 Federica Rachele Danti1
Federica Rachele Danti1 Federica Invernizzi2Isabella Moroni1
Federica Invernizzi2Isabella Moroni1 Barbara Garavaglia2
Barbara Garavaglia2 Nardo Nardocci1
Nardo Nardocci1 Giovanna Zorzi1*
Giovanna Zorzi1*- 1Unit of Child Neurology, Department of Pediatric Neuroscience, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Neurologico Carlo Besta, Milan, Italy
- 2Unit of Medical Genetics and Neurogenetics, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Neurologico C. Besta, Milan, Italy
Paroxysmal exercise-induced neurological symptoms (PENS) encompass a wide spectrum of clinical phenomena commonly presenting during childhood and characteristically elicited by physical exercise. Interestingly, few shared pathogenetic mechanisms have been identified beyond the well-known entity of paroxysmal exercise-induced dyskinesia, PENS could be part of more complex phenotypes including neuromuscular, neurodegenerative, and neurometabolic disease, epilepsies, and psychogenetic disorders. The wide and partially overlapping phenotypes and the genetic heterogeneity make the differential diagnosis frequently difficult and delayed; however, since some of these disorders may be treatable, a prompt diagnosis is mandatory. Therefore, an accurate characterization of these symptoms is pivotal for orienting more targeted biochemical, radiological, neurophysiological, and genetic investigations and finally treatment. In this article, we review the clinical, genetic, pathophysiologic, and therapeutic landscape of paroxysmal exercise induced neurological symptoms, focusing on phenomenology and differential diagnosis.
Introduction
Physical exercise requires a profound adaptation of the whole body to be able to provide the increased energy needs essential for muscle contraction and the physiological functions of vital organs such as the heart, lungs and most importantly the brain. During exercise, based on its intensity and duration, a variety of metabolic pathways are simultaneously activated to produce ATP from different substrates, such as phosphocreatine, carbohydrates, free fatty acids, and branched-chain amino acids (1). Moreover, exercise results in a number of modifications in the synthesis and metabolism of brain neurotransmitters, action potential properties or synaptic transmission that are fundamental for signal transduction both in muscle and neuronal cells (2). Consequently, genetic or acquired defects disturb membrane excitability, synaptic transduction and transport, and storage, mobilization or utilization of the substrates involved in cell metabolism in the brain, muscles or neuromuscular junction (NMJ) (Figure 1). This leads to a wide spectrum of pediatric neurological symptoms characteristically elicited by prolonged physical exertion.
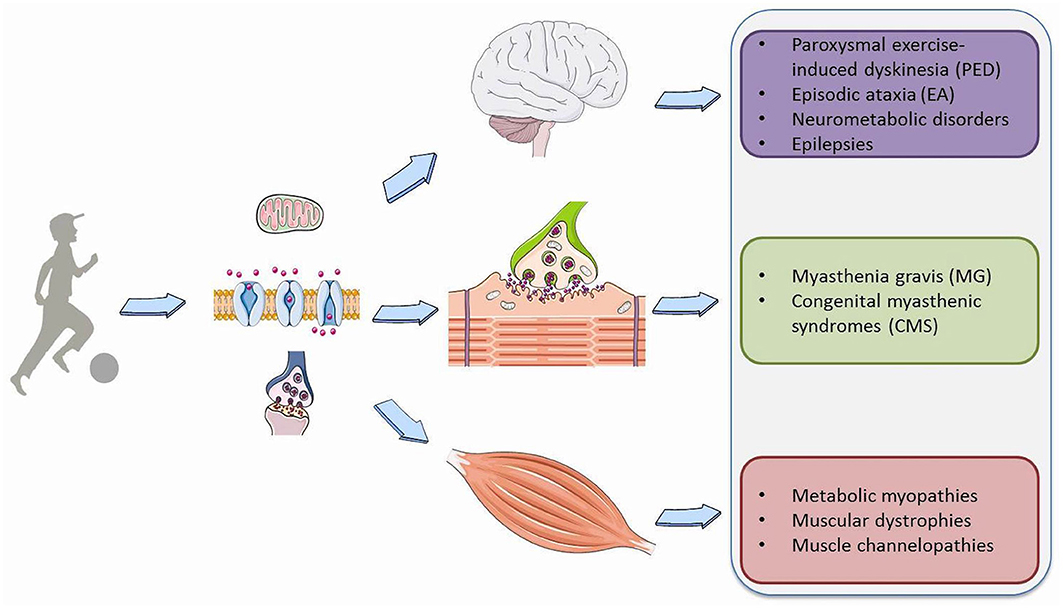
Figure 1. Overview of Paroxysmal exercise-induced neurological symptoms.
We propose to group these phenomena, which are frequently encountered in clinical practice, in a new clinical entity defined as Paroxysmal Exercise-induced Neurological Symptoms (PENS). They range from dyskinesia and ataxia to myalgia, cramping and rhabdomyolysis, myotonia, stiffness or weakness. They could be isolated or part of the phenotype of a number of neuromuscular, neurodegenerative, neurometabolic, epileptic and psychogenetic disorders. The wide number of genes involved in their pathogenesis reflects the high complexity of the causative molecular mechanisms (Table 1). The partial overlap and similarity among reported symptoms and the broad associated genetic heterogeneity make the differential diagnosis difficult and frequently delayed; however, a prompt diagnosis is required since some of these disorders may be treatable. Therefore, an accurate characterization of these symptoms is pivotal for orienting the extensive differential diagnosis through more targeted biochemical, radiological, and neurophysiological investigations and definitive genetic testing.
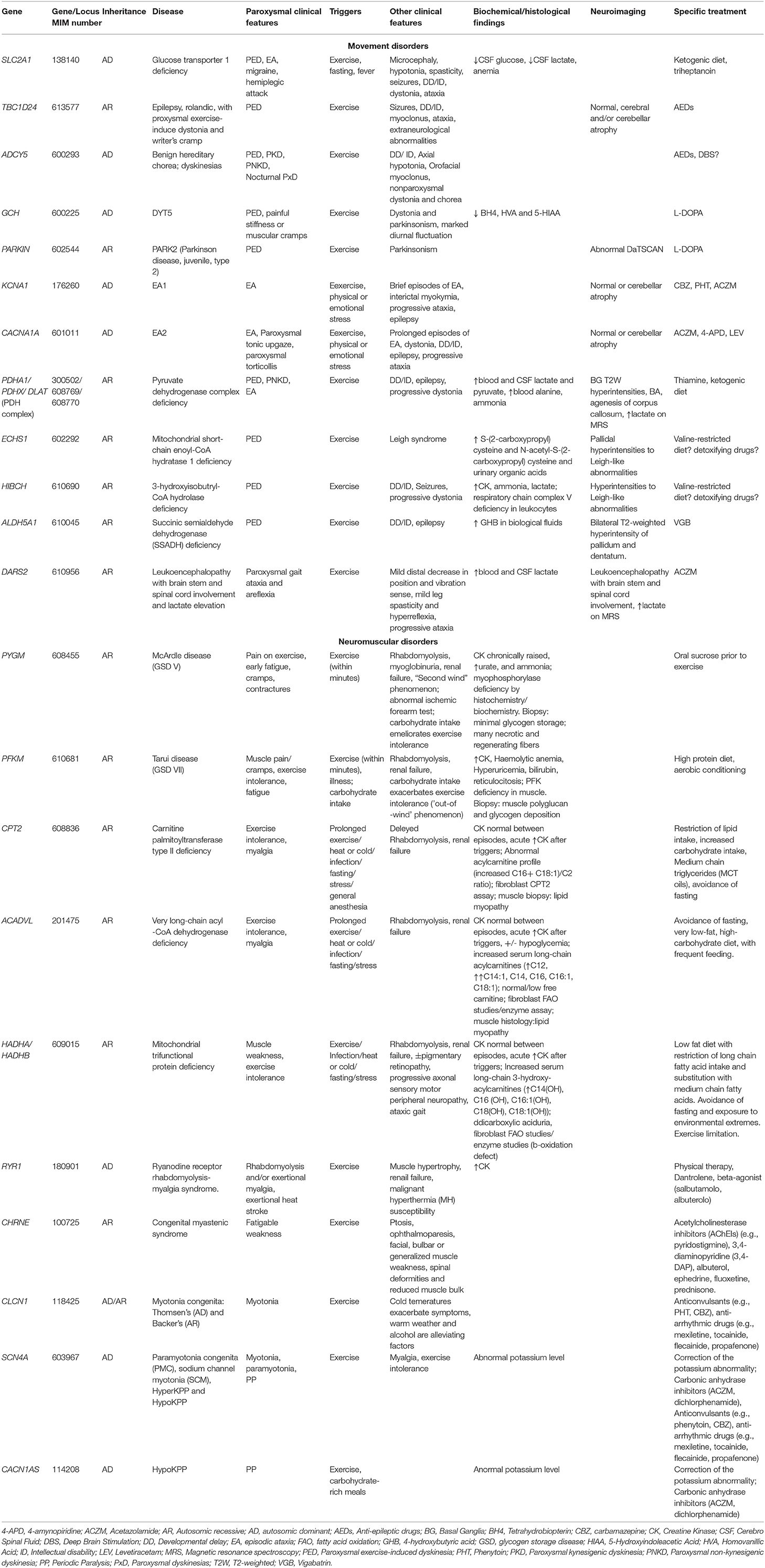
Table 1. Main genetic causes of PENS and related clinical, biochemical/histological/neuroimaging findings and treatments.
This review examines the clinical, genetic, pathophysiologic and therapeutic landscape of PENS, classifying them based on the type of the main paroxysmal symptom and focusing on pathophysiology and differential diagnosis.
Pediatric Paroxysmal Exercise-Induced Neurological Symptoms: Phenotypic and Genotypic Spectrum
Movement Disorders
Paroxysmal Exercise Induced Dyskinesia (PED)
Paroxysmal Exercise Induced Dyskinesias (PED) are genetically heterogeneous conditions characterized by recurrent episodes of sudden, involuntary movement that are typically induced by prolonged exercise (3). They commonly present during infancy or childhood with dystonic or choreic attacks, isolated or in combination, that usually affects the part of the body that is involved in the exercise, such as leg or arm dystonia after prolonged walking or writing. The duration and frequency of PED are very variable, but the attacks usually last 5–30 min and recur daily or a few times per month. PED can be the only or prominent manifestation of a disease or can be associated with other paroxysmal manifestations (epilepsy, migraine) and\or interictal neurological abnormalities. Although rare, they should be promptly recognized since some of them are treatable (4).
