
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurol. , 09 June 2021
Sec. Neuro-Ophthalmology
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.657317
This article is part of the Research Topic Hereditary Optic Neuropathies: A New Perspective View all 11 articles
 Lorenzo Peverelli1,2†
Lorenzo Peverelli1,2† Alessia Catania1†
Alessia Catania1† Silvia Marchet1
Silvia Marchet1 Paola Ciasca3
Paola Ciasca3 Gabriella Cammarata3
Gabriella Cammarata3 Lisa Melzi3
Lisa Melzi3 Antonella Bellino4
Antonella Bellino4 Roberto Fancellu5
Roberto Fancellu5 Eleonora Lamantea1
Eleonora Lamantea1 Mariantonietta Capristo6
Mariantonietta Capristo6 Leonardo Caporali6
Leonardo Caporali6 Chiara La Morgia6,7
Chiara La Morgia6,7 Valerio Carelli6,7
Valerio Carelli6,7 Daniele Ghezzi1,8
Daniele Ghezzi1,8 Stefania Bianchi Marzoli3
Stefania Bianchi Marzoli3 Costanza Lamperti1*
Costanza Lamperti1*Leber's hereditary optic neuropathy (LHON) is due to missense point mutations affecting mitochondrial DNA (mtDNA); 90% of cases harbor the m.3460G>A, m.11778G>A, and m.14484T>C primary mutations. Here, we report and discuss five families with patients affected by symptomatic LHON, in which we found five novel mtDNA variants. Remarkably, these mtDNA variants are located in complex I genes, though without strong deleterious effect on respiration in cellular models: this finding is likely linked to the tissue specificity of LHON. This study observes that in the case of a strong clinical suspicion of LHON, it is recommended to analyze the whole mtDNA sequence, since new rare mtDNA pathogenic variants causing LHON are increasingly identified.
Leber's hereditary optic neuropathy (LHON, OMIM #535000) is one of the most common inherited optic neuropathies causing bilateral loss of central vision. LHON is due to missense point mutations affecting mitochondrial DNA (mtDNA), usually found in the homoplasmic state, leading to mitochondrial dysfunction. Thus, LHON is maternally inherited but characterized by incomplete penetrance even in individuals carrying the same homoplasmic pathogenic mutation and with a clear male predilection (1).
Disease onset usually occurs during the second and third decades of life, but the age of onset can span from 2 to 87 (2). LHON typically presents as a painless, subacute, central vision loss in one eye, sequentially spreading to the other eye in weeks or months. Within 1 year, the large majority of affected patients have the second eye involved. Bilateral simultaneous onset occurs in about 25% of patients. During the acute phase, optic disc hyperemia, peripapillary-telangiectatic vessels, vascular tortuosity, and retinal nerve fiber layer (RNFL) pseudo-edema are often detected, even if these features may be subtle, with a slow progression toward blindness (3, 4).
Fundus changes can be accurately quantified by optical coherence tomography (OCT). In the acute phase, the RNFL initially thickens in the temporal and inferior quadrants and then in the superior and nasal quadrants (5). OCT shows a global RNFL thinning as disease progresses. Visual evoked potentials (VEPs) and pattern electroretinograms (PERG) are typically abnormal, as they reflect optic nerve fiber degeneration (6). The visual prognosis in affected patients is usually poor, also depending on the underlying pathogenic mutation, ranging from individuals declared legally blind, to others who experience spontaneous recovery of visual acuity (7).
Molecular diagnosis currently shows that about 90% of all LHON cases are due to one of three common mtDNA point mutations, at nucleotide positions m.3460, m.11778, and m.14484, defined as “primary mutations.” The diagnostic for other polymorphic variants for specific mtDNA backgrounds (haplogroup J) may be preferentially associated with some of the primary mutations (m.11778, and m.14484) exerting a synergistic modifying role; previously these variants were defined as “secondary mutations” (8). The most common is the m.11778G>A (MT-ND4, p.Arg340His) mutation, which accounts for ~70% of all cases, whereas the m.14484T>C (MT-ND6,p.Met64Val) and m.3460G>A (MT-ND1, p.Ala52Thr) mutations account for ~15% of cases (1). Recently, several rare or private mtDNA variants have been reported and validated, including the uncommon case of unique combinations of mtDNA polymorphic variants leading to a mild defect of complex I activity (9, 10). In view of these considerations, it has been proposed that, in case of evidence of maternal inheritance and clinical/OCT hallmarks of LHON, if none of the common primary mutations is found, the diagnostic gold standard should be the mtDNA complete sequence analysis (10).
