncel.2015.00464
35. Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem. (2003) 85:115–22. doi: 10.1046/j.1471-4159.2003.01642.x
36. Alonso Adel C, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci USA. (2006) 103:8864–9. doi: 10.1073/pnas.0603214103
37. Chételat G. Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nat Rev Neurol. (2013) 9:123–4. doi: 10.1038/nrneurol.2013.21
38. Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease. Acta Neuropathol Comm. (2014) 2:135. doi: 10.1186/s40478-014-0135-5
39. Savory J, Ghribi O, Herman MM. Is amyloid beta-peptide neurotoxic or neuroprotective and what is its role in the binding of metal ions? Neurobiol Aging. (2002) 23:1089–92. doi: 10.1016/s0197-4580(02)00037-4
40. Chételat G, La Joie R, Villain N, Perrotin A, de La Sayette V, Eustache F, et al. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer's disease. Neuroimage Clin. (2013) 2:356–65. doi: 10.1016/j.nicl.2013.02.006
42. Castellani RJ, Perry G. Pathogenesis and disease-modifying therapy in Alzheimer's disease: the flat line of progress. Arch Med Res. (2012) 43:694–8. doi: 10.1016/j.arcmed.2012.09.009
43. Beason-Held LL, Goh JO, An Y, Kraut MA, O'Brien RJ, Ferrucci L, et al. Changes in brain function occur years before the onset of cognitive impairment. J Neurosci. (2013) 33:18008–14. doi: 10.1523/JNEUROSCI.1402-13.2013
44. Vishnu VY. Can tauopathy shake the amyloid cascade hypothesis? Nat Rev Neurol. (2013) 9:356. doi: 10.1038/nrneurol.2013.21-c1
45. Itzhaki RF. Herpes simplex virus type 1 and Alzheimer's disease: increasing evidence for a major role of the virus. Front Aging Neurosci. (2014) 6:202. doi: 10.3389/fnagi.2014.00202
46. Harris SA, Harris EA. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer's disease. J Alzheimers Dis. (2015) 48:319–53. doi: 10.3233/jad-142853
47. Itzhaki RF. Herpes simplex virus type 1 and Alzheimer's disease: possible mechanisms and signposts. Faseb J. (2017) 31:3216–26. doi: 10.1096/fj.201700360
48. Sochocka M, Zwolińska K, Leszek J. The infectious etiology of Alzheimer's disease. Curr Neuropharmacol. (2017) 15:996–1009. doi: 10.2174/1570159x15666170313122937
49. Itzhaki RF. Corroboration of a major role for herpes simplex virus type 1 in Alzheimer's disease. Front Aging Neurosci. (2018) 10:324. doi: 10.3389/fnagi.2018.00324
50. Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, et al. Multiscale analysis of independent Alzheimer's cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. (2018) 99:64–82.e67. doi: 10.1016/j.neuron.2018.05.023
51. Wald A, Corey L. Persistence in the population: epidemiology, transmission. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press (2007).
52. Looker KJ, Magaret AS, May MT, Turner KM, Vickerman P, Gottlieb SL, et al. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS ONE. (2015) 10:e0140765. doi: 10.1371/journal.pone.0140765
53. Enquist LW, Husak PJ, Banfield BW, Smith GA. Infection and spread of alphaherpesviruses in the nervous system. Adv Virus Res. (1998) 51:237–347. doi: 10.1016/s0065-3527(08)60787-3
54. Grinde B. Herpesviruses: latency and reactivation - viral strategies and host response. J Oral Microbiol. (2013) 5. doi: 10.3402/jom.v5i0.22766
55. Cunningham AL, Diefenbach RJ, Miranda-Saksena M, Bosnjak L, Kim M, Jones C, et al. The cycle of human herpes simplex virus infection: virus transport and immune control. J Infect Dis. (2006) 194(Suppl. 1):S11–18. doi: 10.1086/505359
56. Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press (2007).
57. Scheld WM, Whitley RJ, Marra CM. Infection of the Central Nervous System. 4th ed. Philadelphia, PA: Wolters Kluwer Health (2014). p. 907.
58. Ak AK, Mendez MD. Herpes simplex encephalitis. In: StatPearls. Treasure Island, FL: StatPearls Publishing LLC. (2020).
