 Ibrahim Mortada1†
Ibrahim Mortada1† Raymond Farah1†
Raymond Farah1† Sanaa Nabha1
Sanaa Nabha1 David M. Ojcius2
David M. Ojcius2 Youssef Fares1Wassim Y. Almawi3
Youssef Fares1Wassim Y. Almawi3 Najwane Said Sadier1,3*
Najwane Said Sadier1,3*- 1Neuroscience Research Center, Faculty of Medical Sciences, Lebanese University, Beirut, Lebanon
- 2Department of Biomedical Sciences, University of the Pacific, Arthur Dugoni School of Dentistry, San Francisco, CA, United States
- 3College of Health Sciences, Abu Dhabi University, Abu Dhabi, United Arab Emirates
The current treatments for neurodegenerative diseases are mostly symptomatic without affecting the underlying cause of disease. Emerging evidence supports a potential role for immunotherapy in the management of disease progression. Numerous reports raise the exciting prospect that either the immune system or its derivative components could be harnessed to fight the misfolded and aggregated proteins that accumulate in several neurodegenerative diseases. Passive and active vaccinations using monoclonal antibodies and specific antigens that induce adaptive immune responses are currently under evaluation for their potential use in the development of immunotherapies. In this review, we aim to shed light on prominent immunotherapeutic strategies being developed to fight neuroinflammation-induced neurodegeneration, with a focus on innovative immunotherapies such as vaccination therapy.
Introduction
Inflammation of nervous tissue, termed neuroinflammation, occurs in response to diverse cues, such as infection, traumatic brain injury, toxic metabolites, or autoimmunity. Neuroinflammation, which is an important process for maintaining healthy central nervous system (CNS) function following injuries such as physical trauma and infections, is highly regulated due to the lack of regenerative ability of post-mitotic cells of the nervous system. It also constitutes a major component of many neurodegenerative (1) and psychiatric disorders (2).
Inflammatory responses in the CNS are induced by microglia, the resident innate immune cells, and further exacerbated by reactive astrocytes and infiltrating leukocytes (3). They are usually brief and followed by the recruitment of other immune cells to the affected area, clearance of the insulting agent, and tissue repair or scarring, leading to immune resolution. These events together constitute acute neuroinflammation, as opposed to chronic neuroinflammation, which may persist for decades. The majority of neurodegenerative disorders display low-grade chronic neuroinflammation, which can result in collateral damage worse than the original insult (4). In addition to disrupting the neurocircuitry, these conditions ultimately may cause permanent neuronal damage and brain atrophy, which are thought to result from the sustained release of cytotoxic factors by activated microglia, astrocytes and other immune cells, leading to neurodegeneration (4).
Neurodegenerative diseases are characterized by a progressive loss of neurons in several areas of the CNS; and are associated with cognitive, psychiatric, and motor deficits due to atrophy of the affected regions (5). Together, neurodegenerative diseases exert a major global disease burden, with dementia being a public health challenge in many developed countries. As aging is a strong risk factor for the most common neurodegenerative conditions, the global economic and social impact of these diseases on healthcare systems will likely continue to surge significantly in the coming decades due to increasingly aging populations and longer life spans (6). It has been projected that by the year 2050, the population of individuals over the age of 60 will rise from 901 million in 2015 to 2.1 billion people worldwide (7). The increased life expectancy will be accompanied by higher age-related diseases, with the elderly expected to spend most of their later years in ill-health. In fact, a main cause of disability in the elderly is dementia, affecting 44 million people globally, and expected to surpass 135 million people by the year 2050 (8). Over 36 million people worldwide are diagnosed with Alzheimer's disease (AD) or Parkinson's disease (PD), the two most common neurodegenerative disorders. The absence of effective disease-modifying treatments and the failure of most clinical trials for new therapies highlight the need to identify new therapeutic targets to halt disease progression. An important challenge in the developing treatment strategies for most progressive neurodegenerative diseases is their multi-factorial etiology and diverse disease course (9–11). For the most common neurodegenerative diseases, such as AD, PD, amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD), the causes of disease occurrence and progression are not fully known. Moreover, the disease course and severity varies significantly among patients, which complicates the challenge of efficient therapeutic interventions.
Common pathological mechanisms identified in most progressive neurodegenerative diseases involve neurotoxic protein misfolding, oxidative stress, and proteasomal impairment (4, 12). Increasing evidence suggests the presence of causal mechanistic links between toxic misfolded protein assemblies and neurodegeneration. Atypical protein aggregates are currently considered a main feature of most neurodegenerative disorders including PD, ALS, and HD, although their pathological significance is still debated (13).
Most articles in the literature describing immunotherapies for neurodegenerative diseases have typically focused on auto-immune neurodegenerative disorders such as multiple sclerosis (14). Until recently, little was known about immunotherapeutic interventions targeting aging-associated as well as other non-auto-immune neurodegenerative diseases. In this review, we therefore highlight recent immunotherapeutic strategies being developed to treat neuroinflammation-induced neurodegeneration, with a focus on immunotherapies.
Pathogenesis Of Neuroinflammation
Role of Microglia and Astrocytes
Microglia are resident macrophages in the brain and spinal cord and the primary immunocompetent cells in the CNS (15). Microglia constitute ~15% of the total cells in the brain, and their numbers and densities change depending on the brain region (16, 17). In the normal adult brain, microglia usually exist under different phenotypic states depending on the brain activity, plasticity, and response to challenges throughout life. Cutting-edge fate mapping and imaging techniques, along with RNA-seq expression profiling and other complementary technologies, have helped to decipher the origins, functions, and different phenotypes adopted by microglial cells (18, 19). During CNS surveillance, microglia dynamically remodel the structure of their processes and shift from a “surveying” ramified state to reactive amoeboid form in response to disturbances in homeostasis. Cells depart from the surveillance mode and acquire a reactive profile tailored to cope adequately with each specific trigger. This includes chemotactic reorientations, up-regulation of several cell surface proteins, release of numerous secreted factors and other transcriptional adjustments, which can occur in minutes or within a few hours post activation (20). With adjacent microglia surveying the brain parenchyma, studies estimate that the entire neuronal network of the brain can be scanned within hours (20–22). These cells are critical for proper brain development and maintenance of brain homeostasis throughout the life span, and their activation processes seem to be more diverse and dynamic than previously anticipated at all cellular levels (23). Based on the rapidly growing literature and the remaining gaps in microglial incontrovertible status within the CNS, there is an urgent need to create an updated microglial terminology that takes into consideration previous and newly discovered knowledge, including transcriptomic and proteomic profiles in both brain health and disease.
Astrocytes are the most abundant glial cell type in the CNS, providing mechanical and metabolic support to neurons, and helping in the regulation of critical biochemical activities including maintenance of the neural network, ionic and extracellular space volume homeostasis, synaptic plasticity, and blood flow (24). In response to a pathologic insult, astrocytes modify their morphological and functional state and become activated, which can be beneficial (radial glial-like astrocytes) or detrimental (reactive astrocytes) (24). Upon activation, astrocytes promote a pro-inflammatory environment by releasing mediators, such as IL-6 and TNFα. They can also recruit peripheral and CNS immune cells to the site of neuronal injury and degeneration (25). Furthermore, because astrocytes are the gatekeepers of the blood-brain barrier (BBB), they are associated with its pathological breakdown, which occurs during progressive neurodegeneration. The breakdown of BBB is thought to facilitate the entry of peripheral immune cells and blood components, which can exacerbate CNS neuroinflammation and neurodegeneration (26, 27). Nonetheless, the main functions of astrocyte activation and inflammatory responses aim to limit CNS damage and tissue remodeling, and to enhance neuronal recovery. Some studies have highlighted the role of astrocytic degeneration and atrophy in the early stages of neurodegenerative diseases, which may have implications for neuronal loss and disease progression (26).
Mechanisms Of Neuroinflammation Mediated Neurodegeneration
The main feature of neuroinflammation in the CNS is the presence of persistently-activated primary immunocompetent glial cells, microglia, and astrocytes, at the areas of neurodegeneration. The progressive neuronal loss is typically accompanied by the presence of hypertrophic microglia and dystrophic astrocytes. The primary functions of this reactive gliosis were thought to be phagocytic clearance and immune surveillance in the case of activated microglia, and neurotrophic support for astrocytes. This holds true in most cases of acute brain injury or infection, where neuroinflammatory responses are regulated and usually subside when the infection clears or homeostasis is restored at the location of injury. This transient activation of microglia mediates a microenvironment which can promote neuronal survival and recovery of CNS homeostasis (28–30). Acute and regulated microglial activation drives anti-inflammatory and neurotrophic responses, and protects neuronal populations from excitotoxic injury (31, 32). The pro-homeostatic effects of microglia is also evidenced by in vivo studies of microglia depletion and replenishment (33, 34). Thus, it is thought that acute neuroinflammatory responses are beneficial in the CNS, since they halt additional injury and facilitate the survival of neurons (35).