In this report, we present eight patients affected by LHON belonging to five different and unrelated families of European descent, in which we identified five novel mtDNA variants, also providing some functional evidence of possible pathogenicity.
Informed consent for a biological sample collection of fibroblast cell lines and DNA from blood to perform genetic studies was obtained from all patients involved, in agreement with the Declaration of Helsinki. The Ethical Committee of the Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy, approved the study.
We have investigated five families with seven male individuals (patients 1, 3, 4, 5, 6, 7, and 8) and one female (patient 2) clinically affected by typical LHON but lacking any of the primary common mutations. A summary of the demographic, clinical, and genetic data is available in Table 1 (see the Supplementary Material for more detailed information).
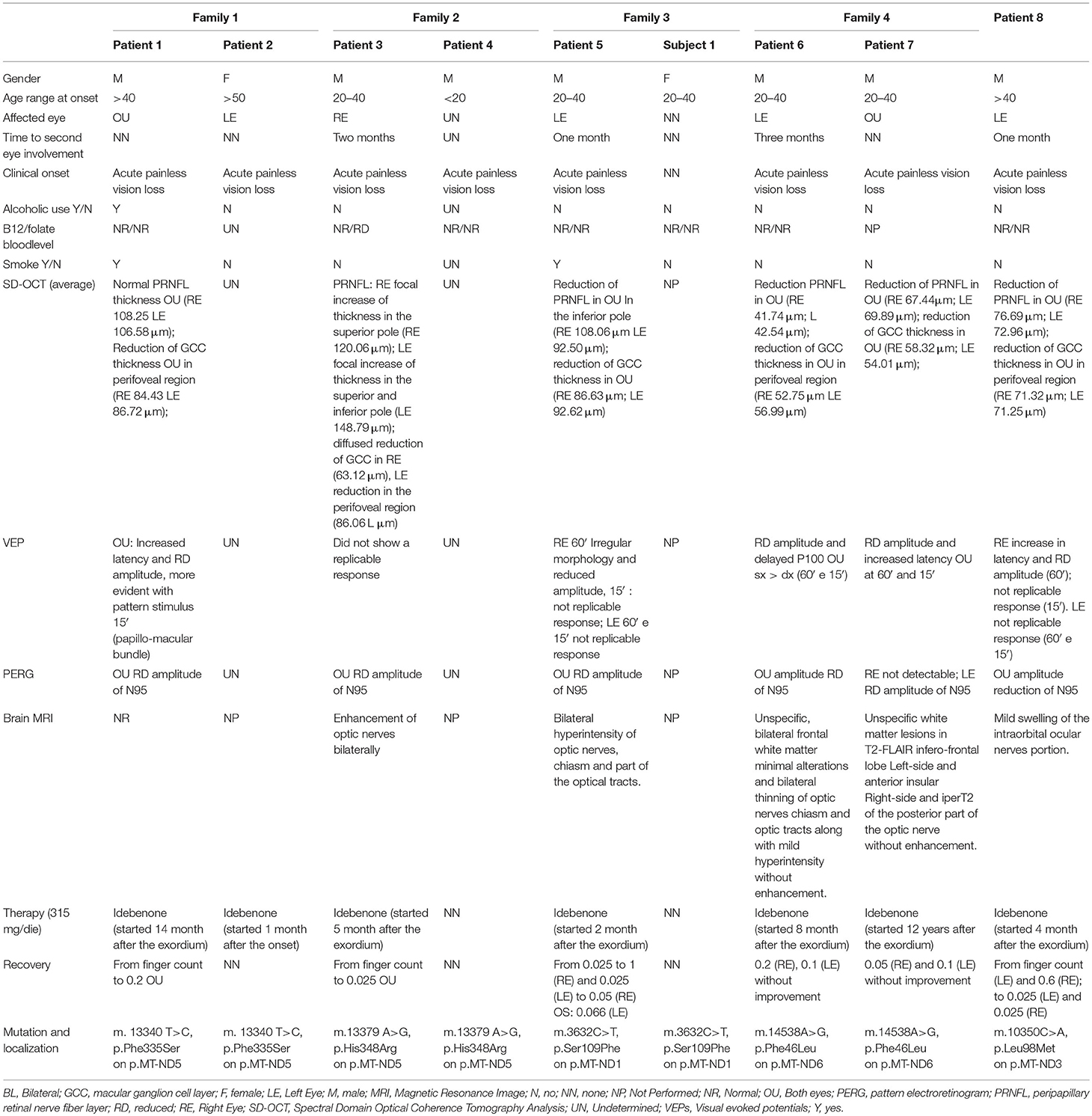
Table 1. Schematic summary of patients'clinical, instrumental, and molecular examinations (see also Supplementary Material 1).