59. Fraser NW, Lawrence WC, Wroblewska Z, Gilden DH, Koprowski H. Herpes simplex type 1 DNA in human brain tissue. Proc Natl Acad Sci USA. (1981) 78:6461–5. doi: 10.1073/pnas.78.10.6461
60. Jamieson GA, Maitland NJ, Wilcock GK, Craske J, Itzhaki RF. Latent herpes simplex virus type 1 in normal and Alzheimer's disease brains. J Med Virol. (1991) 33:224–7. doi: 10.1002/jmv.1890330403
61. Jamieson GA, Maitland NJ, Wilcock GK, Yates CM, Itzhaki RF. Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J Pathol. (1992) 167:365–8. doi: 10.1002/path.1711670403
62. Baringer JR, Pisani P. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol. (1994) 36:823–9. doi: 10.1002/ana.410360605
63. Olsson J, Lövheim H, Honkala E, Karhunen PJ, Elgh F, Kok EH. HSV presence in brains of individuals without dementia: the TASTY brain series. Dis Model Mech. (2016) 9:1349–55. doi: 10.1242/dmm.026674
64. Roizman B, Knipe DM, Whitley R. Herpes simplex viruses. In: Knipe DM, Howley PM, et al., editors. Fields Virology, 5th Edn. Philadelphia, PA: Lippincott, Williams & Wilkins (2007). p. 2501–601.
65. Jennische E, Eriksson CE, Lange S, Trybala E, Bergström T. The anterior commissure is a pathway for contralateral spread of herpes simplex virus type 1 after olfactory tract infection. J Neurovirol. (2015) 21:129–47. doi: 10.1007/s13365-014-0312-0
66. Steiner I. Herpes simplex virus encephalitis: new infection or reactivation? Curr Opin Neurol. (2011) 24:268–74. doi: 10.1097/WCO.0b013e328346be6f
67. Kastrukoff LF, Lau AS, Thomas EE. The effect of mouse strain on herpes simplex virus type 1 (HSV-1) infection of the central nervous system (CNS). Herpesviridae. (2012) 3:4. doi: 10.1186/2042-4280-3-4
68. Margolis TP, LaVail JH, Setzer PY, Dawson CR. Selective spread of herpes simplex virus in the central nervous system after ocular inoculation. J Virol. (1989) 63:4756–61. doi: 10.1128/jvi.63.11.4756-4761.1989
69. LaVail JH, Topp KS, Giblin PA, Garner JA. Factors that contribute to the transneuronal spread of herpes simplex virus. J Neurosci Res. (1997) 49:485–96.
70. Chen SH, Yao HW, Huang WY, Hsu KS, Lei HY, Shiau AL, et al. Efficient reactivation of latent herpes simplex virus from mouse central nervous system tissues. J Virol. (2006) 80:12387–92. doi: 10.1128/jvi.01232-06
71. Yao HW, Ling P, Chen SH, Tung YY, Chen SH. Factors affecting herpes simplex virus reactivation from the explanted mouse brain. Virology. (2012) 433:116–23. doi: 10.1016/j.virol.2012.07.018
72. Yao HW, Ling P, Tung YY, Hsu SM, Chen SH. In vivo reactivation of latent herpes simplex virus 1 in mice can occur in the brain before occurring in the trigeminal ganglion. J Virol. (2014) 88:11264–70. doi: 10.1128/jvi.01616-14
73. Doll JR, Thompson RL, Sawtell NM. Infectious herpes simplex virus in the brain stem is correlated with reactivation in the trigeminal ganglia. J Virol. (2019) 93:e02209-18. doi: 10.1128/jvi.02209-18
74. Mori I, Kimura Y, Naiki H, Matsubara R, Takeuchi T, Yokochi T, et al. Reactivation of HSV-1 in the brain of patients with familial Alzheimer's disease. J Med Virol. (2004) 73:605–11. doi: 10.1002/jmv.20133
75. Mori I, Yokochi T, Koide N, Sugiyama T, Yoshida T, Kimura Y, et al. PCR search for the herpes simplex virus type 1 genome in brain sections of patients with familial Alzheimer's disease. J Clin Microbiol. (2004) 42:936–7. doi: 10.1128/jcm.42.2.936-937.2004
76. Wozniak MA, Shipley SJ, Combrinck M, Wilcock GK, Itzhaki RF. Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer's disease patients. J Med Virol. (2005) 75:300–6. doi: 10.1002/jmv.20271
77. Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J Pathol. (2009) 217:131–8. doi: 10.1002/path.2449