In contrast to the response seen during acute neuronal injury, chronic microglial activation observed in progressive neurodegenerative disorders is morphologically and functionally distinct. Persistent reactive microgliosis has been seen in postmortem brains of patients in all progressive neurodegenerative disorders (4, 36).
While the brain was widely regarded as an immune-privileged site where inflammation only occurred in the context of direct infection or BBB breakdown, it has now been proven that all endogenous CNS cell types express specialized pattern recognition receptors (PRRs) that can trigger an innate immune response to specific host-derived molecules called danger/damage associated molecular pattern (DAMPs) (9, 37, 38). Endogenous DAMPs can directly induce neuroinflammation in the CNS, shifting immunocompetent cells from their beneficial roles to a chronically reactive state, thus contributing to the progression of neurodegeneration (9). This view has changed our understanding and therapeutic approach to neurodegenerative disorders from a neuron-centric perspective of preventing neuronal death or providing trophic support for degenerating neuronal populations. An important common aspect of most progressive neurodegenerative diseases is the presence of persistent insoluble protein aggregates. These aggregates may function as DAMPs to activate PRRs [Toll-like receptors (TLRs); nucleotide-binding domain (NOD) leucine-rich repeat (LRR), and pyrin domain–containing 3 (NLRP3); receptor for advanced glycation end products (RAGE), etc.] on microglia and, thus, they mediate chronic reactive gliosis and neuroinflammation.
Another finding commonly seen in neurodegenerative diseases is an exaggerated release of pro-inflammatory cytokines and chemokines in the CNS such as TNFα and the over expression of IL-1β (39–42). Numerous cytokines and chemokines of the TNF and IL superfamily are also increased in the cerebrospinal fluid (CSF) and serum of patients with neurodegenerative diseases, making them potential markers of disease onset, progression, or treatment efficacy. However, no selective biomarkers have been established for progressive neurodegenerative diseases to this date.
The complement system is an additional major player of the innate immune system that can have a pathogenic role in progressive neurodegenerative diseases (43–46). Specific complement factors and receptors are also up-regulated in the serum and on circulating immune cells of neurodegenerative disease patients, suggesting that complement activation and signaling could be an important link between the CNS and peripheral arms of innate immune system in neurodegenerative diseases (47).
Materials and Methods
A literature search was conducted to investigate the existence of candidate immunotherapies for neurological disorders by using key terms to identify relevant articles on the subject. No date restrictions had been set for the articles to be retrieved from the search. The terms used for the search strategy were grouped into three broad categories based on key concept words “neurological disorders” and “immunotherapy.” Combinations of terms from each category were grouped together and joined with the terms from the other categories to be used in our database search.
Category 1: Alzheimer's disease, Parkinson's disease, Synucleinopathy Amyotrophic lateral sclerosis and Huntington's disease
Category 2: Active immunotherapy, Passive immunotherapy, inflammatory mediators, pattern recognition receptors (PRRs), pathogen-associated molecular patterns (PAMPs).
Eligibility Criteria
Studies of interest consisted of human clinical trial results and results acquired from trials on animal models. The following electronic databases were used: PubMed, OVID Medline, Web of Science, and Google scholar. Articles that did not assess the efficacy of the immunotherapy compared to untreated controls were excluded. All study designs were included; except case series, case reports, reviews, short communications, and letters to the editors. Studies that recruited patients with multiple comorbidities were excluded. In addition, studies conducted in non-English languages were also excluded.
Results: Therapeutic Strategies Targeting Neuroinflammation in The Management Of Neurodegenerative Diseases
Targeting Inflammatory Mediators
TNFα
A major therapeutic strategy is to target the production of secreted and cell surface inflammatory mediators driving neuronal dysfunction and death. Inhibition of soluble TNFα via small-molecule inhibitors or by viral over-expression has proven to be efficient in several experimental models of neurodegenerative disorders, including AD and PD (48, 49). Yet, in a randomized double-blind phase II clinical trial, weekly subcutaneous injections of Etanercept (a TNF inhibitor) did not improve cognition, global function, or behavior in a small group of subjects with mild-to-moderate AD dementia (50). The presence of safe and effective TNFα inhibitors, currently used for systemic inflammatory diseases, renders them potential therapeutic candidates if tested at earlier disease stages or in combination with other anti-inflammatory therapeutics (48).
NLRP3 and Interleukins
Some populations of neurons were found to be susceptible to the chronic production of IL-1β in the CNS (Godoy et al., 2008). Viral over-expression of IL-1β at low levels leads to DA degeneration in the substantia nigra; furthermore, inhibition of NLRP3-driven IL-1β secretion is protective in transgenic AD models (51). Inhibition of the NLRP3 inflammasome can constitute an effective way to block downstream IL-1β production, which is thought to be a master regulator of pro-inflammatory responses in the CNS (52). Tested in a preclinical mouse model of multiple sclerosis, the first small-molecule inhibitor of the NLRP3 inflammasome (MCC950) demonstrated therapeutic efficacy (53). Endogenous ketone bodies were also found to strongly inhibit the NLRP3 inflammasome, suggesting that CNS permeable compounds, such as β-hydroxybutyrate, may have a therapeutic role for neuroinflammatory diseases (54). In experimental models of AD, IL-12, and IL-23 were associated with neutralizing antibodies capable of some therapeutic benefit when given systemically (55). Additional studies are needed to evaluate the efficacy of these mechanisms in other neurodegenerative diseases. As US FDA-approved IL-12 neutralizing drugs are currently used in the management of psoriasis, it may be worthwhile to evaluate these drugs in AD and other neurodegenerative diseases (56). Deposition of complement around plaques and degenerating neurons were also seen in several neurodegenerative disorders (57–59). In models of AD, ALS, and HD, pharmacological inhibition of the receptor for the terminal complement component, C5a, was found to limit disease pathology and progression (60).
Novel research aimed at attenuating neurodegenerative disease pathology examines possible anti-inflammatory and immunomodulatory roles of molecules such as granulocyte-macrophage colony-stimulating factor (GM-CSF), peroxisome proliferator-activated receptor gamma (PPAR-γ) and glucagon-like peptide 1 (GLP-1).
GM-CSF
GM-CSF is an immunomodulatory growth factor and cytokine that is deregulated in neurodegenerative diseases. In fact, GM-CSF elicits its effects on dendritic cells to control the induction and proliferation of regulatory T (Treg) cells, as well as inducing microglial proliferation, ultimately controlling microglial homeostasis and regulating inflammation (61–64). GM-CSF has exhibited an extensive neuroprotective potential in both preclinical and clinical studies in AD. Administration of GM-CSF therapy to AD mouse models successfully attenuates neuroinflammation and cognitive decline by rescuing hippocampal neuronal pathways and enhancing Aβ clearance by recruiting microglia to amyloid plaques (65, 66). Data from clinical trials shows significant cognitive and memory improvement in groups treated with the 127-amino-acid synthetic recombinant form of GM-CSF, Sargamostim (GM-CSF Leukine), when compared to controls. Moreover, phase 2 trials (NCT01409915) involving Sargamostim have deemed it safe and tolerable for all AD patients (67). Sargamostim's immunomodulatory and neuroprotective roles are also being investigated in PD. Preliminary results are promising, as preclinical and phase 1 trials (NCT01882010) have shown significant reduction in PD associated motor symptoms, and that GM-CSF rescues and protects nigrostriatal dopaminergic neurons via Treg induction (68, 69). Additionally, Sargamostim is safe and generally well-tolerated among patients, aside from mild injection site reactions (68).
The efficacy of GM-CSF remains controversial in treating ALS patients due to inconsistencies in clinical results, with some studies showing that GM-CSF does not benefit ALS patients and does not slow down disease progression (70, 71). Current preclinical data suggest that GM-CSF extends the lifespan of transgenic mouse models and has potent anti-inflammatory capabilities by downregulating TNFα and reducing microglial activation (72, 73). Moreover, GM-CSF is neuroprotective; hence, it promotes microglial migration and recruitment to damaged axon segments and preserves large myelinated axons (72, 73). Current clinical studies have yielded conflicting results as disease mechanisms remain to be clearly understood. Aside from being safe and tolerable, the efficacy of GM-CSF in attenuating motor symptoms and alleviating ALS pathology seems rather inconsistent (74, 75). In this regard, GM-CSF has been reported as a potent anti-inflammatory agent in ALS, as it upregulates anti-inflammatory cytokines such as IL-10 and reduces pro-inflammatory cytokines such as TNFβ, monocyte chemoattractant protein-1, interferon-γ, IL-7 and IL-17 (76, 77). Furthermore, Johannessen et al. (77) and Zhang et al. suggest (78) that GM-CSF could play a role in attenuating motor decline and enhancing patient survival. On the other hand, Amirzagar et al. (70)and Nefussy et al. (71) provide evidence that GM-CSF offers no improvement to ALS-related clinical outcomes, with Amirgazar et al. reporting aggravation of disease status and acceleration of disease course, especially in females, in response to GM-CSF treatment.