Fibroblast cultures were obtained from skin biopsies of patients 3, 5, and 6 and from age-matched control subjects. Fibroblast cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplied with 10% fetal calf serum (FCS) at 37°C in a 5% CO2 atmosphere (11).
Trans-mitochondrial cybrids were generated from the fibroblasts of patient 6, as previously reported (12, 13).
Molecular analysis was performed on DNA extracted from peripheral blood lymphocytes of the eight patients and the unaffected individual. According to a standardized protocol (14) the entire mtDNA was PCR-amplified in eight overlapping fragments using a specific set of primer pairs. Each of the eight fragments was then sequenced, with four different “forward” primers, using a 3,100 ABI Prism Automated Sequencer. The mtDNA sequence was finally compared with the revised Cambridge reference mtDNA sequence.
Biochemical evaluation of OXPHOS complex activities was performed as previously described (14) in digitonin-treated skin fibroblasts from normal subjects and patients. The same measurements were performed in patient 6's cybrids and corresponding controls. The cells were cultured in a glucose-rich medium. The specific activity of each complex was normalized to citrate synthase activity, used as a standard mitochondrial mass marker.
The oxygen consumption rate (OCR) was measured in the fibroblasts of patients 3, 5, and 6 and in two controls using a SeaHorse FX-96 apparatus (Bioscience, Copenhagen, Denmark) as described in Invernizzi et al. (15) (data shown in Supplementary Figure 3). Cells were grown in a glucose-rich medium. Data are expressed as the mean ± SD, and comparisons were performed using a non-paired, two-tail Student t-test.
The complete sequence analysis of mtDNA from the eight subjects revealed five putative pathogenic LHON variants, one in each maternal lineage. All of them were absent in the mtDNA sequences from 100 healthy controls (screened and recorded in the in-house database from the Unit of Medical Genetics and Neurogenetics, The Foundation “Carlo Besta” Institute of Neurology). The m.13340 T>C (MT-ND5, p.Phe335Ser) variant was identified in patient 1 and in DNA from an affected first-grade female relative (patient 2) and the m.13379 A>G (MT-ND5, p.His348Arg) variant in patient 3 and his maternal relative (patient 4); both variants affect the ND5 subunit of complex I. Patient 5 and his asymptomatic first degree relative (subject 1) were shown to carry the m.3632C>T (MT-ND1, p.Ser109Phe) variant affecting the ND1 subunit of complex I. The relatives patient 6 and patient 7 carried the m.14538A>G (MT-ND6, p.Phe46Leu) variant in the ND6 subunit of complex I. Finally, we identified the m.10350C>A (MT-ND3,p.Leu98Met) variant in the ND3 subunit of complex I in patient 8. All variants were homoplasmic. The mtDNA haplogroup was determined for each family group using a specific provider (http//www.haplogrep.uibk.ac.at, not reported for privacy purposes, available on request).