78. Letenneur L, Peres K, Fleury H, Garrigue I, Barberger-Gateau P, Helmer C, et al. Seropositivity to herpes simplex virus antibodies and risk of Alzheimer's disease: a population-based cohort study. PLoS ONE. (2008) 3:e3637. doi: 10.1371/journal.pone.0003637
79. Féart C, Helmer C, Fleury H, Béjot Y, Ritchie K, Amouyel P, et al. Association between IgM anti-herpes simplex virus and plasma amyloid-beta levels. PLoS ONE. (2011) 6:e29480. doi: 10.1371/journal.pone.0029480
80. Lovheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F. Reactivated herpes simplex infection increases the risk of Alzheimer's disease. Alzheimers Dement. (2015) 11:593–9. doi: 10.1016/j.jalz.2014.04.522
81. Wu D, Wang C, Pang P, Kong H, Lin Z, Wang H, et al. The association between herpes simplex virus type 1 infection and Alzheimer's disease. J Clin Neurosci. (2020) 82(Pt A):63–70. doi: 10.1016/j.jocn.2020.10.044
82. Marcocci ME, Napoletani G, Protto V, Kolesova O, Piacentini R, Li Puma DD, et al. Herpes simplex virus-1 in the brain: the dark side of a sneaky infection. Trends Microbiol. (2020) 28:808–20. doi: 10.1016/j.tim.2020.03.003
83. Martin C, Aguila B, Araya P, Vio K, Valdivia S, Zambrano A, et al. Inflammatory and neurodegeneration markers during asymptomatic HSV-1 reactivation. J Alzheimers Dis. (2014) 39:849–59. doi: 10.3233/jad-131706
84. De Chiara G, Piacentini R, Fabiani M, Mastrodonato A, Marcocci ME, Limongi D, et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. (2019) 15:e1007617. doi: 10.1371/journal.ppat.1007617
85. Itzhaki RF, Lathe R. Herpes viruses and senile dementia: first population evidence for a causal link. J Alzheimers Dis. (2018) 64:363–6. doi: 10.3233/jad-180266
86. Tzeng NS, Chung CH, Lin FH, Chiang CP, Yeh CB, Huang SY, et al. Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections-a nationwide, population-based cohort study in Taiwan. Neurotherapeutics. (2018) 15:417–29. doi: 10.1007/s13311-018-0611-x
87. Wozniak MA, Frost AL, Preston CM, Itzhaki RF. Antivirals reduce the formation of key Alzheimer's disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS ONE. (2011) 6:e25152. doi: 10.1371/journal.pone.0025152
88. Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer's disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. (2018) 99:56–63.e53. doi: 10.1016/j.neuron.2018.06.030
89. Cairns DM, Rouleau N, Parker RN, Walsh KG, Gehrke L, Kaplan DL. A 3D human brain–like tissue model of herpes-induced Alzheimer's disease. Sci Adv. (2020) 6:eaay8828. doi: 10.1126/sciadv.aay8828
90. De Chiara G, Marcocci ME, Civitelli L, Argnani R, Piacentini R, Ripoli C, et al. APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS ONE. (2010) 5:e13989. doi: 10.1371/journal.pone.0013989
91. Piacentini R, Civitelli L, Ripoli C, Marcocci ME, De Chiara G, Garaci E, et al. HSV-1 promotes Ca2+ -mediated APP phosphorylation and Abeta accumulation in rat cortical neurons. Neurobiol Aging. (2011) 32:2323.e13–26. doi: 10.1016/j.neurobiolaging.2010.06.009
92. Grimaldi LM, Zappalà G, Iemolo F, Castellano AE, Ruggieri S, Bruno G, et al. A pilot study on the use of interferon beta-1a in early Alzheimer's disease subjects. J Neuroinflamm. (2014) 11:30. doi: 10.1186/1742-2094-11-30
93. Tarkowski E, Andreasen N, Tarkowski A, Blennow K. Intrathecal inflammation precedes development of Alzheimer's disease. J Neurol Neurosurg Psychiatry. (2003) 74:1200–5. doi: 10.1136/jnnp.74.9.1200
94. Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, et al. Neuroinflammation and neuronal loss precede Aβ plaque deposition in the hAPP-J20 mouse model of Alzheimer's disease. PLoS ONE. (2013) 8:e59586. doi: 10.1371/journal.pone.0059586
95. Bronzuoli MR, Iacomino A, Steardo L, Scuderi C. Targeting neuroinflammation in Alzheimer's disease. J Inflamm Res. (2016) 9:199–208. doi: 10.2147/JIR.S86958