GLP-1 and PPAR- γ
Despite GLP-1 agonists and PPAR- γ agonists being primarily anti-diabetic drugs, recent findings indicate that these molecules execute neuroprotective and anti-inflammatory functions, which alleviate symptoms of AD and PD as well as rescuing Akt-1 and m-TOR, and insulin signaling in the brain (79–84).
GLP-1 agonists reduce proinflammatory cytokines and reduce disease burden in AD and PD mouse models. The mechanisms detailing the mode of action of these drugs and their pathways have been extensively reviewed (85–87). Nevertheless, recent results obtained from AD and PD clinical trials for GLP-1 agonist drugs have shown minor improvements in disease pathophysiology, if any (88–91). These results are still preliminary and therefore warrant further investigation before proper conclusions can be drawn.
Pre-clinically, PPAR- γ agonists have been shown to reduce neuroinflammation, Aβ-42 load, phosphorylated tau, and synaptophysin, ultimately enhancing spatial memory and motor function in AD mouse models (81–93). Furthermore, two promising PPAR-γ agonists currently under phase II clinical trials, T3D-959 (NCT04251182), and Pioglitazone (NCT00982202), have proven their safety and tolerability among patients (94, 95). Moreover, T3D-959 significantly enhanced cognition and insulin metabolism in AD patients (94). Pre-clinically, Pioglitazone has shown promising results in PD, where it has markedly reduced neuroinflammation and microglial proliferation in multiple PD models; but these results are yet to be validated in clinical trials (96–98).
Targeting the Interaction Between PRRs and DAMPs
PRRs, such as RAGE, Mac1, and TLRs, constitute potential targets for treatment, given the view that DAMPs and misfolded proteins mediate neuroinflammation by interacting with multiple PRRs. Moreover, stimulating PRRs with neuronal DAMPs was found to be associated with the activation of downstream pro-inflammatory pathways, such as NOX2, iNOS, and TNFα (1). Thus, efficient targeting of PRRs involved in each neurodegenerative disease could be a possible strategy to decrease reactive gliosis and chronic self-perpetuating neuroinflammation and neurotoxicity. An inhibitor of RAGE, PF-04494700, showed promising results in pre-clinical models, but results from clinical trials were inconclusive (99). Using transgenic AD models, NLRP3 inhibitors, such as MCC950, showed promising results, providing evidence that NLRP3 is an important intracellular PRR, which detects aggregated misfolded proteins (51).
Vaccine Therapy
The mainstay in vaccine therapy consists of passive and active immunization with either monoclonal antibody infusion, or vaccination with specific antigens that induce adaptive immune responses, respectively. Passive immunization with monoclonal antibodies offers a reduction of the target molecules with a robust dosage management, but the drawbacks of this approach are high cost, frequent administration, and adverse side effects. In contrast, active immunization relies on the use of specific antigens, which induce production of antibodies or the modulation of inflammatory responses (100). Here, we summarize the status of vaccine therapy in neurodegenerative diseases.
Synucleinopathies
Passive Immunization
The treatments for synucleinopathies aim to reduce α-syn accumulation and cell-to-cell transfer, and can be combined with drugs to reduce neuroinflammation, expecting synergistic effects.
Trials in Transgenic Mice
Passive immunization against synucleinopathies relies on different monoclonal antibodies against specific to different regions of α-syn. One such approach consists of using antibodies that target the N-terminal or the central region of α-syn. Rats with nigrostriatal degeneration were subjected to intraperitoneal administration at 2-week intervals with α-syn antibodies during a total 3-month period. Both types of antibodies exerted neuroprotective effects in terms of a reduction in α-syn-induced nigral cell death and a decrease in activated microglia in the substantia nigra, with the antibody targeting the α-syn N-terminal being the most efficient (101). Recently, a new monoclonal antibody, mAb47, has been developed against the protofibril structures of α-syn. This antibody could decrease the intracellular oligomerization of α-syn in vitro (102), and decreased the levels of α-syn protofibrils in the spinal cord in vivo (103). In an attempt to develop a novel passive immunization technique, naturally occurring anti-α-syn autoantibodies, NAbs-α-syn, were purified and isolated from intravenous immune globulin (IVIG) of healthy individuals and used to immunize A53T transgenic PD mouse model. NAbs-α-syn significantly reduced the levels of soluble α-syn, α-syn oligomers, and intracellular phosphorylated α-syn deposits. Ultimately, NAbs-α-syn successfully attenuated memory and motor deficits and neuroinflammation associated with PD (104).
Clinical Trials
Prasinezumab.Prasinezumab, also known as PRX002, a C terminus targeting monoclonal antibody is currently under phase II clinical study, in which targeting α-syn aggregates was shown to significantly reduce free serum α-syn, but without any significant reduction of free CSF α-syn (105, 106). Preclinical results from the mouse monoclonal antibody 9E4, the precursor of PRX002, showed that internalization of the antibody-α-syn aggregates complex occurs via the Fc-γ receptors on the surface of microglial cells, leading to a reduction in α-syn concentration (106). Results of the phase I clinical trial showed that PRX002 antibody levels increased in patient CSF in a dose-dependent manner, which might indicate that PRX002 might target α-syn extracellular aggregates in the CNS (105). PRX002 was shown to be safe and well-tolerated, and does not cause immunogenicity in the host (105, 107).
BIIB054.BIIB054 is an N-terminus targeting monoclonal antibody currently under phase II clinical study that preferentially targets α-syn aggregates (108). The current study showed that BIIB054 formed complexes with serum α-syn and described it as a safe and tolerable candidate monoclonal antibody against α-syn (109).
Active Immunization
Mice vaccinated with recombinant human α-syn produced high affinity α-syn antibodies mainly targeting the C-terminus of α-syn (110). Interestingly, the immunized mice showed reduced accumulation of α-syn in neuronal cell bodies and synapses in the temporal cortex and a higher conservation of synaptophysin-positive nerve terminals (110). It was proposed that α-syn is degraded by lysosomal processing. Immunization against α-syn has also been explored in a rat model of PD based on the viral delivery of human α-syn into the nigrostriatal pathway. Interestingly, this form of active immunization resulted in an adaptive immune response mediated by CD4–positive, Foxp3–positive cells that successfully infiltrated the nigrostriatal system. As a result, decreased α-syn inclusions in the substantia nigra were observed in the experimental group, indicating that the observed adaptive immune responses possess therapeutic potential (111). Moreover, AFFITOPE consists of active vaccination with short α-syn peptides, which has provided promising results in-vivo. AFFITOPE successfully generated anti-α-syn antibodies that infiltrated the CSF and plasma in high titres, targeted α-syn aggregates, and reduced neuronal degeneration by attenuating oligomeric α-syn accumulation in neurons (112–114). AFFITOPE is currently undergoing extensive phase I clinical trials that are yielding promising results displaying its safety and tolerability among patients (114).
Overall, the outcomes from preclinical studies investigating both active and passive immunization strategies in several models of α-synucleinopathies are considered as promising.
Amyloid β and Tau
Passive Immunization
Passive immunization against AD is currently the most advanced immunotherapy under development.
Amyloid β Targeting Therapies
Trials in Transgenic Mice.An anti-Aβ antibody targeting the Aβ 31–35 sequence showed positive immunological results and reduction of disease pathology in transgenic mice. The antibody effectively bound to Aβ-42 species and attenuated their toxicity and prevented Aβ-42 induced cell death as well as restoring hippocampal synaptic plasticity. Furthermore, anti Aβ31–35 antibody rescued spatial learning and memory, which makes targeting the Aβ31–35 sequence a promising candidate for future investigation in the quest for passive immunization therapies for AD (115).
Clinical Trials
Bapineuzumab.The first studied candidate monoclonal antibody was bapineuzumab, which targets the Aβ N-terminus and possesses a higher affinity for deposited amyloid plaques than soluble Aβ monomers (116, 117). However, multiple studies and a meta-analysis conducted by Abushouk et al. assessing the potential use of bapineuzumab in the treatment of AD, reported significant association with severe adverse events and no enhancement of cognitive decline, and therefore concluded that bapineuzumab is not recommended for use in the treatment of AD and clinical trials have since then been discontinued (117–123).
Solaneuzumab.Solanezumab is a monoclonal antibody that targets the mid domain of the Aβ peptide and selectively binds to monomeric, soluble Aβ (123–126). However, due to its failure in reducing Aβ burden and in light of recent shortcomings in phase III trials that revealed that solaneuzumab did not improve cognitive decline in patients with mild AD, current trials were terminated due to less and less promising results (127, 128).
Gantenerumab.Gantenerumab is a monoclonal antibody that targets aggregated Aβ in the brain. It targets the N terminus as well as the mid-domain of the Aβ peptide, and shows high affinity for aggregated amyloid β species. The evaluation of this candidate in a phase III clinical trial indicated a dosage–dependent reduction of Aβ plaques in the brain below threshold for healthy levels (129, 130). Moreover, it has been shown to ameliorate mental status and decrease cognitive decline in patients, especially in early stages of the disease (130, 131).