All the identified variants affected evolutionarily highly conserved base pairs within the mitochondrial complex I subunits. More precisely, p.Phe335 and p.His348 are conserved in 97 and 94%, respectively, of more than 5,000 p.MT-ND5 sequences from protists to humans (16); from the same database, p.Ser109 results conserved in 80%, and p.Leu98 in 94% homolog sequences. However, p.Phe46 is only conserved in 16% of more than 5,000 p.MT-ND6 sequences. All the affected amino acid residues are located in transmembrane domains or adjacent to them. These variants are reported as probably damaging by most of the commonly used prediction software (Supplementary Table 1), although the predictions were not fully consistent; notably, the same results were observed even for other known LHON mutations.
The mitochondrial respiratory chain activity in fibroblasts from three unrelated patients (patients 3, 5, and 6) harboring three different variants (p.His348Arg in MT-ND5, p.Ser109Phe in MT-ND1, and p.Phe46Leu in MT-ND6) showed a significant reduction in complex I activity in the fibroblasts from patients 5 and 6 (compared with age and sex-matched controls), whereas in patient 3 there was only a slight, nonsignificant reduction (Figure 1A). A statistically significant reduction of complex I activity was confirmed in cybrids of patient 6 compared with controls (Figure 1B).
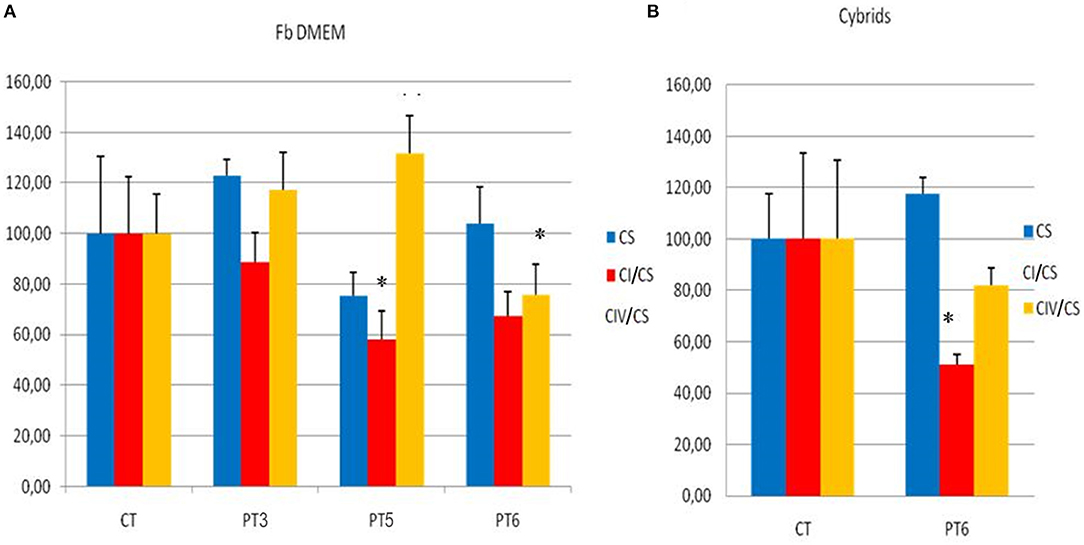
Figure 1. Mitochondrial respiratory chain activities on fibroblasts and cybrids. (A) Mitochondrial respiratory chain activities of CI and CIV complexes in Patient 3 (PT3), Patient 4 (PT4), Patient 6 (PT6) fibroblasts, and mediated controls (six different fibroblast cell lines) cultured in glucose-rich DMEM medium. (B) Mitochondrial respiratory chain activities of CI and CIV complexes in Patient 6 cybrids cultured in glucose-rich DMEM (PT6) and 5 mediated trans-mitochondrial cybrids controls (CT) grown in the same medium. Enzymatic activities (nmol/min/mg of protein) are expressed in percentage compared to controls and normalized with citrate syntase (CS) activity (blue column). CI, complex I (red column); CIV, complex IV (orange column). Student's t-test *p ≤ 0.05.
Finally, the oxygen consumption rate (OCR) failed to show any clear difference in mitochondrial respiration rate in all patients analyzed (patients 3, 5, and 6) (see Supplementary Figure 3). SeaHorse respirometry was not performed on cybrids.
In this study, we report five putatively pathogenic mtDNA variants, all affecting ND subunits of complex I, in 8 patients form 5 unrelated families presenting with a classical LHON phenotype.