96. Jäkel S, Dimou L. Glial cells and their function in the adult brain: a journey through the history of their ablation. Front Cell Neurosci. (2017) 11:24. doi: 10.3389/fncel.2017.00024
97. Kimelberg HK. Functions of mature mammalian astrocytes: a current view. Neuroscientist. (2010) 16:79–106. doi: 10.1177/1073858409342593
98. Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci. (2013) 16:668–76. doi: 10.1038/nn.3390
99. Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol. (2015) 36:605–13. doi: 10.1016/j.it.2015.08.008
100. Fu H, Zhao Y, Hu D, Wang S, Yu T, Zhang L. Depletion of microglia exacerbates injury and impairs function recovery after spinal cord injury in mice. Cell Death Dis. (2020) 11:528. doi: 10.1038/s41419-020-2733-4
101. Kim YS, Jung HM, Yoon BE. Exploring glia to better understand Alzheimer's disease. Anim Cells Syst. (2018) 22:213–8. doi: 10.1080/19768354.2018.1508498
102. Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. (2012) 87:10–20. doi: 10.1016/j.brainresbull.2011.10.004
103. Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. (2009) 132(Pt 2):288–95. doi: 10.1093/brain/awn109
104. Matejuk A, Ransohoff RM. Crosstalk between astrocytes and microglia: an overview. Front Immunol. (2020) 11:1416. doi: 10.3389/fimmu.2020.01416
105. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. (2020) 9:42. doi: 10.1186/s40035-020-00221-2
106. Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. (2009) 29:11982–92. doi: 10.1523/jneurosci.3158-09.2009
107. Yang CN, Shiao YJ, Shie FS, Guo BS, Chen PH, Cho CY, et al. Mechanism mediating oligomeric Aβ clearance by naïve primary microglia. Neurobiol Dis. (2011) 42:221–30. doi: 10.1016/j.nbd.2011.01.005
108. Simpson DSA, Oliver PL. ROS generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants. (2020) 9:743. doi: 10.3390/antiox9080743
109. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. (2008) 28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
110. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. (2018) 4:575–90. doi: 10.1016/j.trci.2018.06.014
111. Ismail R, Parbo P, Madsen LS, Hansen AK, Hansen KV, Schaldemose JL, et al. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer's disease: a longitudinal PET study. J Neuroinflamm. (2020) 17:151. doi: 10.1186/s12974-020-01820-6
112. Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer's disease. J Neuroinflamm. (2005) 2:9. doi: 10.1186/1742-2094-2-9
113. Quintanilla RA, Orellana DI, González-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. (2004) 295:245–57. doi: 10.1016/j.yexcr.2004.01.002
114. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
115. Hanslik KL, Ulland TK. The role of microglia and the Nlrp3 inflammasome in Alzheimer's disease. Front Neurol. (2020) 11: 570711. doi: 10.3389/fneur.2020.570711
116. Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. (2015) 25:308–15. doi: 10.1016/j.tcb.2014.12.009
117. Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. J Cell Sci. (2017) 130:3955–63. doi: 10.1242/jcs.207365
118. Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. (2019) 575:669–73. doi: 10.1038/s41586-019-1769-z
119. Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, et al. Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer's disease. Cell Death Dis. (2014) 5:e1382. doi: 10.1038/cddis.2014.348
120. Liu L, Chan C. IPAF inflammasome is involved in interleukin-1β production from astrocytes, induced by palmitate; implications for Alzheimer's disease. Neurobiol Aging. (2014) 35:309–21. doi: 10.1016/j.neurobiolaging.2013.08.016
121. Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. (2003) 9:453–7. doi: 10.1038/nm838
122. Kaur D, Sharma V, Deshmukh R. Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer's disease. Inflammopharmacology. (2019) 27:663–77. doi: 10.1007/s10787-019-00580-x
123. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
124. Acosta C, Anderson HD, Anderson CM. Astrocyte dysfunction in Alzheimer disease. J Neurosci Res. (2017) 95:2430–47. doi: 10.1002/jnr.24075
125. ElAli A, Rivest S. Microglia in Alzheimer's disease: a multifaceted relationship. Brain Behav Immun. (2016) 55:138–50. doi: 10.1016/j.bbi.2015.07.021
126. Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. (2016) 53:1181–94. doi: 10.1007/s12035-014-9070-5
127. Ulrich JD, Holtzman DM. TREM2 function in Alzheimer's disease and neurodegeneration. ACS Chem Neurosci. (2016) 7:420–7. doi: 10.1021/acschemneuro.5b00313
128. Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer's disease. Mol Neurodegener. (2018) 13:66. doi: 10.1186/s13024-018-0298-9
129. Moore Z, Taylor JM, Crack PJ. The involvement of microglia in Alzheimer's disease: a new dog in the fight. Br J Pharmacol. (2019) 176:3533–43. doi: 10.1111/bph.14546
130. Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflamm. (2012) 9:151. doi: 10.1186/1742-2094-9-151
131. Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science. (1976) 193:415–7. doi: 10.1126/science.180605
132. Kraska A, Santin MD, Dorieux O, Joseph-Mathurin N, Bourrin E, Petit F, et al. In vivo cross-sectional characterization of cerebral alterations induced by intracerebroventricular administration of streptozotocin. PLoS ONE. (2012) 7:e46196. doi: 10.1371/journal.pone.0046196
133. Chen Y, Liang Z, Blanchard J, Dai CL, Sun S, Lee MH, et al. A non-transgenic mouse model (icv-STZ mouse) of Alzheimer's disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol Neurobiol. (2013) 47:711–25. doi: 10.1007/s12035-012-8375-5
134. Nazem A, Sankowski R, Bacher M, Al-Abed Y. Rodent models of neuroinflammation for Alzheimer's disease. J Neuroinflamm. (2015) 12:74. doi: 10.1186/s12974-015-0291-y
135. Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. (1996) 10:816–25. doi: 10.1101/gad.10.7.816
136. Sundaram JR, Chan ES, Poore CP, Pareek TK, Cheong WF, Shui G, et al. Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J Neurosci. (2012) 32:1020–34. doi: 10.1523/JNEUROSCI.5177-11.2012
137. St George-Hyslop PH. Genetic factors in the genesis of Alzheimer's disease. Ann N Y Acad Sci. (2000) 924:1–7. doi: 10.1111/j.1749-6632.2000.tb05552.x
138. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. (2003) 39:409–21. doi: 10.1016/s0896-6273(03)00434-3
139. Grimaldi A, Brighi C, Peruzzi G, Ragozzino D, Bonanni V, Limatola C, et al. Inflammation, neurodegeneration and protein aggregation in the retina as ocular biomarkers for Alzheimer's disease in the 3xTg-AD mouse model. Cell Death Dis. (2018) 9:685. doi: 10.1038/s41419-018-0740-5
140. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. (2015) 18:1584–93. doi: 10.1038/nn.4132
141. Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. (2019) 10:3758. doi: 10.1038/s41467-019-11674-z
142. Hori Y, Takeda S, Cho H, Wegmann S, Shoup TM, Takahashi K, et al. A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid β in the brain in a transgenic model of Alzheimer disease. J Biol Chem. (2015) 290:1966–78. doi: 10.1074/jbc.M114.586602
143. Zhang C, Griciuc A, Hudry E, Wan Y, Quinti L, Ward J, et al. Cromolyn reduces levels of the Alzheimer's disease-associated amyloid β-protein by promoting microglial phagocytosis. Sci Rep. (2018) 8:1144. doi: 10.1038/s41598-018-19641-2
144. Jana M, Palencia CA, Pahan K. Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer's disease. J Immunol. (2008) 181:7254–62. doi: 10.4049/jimmunol.181.10.7254