Crenezumab.Crenezumab is a monoclonal antibody targeting the mid-domain of Aβ with reduced effector function, and it binds to Aβ oligomers, fibrils, and plaques, limiting aggregation and facilitating disaggregation (132). One advantage of this antibody is that it is an IgG4 antibody, which shows reduced pro-inflammatory activity and limited risk of vasogenic edema (132–134). Despite evidence of tolerability of Crenezumab among patients and Aβ plaque reduction (135), overall cognitive decline and disease pathology were not improved, and therefore the efficacy of the treatment remains in question (136, 137). After launching a phase III investigation for Crenenzumab's potential use as treatment or preventive therapy for patients suffering from familial AD (138), an interim analysis, showed that it was unlikely to meet primary endpoints. Clinical trials involving Crenezumab have been since then terminated.
Aducanumab.Aducanumab is a monoclonal antibody that is currently under phase III testing. It can bind the N-terminal of both soluble and insoluble Aβ species, with recent evidence emphasizing its strict high affinity binding to aggregated and pathogenic forms of Aβ (139). Studies conducted on aducanumab collectively agree on the tolerability of the antibody and report minimal treatment-induced adverse effects. In addition, aducanumab has been shown to infiltrate different brain regions and reduce Aβ plaques as well as slowing disease progression (140–142). Trials involving Aducanumab have been since then terminated due to futility of analysis.
Ponezumab.Ponezumab is a monoclonal antibody that targets soluble amyloid beta by binding to the C-terminal of the sequence (143). It is speculated to reduce CNS amyloid beta species by sequestration of free blood Aβ and therefore shifting the blood-brain Aβ equilibrium toward the blood (144, 145). In the study conducted by Landen et al., the tolerability and effectiveness of treatment with ponezumanb was assessed. It was found that ponezumab had a reduced likelihood of inducing adverse events and was therefore tolerated in patients of the study group. On the other hand, ponezumab showed no effect on cognitive abilities in patients and did not reduce the severity of disease pathology (146–148). Notably, ponezumab showed an increase of free Aβ species in the blood without infiltrating the CSF. This increase was explained by the mode of action of ponezumab, which was discussed above (147). Based on the above, ponezumab clinical trials have been discontinued.
Tau Targeting Therapies
Trials on Transgenic Mice.CBTAU-22.1 In a novel approach, and in an effort to enhance the therapeutic potential of a naturally occurring tau specific human antibody CBTAU-22.1, van Ameijde et al. successfully improved the affinity of CBTAU-22.1 through random mutagenesis and a variant, dmCBTAU-22.1, was generated. In vitro, dmCBTAU-22.1 reduced pathological tau aggregation, seeding and spread. In vivo, P301L transgenic mice were treated with dmCBTAU-22.1, which successfully reduced tau paired helical filaments as compared to controls (149).
43D, 77E9, and Antibody D.Mouse monoclonal antibody 43D, which targets the N terminal projection of tau, produced the most pronounced results of the three. Triple transgenic (3xTG) mice treated with antibody 43D exhibited reduced total tau, p-tau, p-tau seeding and propagation, as well as attenuation of the amyloid beta and amyloid precursor protein load in the hippocampus (150, 151). Moreover, antibody 77E9 exhibited similar effects as 43D in attenuating disease pathology, excluding the effects of 43D seen on p-tau and amyloid beta (151). Dai et al. evaluated the effect of a combined dose of 77E9 and 43D antibodies, but the results were less effective than the sole use of each antibody (151). Both antibodies reduced short-term and spatial memory impairments in 3xTG mice (151). The third of these antibodies, antibody D, is a tau antibody that recognizes the central region of tau protein. In the context of passive immunization, antibody D reduced tau seeding and propagation of tau pathology to different brain regions (152).
Anti-Phosphorylated Tau Antibodies.Passive immunization with specific monoclonal antibodies targeting phosphorylated tau is another promising approach to treat AD; however, their use is currently limited to test animals. D'Abramo investigated the use of three anti-phosphorylated tau antibodies: RZ3, CP13, and PG5 as antibody therapies for AD in JNLP3 transgenic mice. Of these three antibodies, CP13, which targets phosphorylated Serine residue 202 of tau, was the only one to successfully reduce insoluble or soluble tau species in the cortex and hindbrain of transgenic mice (153).
TOMA.Gerson et al. was the only team that investigated the use of anti-tau oligomer antibodies in treatment of PD. Using A53T transgenic mice overexpressing mutated α-synuclein, they found that the use of tau oligomer-specific monoclonal antibody (TOMA) significantly reduced toxic tau oligomers, which in turn reduced α-synuclein oligomers with fibril-like characteristics as well as reducing Lewy Body structures. Ultimately, TOMA protected the immunized mice from cognitive and motor deficits related to PD (154). Moreover, TOMA was also tested on different AD mouse models, JNLP3, Htau mice and Tg2576, and yielded promising results. TOMA successfully targeted toxic oligomeric tau species, reduced memory deficits and cognitive decline, ultimately slowing down disease progression (155–157).
Clinical Trials
Monoclonal antibodies targeting tau protein have started to make the leap from animal testing to clinical trials. From these, C2N 8E12, gosuranemab, zagotenemab, and semorinemab are worthy of mention, as they are currently being assessed for clinical tolerability in phase II trials. Preliminary safety and tolerability results are promising thus far; however, information on these treatments remains rudimentary at best, hence further research is required to evaluate their clinical efficacy and safety (158–162).
Active Immunization
Amyloid β Targeting Therapies
Amyloid β Targeting Therapies.
AN-1792 The first vaccine tested in humans (starting in 2000), called AN-1792, consisted of a full-length pre-aggregated amyloid peptide (Aβ1–42). AN-1792 successfully cleared Aβ40, Aβ42 and Aβ43 from AD plaques. This vaccine, however, led to severe side effects, including aseptic meningoencephalitis, in ~6% of AD patients (100, 163–167). Due to these side effects, AN-1792 trials have since been terminated.
ACI-24.
ACI-24 is a liposome-based vaccine designed to induce an antibody response against aggregated Aβ peptides. In preclinical studies, repeated subcutaneous injection of ACI-24 into AD transgenic mice yielded high titers of anti-Aβ antibodies, reducing the concentration of insoluble Aβ1-40, and Aβ1-42 (168). Preliminary phase I results are promising, with ACI-24 being well-tolerated among patients, additionally, the vaccine has successfully triggered an immune response in patients (169). ACI-24 is currently under phase II clinical trial for safety, tolerability and immunogenicity for its use on patients with AD.
CAD 106.
CAD106 is an anti-Aβ vaccine generated from multiple copies of the Aβ1–6 peptide. In a phase II study, CAD106 was well-tolerated in the study group with a minimal number of treatment related adverse effects, which did not negatively affect treatment tolerability and safety. CAD106 successfully increased anti-Aβ antibody titers, which successfully targeted and cleared Aβ species. Moreover, the treatment did not cause occurrences of meningoencephalitis, autoimmune disease or CNS inflammation (170–172).
ACC-001.
ACC-001, constituted of N terminal (Aβ1-7) peptide, is another vaccine that has successfully undergone phase II trials. ACC-001 was shown to be well-tolerated by patients enrolled in the study, and to induce a robust immune response mediated by anti-Aβ IgG. Coupling ACC-001 with QS-1 as an adjuvant further increased the robustness of the observed immune response (173–176).
ABvac40.
ABvac40 is a vaccine formulated from repeats generated from C-terminus fragments of Aβ40. It has successfully generated specific anti-Aβ40 antibodies in patients with mild to moderate AD. Initial tolerability and safety tests have been successful, with minor injection site reactions and adverse events being recorded; moreover, treatment did not trigger vasogenic edema or microhemorrhage. Based on the promising phase I results, ABvac40 has moved to phase II clinical trial testing in which its efficacy and impact on cognition will be assessed (177).
Tau Targeting Therapies
Trials in Transgenic Mice.
Tau379–408[P-Ser396, 404] In a different approach, the effectiveness of phosphorylated tau peptide, Tau379–408[P-Ser396, 404], as an immunogen was tested in 3xTG mice. This study demonstrated that phosphorylated tau peptide vaccine triggered the generation of anti-tau antibodies which led to reduction of pathological tau aggregates, clearance of both soluble and insoluble tau species, and the reduction of Aβ deposition (178). These preliminary results show the potential use of tau peptide in active immunization against AD pathology and the need to assess the potential clinical application of these vaccines through clinical trials.
Clinical Trials
AADvac1.