All of them are not present in diverse genomic databases (Mitomap, Ensembl, and Genome Browser) and were not present in the mtDNA from 100 healthy controls, suggesting a pathogenic association of these variants with LHON.
As further support to our hypotheses of pathogenicity, all these variants are extremely rare also in the set of 195,983 individuals from HelixMTdb (17) and 51,836 individuals from GenBank databases. In particular, m.3632C>T has been found only once in the heteroplasmic state in HelixMTdb and not once in GenBank databases; m.10350C>A has been found in the homoplasmic state only once in HelixMTdb and no individuals in Genbank; m.13340T>C has been found twice in the homoplasmic state and twice in the heteroplasmic state in HelixMTdb and in no individual from GenBank; m.13379A>G was not found in HelixMTdb nor in GenBank; finally, m.14538A>G was found once in the homoplasmic state in HelixMTdb and was not found in GenBank.
In silico prediction of pathogenicity using online resources classified the m.13340T>C, m.13379A>G, and m.3632C>T variants as functionally “deleterious”; the scores of pathogenicity were less consistent for the m.10350C>A variant and to a greater extent for the m.14538A>G variant. Four of them (m.13340 T>C, m.3632C>T, m.14538A>G, and m.10350C>A) have not previously been reported. Interestingly, the m.13379 A>G (MT-ND5) variant has been very recently found in an unrelated patient affected by typical LHON disease (18), thus representing an additional proof of its probable pathogenicity.
To further validate the pathogenicity of the mtDNA variants identified, we tested biochemical activity of respiratory complexes on both fibroblasts (from three patients) and cybrids (from one patient). We observed, to a variable extent, a reduction of complex I activity (Figure 1). SeaHorse respirometry, however, failed to show defective oxygen consumption. These results are not surprising; in fact, there is much evidence that even confirmed pathogenic LHON mutations may not display any detectable respiratory chain defect (19–22). High-resolution respirometry performed on fibroblasts from the recently described patient carrying the m.13379 A>G variant, gave ambiguous results, as well (15). The authors also report a high level of ROS production and a reduction of mitochondrial membrane potential (15).
It is worth considering that LHON is a tissue-specific disease, involving retinal ganglion cells and the optic nerve. Thus, the biochemical assessments performed on skin fibroblasts, a tissue not involved in the disease phenotype, may not necessarily mirror the respiratory activity of the tissue targeted by the disease. Besides, even for the three best-known primary mutations there is no validated biochemical method to prove their pathogenicity on fibroblasts.
The possible contribution of nuclear gene variants as molecular modifiers for the development of symptomatic LHON is not yet established and currently represents a challenging topic, worth further exploration. However, it has been previously reported that specific mitochondrial haplogroups, such as the haplogroup J, exert the role of penetrance modifiers in LHON (23). In fact, both LHON primary mutation m.11778G>A/MT-ND4 and m.14484T>C/MT-ND6, when occurring on either a J1c or J2b mtDNA haplogroups, have increased risk of being symptomatic (23). In our cohort, one of the above-mentioned haplogroups was detected in patients 3 and 4. Notably, patient 3, despite Idebenone therapy, has been treated for more than 1 year but has not improved his visual function, supporting previous reports (23). Idebenone was administered to all affected patients (with exclusion of patient 4). All but patient 7, for whom therapy was started 12 years after the clinical onset due to misdiagnosis, received therapy roughly within the first year after onset. In five out of seven cases, idebenone treatment has been continued for more than 2 years with the exception of patient 2 (treated for slightly longer than 1 year) and patient 8, who has been treated for 5 months at the time of this study. We observed improvement in visual acuity in three out of six patients, stability in two, and a clear worsening in one (Table 1). In summary, therapy effectiveness for patients harboring these five novel mtDNA variants is in line with previously reported data on LHON due to “classic” mutations (1). Since the two patients in whom idebenone was ineffective carry the same m.14538A>G, we may speculate that this variant could be less responsive to therapy.