145. Han D, Tian Y, Zhang M, Zhou Z, Lu J. Prevention and treatment of experimental autoimmune encephalomyelitis with recombinant adeno-associated virus-mediated alpha-melanocyte-stimulating hormone-transduced PLP139-151-specific T cells. Gene Ther. (2007) 14:383–95. doi: 10.1038/sj.gt.3302862
146. Carniglia L, Ramírez D, Durand D, Saba J, Caruso C, Lasaga M. [Nle4, D-Phe7]-α-MSH inhibits toll-like receptor (TLR)2- and TLR4-induced microglial activation and promotes a M2-like phenotype. PLoS ONE. (2016) 11:e0158564. doi: 10.1371/journal.pone.0158564
147. Giuliani D, Bitto A, Galantucci M, Zaffe D, Ottani A, Irrera N, et al. Melanocortins protect against progression of Alzheimer's disease in triple-transgenic mice by targeting multiple pathophysiological pathways. Neurobiol Aging. (2014) 35:537–47. doi: 10.1016/j.neurobiolaging.2013.08.030
148. Morihara T, Teter B, Yang F, Lim GP, Boudinot S, Boudinot FD, et al. Ibuprofen suppresses interleukin-1beta induction of pro-amyloidogenic alpha1-antichymotrypsin to ameliorate beta-amyloid (Abeta) pathology in Alzheimer's models. Neuropsychopharm. (2005) 30:1111–20. doi: 10.1038/sj.npp.1300668
149. Walker D, Lue L-F. Anti-inflammatory and immune therapy for Alzheimer's disease: current status and future directions. Curr Neuropharmacol. (2007) 5:232–43. doi: 10.2174/157015907782793667
150. Zhang C, Wang Y, Wang D, Zhang J, Zhang F. NSAID exposure and risk of Alzheimer's disease: an updated meta-analysis from cohort studies. Front Aging Neurosci. (2018) 10. doi: 10.3389/fnagi.2018.00083
151. Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, et al. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer's disease model. J Immunol. (2011) 187:6539–49. doi: 10.4049/jimmunol.1100620
152. Shi JQ, Shen W, Chen J, Wang BR, Zhong LL, Zhu YW, et al. Anti-TNF-α reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. (2011) 1368:239–47. doi: 10.1016/j.brainres.2010.10.053
153. Lokensgard JR, Hu S, Sheng W, van Oijen M, Cox D, Cheeran MC, et al. Robust expression of TNF-alpha, IL-1beta, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J Neurovirol. (2001) 7:208–19. doi: 10.1080/13550280152403254
154. Villalba M, Hott M, Martin C, Aguila B, Valdivia S, Quezada C, et al. Herpes simplex virus type 1 induces simultaneous activation of Toll-like receptors 2 and 4 and expression of the endogenous ligand serum amyloid A in astrocytes. Med Microbiol Immunol. (2012) 201:371–9. doi: 10.1007/s00430-012-0247-0
155. Marques CP, Hu S, Sheng W, Cheeran MC, Cox D, Lokensgard JR. Interleukin-10 attenuates production of HSV-induced inflammatory mediators by human microglia. Glia. (2004) 47:358–66. doi: 10.1002/glia.20045
156. Aravalli RN, Hu S, Rowen TN, Palmquist JM, Lokensgard JR. Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J Immunol. (2005) 175:4189–93. doi: 10.4049/jimmunol.175.7.4189
157. Aravalli RN, Hu S, Lokensgard JR. Inhibition of toll-like receptor signaling in primary murine microglia. J Neuroimmune Pharmacol. (2008) 3:5–11. doi: 10.1007/s11481-007-9097-8
158. Wang JP, Bowen GN, Zhou S, Cerny A, Zacharia A, Knipe DM, et al. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J Virol. (2012) 86:2273–81. doi: 10.1128/JVI.06010-11
159. Marques CP, Cheeran MC, Palmquist JM, Hu S, Urban SL, Lokensgard JR. Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J Immunol. (2008) 181:6417–26. doi: 10.4049/jimmunol.181.9.6417
160. Conrady CD, Zheng M, van Rooijen N, Drevets DA, Royer D, Alleman A, et al. Microglia and a functional type I IFN pathway are required to counter HSV-1-driven brain lateral ventricle enlargement and encephalitis. J Immunol. (2013) 190:2807–17. doi: 10.4049/jimmunol.1203265
161. Carr DJ, Veress LA, Noisakran S, Campbell IL. Astrocyte-targeted expression of IFN-alpha1 protects mice from acute ocular herpes simplex virus type 1 infection. J Immunol. (1998) 161:4859–65.