Heterozygous mutations in the SLC2A1 (solute carrier family two, member one) gene encoding for the glucose transporter gene 1 (GLUT1) are the main causative defect of PED (5, 6). These mutations can be isolated or in the context of a more complex phenotype (GLUT1 deficiency syndrome, GLUT1-DS), which include epilepsy, ataxia, spasticity, dystonia, intellectual disability and other paroxysmal events such as migraine, hemiplegic attacks or episodic ataxia (EA) (7). Typically, the onset is during childhood in otherwise normal subjects or those with a mild interictal neurological abnormality or a history of epilepsy with generalized seizures. Attacks are predominantly dystonic or choreo-dystonic with a focal or unilateral distribution and generalization is rare (5–9). Besides exercise, fasting or fever are also reported as triggering factors due to energy metabolism defect (3, 7). Glucose is the essential substrate for brain energy metabolism, especially in infants and children where the brain can consume as much as 80% of the body's total glucose supply in the resting state (9). GLUT1 is constitutively expressed in most tissues but predominantly expressed in erythrocytes, brain endothelial cells, and astrocytes and is fundamental for the passage of glucose across the blood–brain barrier and cell membranes. Consequently, a low concentration of glucose in cerebrospinal fluid (CSF) and a decreased CSF to blood glucose ratio are the best biochemical clues to diagnosis, although they are not constantly found. Molecular analysis of the SLC2A1 gene confirms the diagnosis (Figure 2). The mutation type and resulting residual GLUT1 activity are responsible for the heterogeneity and severity of the phenotype (10). Exon 4, which encodes the fourth transmembrane domain of GLUT1, has been suggested as a vulnerable region of the protein, and mutational hotspots have also been reported (11). The ketogenic diet is the gold-standard therapy for epilepsy and paroxysmal and non-paroxysmal movement disorder (12), however, the dietary constraints and side effects may be poorly tolerated and inefficacy has also been reported (13). Treatment with triheptanoin has resulted in a dramatic and sustained reduction of non-epileptic paroxysmal events in a group of GLUT1-DS patients who were not on the ketogenic diet (13).
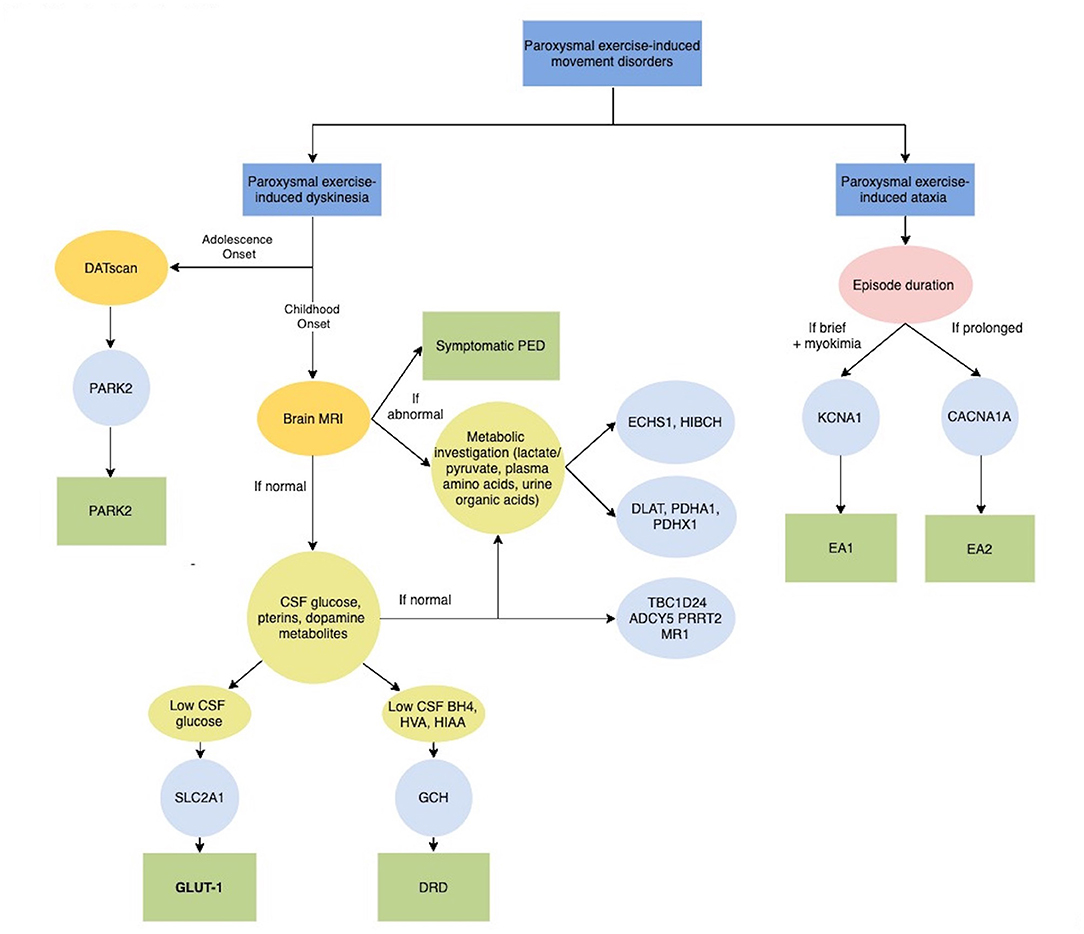
Figure 2. Diagnostic algorithm for Paroxysmal exercise-induced movement disorders. ADCY5, Adenylate Cyclase 5; BH4, tetrahydrobiobterin; CACNA1A, Calcium Channel, Voltage-Dependent, P/Q Type, Alpha-1a Subunit; CSF, Cerebrospinal fluid; DATscan, Dopamine transporter (DAT) single photon emission computerized tomography (SPECT) imaging technique; DLAT, Dihydrolipoamide acetyltransferase; DRD, Dopa-Responsive Dystonia; EA1, Episodic Ataxia, Type 1; EA2, Episodic Ataxia, Type 2; ECHS1, Short-chain enoyl-CoA hydratase; GLUT-1, Glucose Transport Defect, Blood-Brain Barrier, Deficiency Syndrome 1; GCH, GTP Cyclohydrolase I; HIAA, 5-hydroxyindolacetic acid; HIBCH, 3-Hydroxyisobutyryl-CoA hydrolase; HVA, Homovanalinaic Acid; KCNA1, Potassium Channel, Voltage-Gated, Member 1; MRI, Magnetic Resonance Imaging; MR-1, Myofibrillogenesis Regulator 1; PARK2, Parkinson disease-2; PDHA1, Pyruvate dehydrogenase E1-alpha deficiency; PDHX1, Pyruvate Dehydrogenase Complex Component X; PED, Paroxysmal exercise-induced dyskinesia; PRRT2, Proline-Rich Transmembrane Protein 2; SLC2A1, Solute Carrier Family 2 (Facilitated Glucose Transporter), Member 1; TBC1D24, Tbc1 Domain Family, Member 24.
Bi-allelic mutations in the TBC1D24 gene, which encodes for a presynaptic protein involved in vesicle tracking, have been described in a wide range of neurological disorders that include epileptic syndromes, DOORS syndrome (deafness, onychodystrophy, osteodystrophy, intellectual disability and seizures) or syndromic and non-syndromic deafness (14). Recently PED as a prominent clinical feature associated with epilepsy has been reported in few patients (15–17). Episodes starts usually between 1 and 2 years of age and are characterized by dystonia triggered by exercise or fatigue; patients present with episodes of trunk bending laterally or backward without clear discomfort, some other cases have arm dystonia after writing or performing motor tasks. The phenomenology of the movement disorder has also be reported as hyperkinetic (tremor or fine myoclonus), involving the oromandibular region or the voice after prolonged chewing or singing (17). Episodes last seconds to hours and are alleviated or ceased by resting. No specific treatment results in marked benefit, although a possible effect of acetazolamide (17) and carbamazepine (16) has been suggested. Therefore, avoidance of precipitating factors remains the most effective therapeutic strategy. For cases with long follow-up, a reduction of the severity of the paroxysms although persisting into adulthood has been documented (15).
Mutations in ADCY5, which encodes for adenylate cyclase type 5 (AC5), a membrane-bound enzyme highly expressed in striatal neurons, were first described in a large dominant kindred with affected cases presenting with an early-onset hyperkinetic movement disorder defined Familial Dyskinesia with Facial Myokymia (18). The spectrum of ADCY5- related movement disorder have been gradually expanded to include NKX2-1 negative patients with benign hereditary chorea (19), childhood-onset dystonia, chorea and myoclonus and patients diagnosed with dyskinetic cerebral palsy. The hallmarks of the disease are episodic exacerbations of baseline movement during sleep and when awake (20, 21). Delayed motor and/or language milestones with or without axial hypotonia are frequently reported. The paroxysmal dyskinesias are mostly generalized, with rapid dystonic and ballistic movements, and can last for hours and are sometimes painful. Different triggers, even in the same patient, can provoke the paroxysms, including, but not exclusively, physical exercise (21). Although patients have a chronic, sometimes slowly progressive, movement disorder, they usually complain the paroxysmal dyskinesias being the most disabling symptoms. The antiepileptic drug, acetazolamide, benzodiazepines, tetrabenazine, propranolol have been tested with little or no benefit; recently pallidal deep brain stimulation improved both the chronic and paroxysmal movement disorder in few patients (22). The disease is transmitted with an autosomal dominant mode of inheritance and the majority of reported ADCY5 mutations are located in exons 2 and 10, at residue 418 and 1029, respectively (20, 21).
Moreover, exercise is reported to be a fairly frequent trigger in patients carrying PRRT2 and MR-1 mutations (3), which are more typically responsible for paroxysmal kinesigenic dyskinesias (PKD) and paroxysmal non-kinesigenic dyskinesias (PNKD) (Figure 2) (11).
In rare cases, PED can occur as the first symptom of juvenile parkinsonism and DOPA-responsive dystonia (DRD) due to PARK2 and GCH1 mutations respectively, supporting the role of neurotransmitter deficiency, together with brain energy deficiency, in the pathophysiology of PED (Figure 2).
The E3 ubiquitin ligase PARKIN (encoded by PARK2) works together with PINK1 (a mitochondrial-targeted kinase encoded by PARK6) in a common pathway to remove dysfunctional mitochondria by autophagy. Therefore PARK2 and PARK6 mutations result in the accumulation of dysfunctional mitochondria that release reactive oxygen species (23). The mean onset of juvenile parkinsonism is around age 30 years, but adolescent cases are recognized and PED, typically presenting with leg or foot dystonic posturing elicited by prolonged walking is particularly frequent in this age range (24, 25). GCH1 codes for GTP cyclohydrolase I, a rate limiting enzyme in the synthesis of tetrahydrobiobterin (BH4) from GTP. Autosomal dominant GCH deficiency, commonly known as Dopa-responsive dystonia, Segawa disease, or DYT5, is defined by the association of dystonia, tremor or bradykinesia, marked diurnal fluctuation, specific CSF pattern (low levels of BH4, homovanillic acid, and 5-hydroxyindolacetic acid) and sustained response to L-dopa administration (Figure 2) (24, 26). PED is often the presenting symptom of the disease; notably, together with abnormal posturing, patients frequently complain of painful stiffness or muscular cramps, suggesting a differential diagnosis among neuromuscular disorders. As the disease progresses dystonia involves other body regions and parkinsonian signs also become evident, but there are reported patients in which PED remained the unique manifestation (27).
Paroxysmal Exercise Induced Ataxia
Episodic ataxia (EA) comprises a heterogeneous group of genetic conditions characterized by recurrent episodes of imbalance, incoordination and postural instability (28). EA can result from mutations in several genes that play important roles in the nervous system. Physical exercise has been reported to trigger episodes of ataxia due to mutations in KCNA1 and CACNA1A, which are responsible for episodic ataxia types 1 and 2, respectively (Figure 2). These genes encode for proteins that are involved in the transport of ions across cell membranes and play important roles in excitatory neurotransmission (28). As for PED, exercise-induced episodes are usually associated with a variety of other paroxysmal manifestations and persistent neurological symptoms.