Tau active immunization approaches represent another path under exploration. AADvac1 is a new vaccine that can specifically recognize pathological tau oligomers. Active immunotherapy with this vaccine decreased the extent of oligomers and of neurofibrillary pathology in the brains of transgenic rats (14). The formulation comprises a synthetic peptide from the tau aggregation domain. AADvac1 successfully triggered anti-tau antibody production, and adverse effects were limited to injection site reactions. Moreover, preliminary data indicated a reduction in cognitive decline in patients undergoing the treatment. A phase II clinical trial involving AADvac1 has been launched to assess its efficacy (179–181).
ACI-35.
ACI-35 is a liposomal-based vaccine consisting of a synthetic tau peptide phosphorylated on pathological residues S396 and S404 thus resembling the phospho-epitope of tau. The evaluation in test animals, wild-type C57BL/6, and P301L, indicated a positive immunogenic activity specifically targeting pathological phosphorylated tau species with no neurological side effects or inflammation of neural tissue (182). Preliminary findings from phase I trial indicated that the treatment is tolerable and safe, but was not efficiently immunogenic; therefore, a second-generation vaccine, ACI-35.030, has been generated. ACI-35.030 is 5 times more immunogenic than its predecessor and will replace ACI-35 in the phase I/II safety and tolerability clinical trial (183).
Superoxide Dismutase 1
Active Immunization
Superoxide dismutase 1 (SOD1), an enzyme known for its role in relieving oxidative stress, has been studied thoroughly as a target for mutations in ALS. The cause of 20% of familial ALS cases has been attributed to mutated SOD1 (184). Currently, there is no consensus on mutant SOD1's pathological function in ALS, but evidence suggests that mutant SOD1 might interfere with cellular metabolism (185–189) and oxidative stress pathways (190), and might cause metal dyshomeostsis (191–194). Takeuchi et al. have investigated the efficiency of a vaccine targeting extracellular SOD1 in immunizing transgenic mice against ALS (195). In their experiment, they tested the efficiency of mutant SOD1 (G93A mutant SOD1) and wild-type SOD1 (wt-SOD1) vaccines in relieving the symptoms and reducing the pathogenicity of ALS. Takeuchi et al. generated the vaccine based on these two types of SOD1 due to the diversity of mutations that typically affect this enzyme in ALS (196–198). Upon inoculation, wt-SOD1 vaccine extended life expectancy and delayed disease onset, while both wt-SOD1 and mutant SOD1 vaccinations reduced anterior horn motor neuron loss (195). A similar vaccine was modeled based on mutant SOD1 and tested on G37R SOD1 and G93A SOD1 transgenic mice and resulted in significant life span extension and delay in disease onset in mice that do not suffer from an overexpression of the mutant SOD1 phenotype, i.e. G37R SOD1 (199).
Passive Immunization
Antibody therapy has been investigated against SOD1 mutant oligomers. Passive immunization of SOD1-G93A mice with anti-SOD1 antibody (W20) targeting toxic soluble SOD1 oligomers protected motor neurons from apoptosis and extended their survival. This was achieved by reducing neurotoxic SOD1 oligomer aggregates and substantially inhibiting gliosis and neuroinflammation in the spinal cords and brain stems of transgenic mice. Ultimately, W20 improved motor neuron survival and motor performance (200).
C9orf72
Passive Immunization
C9orf72 is a protein coding gene involved in endosomal trafficking (201). Mutation of the hexanucleotidic GGGGCC (G4C2) intronic repeat, upstream the C9orf72 coding sequence, leads to the expansion of this repeat from 2 to 22 copies to 700–1,600 copies (202, 203). The G4C2 expansion can be translated by repeat-associated non-ATG (RAN) translation to generate dipeptide repeat protein aggregates (DPR): poly GA, poly GP, poly GR, poly PA, and poly PR (204, 205). Poly GA has been described as an abundant and cytotoxic DPR in brain and spinal cord neuronal inclusions in ALS patients (205–207). The exact role of Poly GA and its contribution to disease pathology remains under study. It was shown to act as a nucleation seed to promote DPR aggregation, it enhanced G4C2 repeat expression and translation, and it could be transmitted between neurons (Qihui 207). This indicates that poly GA potentially aids in the spread of disease pathology in affected individuals. Poly GA causes reduced dendritic branching, proteasomal inhibition, apoptosis, and endoplasmic reticulum stress (206, 207). Zhou et al. investigated poly GA as a candidate target for immunotherapy for ALS. Treatment of poly GA transfected cells with anti-GA antibody showed a significant decrease in poly-GA seeding, aggregation and spread, suggesting the potential application of anti-poly GA antibodies as immunotherapies for ALS (208). Recently, Nguyen et al. investigated the potential use of anti-GA RAN antibodies in treating Frontotemporal Dementia (FTD) and ALS. Herein, passive immunization of FTD/ALS C9-BAC transgenic mice with anti-GA RAN antibodies, α-GA1, α-GP1, and α-GA2, has effectively reduced neuroinflammation and neurodegeneration in the anterior and posterior horns of the lumbar spine (209). Moreover, anti-GA RAN antibodies successfully rescued proteasome activity and subsequently reduced pathogenic poly-GA aggregates. Collectively, these changes significantly reduced physical and behavioral pathology, as well as survival, thus anti-GA RAN antibodies qualify as promising candidate therapies for ALS and FTD that merit further assessment in clinical trials (209).
Active Immunization
Novel findings indicate the possible use of poly-GA coupled with ovalbumin, OVA-(GA)10, as an active immunization agent. OVA-(GA)10 successfully elicited a robust immune response and induced an increase in anti-GA antibody titres in transgenic GA-CFP mice (210). Moreover, the produced anti-GA antibodies exhibited a high specificity to poly-GA aggregates resulting in their reduction in the brains of transgenic mice, ultimately reducing motor deficits and neurodegeneration (210). OVA-(GA)10 also attenuated neuroinflammation by normalizing the number of activated microglial cells and macrophages and reversing TDP-43 cytoplasmic mis-localization in the spinal cord. These findings corroborate the possible application of OVA-(GA)10 as a candidate vaccine to reduce disease severity and pathology in ALS and FTD (210).
Mutant Huntingtin Protein
Active Immunization
Current reports on active and passive immunization against mutant Huntingtin (m-Htt) proteins remain inconclusive. Results obtained for active immunization are preliminary. A study conducted on a candidate plasmid vaccine administered to HDR6/2 mice proved to have no effect on the number of m-Htt aggregates (211). Another study examined the use of peptide, protein, and DNA plasmid vaccines against m-Htt and detected a robust antibody production after vaccination with a combination of three non-overlapping HTT exon1 peptides. However, this study did not assess the efficacy of the developed vaccine in reducing m-Htt aggregates (212).
Discussion
Emerging evidence supports the use of immunotherapeutic agents in the management of neurodegenerative diseases. Our understanding of immunomodulatory mechanisms in the CNS has greatly evolved in recent years. In this work, we discussed the pathogenesis of neuroinflammation and its role in neurodegeneration. The switch from a succinct limited immune response to a sustained chronic response appears to be the initiating event in disease onset. Understanding the exact mechanism behind this switch is essential to halt its progression and prevent occurrence of disease. Increased release of cytokines and chemokines by overactive immune cells further exacerbates the damage and accentuates neurodegeneration. We then discussed immunotherapeutic modalities that could revolutionize the management of so far intractable neurodegenerative diseases, with a focus on passive and active therapies targeting hallmark disease biomarkers due to the availability of extensive reviews covering neuroinflammatory targets. Vaccination therapy is an interesting approach as vaccines are considered main players in preventive medicine and public health (Figure 1, Table 1). More research on the pathophysiology and structures of Aβ, tau, and α-syn will bring us a step forward toward tailoring stronger vaccines capable of mounting specific immune responses against the accumulation of these proteins. Of major importance is the avoidance of serious secondary adverse events such as meningoencephalitis or possibly the induction of seizures upon introducing the abnormal protein aggregates. In this regard, Aβ42-specific T-cell activation has been demonstrated to cause multiple undesirable effects in AD patients including meningoencephalitis which could potentially exacerbate patient health status (213, 214). Since Aβ active immunization introduces Aβ peptides into the blood stream, these could serve as an immunogenic activator of an anti-Aβ T-cell mediated immune response. A suggested mechanism to bypass this undesirable response was to design a chimeric Aβ vaccine where Aβ immunogen presentation is carried out by Norovirus particles. This approach significantly enhanced immunogenicity and antibody production without Aβ42-specific T-cell activation (215, 216). Another suggested mechanism to bypass Aβ42-specific T-cell activation was by using peptide fragments as immunogenic particles for active immunization against AD instead of full-length peptides (171, 173). These findings further substantiate the necessity for future investigation to focus on the type of immunogenic particle, mode of delivery, and antigenic presentation of the immunogen by the vaccine.