The peak age of disease onset was between the second and the third decades of life, and it is unusual for LHON patients to experience vision loss beyond 50 years of age, unless triggering factors have favored the disease onset as recently proposed (24). In relation to this observation, it is worth noticing that patients 1, 2, and 8 had late onset vision loss, the first around 50 and the third when he was almost 60. We may speculate that the variants in family 1 and patient 8 (m.13340 T>C and m.10350C>A) could be linked to phenotypes with later onset compared with classic LHON. Patient 1 was a heavy smoker and LHON vision loss occurred after at least 20 years of tobacco smoking and alcohol consumption (25, 26). However, familiar history supports pathogenicity of the identified variant, as a first-grade female relative (patient 2) affected by later onset LHON was found to be also positive for this variant. In patient 1 smoking and alcohol consumption could have contributed to accelerating the clinical onset. However, in patient 8, neither smoking nor alcohol abuse was reported; in this case, the identified variant (m.10350C>A) may be independently related to a later onset disease phenotype, or it could represent an additional predisposing factor within a specific genetic background or some other underrecognized triggering environmental/epigenetic influence. The latter is the most probable hypothesis, since we have an overall lower evidence of pathogenicity.
Possible variants on nuclear DNA have not been investigated in our group of patients because, with the exclusion of the recently reported DNAJC30 gene (27), all the other nuclear genes have been associated with complex phenotypes rather than isolated Leber's hereditary optic neuropathy. DNAJC30 has a “relevant” prevalence only in the Eastern European populations (none of our patients has such origins); moreover, it shows a recessive mode of inheritance that is not compatible with some of our pedigrees.
In conclusion, our study aims to emphasize the diagnostic relevance of running the whole mtDNA sequence analysis whenever LHON is documented on the clinical/OCT ground and the common mtDNA mutations are not detected, since rare or private mtDNA variants may be found (9). Besides, thanks to the advancement of the sequencing technologies, NGS is now widely available in most of the laboratories for mtDNA screening and is expected to become the method of choice for genetic analysis on mtDNA since it allows a rapid and cost-effective sequencing of the whole mtDNA with concurrent accurate quantification of heteroplasmy levels for point mutations (28). Remarkably, also, in our case series the novel mtDNA variants invariably affected ND subunits of complex I, and functional studies support a slight but detectable biochemical defect of complex I function in some of them. However, in all of them, we found relevant biological (low frequency of the identified variants in general population, evolutionary conservation, key location within complex I, and pathogenicity prediction) or clinical (typical course of disease and neuro-ophthalmological findings, family history) plausibility in support of their possible pathogenicity or potential contribution to pathogenicity in combination with other not yet determined genetic or extrinsic factors. Validation in further families will be necessary to consolidate the pathogenic role of these novel mtDNA variants in LHON. The possible role of other nuclear genes or environmental factors as disease modifiers needs to be further explored in future studies.
The original contributions presented in the study are publicly available. This data can be found here: https://zenodo.org/record/4683798#.YHfsV-gzaUk.
The studies involving human participants were reviewed and approved by the Ethical Committee of the Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan. The patients/participants provided their written informed consent to participate in this study.
LP and AC performed neurologic evaluations and wrote the paper. SM, EL, and MC performed in vitro studies. RF helped with patients selection. PC, GC, and LM performed all neuro-ophthalmological evaluations and contributed to manuscript preparation. AB, LC, and CLM assisted in data analysis and manuscript preparation. VC, DG, SB, and CL led the overall effort. All authors contributed to the article and approved the submitted version.
This study was supported by Horizon2020 through E-Rare project GENOMIT (grant to CL) and Italian Ministry of Health (RF-200 to CL). CL, AC, SM, DG, and EL are members of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO-NMD).
CL and VC are involved in LHON clinical trials with Santhera and GenSight Pharmaceuticals, serving also as consultants.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.657317/full#supplementary-material
LHON, Leber hereditary optic neuropathy; RNFL, retinal nerve fiber layer; OCT, optical coherence tomography; VEPs, Visual evoked potentials; PERG, pattern electroretinogram; DMEM, Dulbecco's modified Eagle's medium; FCS, fetal calf serum; OCR, Oxygen Consumption Rate; CS, citrate synthase; SD-OCT, Spectral Domain OCT Analysis; ctrls, controls.
1. Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res. (2011) 30:81–114. doi: 10.1016/j.preteyeres.2010.11.002
2. Riordan-Eva P, Sanders MD, Govan GG, Sweeney MG, Da Costa J, Harding AE. The clinical features of Leber's hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. (1995) 2(118 Pt):319–37. doi: 10.1093/brain/118.2.319
3. Carelli V, Chan DC. Mitochondrial DNA: impacting central and peripheral nervous systems. Neuron. (2014) 84:1126–42. doi: 10.1016/j.neuron.2014.11.022
4. La Morgia C, Carbonelli M, Barboni P, Sadun AA, Carelli V. Medical management of hereditary optic neuropathies. Front Neurol. (2014) 5:141. doi: 10.3389/fneur.2014.00141
5. Barboni P, Carbonelli M, Savini G, Ramos CV, Carta A, Berezovsky A, et al. Natural history of Leber's hereditary optic neuropathy: longitudinal analysis of the retinal nerve fiber layer by optical coherence tomography. Ophthalmology. (2010) 117:623–7. doi: 10.1016/j.ophtha.2009.07.026
6. Ziccardi L, Sadun F, De Negri AM, Barboni P, Savini G, Borrelli E, et al. Retinal function and neural conduction along the visual pathways in affected and unaffected carriers with Leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci. (2013) 54:6893–901. doi: 10.1167/iovs.13-12894
7. Kirkman MA, Korsten A, Leonhardt M, Dimitriadis K, De Coo IF, Klopstock T, et al. Quality of life in patients with Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci. (2009) 50:3112–5. doi: 10.1167/iovs.08-3166
8. Caporali L, Maresca A, Capristo M, Del Dotto V, Tagliavini F, Valentino ML, et al. Incomplete penetrance in mitochondrial optic neuropathies. Mitochondrion. (2017) 36:130–7. doi: 10.1016/j.mito.2017.07.004
9. Achilli A, Iommarini L, Olivieri A, Pala M, Hooshiar Kashani B, Reynier P, et al. Rare primary mitochondrial DNA mutations and probable synergistic variants in Leber's hereditary optic neuropathy. PLoS One. (2012) 7:e42242. doi: 10.1371/journal.pone.0042242
10. Caporali L, Iommarini L, La Morgia C, Olivieri A, Achilli A, Maresca A, et al. Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance Leber's hereditary optic neuropathy. PLoS Genet. (2018) 14:e1007210. doi: 10.1371/journal.pgen.1007210
11. Munaro M, Tiranti V, Sandonà D., Lamantea E, Uziel G, Bisson R, et al. A single cell complementation class is common to several cases of cytochrome c oxidase-defective Leigh's syndrome. Hum Mol Genet. (1997) 6:221–8. doi: 10.1093/hmg/6.2.221
12. King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. (1989) 246:500–3. doi: 10.1126/science.2814477
13. King MP, Attardi G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. (1996) 264:304–13. doi: 10.1016/S0076-6879(96)64029-4
14. Bugiani M, Invernizzi F, Alberio S, Briem E, Lamantea E, Carrara F, et al. Clinical and molecular findings in children with complex I deficiency. Biochim Biophys Acta. (2004) 1659:136–47. doi: 10.1016/j.bbabio.2004.09.006
15. Invernizzi F, D'Amato I, Jensen PB, Ravaglia S, Zeviani M, Tiranti V. Microscaleoxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion. (2012) 12:328–35. doi: 10.1016/j.mito.2012.01.001
16. Martín-Navarro A, Gaudioso-Simón A, Álvarez-Jarreta J, Montoya J, Mayordomo E, Ruiz-Pesini E. Machine learning classifier for identification of damaging missense mutations exclusive to human mitochondrial DNA-encoded polypeptides. BMC Bioinformatics. (2017) 18:158. doi: 10.1186/s12859-017-1562-7
17. Bolze A, Mendez F, White S, Tanudjaja F, Isaksson M, Jiang R, et al. A catalog of homoplasmic and heteroplasmic mitochondrial DNA variants in humans. bioRxiv [Preprint]. (2020). doi: 10.1101/798264