Episodic ataxia type 1 (EA1) is characterized by brief episodes of ataxia with myokimia and later, the development of cerebellar signs. Onset is usually before the age of 10 years. Ataxic symptoms such as dizziness, unsteady, wide-based gait, incoordination, and dysarthria are often accompanied by weakness, stiffness, headache, nausea, vomiting, and visual disturbance. Myokimia usually involves the periocular or perioral region or of the distal extremities. It can be sometimes be misinterpreted as tremor or chorea, making the differential diagnosis between PED and EA often difficult. Physical exertion, emotional stress and environmental temperature are the usual triggers for attacks. Episodes are usually brief, but the severity may range from inability to stand and walk to independent walking. The frequency of attacks is variable, daily to monthly, and tends to decrease during adulthood. Up to 20% of EA1 patients develop persistent cerebellar signs. Carbamazepine is the most useful drug in EA1, but some benefits have been reported with valproic acid and acetazolamide (28, 29). EA1 is caused by heterozygous mutations in KCNA1, which encodes the a1 subunit of a neuronal voltage-gated potassium channel, Kv1.1, that is highly expressed in the interneurons and Purkinje cells in the cerebellum and plays an important role in the inhibition of the cerebellar outputs. Around 30 KCNA1 variants have been identified so far and the majority of them are missense mutations (30).
Episodic ataxia 2 (EA2), the most common subtype of EA, is a well-characterized genetic condition in which patients experience prolonged episodes of incoordination from early childhood, vertigo, dysarthria and often general weakness (31). Similarly to EA1, common triggers are exercise, physical or emotional stress but paroxysms can also arise spontaneously. Episodes are highly disabling and can last several days. As the disease progresses, interictal ataxia and nystagmus appear. Many cases with atypical features such as late onset, or paroxysmal tonic upward gaze in infancy or the appearance of dystonia, epilepsy and cognitive impairment are reported. Acetazolamide is the drug of choice to prevent attacks, both in adults and children. In cases of unresponsiveness or side effects, 4-aminopyridine has been demonstrated to be effective in a controlled trial (32). EA2 is caused by heterozygous mutations in CACNA1A, which encodes the a1A subunit of a neuronal voltage-gated calcium channel, Cav2. This channel, widely expressed in the Purkinje and granule cells of the cerebellum, mediates calcium entry and firing of the cells (28).
Neurometabolic Disorders
PED has been recently associated with the deficiency of a number of mitochondrial enzymes involved in energy production and branched-chain amino acids (BCAA; leucine, isoleucine, and valine) catabolism. They include Pyruvate dehydrogenase (PDH) complex, short-chain enoyl-CoA hydratase (ECSH1) and 3-Hydroxyisobutyryl-CoA hydrolase (HIBCH) (Figure 2) (33, 34).
PDH complex catalyses the oxidative decarboxylation of pyruvate with the production of acetyl-CoA; therefore, it connects the glycolytic pathway to the Krebs cycle and plays a central role in glucose metabolism in fed and fasting states. PDH complex is composed of three catalytic subunits: pyruvate dehydrogenase (PDH; E1, a heterotetramer of 2 subunits encoded by PDHA1 and PDHB1 genes), dihydrolipoamide acetyltransferase (E2, encoded by DLAT gene), and dihydrolipoamide dehydrogenase (E3, encoded by DLD gene), and of an additional component, the E3-binding protein (encoded by PDHX1) (35, 36). PDHA1, DLAT, and PDHX1 mutations have been linked to continuously expanding phenotypes inherited with X-linked (PDHA1 mutations, representing the main cause of PDH deficiency) or autosomal recessive pattern (DLAT and PDHX1 mutation). Clinical findings range from severe infantile lactic acidosis to milder chronic neurological disorders including intermittent and recurrent acute neurological symptoms such as episodic ataxia, peripheral weakness, and movement disorders such as PED and PNKD (36–38). In few patients recurrent dystonic or hemidystonic attacks have been described; they are triggered by prolonged walking and running and occur as a unique clinical manifestation or within complex neurological phenotypes (39–41). Elevated serum lactate and bilateral pallidal hyperintensities, typical hallmarks of the disease, could be absent, particularly at onset (39). PDH deficiency is a potentially treatable disorder, partially responding to ketogenic diet and thiamine supplementation (42).
ECHS1 and HIBCH enzymes play a major role in the valine catabolic pathway and fatty acids degradation. Valine is essential for regulating protein synthesis, neurotransmission, and energy production (43). Moreover, recessive mutations in those genes generate secondary deficiencies in the mitochondrial oxidative phosphorylation system and PDH complex. Interestingly, specific dietary regimens for reducing the workload on the valine catabolic pathway, associated with detoxifying drugs, such as cysteamine and N- acetylcysteine, for replenishing intramitochondrial glutathione are a potential treatment strategy for both ECSH1 and HIBCH defects (42).
Recessive mutations in ECHS1 have been reported as a novel cause of Leigh syndrome or atypical forms with later onset (34). Interestingly recent reports widened the clinical range of ECHS1-related disorders including few normally developing unrelated children with paroxysmal and frequently asymmetric dystonic episodes in the lower limbs lasting few minutes; most of them harbored the c.518C>T (p.A173V) variant (44–47). Elevated S-(2-carboxypropyl)cysteine and N-acetyl-S-(2-carboxypropyl)cysteine and urinary excretion of organic acids (2,3-dihydroxy-2-methylbutyric acid, 3-MGA, 3-HIVA, and EMA) and T2-weighted bilateral abnormalities at the level of the globus pallidus have been suggested in diagnosis, even in the mildest form (34, 44).
Similarly, autosomal recessive HIBCH mutations have been reported as a rare cause of early-infantile mitochondrial encephalopathy with progressive dystonia. A milder phenotype was identified in a 6-year-old girl suffering from isolated exercise-induced dystonic posturing in both lower extremities and the waist associated with bilateral symmetrical hyperintensities in the globus pallidus on brain MRI. Interictal neurological examination was unremarkable. Laboratory findings showed elevated serum creatine kinase (CK), ammonia, lactate and respiratory chain complex V deficiency in blood peripheral leukocytes. The patient demonstrated marked clinical and neuroradiological improvements after treatment with a low-valine diet (48). An increase of 3-hydroxy-isobutyryl-carnitine has been reported as a distinguishing finding of HIBCH from ECSH1 deficiency (33, 34).
PED has been described in two Italian siblings with mutations in ALDH5A1 encoding for another mitochondrial enzyme, Succinic semialdehyde dehydrogenase (SSADH) that results in aberrant metabolism of the neurotransmitter GABA, the main inhibitory neurotransmitter of the central nervous system (49). They presented with early onset developmental delay and ID associated with dystonic postures of upper limbs and seizures. During adolescence, they exhibited PED that responds to vigabatrin which irreversibly inhibits GABA transaminase, and brain MRI showed bilateral T2-weighted hyperintensity of pallidum and dentatum. This rare autosomal recessive disorder exhibits a highly phenotypic heterogeneity, encompassing varying degrees of developmental delay, hypotonia, ataxia, seizures and movement disorder such as dystonia or choreoatetosis. Elevations of 4-hydroxybutyric acid (GHB) in biological fluids could guide the diagnosis. Treatment with vigabatrin resulted in mixed outcomes (50, 51).
Juvenile onset exercise-induced paroxysmal gait ataxia and areflexia of the upper and lower extremities are reported in a single case an atypical phenotype associated with a novel homozygous mutation c.1825C>T (p.R609W) in the DARS2 gene. Heterozygous mutations in this gene, encoding a mitochondrial aspartyl-tRNA synthetase, are typically responsible for a more severe presentation defined as leukoencephalopathy with brain stem and spinal cord involvement and brain lactate elevation (LBSL) (52). The patient disclosed a brain MRI compatible with several major and supportive MRI criteria for LBSL and lactate peak in spectroscopy but permanent cerebellar ataxia or gait spasticity, hallmarks for this disease, were absent (53). Acetazolamide was effective in achieving good control of attacks (52).
Neuromuscular Disorders
Exercise may induce a wide range of symptoms that point to a primary impairment of muscles or NMJ. These symptoms can be clustered into two distinct clinical categories, named Exercise intolerance and Paroxysmal exercise induced stiffness or weakness.
Exercise intolerance includes a variable combination of fatigue, muscle cramps and myalgia that causes inability or a decreased ability to perform physical exercise at the normally expected level based on age and sex. It manifests in metabolic myopathies as a consequence of a failure to meet the energy requirements of a motor task, and in addition in Myasthenia Gravis and Congenital Myasthenic Syndromes (Figure 3). Furthermore, exercise could induce paroxysmal weakness or stiffness in patients with Myotonic Dystrophies and Primary Muscle Channelopathies that include Non-dystrophic Myotonias and Periodic Paralysis (Figure 4).
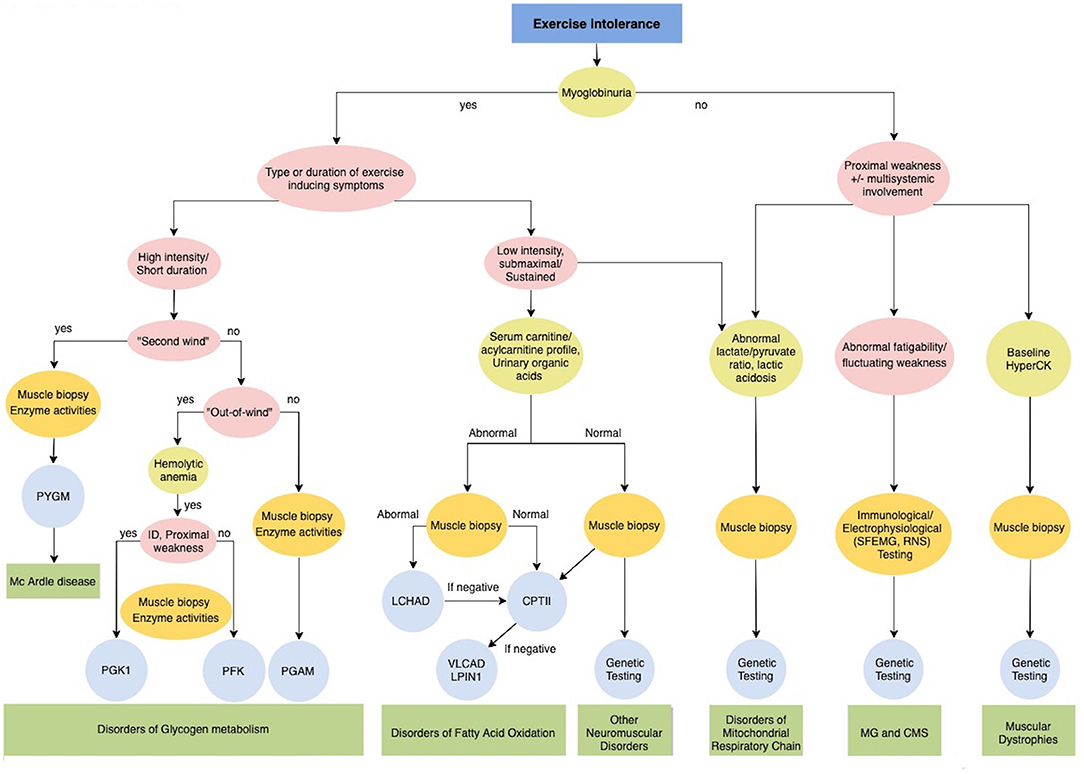
Figure 3. Diagnostic algorithm for Exercise intolerance. CMS, Congenital myasthenic syndromes; CPTII, carnitine palmitoyltransferase II; ID, intellectual disability; LCHAD Long-chain 3-hydroxyacyl-CoA dehydrogenase; LPIN1, Phosphatidate Phosphatase; MG, Myasthenia gravis; PFKM, phosphofructokinase; PGAM, Phosphoglycerate mutase; PGK1, Phosphoglycerate kinase 1; PYGM, phosphorylase; VLCAD, very long-chain long-chain acyl-CoA dehydrogenase.