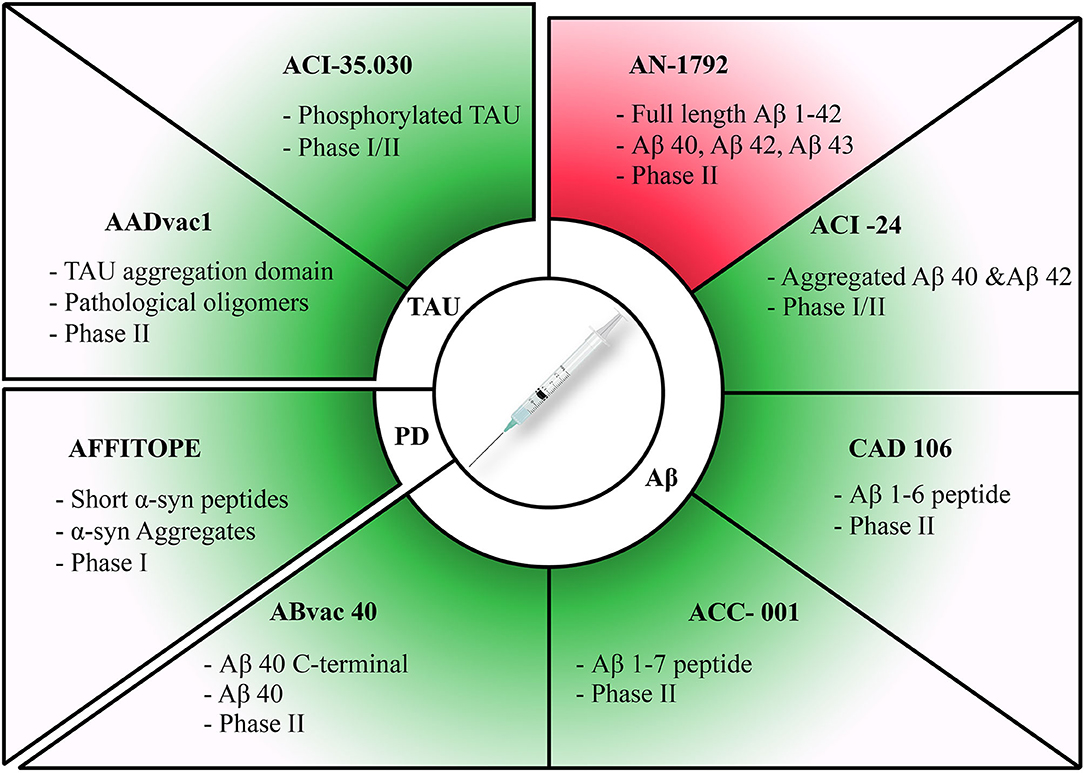
Figure 1. Summary of active immunization therapies assessed in clinical trials for different diseases. Discontinued trials highlighted in red and ongoing trials highlighted in green. Therapies have been categorized according to each pathology they target. The target of each therapy along with its current clinical trial phase have been stated for each therapy.
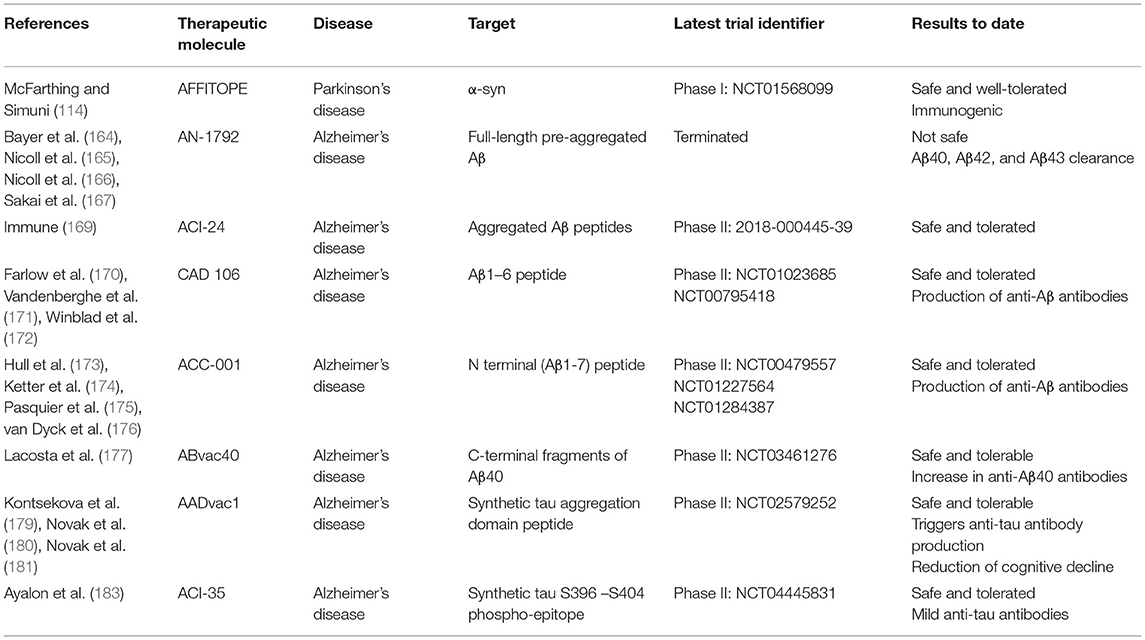
Table 1. Study characteristics of active immunotherapy clinical trials.
The mode of administration of the vaccine represents another important consideration for ameliorating efficacy. Recently, there has been a focus on passive immunization with monoclonal antibodies, which may represent a safer approach than active immunization (Figure 2, Table 2). Overall, therapeutic antibodies are one of the fastest growing areas in the pharmaceutical industry for the treatment of cancer, autoimmune disorders, and now also for neurodegenerative disorders. Furthermore, another pivotal factor related to peptide based vaccines is the choice of the carrier protein which has been shown to impact the duration and extent of the elicited humoral immune response (217, 218).
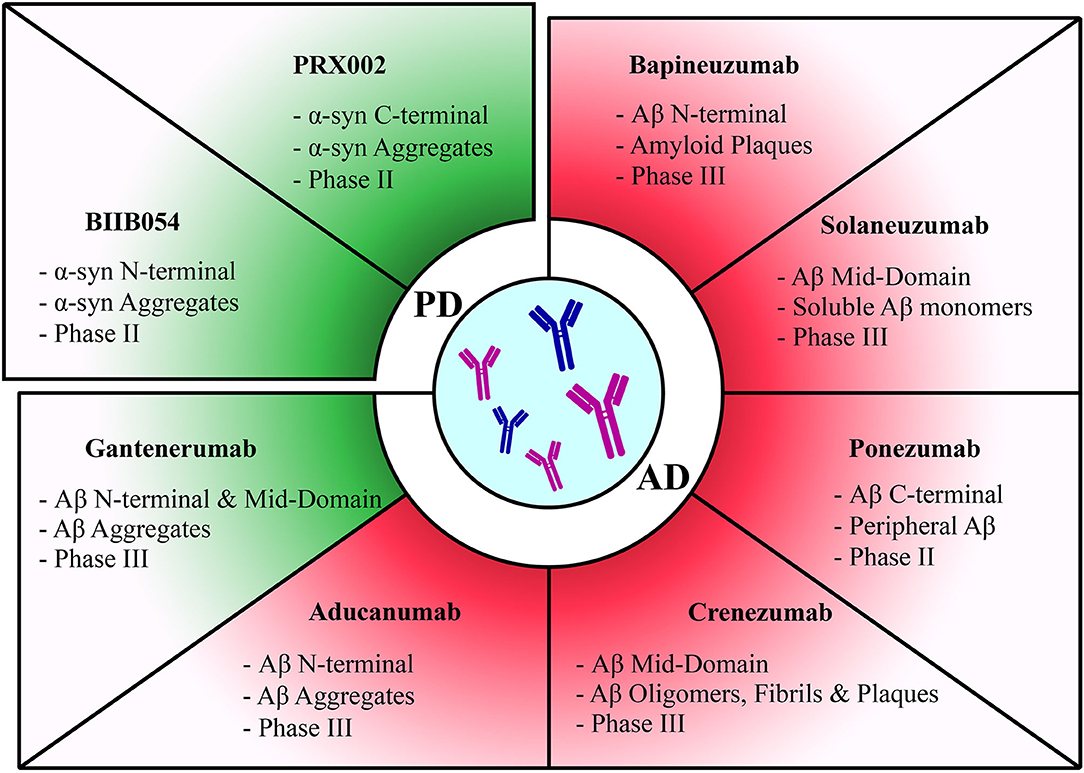
Figure 2. Summary of passive immunization therapies assessed in clinical trials for different diseases. Discontinued trials highlighted in red and ongoing trials highlighted in green. Therapies have been categorized according to each pathology they target. The target of each therapy along with its current clinical trial phase have been stated for each therapy.