162. Reinert LS, Lopušná K, Winther H, Sun C, Thomsen MK, Nandakumar R, et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun. (2016) 7:13348. doi: 10.1038/ncomms13348
163. Mørk N, Kofod-Olsen E, Sørensen KB, Bach E, Ørntoft TF, Østergaard L, et al. Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun. (2015) 16:552–66. doi: 10.1038/gene.2015.46
164. Webre JM, Hill JM, Nolan NM, Clement C, McFerrin HE, Bhattacharjee PS, et al. Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J Biomed Biotechnol. (2012) 2012:612316. doi: 10.1155/2012/612316
165. Kollias CM, Huneke RB, Wigdahl B, Jennings SR. Animal models of herpes simplex virus immunity and pathogenesis. J Neurovirol. (2015) 21:8–23. doi: 10.1007/s13365-014-0302-2
166. Shimeld C, Whiteland JL, Williams NA, Easty DL, Hill TJ. Reactivation of herpes simplex virus type 1 in the mouse trigeminal ganglion: an in vivo study of virus antigen and immune cell infiltration. J Gen Virol. (1996) 77:2583–90. doi: 10.1099/0022-1317-77-10-2583
167. Shimeld C, Easty DL, Hill TJ. Reactivation of herpes simplex virus type 1 in the mouse trigeminal ganglion: an in vivo study of virus antigen and cytokines. J Virol. (1999) 73:1767–73. doi: 10.1128/jvi.73.3.1767-1773.1999
168. Theil D, Derfuss T, Paripovic I, Herberger S, Meinl E, Schueler O, et al. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am J Pathol. (2003) 163:2179–84. doi: 10.1016/s0002-9440(10)63575-4
169. Verjans GM, Hintzen RQ, van Dun JM, Poot A, Milikan JC, Laman JD, et al. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci USA. (2007) 104:3496–501. doi: 10.1073/pnas.0610847104
170. Doll JR, Hoebe K, Thompson RL, Sawtell NM. Resolution of herpes simplex virus reactivation in vivo results in neuronal destruction. PLoS Pathog. (2020) 16:e1008296. doi: 10.1371/journal.ppat.1008296
171. Laval K, Vernejoul JB, Van Cleemput J, Koyuncu OO, Enquist LW. Virulent pseudorabies virus infection induces a specific and lethal systemic inflammatory response in mice. J Virol. (2018) 92:e01614-18. doi: 10.1128/JVI.01614-18
172. Laval K, Van Cleemput J, Vernejoul JB, Enquist LW. Alphaherpesvirus infection of mice primes PNS neurons to an inflammatory state regulated by TLR2 and type I IFN signaling. PLoS Pathog. (2019) 15:e1008087. doi: 10.1371/journal.ppat.1008087
173. Pomeranz LE, Reynolds AE, Hengartner CJ. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev. (2005) 69:462–500. doi: 10.1128/mmbr.69.3.462-500.2005
174. Laval K, Enquist LW. The neuropathic itch caused by pseudorabies virus. Pathogens. (2020) 9:254. doi: 10.3390/pathogens9040254
175. van den Pol AN, Ding S, Robek MD. Long-distance interferon signaling within the brain blocks virus spread. J Virol. (2014) 88:3695–704. doi: 10.1128/jvi.03509-13
176. Laval K, Maturana CJ, Enquist LW. Mouse footpad inoculation model to study viral-induced neuroinflammatory responses. J Vis Exp. (2020) 160:e61121. doi: 10.3791/61121
Keywords: neuroinflammation, Alzheimer's disease, pathogenesis, peripheral nervous system, herpesvirus
Citation: Laval K and Enquist LW (2021) The Potential Role of Herpes Simplex Virus Type 1 and Neuroinflammation in the Pathogenesis of Alzheimer's Disease. Front. Neurol. 12:658695. doi: 10.3389/fneur.2021.658695
Received: 27 January 2021; Accepted: 10 March 2021;
Published: 06 April 2021.
Edited by:
Claudia Kimie Suemoto, University of São Paulo, BrazilReviewed by:
Eliana Cristina De Brito Toscano, Federal University of Minas Gerais, BrazilAntonio Giuliano Zippo, National Research Council (CNR), Italy
Copyright © 2021 Laval and Enquist. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathlyn Laval, a2F0aGx5bi5sYXZhbCYjeDAwMDQwO3VnZW50LmJl