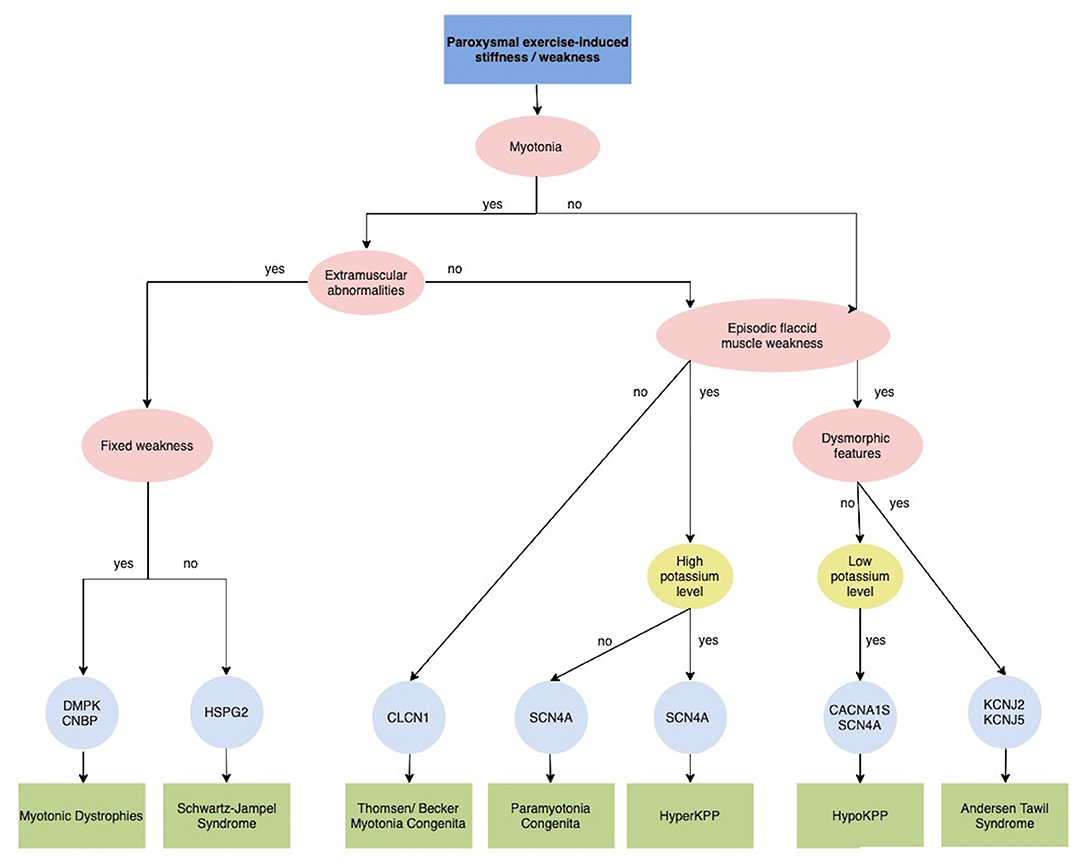
Figure 4. Diagnostic algorithm for Paroxysmal exercise-induced stiffness/weakness. CACNA1S, calcium voltage-gated channel subunit alpha1 S; CLCN1, chloride channel 1; CNBP, CCHC-type zinc finger, nucleic acid binding protein; DMPK, dystrophia myotonica protein kinase; HSPG2, heparan sulfate proteoglycan of the basement membrane; HyperPP, Hyperkalemic periodic paralysis; HypoPP, Hypokalemic periodic paralysis; KCNJ2, potassium voltage-gated channel subfamily J member 2; KCNJ5, potassium voltage-gated channel subfamily J member 5; SCN4A, sodium voltage-gated channel alpha subunit 4.
Detailed clinical history must include previous symptoms, anatomical distribution and sequence progression pattern, associated features and family history. In particular, a history of dark colored urine or myoglobinuria should be acquired. Myoglobinuria as a sign of rhabdomyolysis strongly suggests a metabolic myopathy (Figure 3). Specific investigation of the intensity and duration of exercise and factors which worsen or improve symptoms are fundamental for orienting etiological definition. Neurological and general examination should assess dynamic and static symptoms and multisystemic involvement.
An evaluation must involve blood testing (CK, acylcarnitine profile, lactate/pyruvate, amino acids), urine organic acids, exercise testing, neurophysiological, muscle biopsy for histology, ultrastructure, enzyme testing, eventually brain MRI/spectroscopy, and targeted or untargeted genetic testing (54, 55). If all studies are normal, a psychogenic cause should be considered (56).
Metabolic Myopathies
Metabolic myopathies are a heterogeneous group of rare genetic disorders characterized by muscle dysfunction secondary to an acute bioenergetic crisis. This is due to defects in the biochemical pathways of storage, mobilization and utilization of the substrates involved in muscle cell metabolism (57, 58).
Metabolic myopathies typically present during childhood with a wide clinical spectrum including acute forms of exercise intolerance with the normal interictal clinical examination; or progressive forms characterized by proximal muscle weakness frequently combined with multisystem involvement (59–61). Three main types of metabolic myopathies are distinguished according to the impaired metabolic pathway: (1) disorders of glycolytic and glycogenolytic metabolism; (2) disorders of fatty acid metabolism; and (3) disorders of the mitochondrial respiratory chain. Establishing the type of activity that produces the acute phenotype is essential for better orienting toward the impaired metabolic pathway (Figure 3) (58, 60).
More specifically, glycogen breakdown and glycolysis defects manifest with episodes of exercise intolerance, muscle pain, contractures and hyperCKemia early after (usually within 5 min) short intense, isometric activity such as running or lifting heavy objects. Basal CK level is usually elevated and examination is usually normal in children, while interictal weakness can emerge in adults. Distinguishing features can help narrow the diagnosis (60, 62). In particular, a marked improvement in the exercise tolerance about 10 min into aerobic, dynamic, large muscle mass exercise followed by the ability to continue exercise without difficulty (“second wind” phenomenon) directs diagnosis toward muscle phosphorylase deficiency or McArdle's disease (Glycogen storage disease, or GSD, type V), the most common disorder of carbohydrate metabolism (63). Non-ischemic forearm exercise testing is abnormal, showing only an increase in ammonia and constant levels of lactic acid and pyruvate. The absence of myophosphorylase and muscle biopsy and genetic mutation analysis confirmed the diagnosis; screening of the common mutations (p.R50X, p.W798R, p.G205S in Caucasians and p.Phe710del in Japanese) in PYGM should be evaluated (63).
Glucose or sucrose intake usually ameliorates symptoms in McArdle's disease, while exacerbating them in PFK deficiency (“out-of-wind” phenomenon of Tarui's disease or GSDVII), the second most common glycolytic disorder. Haemolytic anemia has been described in patients with PFKM but also in PGK1 mutations; in the latter encephalopathy and hyperammonaemia are typically reported. Other less frequent muscle glycogenosis with acute exercise-induced phenotype are due to distal glycolytic defects- GSD IX, X and XI (PHKA1, PGAM and LDHA mutations respectively), characterized by abnormal forearm exercise test (reduced increased of lactate levels) (61). Skin rush during summer or uterine stiffness during pregnancy orient toward LDHA mutations; bifid uvula and hepatopathy are described in Phosphoglucomutases 1 (PGM1)-associated myopathy (GSD XIV); recessive mutations in the ENO3 gene cause GSD XIII or muscle β-enolase deficiency; phenotype elicited by strenuous exertion and isometric muscle activity is usually milder than GSD V and “second wind” phenomenon is not observed (60–62, 64). Exercise intolerance could occur even in other GSD in the context of prominent progressive neuromuscular features such as Pompe's disease due to GAA mutation; however proximal weakness, fixed contractures, and cardio-respiratory involvement; GSD III and IV distal muscle weakness and liver involvement. If suggesting clinical key features are identified, single gene testing should be undertaken; otherwise, muscle biopsy for histochemical and enzymatic testing could direct diagnosis (60–62). Muscle biopsy is typically unremarkable in GSDV, while increased glycogen concentration by periodic acid-Schiff (PAS) staining and electronic microscopy can be disclosed in GSDII (65). Currently, there is no specific treatment for glycogen storage diseases that results in exercise-induced symptoms, apart from avoiding intense physical exercise and treating symptoms. A low dose of creatine monohydrate may provide a mild benefit in PYGM mutations. Immediate pre-exercise carbohydrate improves symptoms in the glycogenolytic defects (such as GSDV) but can exacerbate symptoms in glycolytic defects (such as GSDVII) (63).
Long chain fatty acids represent the main key energy source for muscle during prolonged, low intensity activity. Recurrent episodes of myalgia, fatigue, rhabdomyolysis precipitated by prolonged sub maximal aerobic exercise (or after other triggering events such as fasting, illness or emotional stress), should lead clinicians to consider defects of fatty acid metabolism (60, 62). The most common is Carnitine palmitoyltransferase II (CPTII) enzyme deficiency, which is due to recessive mutations in CPT2 gene. This gene is part of the long chain fatty acids transportation system through the mitochondrial membrane and more than 30 pathogenic mutations have been identified, with p.S113L identified as the most common (66). Investigations usually disclose abnormal serum carnitine/acylcarnitine profile, with increased long-chain acylcarnitine. Ictal serum CK can be markedly elevated while the basal one is normal, as well as neurological examination. Very-long-chain acyl-CoA dehydrogenase (VLCAD) catalyses the initial step of mitochondrial beta-oxidation of long-chain fatty acids. Its deficiency is caused by recessive mutations in the ACADVL gene, which manifest in an infantile form complicated by severe cardiomyopathy or with an adult-onset phenotype clinically indistinguishable from adult CPTII deficiency. Plasma acylcarnitines pattern during periods of metabolic stress shows increased long chain intermediate, likewise to long-chain 3-hydroxyacylCoA dehydrogenase (LCHAD) deficiency due to mutation in the mitochondrial Trifunctional Protein (MTP). This is a very rare fatty acid oxidation (FAO) defect in which myoglobinuria is often associated with life-threatening respiratory distress and/or axonal sensory-motor neuropathy (62). Muscle biopsy is usually uninformative in CPTII and VLCAD, while in LCHAD a chronic denervation or rarely lipid storage could be disclosed (61). Therapeutic recommendations include the avoidance of precipitating factors, such as prolonged fasting, aerobic exercise, infections, exposure to cold and intensive saline, glucose solution administration, and monitoring of electrolytes, kidney and cardiac functions in the case of rhabdomyolysis.