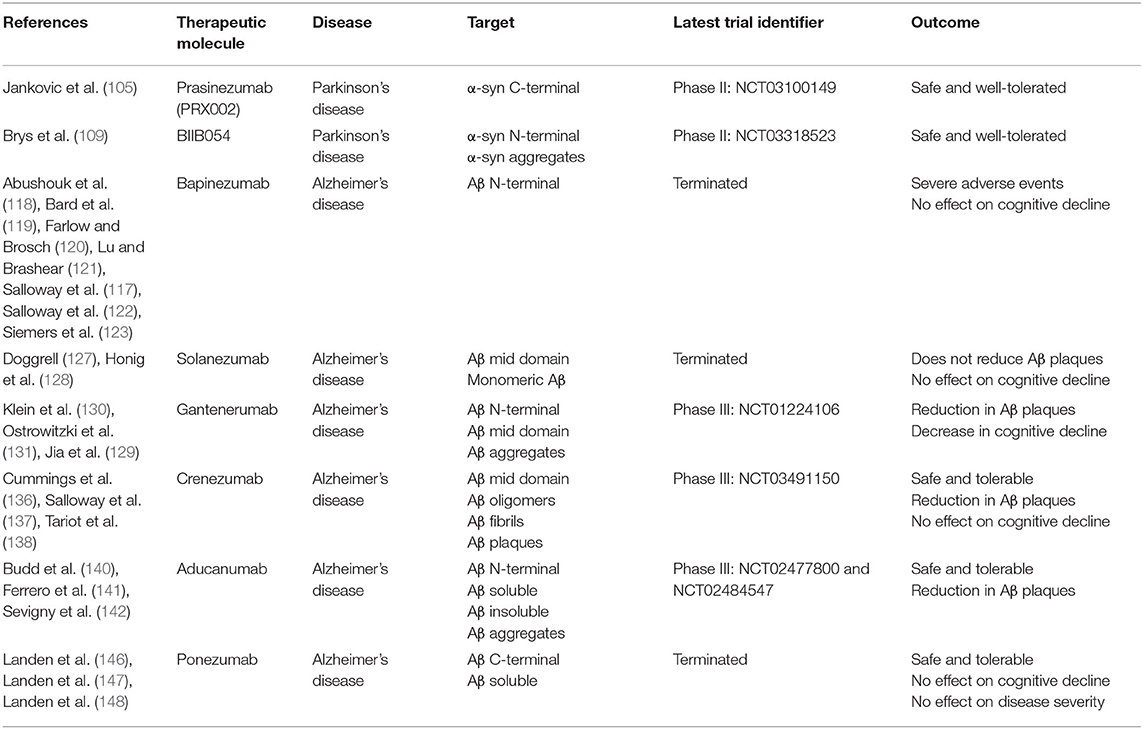
Table 2. Study characteristics of passive immunotherapy clinical trials.
The search for novel targets for the administered passive and active immunization therapies might provide new strategies for treatment. In a novel approach, Thomas et al. examined the possibility of indirectly targeting dysfunctional tau peptide by passively immunizing transgenic mice against BIN1 (bridging integrator 1) gene product (219). The gene BIN1 has been associated with AD, and its product, Myc box-dependent-interacting protein 1, has been shown to co-localize and interact with tau and enhance its pathogenicity by promoting tau release and neurotoxicity (220–223). Anti-BIN1 antibody reduced p-tau species and increased survival in P301S transgenic mice (219). In light of these findings, immunotherapeutic strategies might benefit from exploring novel targets to indirectly reduce disease pathology rather than exclusively target conventional disease biomarkers, which have so far provided limited promise and success.
Understanding the different steps of neuroinflammation provided useful insights into possible options to break the chain of events leading to disease. Modulating the innate immune system remains a plausible approach in managing neurodegeneration, similarly to several other autoimmune and inflammatory diseases. Nevertheless, new approaches currently under investigation could become pivotal therapeutic options in the future. Recent reports suggested that microglial modulation by the gut microbiota can be an exciting novel therapeutic target (224). In fact, PD patients were found to have an altered gut microbiota compared to matched healthy cohorts (225). Furthermore, dysregulation in sleep patterns is seen in several neurodegenerative diseases, and recent data support the view that altered sleep can affect neuroinflammation (226). The exact mechanisms by which dysregulated sleep modulates pro- or anti-inflammatory responses in the CNS are not fully understood yet. Lastly, the effects of systemic immune responses in modulating acute and chronic neuroinflammation are being elucidated. The recently discovered lymphatic vessels in the brain which can transport fluid and immune cells from the CSF constitute another relevant finding (227). These CNS lymphatic networks interconnected to deep cervical lymph nodes could play a role as peripheral immune mediators in regulating neuroinflammatory responses.
The burden of neurological disease is growing globally, and neurodegenerative disorders represent major unmet medical needs and costs to healthcare systems worldwide. The challenges of translating scientific advances into new therapies in neurology are increasing, which are partly due to the lack of patients' willingness to participate in clinical trials and the complexity of developing neurotherapeutics due to a paucity of validated biomarkers (228), longer duration of clinical trials, and higher failure rates due to lack of efficacy (229). Examples of factors determining the ability of a therapeutic candidate to be considered as a potential successful drug include efficient delivery of this candidate, with the appropriate dosage to act on the intended cell or tissue for a specific duration of time; obviously when the target is within the CNS, difficulties and uncertainties arise due the complexed nature of the brain, the intricacies of models mimicking human neurological diseases and the poor functional outcome measures (230). Nevertheless, in searching for more efficient treatments, immunotherapy targeting abnormal protein aggregates or inflammatory molecules is emerging as a promising therapeutic strategy. Our basic understanding of innate immune responses in the CNS during healthy aging and in neurodegenerative diseases has largely progressed in the last two decades. Yet, significant knowledge gaps still exist in understanding the full mechanisms of beneficial and pathologic neuroinflammatory responses tilting the balance toward healthy or diseased aging. With the emergence of additional insights into neuron–glial and glial–glial interactions in the CNS, targeted and potentially more effective therapeutic strategies can be attained. So far, late intervention seems to be the most important cause for treatment failure in several trials, meaning that to battle these diseases, the treatment essentially needs to be started at an early stage. Thus, in addition to the development of more efficacious drugs, better diagnostic strategies are warranted to diagnose these disorders at a time when there still has been no or only limited damage to the CNS. Recent research efforts surrounding neurological diseases are directed toward discovering valid disease biomarkers from body fluids; and good candidates (other than blood and CSF) offering promise as a biomarker pool for neurological disease diagnosis and monitoring, are urine and saliva (231). Finally, limitations in our understanding of the interplay between the innate response in the CNS and systemic immunity are challenges that need to be overcome. The innate immune system, therefore, provides exciting opportunities for disease-modifying treatments in the CNS that are both innovative and feasible. As our knowledge of the precise underling immune mechanisms advances, more effective therapies will be developed in managing these so far intractable neurodegenerative diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This project has been funded by the College of Health Sciences, Abu Dhabi University, Abu Dhabi, United Arab Emirates.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gordon R, Woodruff TM. Chapter 3 - neuroinflammation as a therapeutic target in neurodegenerative diseases A2 - Baekelandt, Veerle. In: Lobbestael E, editor. Disease-Modifying Targets in Neurodegenerative Disorders. Academic Press (2017). p. 49–80. doi: 10.1016/B978-0-12-805120-7.00003-8
2. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. (2014) 15:300–12. doi: 10.1038/nrn3722
3. Chen SH, Oyarzabal EA, Santos J, Wang Q, Jiang L, Hong JS. Chapter 18 - neuroinflammation in neurological dysfunction and degeneration A2 - aschner, michael. In: Costa LG, editor. Environmental Factors in Neurodevelopmental and Neurodegenerative Disorders. Boston, MA: Academic Press (2015). p. 385–407. doi: 10.1016/B978-0-12-800228-5.00018-2
4. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. (2007) 8:57–69. doi: 10.1038/nrn2038
5. Cummings JL, Pillai JA. Neurodegenerative Diseases: Evolving Unifying Principles. Oxford: Oxford University Press. doi: 10.1093/med/9780190233563.003.0001
6. Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. (2014) 13:1045–60. doi: 10.1016/S1474-4422(14)70117-6
7. United Nations Population Division. World Population Ageing 2015. (2015). Retrieved from http://www.un.org/en/development/desa/population/publications/pdf/ageing/WPA2015_Report.pdf
8. Prince M, Albanese E, Guerchet M, Prina M. World Alzheimer Report 2014: Dementia and Risk Reduction: An Analysis of Protective and Modifiable Factors. London (2014). Retrieved from: https://www.alz.co.uk/research/WorldAlzheimerReport2014.pdf~
9. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. (2014) 14:463–77. doi: 10.1038/nri3705
10. Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, et al. Missing pieces in the Parkinson's disease puzzle. Nat Med. (2010) 16:653–61. doi: 10.1038/nm.2165
11. Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. (2007) 30:244–50. doi: 10.1016/j.tins.2007.03.009
12. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. (2006) 443:787–95. doi: 10.1038/nature05292
13. Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET, et al. Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov. (2015) 14:759. doi: 10.1038/nrd4593
14. Liang Z, Zhao Y, Ruan L, Zhu L, Jin K, Zhuge Q, et al. Impact of aging immune system on neurodegeneration and potential immunotherapies. Progr Neurobiol. (2017) 157 (Suppl. C):2–28. doi: 10.1016/j.pneurobio.2017.07.006
15. Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. (2011) 11:775–87. doi: 10.1038/nri3086
16. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. (2014) 32:367–402. doi: 10.1146/annurev-immunol-032713-120240