18. Krylova TD, Sheremet NL, Tabakov VY, Lyamzaev KG, Itkis YS, Tsygankova PG, et al. Three rare pathogenic mtDNA substitutions in LHON patients with low heteroplasmy. Mitochondrion. (2020) 50:139–44. doi: 10.1016/j.mito.2019.10.002
19. Brown MD, Trounce IA, Jun AS, Allen JC, Wallace DC. Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber's hereditary optic neuropathy mitochondrial DNA mutation. J Biol Chem. (2000) 275:39831–6. doi: 10.1074/jbc.M006476200
20. Hofhaus G, Johns DR, Hurkoi O, Attardi G, Chomyn A. Respiration and growth defects in transmitochondrial cell lines carrying the 11778 mutation associated with Leber's hereditary optic neuropathy. J Biol Chem. (1996) 271:13155–61. doi: 10.1074/jbc.271.22.13155
21. Majander A, Huoponen K, Savontaus M, Nikoskelainen EK, Wikstrom M. Electron transfer properties of NADH-ubiquinone the rarity of symptoms outside the optic pathways. The reductase in the NA1/3460 and the ND4/11778 mutations of the Leber hereditary optic neuroretinopathy (LHON). FEBS Lett. (1991) 292:289–92 doi: 10.1016/0014-5793(91)80886-8
22. Cock HR, Cooper JM, Schapira AHV. The 14484 ND6 mtDNA mutation in Leber hereditary optic neuropathy does not affect fibroblast complex I activity. Am J Hum Genet. (1995) 57:1501–2.
23. Carelli V, Achilli A, Valentino ML, Rengo C, Semino O, Pala M, et al. Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am J Hum Genet. (2006) 78:564–74. doi: 10.1086/501236
24. Carelli V, d'Adamo P, Valentino ML, La Morgia C, Ross-Cisneros FN, Caporali L, et al. Parsing the differences in affected with LHON: genetic versus environmental triggers of disease conversion. Brain. (2016) 139(Pt 3):e17. doi: 10.1093/brain/awv339
25. Kirkman MA, Yu-Wai-Man P, Korsten A, Leonhardt M, Dimitriadis K, De Coo IF, et al. Gene-environment interactions in Leber hereditary optic neuropathy. Brain. (2009) 132(Pt 9):2317–26 doi: 10.1093/brain/awp158
26. Carelli V, Franceschini F, Venturi S, Barboni P, Savini G, Barbieri G, et al. Grand rounds: could occupational exposure to n-hexane and other solvents precipitate visual failure in leber hereditary optic neuropathy? Environ Health Perspect. (2007) 115:113–5. doi: 10.1289/ehp.9245
27. Wiggs JL. DNAJC30 biallelic mutations extend mitochondrial complex I-deficient phenotypes to include recessive Leber's hereditary optic neuropathy. J Clin Invest. (2021) 131:e147734. doi: 10.1172/JCI147734
Keywords: Leber optic atrophy, mitochondrial respiratory chain, complex I, LHON, transmitochondrial cybrids
Citation: Peverelli L, Catania A, Marchet S, Ciasca P, Cammarata G, Melzi L, Bellino A, Fancellu R, Lamantea E, Capristo M, Caporali L, La Morgia C, Carelli V, Ghezzi D, Bianchi Marzoli S and Lamperti C (2021) Leber's Hereditary Optic Neuropathy: A Report on Novel mtDNA Pathogenic Variants. Front. Neurol. 12:657317. doi: 10.3389/fneur.2021.657317
Received: 22 January 2021; Accepted: 07 April 2021;
Published: 09 June 2021.
Edited by:
Michael S. Lee, University of Minnesota Twin Cities, United StatesReviewed by:
Eduardo Ruiz-Pesini, University of Zaragoza, SpainCopyright © 2021 Peverelli, Catania, Marchet, Ciasca, Cammarata, Melzi, Bellino, Fancellu, Lamantea, Capristo, Caporali, La Morgia, Carelli, Ghezzi, Bianchi Marzoli and Lamperti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Costanza Lamperti, Y29zdGFuemEubGFtcGVydGlAaXN0aXR1dG8tYmVzdGEuaXQ=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.