Mitochondrial myopathies encompass a broad and heterogeneous group of rare genetic progressive conditions caused by mutations in genes in the nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) that impair the oxidative phosphorylation (OXPHOS) in the mitochondria (67, 68). The consequence is a deficit in energy production in the form of adenosine triphosphate (ATP), with muscles being the most affected tissue. Mitochondrial myopathies rarely manifest with acute myalgia and myoglobinuria, such as in complex I, III (cytochrome b) and IV (cytochrome c oxidase or COX) (61, 68, 69). However, recurrent exercise intolerance and premature fatigue are frequently reported in mitochondrial myopathies. Comparable to fatty acid oxidation defects, symptoms usually manifest after prolonged submaximal leisurely activity or during fasting, intercurrent illnesses or fever. Exercise induced symptoms may manifest as isolated features or associated with progressive weakness and multisystemic involvement (65). Muscle weakness typically affects extraocular muscles (ptosis, ophthalmoparesis, or both), but frequently spreads to bulbar, limb or axial muscles. Identification of an accompanying multisystem disease, variably including cognitive impairment, encephalopathy, peripheral neuropathy, epilepsy, stroke-like events, cardiomyopathy, hepatopathy and nephropathy, gastrointestinal dysmotility, diabetes, or maternal inheritance pattern increases clinical suspicion of mitochondrial disease (65, 69). Exercise intolerance or rhabdomyolysis could be part of the classic maternally inherited mitochondrial encephalomyopathies such as MELAS, MERRF, MNGIE or Kearns-Sayre Syndrome/C-PEO (61) or secondary to a number of mitochondrial dysfunction, such as complex III (cytochrome b) and IV (cytochrome c oxidase or COX) such us tRNA mutations and Coenzyme Q10 deficiencies (61, 68). Notably, the latter form responds to CoQ10 supplementation (60). Laboratory investigations should include serum lactate and pyruvate (increased ratio) and CK (normal or mildly elevated), muscle biopsy (typically displaying ragged red fibers (RRF), COX-negative fibers), and measurement of the activities of respiratory chain complexes (61).
The deficiency of muscle-specific phosphatidic acid phosphatase (LPIN1) regulates lipid metabolism and the mitochondrial respiratory chain. It is recognized as a common cause of AR recurrent and life-threatening acute rhabdomyolysis of childhood-onset, sometimes triggered by exercise but mainly by fever (70).
Rarely, the muscular dystrophies caused by DMD mutations (71, 72), limb girdle muscular dystrophy due to FKRP, ANO5, CPN3 (73), CAV3 (74), DYSF (75) and sarcoglycanopathies (76), and channelopathies secondary to RYR1 (77, 78) mutations, can present in a “pseudometabolic fashion” (Figure 3) (60). Dystrophin, encoded by the DMD gene, is a component of the dystrophin-associated glycoprotein complex which accumulates at the NMJ and synapses and has a structural function in stabilizing the sarcolemma and synaptic transmission. Patients carrying DMD mutations and exhibiting this particular phenotype presented with or without fixed muscle weakness or calf hypertrophy and frequently carried exon deletions throughout the gene or particularly mutations involving the proximal third of the rod domain (71, 72). Rhabdomyolysis triggered by exercise, heat, or infections and a mild proximal weakness and ptosis is detected in some patients carrying heterozygous RYR1mutations, encoding for the principal sarcoplasmic reticulum calcium release channel, with a fundamental role in excitation-contraction coupling. RYR1 mutations are a common cause of neuromuscular disease, ranging from various congenital myopathies to the malignant hyperthermia (MH) susceptibility. It is therefore important to these identify patients (77, 78). Finally, specific polymorphic variants in a number of genes have been reported in association with increased susceptibility to exertional rhabdomyolysis, together with exceptional athletic abilities and including ACE, ACTN3, CCL2 and CCR2 genes (35).
Myasthenia Gravis and Congenital Myasthenic Syndromes
An abnormal fatigability or fluctuating muscle weakness that worsens after exertion suggests a suspect of myasthenia gravis (MG) or congenital myasthenic syndromes (CMS) (Figure 3). They are respectively autoimmune and genetic diseases caused by impairment of synaptic transmission in the NMJ (79).
MG is an autoimmune disease caused by antibodies directed against key molecules at the NMJ, such as the nicotinic acetylcholine receptor (AChR), muscle-specific kinase (MuSK), and low-density lipoprotein receptor-related protein 4 (Lrp4), or agrin in the postsynaptic membrane. Those autoantibodies alter densities or function of AChRs and neuromuscular transmission. Consequently, they result in a localized or generalized fluctuating weakness that primarily affects ocular muscle with ptosis and diplopia (80). Fatigability can be elicited during examination by prolonged upgaze or repetitive movements; improvement of ptosis after the application of ice in a latex glove to the eyes can help in distinguishing myasthenia from other causes of ptosis (79). if suspected in a clinical setting, they are confirmed by positive antibody tests and electrophysiological tests, including single-fiber electromyography (SFEMG), and repetitive nerve stimulation (RNS), but routine nerve conduction tests and EMG are not usually informative. Good prognosis is obtained with adequate and prompt symptomatic treatment with AChE inhibitor (pyridostigmine), thymectomy and immunomodulatory treatments including corticosteroids, immunoglobulin (IVIg), plasma exchange and immunosuppressants being mycophenolate mofetil and azathioprine the most commonly used (80–82). Although onset ranges from birth to adulthood, MG during childhood is very infrequent, and the diagnosis is challenging because of the higher percentage of seronegative patients and the possible differential diagnosis of CMS (81).
CMS comprise a group of rare early onset hereditary disorders caused by mutations in several genes that code for constitutive proteins of the NMJ and are then characterized by impaired neuromuscular transmission (83, 84). The phenotype is typically dominated by fatigable muscle weakness, frequently combined with ptosis, ophthalmoparesis, facial, bulbar or generalized muscle weakness, spinal deformities and reduced muscle bulk; cognitive disability, dysmorphism, neuropathy, or epilepsy are occasional (85). The diagnosis is secured by SFEMG or RNS demonstrating a neuromuscular transmission defect (Figure 3). Mutation in more than 30 causative genes has been identified, mostly harbored in an autosomal recessive pattern (84). CMS are classified according to the location of the mutated protein as presynaptic, synaptic or postsynaptic ant, but new gene coding for ubiquitous proteins have been identified recently, including those involved in the glycosylation pathways and the synthesis of propyloligopeptidase (84). The most common causative gene is CHRNE, which accounts for 30–50% of the CMS cases, followed by RAPSN, COLQ, DOK7, CHAT, and GFPT1 (85). Genotype-phenotype correlations are difficult. However, some peculiar phenotypic aspects may point toward a specific genetic defect, such as episodic apneas (CHAT, RAPSN), tongue atrophy and stridor and vocal cord paralysis in neonates or infants (DOK7), congenital contractures (RAPSN, AChR δ or γ subunit, CHAT), limb-girdle and axial distribution of weakness (DOK7, GFPT1, DPAGT1, ALG2, ALG14), facial dysmorphism (SYB1, RAPSN, SCN4A, COLQ), seizures or ID (DPAGT1). Interestingly, a mutation in SCN4A, encoding for a post-synaptic sodium channel responsible for the generation of membrane action potentials, have been linked to CMS but also to hyperkalaemic or hypokalaemic periodic paralysis, myotonia presentation and congenital myopathy or fetal akinesia (86). The main symptomatic treatment in most CMS subtypes, such as AChR deficiency, fast-channel syndrome and rapsyn deficiency is the AChE inhibitor (pyridostigmine), which prolongs synaptic response to Ach. It is, however, ineffective or dangerous in CMS due to mutations in COLQ, DOK7 and CHRNA1. 3,4-Diaminopyridine, a potassium channel blocker that enables the release of ACh from the presynaptic terminal, is used as an adjunctive treatment with pyridostigmine and ChAT CMS could theoretically worsen in response. Adrenergic agonists are effective in DOK7 and AChE deficiency through stabilization of the postsynaptic membrane. Long-lived open-channel blockers of the AChR ion channel (fluoxetine and quinidine) are used in the slow-channel syndrome (83). Therefore, genetic characterization of CMS is central for selecting the adequate tailored treatment (83–85).
Primary Muscle Channellopathies
Primary muscles channelopathies are a group of inherited disorders caused by mutations in genes encoding sodium channel (SCN4A), chloride channel (CLCN1), calcium channel (CACNA1S), or potassium channel (KCNJ2 and KCNJ18) (87). They comprise two major categories: non-dystrophic myotonia (NDMs) and periodic paralysis (PP) (88). Despite different pathophysiology, the cardinal clinical manifestations of these disorders are muscle weakness or stiffness elicited or worsened by exercise.
Myotonia, the cardinal features of NDMs, is defined by an involuntary muscle contraction that persists for several seconds after cessation of voluntary effort and causes stiffness, impaired mobility and rarely pain. Myotonia can involve the eye, facial and jaw muscles as well as the arms and legs. Myotonic stiffness decreased with continued voluntary muscle activity (warm-up phenomenon), but sometimes myotonic stiffness may paradoxically worsen by repeated muscle activity (paramyotonia).
The NDMs include myotonia congenita (MC) due to mutations in the skeletal muscle chloride channel gene CLCN1, paramyotonia congenita (PMC) and sodium channel myotonia (SCM), both of the latter are caused by mutations in the skeletal muscle sodium channel gene SCN4A encoding Nav1.4. Chloride channel myotonia can be inherited in a recessive or dominant pattern (Becker's and Thomsen's myotonia, respectively), while myotonia due to sodium channel mutations is always autosomal dominant (89). Interestingly, exercise improves symptoms in MC, while it worse them in SCN4A-related disorders. In individuals with Thomsen disease, myotonia, associated muscle rigidity, and muscle hypertrophy may become apparent from infancy to approximately 2–3 years of age, while in those with Becker disease, age of onset is usually later (89). In severe myotonic disorders, patients develop a body-builder-like appearance secondary to continuous myotonic contractions.
The absence of severe fixed weakness or muscle wasting distinguishes NDMs from dystrophic myotonias, such as myotonic dystrophies type 1 and type 2 (DM1 and DM2 due to DMPK and CNBP mutations, respectively), which present with both progressive muscle weakness and multisystem involvement (Figure 4) (87). Schwartz-Jampel Syndrome should be considered in the differential diagnosis in patients with severe myotonia not associated with the warm-up phenomenon and dysmorphic features, such as short stature, muscular hypertrophy, joint contractures, bone dysplasia, ocular and facial abnormalities. This condition is caused by a loss-of-function mutation in the HSPG2 gene, which encodes perlecan, a major component of basement membranes (90).
PMC is characterized by episodes of sustained myotonia that, unlike other forms, worsen by exercise and repeated movements and exposure to cold temperatures. Stiffness most commonly affects the muscles in the neck, face, arms and hands. Intermittent periods of muscle weakness (flaccid paresis) have also been reported (89).
The intensity and the duration of the myotonic stiffness are variable and may also depend upon other trigger factors such as temperature, emotional stress, and concomitant medical conditions. Especially in children, myotonia causing rigidity and stiffness of a body segment after a voluntary movement can be difficult to distinguish from paroxysmal dyskinesia (91). The first patient reported in the literature with paroxysmal dyskinesias was defined as having myotonia congenita (92). Treatment of myotonia includes pharmacological agents (mexiletine at the first line, but also lamotrigine, carbamazepine and acetazolamide) as well as dietary and lifestyle precautions (93).
The PPs are a group of skeletal muscle channelopathies characterized by intermittent attacks of muscle weakness often associated with altered serum potassium levels (Figure 4). Primary periodic paralysis (PP) include hypokalemic paralysis (HypoKPP), hyperkalemic paralysis (HyperKPP), and Andersen-Tawil syndrome (ATS) (94). They are caused by mutations in the skeletal muscle sodium, calcium, and potassium channels and have an autosomal dominant inheritance. Despite genetic heterogeneity, all forms share a common final mechanistic pathway, aberrant depolarization and muscle fiber unexcitability (89). Typically, attacks of paralysis are provoked by exercise or periods of rest (minutes to hours) after vigorous exercise but also infection, stress, fatigue, menses can trigger the attacks. Onset is during the first or second decade and the frequency of episodes is variable but tends to reduce over time. Between episodes patients are neurologically normal until adulthood, when they may develop progressive proximal fixed weakness.
HypoKPP, the most common form of PPs, is caused by mutations in the alpha subunits of either the skeletal muscle L-type calcium channel gene CACN1AS (HypoKPP1) or the skeletal muscle sodium channel gene SCN4A (HypoKPP2) (94). Patients with HypoKPP tend to have prolonged and severe attacks of weakness (several hours to days or weeks), focal or generalized, usually sparing facial and respiratory muscles. Symptom onset is typically in the second decade. Besides exercise, carbohydrate-rich meals are a typical trigger (95). During episodes, potassium levels are constantly reduced (<3.0 mM) and potassium supplements are part of the treatment regime, while the reverse is true for HyperPP where serum potassium levels are usually elevated and attacks can be triggered by potassium and ameliorated by glucose.
Patients with HyperKPP exhibit an earlier onset. Attacks are characterized by generalized weakness lasting hours, sometimes with fatal involvement of bulbar muscle. Electrical and clinical myotonia is a useful diagnostic pointer as it is often associated with HyperKPP and not found in HypoKPP. HyperKPP is caused by mutations in SCN4A, as well as the allelic disorder paramyotonia congenital.
ATS is characterized by the clinical triad of episodic flaccid muscle weakness, ventricular arrhythmias and prolonged QT interval and dysmorphic features. These include short stature, scoliosis, low-set ears, hypertelorism, broad nasal bridge, micrognathia, clino- or syndactyly, and toes joined at the base. Myotonia is not present. Patients may exhibit either hypokaliemic or hyperkaliemic paralysis. Around 60% of ATS patients carried a mutation in the potassium inward rectifier KCNJ2 gene on chromosome 17q24 (87, 95).
Epilepsy
Although epilepsy is characterized by recurrent, unprovoked seizures, a number of endogenous or exogenous precipitant factors have been identified. They include sleep deprivation, intercurrent illnesses, menstrual cycle, alcohol intake, protracted fasting and photic stimulation. Physical exhaustion due to exercise or sports activity has also been considered. Its causative role has primarily been attributed to hyperventilation, hypoxia, hypercapnia, hyperthermia, hypoglicemia and hyponatremia (96, 97). There are very few reports of adult patients with clear exercise-induced seizures, and interestingly most of them demonstrated temporal lobe partial epilepsy (98, 99). Report of pediatric cases are even more scarce (100), but the possibility that a paroxysmal event precipitated by exercise may be epileptic in nature has to be taken into account in the differential diagnosis of PENS.
Psychogenic Disorders
The term psychogenetic or functional disorders encompass symptoms and disturbances of motor function that are not explained by organic conditions (101).
Psychogenetic disorders in adult neurological patients are well-described and characterized in the literature, but in recent years functional phenomenology in pediatric neurology has emerged as an important topic frequently encountered in clinical practice (101).
Psychogenic paroxysmal movement disorders are characterized by episodes that usually have multiple or inconsistent triggers in respect of primary paroxysmal dyskinesias; onset of the clinical symptoms is often acute following a stressor life event. The duration of episodes is also variable and patients often present with other unexplained somatic symptoms (102).
Common functional symptoms are also weakness, fatigue, paralysis, myalgias and cramps, which may lead clinicians to initially suspect a possible neuromuscular disorder (56). The diagnosis of a functional neuromuscular disorder is based upon the inconsistency of the clinical symptoms and the neurological examination, with the support, whenever appropriate, of medical tests.
Discussion
A wide range of neurological symptoms often encountered in clinical practice are typically elicited by physical exercise. Unfortunately, being paroxysmal and not easily observed during an examination, the characterization of PENS is based on a clinical description from parents and patients; however, the partial resemblance among symptoms and the extensive genetic heterogeneity associated make the differential diagnosis frequently difficult and delayed. However, reaching a diagnosis is mandatory due to the availability of specific treatment. We suggest practical algorithms which could help orient the diagnosis.
The first step consists of correctly recognizing the clinical phenomenology and classifying it within three main categories of PENS: paroxysmal moment disorder, exercise intolerance or a paroxysmal exercise induced stiffness /weakness; and we propose a flowchart for each clinical entity (Figures 2–4).
An accurate assessment of the age at onset, type and duration of the triggering exercise, the presence of non-paroxysmal neurological symptoms and/or multisystemic involvement are fundamental for guide a multi-step path that requires the support of neuroradiologic, electrophysiological and laboratory investigations.
While in adolescents a PARK2 should be investigated through a DATscan, in childhood Brain MRI is fundamental to exclude symptomatic PED and investigate specific abnormalities in basal ganglia that can direct genetic testing for neurometabolic disorders. In patients with normal brain MRI, a lumbar puncture would be crucial for exploring DRD and GLUT1 syndrome (Figure 2). In patients with exercise intolerance particular clinical key signs or symptoms, such as myoglobinuria, “second wind phenomenon,” the presence of baseline neurological or multisystemic involvement and the type of duration of exercise inducing those symptoms are the most relevant information for address diagnostic suspicion of metabolic myopathies rather than other neuromuscular disorders, including myasthenia and muscular dystrophies (Figure 3). In most cases, histological and immunohistochemical analysis on muscle biopsy is fundamental to addressing specific genetic testing. Finally, the clinical and/or electrophysiological evidence of myotonia or paramyotonia, together with the detection of other muscular or extramuscular features and altered serum potassium levels could expedite the diagnostic process (Figure 4). In some cases, such as EAs or Myotonic Dystrophies, the characterization of the specific motor phenomenon in the context of a positive familial history could directly suggest specific genetic confirmation. If a defined etiological diagnosis cannot be reached after extensive work-up, especially when the paroxysmal phenomenon does not fit properly into a precise category, the possibility of a psychogenic disorder has to be ruled out.
Intriguingly, despite the wide clinical and genetic heterogeneity described herein, it is possible to identify some common pathogenic mechanisms responsible for these episodic neurological disorders. The defects in energy metabolism that impair the ability to respond to increased energy needs in the brain and muscles during exercise appear to be crucial pathogenic mechanisms for PED due to SLC2A1 mutations and mitochondrial disorders (including defect of PDH complex, ECSH1 and HIBCH) and metabolic myopathies. Secondly, a number of PENS grouped as “channelopathies,” due to mutations in either central and muscular ion channels or their interacting proteins (103, 104). These mutations result in the alteration of action potential properties or synaptic transmission; several stressors, including exercise, can prompt an unstable and symptomatic state. Consistent with the distribution of ion channels throughout the human body and their resulting hyper- or hypoexcitability, channelopathies manifest with a variety of PENS, including movement disorders and myotonia, myasthenia and periodic paralysis (86, 104). Moreover, mutations in several PENS-associated genes such as PRRT2, MR-1, ADCY5 and TBC1D24 results in alterations of the synaptic function and regulation of vesicle trafficking and neurotransmitter release (105). Finally, impaired neurotransmission due to the defect of dopamine and GABA metabolism (PARKIN, GCH and ALDH5A1 mutations) is responsible for exercise- induced movement disorders.
Finally, we propose a new clinical entity named PENS and reviewed clinical features, genetic and principles of treatment mainly focusing on phenomenology and clues for differential diagnosis. Given the overlapping clinical features and molecular bases, considering PENS as a whole would help consider the entire range of differential diagnosis.
Author Contributions
GZ, FD, NN, and BG contributed to conception and design of the study. GZ and FD wrote the first draft of the manuscript. IM and FI wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Figure 1 was created using Servier Medical Art templates, which were licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com.
References
1. Hargreaves M, Spriet LL. Skeletal muscle energy metabolism during exercise. Nat Metab. (2020) 2:817–28. doi: 10.1038/s42255-020-0251-4
2. Taylor JL, Amann M, Duchateau J, Meesen R, Rice CL. Neural contributions to muscle fatigue. Med Sci Sport Exerc. (2016) 48:2294–306. doi: 10.1249/MSS.0000000000000923
3. Erro R, Sheerin UM, Bhatia KP. Paroxysmal dyskinesias revisited: a review of 500 genetically proven cases and a new classification. Mov Disord. (2014) 29:1108–16. doi: 10.1002/mds.25933
4. Latorre A, Bhatia KP. Treatment of paroxysmal dyskinesia. Neurol Clin. (2020) 38:433–47. doi: 10.1016/j.ncl.2020.01.007
5. Suls A, Dedeken P, Goffin K, Van Esch H, Dupont P, Cassiman D, et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. (2008) 131:1831–44. doi: 10.1093/brain/awn113
6. Zorzi G, Castellotti B, Zibordi F, Gellera C, Nardocci N. Paroxysmal movement disorders in GLUT1 deficiency syndrome. Neurology. (2008) 71:146–8. doi: 10.1212/01.wnl.0000316804.10020.ba
7. Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep. (2013) 13:342. doi: 10.1007/s11910-013-0342-7
8. Mongin M, Mezouar N, Dodet P, Vidailhet M, Roze E. Paroxysmal exercise-induced dyskinesias caused by GLUT1 deficiency syndrome. Tremor Other Hyperkin Mov. (2016) 2016:1–2. doi: 10.5334/tohm.328
9. De Giorgis V, Veggiotti P. GLUT1 deficiency syndrome 2013: current state of the art. Seizure. (2013) 22:803–11. doi: 10.1016/j.seizure.2013.07.003
10. Wang D, Pascual JM, Yang H, Engelstad K, Jhung S, Sun RP, et al. Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol. (2005) 57:111–8. doi: 10.1002/ana.20331
11. Gardiner AR, Jaffer F, Dale RC, Labrum R, Erro R, Meyer E, et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain. (2015) 138:3567–80. doi: 10.1093/brain/awv310
12. Leen WG, Mewasingh L, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA. Movement disorders in GLUT1 deficiency syndrome respond to the modified Atkins diet. Mov Disord. (2013) 28:1439–42. doi: 10.1002/mds.25515
13. Hainque E, Meneret A, Gras D, Atencio M, Luton MP, Barbier M, et al. Transition from ketogenic diet to triheptanoin in patients with GLUT1 deficiency syndrome. J Neurol Neurosurg Psychiatry. (2020) 91:444–5. doi: 10.1136/jnnp-2019-321694
14. Gnidovec B, Repetto GM, Doummar D, Lees MM, Rosenfeld JA, Striano P. TBC1D24 genotype – phenotype correlation Epilepsies and other neurologic features. Neurology. (2016) 87:77–87. doi: 10.1212/WNL.0000000000002807
15. Lüthy K, Mei D, Fischer B, De Fusco M, Swerts J, Paesmans J, et al. TBC1D24-TLDc-related epilepsy exercise-induced dystonia: rescue by antioxidants in a disease model. Brain. (2019) 142:2319–35. doi: 10.1093/brain/awz175
16. Steel D, Heim J, Kruer MC, Sanchis-Juan A, Raymond LF, Eunson P, et al. Biallelic mutations of TBC1D24 in exercise-induced paroxysmal dystonia. Mov Disord. (2020) 35:372–3. doi: 10.1002/mds.27981
17. Zimmern V, Riant F, Roze E, Ranza E, Lehmann-Horn F, de Bellescize J, et al. Infantile-onset paroxysmal movement disorder and episodic ataxia associated with a TBC1D24 mutation. Neuropediatrics. (2019) 50:308–12. doi: 10.1055/s-0039-1688410
18. Chen YZ, Matsushita MM, Robertson P, Rieder M, Girirajan S, Antonacci F, et al. Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol. (2012) 69:630–5. doi: 10.1001/archneurol.2012.54
19. Morgan JC, Kurek JA, Davis J, Sethi KD. ADCY5 mutations are another cause of benign hereditary chorea. Neurology. (2016) 86:978–9. doi: 10.1212/WNL.0000000000002479
20. Carecchio M, Mencacci NE, Iodice A, Pons R, Panteghini C, Zorzi G, et al. ADCY5-related movement disorders: frequency, disease course and phenotypic variability in a cohort of paediatric patients. Parkinsonism Relat Disord. (2017) 41:37–43. doi: 10.1016/j.parkreldis.2017.05.004
21. Friedman JR, Méneret A, Chen D-H, Trouillard O, Vidailhet M, Raskind WH, et al. ADCY5 mutation carriers display pleiotropic paroxysmal day and nighttime dyskinesias. Mov Disord. (2016) 31:147–8. doi: 10.1002/mds.26494
22. de Almeida Marcelino AL, Mainka T, Krause P, Poewe W, Ganos C, Kühn AA. Deep brain stimulation reduces (nocturnal) dyskinetic exacerbations in patients with ADCY5 mutation: a case series. J Neurol. (2020) 267:3624–31. doi: 10.1007/s00415-020-09871-8
23. Barodia SK, Creed RB, Goldberg MS. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res Bull. (2017) 133:51–9. doi: 10.1016/j.brainresbull.2016.12.004
24. Erro R, Stamelou M, Ganos C, Skorvanek M, Han V, Batla A, et al. The clinical syndrome of paroxysmal exercise-induced dystonia: diagnostic outcomes and an algorithm. Mov Disord Clin Pract. (2014) 1:57–61. doi: 10.1002/mdc3.12007
25. Wasner K, Grünewald A, Klein C. Parkin-linked Parkinson's disease: from clinical insights to pathogenic mechanisms and novel therapeutic approaches. Neurosci Res. (2020) 159:34–9. doi: 10.1016/j.neures.2020.09.001
26. Ng J, Papandreou A, Heales SJ, Kurian MA. Monoamine neurotransmitter disorders - Clinical advances and future perspectives. Nat Rev Neurol. (2015) 11:567–84. doi: 10.1038/nrneurol.2015.172
27. Dale RC, Melchers A, Fung VS, Grattan-Smith P, Houlden H, Earl J. Familial paroxysmal exercise-induced dystonia: atypical presentation of autosomal dominant GTP-cyclohydrolase 1 deficiency. Dev Med Child Neurol. (2010) 52:583–6. doi: 10.1111/j.1469-8749.2010.03619.x
28. Choi K-D, Choi J-H. Episodic ataxias: clinical and genetic features. J Mov Disord. (2016) 9:129–35. doi: 10.14802/jmd.16028
29. Graves TD, Cha YH, Hahn AF, Barohn R, Salajegheh MK, Griggs RC, et al. Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain. (2014) 137:1009–18. doi: 10.1093/brain/awu012
30. Paulhus K, Ammerman L, Glasscock E. Clinical spectrum of KCNA1 mutations: new insights into episodic ataxia and epilepsy comorbidity. Int J Mol Sci. (2020) 21:2802. doi: 10.3390/ijms21082802
31. Jen JC, Wan J. Episodic Ataxias. 1st ed. Elsevier B.V. (2018). doi: 10.1016/B978-0-444-64076-5.00033-8
32. Strupp M, Kalla R, Claassen J, Adrion C, Mansmann U, Klopstock T, et al. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology. (2011) 77:269–75. doi: 10.1212/WNL.0b013e318225ab07
33. Garone G, Capuano A, Travaglini L, Graziola F, Stregapede F, Zanni G, et al. Clinical and genetic overview of paroxysmal movement disorders and episodic ataxias. Int J Mol Sci. (2020) 21:3603. doi: 10.3390/ijms21103603
34. Marti-Sanchez L, Baide-Mairena H, Marcé-Grau A, Pons R, Skouma A, López-Laso E, et al. Delineating the neurological phenotype in children with defects in the ECHS1 or HIBCH gene. J Inherit Metab Dis. (2021) 44:401–14. doi: 10.1002/jimd.12288
35. Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem. (2014) 289:16615–23. doi: 10.1074/jbc.R114.563148
36. Erro R, Bhatia KP. Unravelling of the paroxysmal dyskinesias. J Neurol Neurosurg Psychiatry. (2019) 90:227–34. doi: 10.1136/jnnp-2018-318932
37. Patel KP, O'Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. (2012) 106:385–94. doi: 10.1016/j.ymgme.2012.03.017
38. Cordeiro D, Bullivant G, Siriwardena K, Evans A, Kobayashi J, Cohn RD, et al. Genetic landscape of pediatric movement disorders and management implications. Neurol Genet. (2018) 4:265. doi: 10.1212/NXG.0000000000000265
39. Castiglioni C, Verrigni D, Okuma C, Diaz A, Alvarez K, Rizza T, et al. Pyruvate dehydrogenase deficiency presenting as isolated paroxysmal exercise induced dystonia successfully reversed with thiamine supplementation. Case report and mini-review. Eur J Paediatr Neurol. (2015) 19:497–503. doi: 10.1016/j.ejpn.2015.04.008
40. Friedman J, Feigenbaum A, Chuang N, Silhavy J, Gleeson JG. Pyruvate dehydrogenase complex-E2 deficiency causes paroxysmal exercise-induced dyskinesia. Neurology. (2017) 89:2297–8. doi: 10.1212/WNL.0000000000004689
41. Méneret A, Roze E. Paroxysmal movement disorders: an update. Rev Neurol. (2016) 172:433–45. doi: 10.1016/j.neurol.2016.07.005
42. Galosi S, Nardecchia F, Leuzzi V. Treatable inherited movement disorders in children: spotlight on clinical and biochemical features. Mov Disord Clin Pract. (2020) 7:154–66. doi: 10.1002/mdc3.12897
43. Holeček M. Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr Metab. (2018) 15:33. doi: 10.1186/s12986-018-0271-1
44. Olgiati S, Skorvanek M, Quadri M, Minneboo M, Graafland J, Breedveld GJ, et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov Disord. (2016) 31:1041–8. doi: 10.1002/mds.26610
45. Mahajan A, Constantinou J, Sidiropoulos C. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: case presentation and initial benefit of intervention. J Neurol. (2017) 264:185–7. doi: 10.1007/s00415-016-8381-z
46. Masnadaa S, Parazzinib C, Binic P, Barbarinic M, Albertid L, ValenteeLuisa M, et al. Phenotypic spectrum of short-chain enoyl-Coa hydratase-1 (ECHS1) deficiency. Eur J Paediatr Neurol. (2020) 28:151–58. doi: 10.1016/j.ejpn.2020.07.007
47. Illsinger S, Korenke GC, Boesch S, Nocker M, Karall D, Nuoffer JM, et al. Paroxysmal and non-paroxysmal dystonia in 3 patients with biallelic ECHS1 variants: expanding the neurological spectrum and therapeutic approaches. Eur J Med Genet. (2020) 63:104046. doi: 10.1016/j.ejmg.2020.104046
48. Xu Y, Zhang J, Yu K, Feng F, Sun X, Li C, et al. A therapeutic regimen for 3-hydroxyisobutyryl-CoA hydrolase deficiency with exercise-induced dystonia. Eur J Paediatr Neurol. (2019) 23:755–9. doi: 10.1016/j.ejpn.2017.11.004
49. Leuzzi V, Di Sabato ML, Deodato F, Rizzo C, Boenzi S, Carducci C, et al. Vigabatrin improves paroxysmal dystonia in succinic semialdehyde dehydrogenase deficiency. Neurology. (2007) 68:1320.LP−1. doi: 10.1212/01.wnl.0000259537.54082.6d
50. Malaspina P, Roullet JB, Pearl PL, Ainslie GR, Vogel KR, Gibson KM. Succinic semialdehyde dehydrogenase deficiency (SSADHD): pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism. Neurochem Int. (2016) 99:72–84. doi: 10.1016/j.neuint.2016.06.009
51. Gahr M, Connemann BJ, Schönfeldt-Lecuona CJ, Freudenmann RW. Succinat-Semialdehyd-dehydrogenase-mangel: eine vererbbare neurometabolische Erkrankung. Fortschritte der Neurol Psychiatr. (2013) 81:154–61. doi: 10.1055/s-0032-1330544
52. Synofzik M, Schicks J, Lindig T, Biskup S, Schmidt T, Hansel J, et al. Acetazolamide-responsive exercise-induced episodic ataxia associated with a novel homozygous DARS2 mutation. J Med Genet. (2011) 48:713–5. doi: 10.1136/jmg.2011.090282
53. Scheper GC, van der Klok T, van Andel RJ, van Berkel CGM, Sissler M, Smet J, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. (2007) 39:534–9. doi: 10.1038/ng2013
54. Darras BT, Friedman NR. Metabolic myopathies: a clinical approach; part I. Pediatr Neurol. (2000) 22:87–97. doi: 10.1016/S0887-8994(99)00133-2
55. McDonald CM. Clinical approach to the diagnostic evaluation of hereditary and acquired neuromuscular diseases. Phys Med Rehabil Clin N Am. (2012) 23:495–563. doi: 10.1016/j.pmr.2012.06.011
56. Torricelli RPJE. Acute muscular weakness in children. Arq Neuropsiquiatr. (2017) 75:248–54. doi: 10.1590/0004-282x20170026
57. Quinlivan R, Jungbluth H. Myopathic causes of exercise intolerance with rhabdomyolysis. Dev Med Child Neurol. (2012) 54:886–91. doi: 10.1111/j.1469-8749.2012.04320.x
58. Lilleker JB, Keh YS, Roncaroli F, Sharma R, Roberts M. Metabolic myopathies: a practical approach. Pract Neurol. (2018) 18:14–26. doi: 10.1136/practneurol-2017-001708
59. Fernandes PM, Davenport RJ. How to do it: Investigate exertional rhabdomyolysis (or not). Pract Neurol. (2019) 19:43–8. doi: 10.1136/practneurol-2018-002008
60. Nance JR, Mammen AL. Diagnostic evaluation of rhabdomyolysis. Muscle Nerve. (2015) 51:793–810. doi: 10.1002/mus.24606
61. Toscano A, Barca E, Musumeci O. Update on diagnostics of metabolic myopathies. Curr Opin Neurol. (2017) 30:553–62. doi: 10.1097/WCO.0000000000000483
62. Scalco RS, Gardiner AR, Pitceathly RD, Zanoteli E, Becker J, Holton JL, et al. Rhabdomyolysis: a genetic perspective. Orphanet J Rare Dis. (2015) 10:51. doi: 10.1186/s13023-015-0264-3
63. Tarnopolsky MA. Myopathies related to glycogen metabolism disorders. Neurotherapeutics. (2018) 15:915–27. doi: 10.1007/s13311-018-00684-2
64. Musumeci O, Brady S, Rodolico C, Ciranni A, Montagnese F, Aguennouz M, et al. Recurrent rhabdomyolysis due to muscle β-enolase deficiency: very rare or underestimated? J Neurol. (2014) 261:2424–8. doi: 10.1007/s00415-014-7512-7
65. Berardo A, Dimauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep. (2010) 10:118–26. doi: 10.1007/s11910-010-0096-4
66. Joshi PR, Deschauer M, Zierz S. Carnitine palmitoyltransferase II (CPT II) deficiency: genotype–phenotype analysis of 50 patients. J Neurol Sci. (2014) 338:107–11. doi: 10.1016/j.jns.2013.12.026
67. Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Prim. (2016) 2:16080. doi: 10.1038/nrdp.2016.80
68. de Barcelos IP, Emmanuele V, Hirano M. Advances in primary mitochondrial myopathies. Curr Opin Neurol. (2019) 32:715–21. doi: 10.1097/WCO.0000000000000743
69. Milone M, Wong L-J. Diagnosis of mitochondrial myopathies. Mol Genet Metab. (2013) 110:35–41. doi: 10.1016/j.ymgme.2013.07.007
70. Zeharia A, Shaag A, Houtkooper RH, Hindi T, de Lonlay P, Erez G, et al. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am J Hum Genet. (2008) 83:489–94. doi: 10.1016/j.ajhg.2008.09.002
71. Veerapandiyan A, Shashi V, Jiang Y-H, Gallentine WB, Schoch K, Smith EC. Pseudometabolic presentation of dystrophinopathy due to a missense mutation. Muscle Nerve. (2010) 42:975–9. doi: 10.1002/mus.21823
72. Liewluck T, Tian X, Wong L-J, Pestronk A. Dystrophinopathy mimicking metabolic myopathies. Neuromuscul Disord. (2015) 25:653–7. doi: 10.1016/j.nmd.2015.04.001
73. Lahoria R, Milone M. Rhabdomyolysis featuring muscular dystrophies. J Neurol Sci. (2016) 361:29–33. doi: 10.1016/j.jns.2015.12.013
74. Scalco RS, Gardiner AR, Pitceathly RDS, Hilton-Jones D, Schapira AH, Turner C, et al. CAV3 mutations causing exercise intolerance, myalgia and rhabdomyolysis: expanding the phenotypic spectrum of caveolinopathies. Neuromuscul Disord. (2016) 26:504–10. doi: 10.1016/j.nmd.2016.05.006
75. Moody S, Mancias P. Dysferlinopathy presenting as rhabdomyolysis and acute renal failure. J Child Neurol. (2013) 28:502–5. doi: 10.1177/0883073812444607
76. Xie Z, Hou Y, Yu M, Liu Y, Fan Y, Zhang W, et al. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J Rare Dis. (2019) 14:43. doi: 10.1186/s13023-019-1021-9
77. Dlamini N, Voermans NC, Lillis S, Stewart K, Kamsteeg E-J, Drost G, et al. Mutations in RYR1 are a common cause of exertional myalgia and rhabdomyolysis. Neuromuscul Disord. (2013) 23:540–8. doi: 10.1016/j.nmd.2013.03.008
78. Scalco RS, Snoeck M, Quinlivan R, Treves S, Laforét P, Jungbluth H, et al. Exertional rhabdomyolysis: physiological response or manifestation of an underlying myopathy? BMJ Open Sport Exerc Med. (2016) 2:e000151. doi: 10.1136/bmjsem-2016-000151
79. Spillane J, Beeson DJ, Kullmann DM. Myasthenia and related disorders of the neuromuscular junction. J Neurol Neurosurg Psychiatry. (2010) 81:850–7. doi: 10.1136/jnnp.2008.169367
80. Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM. Myasthenia gravis. Nat Rev Dis Prim. (2019) 5:30. doi: 10.1038/s41572-019-0079-y
81. Ciafaloni E. Myasthenia gravis and congenital myasthenic syndromes. Contin Lifelong Learn Neurol. (2019) 25:1767–84. doi: 10.1212/CON.0000000000000800
82. Pasnoor M, Dimachkie MM, Farmakidis C, Barohn RJ. Diagnosis of myasthenia gravis. Neurol Clin. (2018) 36:261–74. doi: 10.1016/j.ncl.2018.01.010
83. Engel AG, Shen X-M, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. (2015) 14:420–34. doi: 10.1016/S1474-4422(14)70201-7
84. Vanhaesebrouck AE, Beeson D. The congenital myasthenic syndromes: expanding genetic and phenotypic spectrums and refining treatment strategies. Curr Opin Neurol. (2019) 32:696–703. doi: 10.1097/WCO.0000000000000736
85. Finsterer J. Congenital myasthenic syndromes. Orphanet J Rare Dis. (2019) 14:57. doi: 10.1186/s13023-019-1025-5
86. Matthews E, Balestrini S, Sisodiya SM, Hanna MG. Muscle and brain sodium channelopathies: genetic causes, clinical phenotypes, and management approaches. Lancet Child Adolesc Heal. (2020) 4:536–47. doi: 10.1016/S2352-4642(19)30425-0
87. Phillips L, Trivedi JR. Skeletal muscle channelopathies. Neurotherapeutics. (2018) 15:954–65. doi: 10.1007/s13311-018-00678-0
88. Al-Ghamdi F, Darras BT, Ghosh PS. Spectrum of nondystrophic skeletal muscle channelopathies in children. Pediatr Neurol. (2017) 70:26–33. doi: 10.1016/j.pediatrneurol.2017.02.006
89. Morales F, Pusch M. An up-to-date overview of the complexity of genotype-phenotype relationships in myotonic channelopathies. Front Neurol. (2020) 10:1404. doi: 10.3389/fneur.2019.01404
90. Miller TM. Differential diagnosis of myotonic disorders. Muscle Nerve. (2008) 37:293–9. doi: 10.1002/mus.20923
91. Prior DE, Ghosh PS. Clinical reasoning: a 10-year-old girl with muscle stiffness. Neurology. (2020) 95:e773–8. doi: 10.1212/WNL.0000000000010026
92. Kato N, Sadamatsu M, Kikuchi T, Niikawa N, Fukuyama Y. Paroxysmal kinesigenic choreoathetosis: from first discovery in 1892 to genetic linkage with benign familial infantile convulsions. Epilepsy Res. (2006) 70:174–84. doi: 10.1016/j.eplepsyres.2006.02.009
93. Jitpimolmard N, Matthews E, Fialho D. Treatment updates for neuromuscular channelopathies. Curr Treat Options Neurol. (2020) 22:34. doi: 10.1007/s11940-020-00644-2
94. Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. (2006) 129:8–17. doi: 10.1093/brain/awh639
95. Statland JM, Fontaine B, Hanna MG, Johnson NE, Kissel JT, Sansone VA, et al. Review of the diagnosis and treatment of periodic paralysis. Muscle Nerve. (2018) 57:522–30. doi: 10.1002/mus.26009
96. Arida RM. Physical exercise and seizure activity. Biochim Biophys Acta - Mol Basis Dis. (2021) 1867:165979. doi: 10.1016/j.bbadis.2020.165979
97. Pimentel J, Tojal R, Morgado J. Epilepsy and physical exercise. Seizure. (2015) 25:87–94. doi: 10.1016/j.seizure.2014.09.015
98. Kamel JT, Badawy RAB, Cook MJ. Exercise-induced seizures and lateral asymmetry in patients with temporal lobe epilepsy. Epilepsy Behav Case Reports. (2014) 2:26–30. doi: 10.1016/j.ebcr.2013.12.004
99. Ogunyemi AO, Gomez MR, Klass DW. Seizures induced by exercise. Neurology. (1988) 38:633–4. doi: 10.1212/WNL.38.4.633
100. Schmitt B, Thun-Hohenstein L, Vontobel H, Boltshauser E. Seizures induced by physical exercise: report of two cases. Neuropediatrics. (1994) 25:51–3. doi: 10.1055/s-2008-1071584
101. Harris SR. Psychogenic movement disorders in children and adolescents: an update. Eur J Pediatr. (2019) 178:581–5. doi: 10.1007/s00431-019-03317-8
102. Ganos C, Aguirregomozcorta M, Batla A, Stamelou M, Schwingenschuh P, Münchau A, et al. Psychogenic paroxysmal movement disorders - clinical features anddiagnostic clues. Park Relat Disord. (2014) 20:41–6. doi: 10.1016/j.parkreldis.2013.09.012
104. Ryan DP, Ptáček LJ. Episodic neurological channelopathies. Neuron. (2010) 68:282–92. doi: 10.1016/j.neuron.2010.10.008
Keywords: paroxysmal dyskinesia, episodic ataxia, exercise intolerance, exercise, pediatric
Citation: Danti FR, Invernizzi F, Moroni I, Garavaglia B, Nardocci N and Zorzi G (2021) Pediatric Paroxysmal Exercise-Induced Neurological Symptoms: Clinical Spectrum and Diagnostic Algorithm. Front. Neurol. 12:658178. doi: 10.3389/fneur.2021.658178
Received: 25 January 2021; Accepted: 21 April 2021;
Published: 01 June 2021.
Edited by:
Anna De Rosa, Università di Napoli Federico II, ItalyReviewed by:
Marta Ruiz-Lopez, University Hospital Fundación Jiménez Díaz, SpainMarianna Amboni, University of Salerno, Italy
Copyright © 2021 Danti, Invernizzi, Moroni, Garavaglia, Nardocci and Zorzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanna Zorzi, Z2lvdmFubmEuem9yemlAaXN0aXR1dG8tYmVzdGEuaXQ=