17. Schafer DP, Stevens B. Microglia function in central nervous system development and plasticity. Cold Spring Harb Perspect Biol. (2015) 7:a020545. doi: 10.1101/cshperspect.a020545
18. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
19. Tremblay M. A diversity of cell types, subtypes and phenotypes in the central nervous system: the importance of studying their complex relationships. Front Cell Neurosci. (2020) 14:628347. doi: 10.3389/fncel.2020.628347
20. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. (2007) 10:1387–94. doi: 10.1038/nn1997
21. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
22. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
23. Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol. (2017) 79:619–43. doi: 10.1146/annurev-physiol-022516-034406
24. Mattotti M, Alvarez Z, Ortega JA, Planell JA, Engel E, Alcántara S. Inducing functional radial glia-like progenitors from cortical astrocyte cultures using micropatterned PMMA. Biomaterials. (2012) 33:1759–70. doi: 10.1016/j.biomaterials.2011.10.086
25. Hennessy E, Griffin EW. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1beta and tnf-alpha. J Neurosci. (2015) 35:8411–22. doi: 10.1523/JNEUROSCI.2745-14.2015
26. Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Rev. Neurol. (2006) 2:679. doi: 10.1038/ncpneuro0355
27. Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. (2008) 57:178–201. doi: 10.1016/j.neuron.2008.01.003
28. Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. (2007) 147:867–83. doi: 10.1016/j.neuroscience.2007.02.055
29. Napoli I, Neumann H. Protective effects of microglia in multiple sclerosis. Exp Neurol. (2010) 225:24–8. doi: 10.1016/j.expneurol.2009.04.024
30. Simard AR, Rivest S. Neuroprotective effects of resident microglia following acute brain injury. J Comparat Neurol. (2007) 504:716–29. doi: 10.1002/cne.21469
31. Persson M, Brantefjord M, Hansson E, Rönnbäck L. Lipopolysaccharide increases microglial GLT-1 expression and glutamate uptake capacity in vitro by a mechanism dependent on TNF-α. Glia. (2005) 51:111–20. doi: 10.1002/glia.20191
32. Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, et al. Protective autoimmunity: interferon-γ enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem. (2005) 92:997–1009. doi: 10.1111/j.1471-4159.2004.02954.x
33. Gowing G, Vallières L, Julien JP. Mouse model for ablation of proliferating microgliain acute CNS injuries. Glia. (2006) 53:331–7. doi: 10.1002/glia.20288
34. Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. (2005) 11:146–52. doi: 10.1038/nm1177
35. Biber K, Owens T, Boddeke E. What is microglia neurotoxicity (Not)? Glia. (2014) 62:841–54. doi: 10.1002/glia.22654
36. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
37. Maslanik T, Mahaffey L, Tannura K, Beninson L, Greenwood BN, Fleshner M. The inflammasome and danger associated molecular patterns (DAMPs) are implicated in cytokine and chemokine responses following stressor exposure. Brain Behav Immun. (2013) 28:54–62. doi: 10.1016/j.bbi.2012.10.014
38. Minkiewicz J, Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. (2013) 61:1113–21. doi: 10.1002/glia.22499
39. Mrak RE, Griffin WST. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. (2005) 26:349–54. doi: 10.1016/j.neurobiolaging.2004.05.010
40. Gordon R, Anantharam V, Kanthasamy AG, Kanthasamy A. Proteolytic activation of proapoptotic kinase protein kinase Cδ by tumor necrosis factor α death receptor signaling in dopaminergic neurons during neuroinflammation. J Neuroinflammation. (2012) 9:82. doi: 10.1186/1742-2094-9-82
41. McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. (2008) 5:45. doi: 10.1186/1742-2094-5-45
42. Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson's disease. Brain. (2008) 131:1880–94. doi: 10.1093/brain/awn101
43. Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, et al. Neurodegeneration by activation of the microglial complement–phagosome pathway. J Neurosci. (2014) 34:8546–56. doi: 10.1523/JNEUROSCI.5002-13.2014
44. Farkas I, Takahashi M, Fukuda A, Yamamoto N, Akatsu H, Baranyi L, et al. Complement C5a receptor-mediated signaling may be involved in neurodegeneration in Alzheimer's disease. J Immunol. (2003) 170:5764–71. doi: 10.4049/jimmunol.170.11.5764
45. Jacob A, Alexander JJ. Complement and blood–brain barrier integrity. Mol Immunol. (2014) 61:149–52. doi: 10.1016/j.molimm.2014.06.039
46. Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM. The role of the complement system and the activation fragment C5a in the central nervous system. Neuromol Med. (2010) 12:179–92. doi: 10.1007/s12017-009-8085-y
47. Mantovani S, Gordon R, Macmaw J, Pfluger C, Henderson R, Noakes P, et al. Elevation of the terminal complement activation products C5a and C5b-9 in ALS patient blood. J Neuroimmunol. (2014) 276:213–8. doi: 10.1016/j.jneuroim.2014.09.005
48. Barnum CJ, Chen X, Chung J, Chang J, Williams M, Grigoryan N, et al. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro® 1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J Parkinson Dis. (2014) 4:349–360. doi: 10.3233/JPD-140410
49. Detrait ER, Danis B, Lamberty Y, Foerch P. Peripheral administration of an anti-TNF-alpha receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-alpha levels and memory deficits in mice. Neurochem Int. (2014) 72:10–3. doi: 10.1016/j.neuint.2014.04.001
50. Butchart J, Brook L, Hopkins V, Teeling J, Puntener U, Culliford D, et al. Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology. (2015) 84:2161–8. doi: 10.1212/WNL.0000000000001617
51. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer' s disease and contributes to pathology in APP/PS1 mice. Nature. (2013) 493:674–8. doi: 10.1038/nature11729
52. Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson's disease. J Neuroinflammation. (2008) 5:8. doi: 10.1186/1742-2094-5-8
53. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. (2015) 21:248–55. doi: 10.1038/nm.3806
54. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite [beta]-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. (2015) 21:263–9. doi: 10.1038/nm.3804
55. Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat Med. (2012) 18:1812–9. doi: 10.1038/nm.2965
56. Papatriantafyllou M. Immunological bullets against Alzheimer's disease. Nat Rev Drug Discov. (2013) 12:24. doi: 10.1038/nrd3919
57. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. (2000) 21:383–421. doi: 10.1016/S0197-4580(00)00124-X
58. McGeer PL, McGeer EG. Inflammation of the brain in Alzheimer's disease: implications for therapy. J Leukoc Biol. (1999) 65:409–15. doi: 10.1002/jlb.65.4.409
59. Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA. (2002) 99:10837–42. doi: 10.1073/pnas.162350199
60. Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5–C5a receptor axis. Mol Immunol. (2011) 48:1631–42. doi: 10.1016/j.molimm.2011.04.014
61. Bhattacharya P, Budnick I, Singh M, Thiruppathi M, Alharshawi K, Elshabrawy H, et al. Dual role of GM-CSF as a pro-inflammatory and a regulatory cytokine: implications for immune therapy. J Interfer Cytok Res. (2015) 35:585–99. doi: 10.1089/jir.2014.0149
62. Chitu V, Biundo F, Shlager GGL, Park ES, Wang P, Gulinello ME, et al. Microglial homeostasis requires balanced CSF-1/CSF-2 receptor signaling. Cell Rep. (2020) 30:3004–19.e05. doi: 10.1016/j.celrep.2020.02.028
63. Dikmen HO, Hemmerich M, Lewen A, Hollnagel JO, Chausse B, Kann O. GM-CSF induces noninflammatory proliferation of microglia and disturbs electrical neuronal network rhythms in situ. J Neuroinflammation. (2020) 17:235. doi: 10.1186/s12974-020-01903-4
64. Esen N, Kielian T. Effects of low dose GM-CSF on microglial inflammatory profiles to diverse pathogen-associated molecular patterns (PAMPs). J Neuroinflammation. (2007) 4:10. doi: 10.1186/1742-2094-4-10
65. Kiyota T, Machhi J, Lu Y, Dyavarshetty B, Nemati M, Yokoyama I, et al. Granulocyte-macrophage colony-stimulating factor neuroprotective activities in Alzheimer's disease mice. J Neuroimmunol. (2018) 319:80–92. doi: 10.1016/j.jneuroim.2018.03.009
66. Sanchez-Ramos J, Song S, Sava V, Catlow B, Lin X, Mori T, et al. Granulocyte colony stimulating factor decreases brain amyloid burden and reverses cognitive impairment in Alzheimer's mice. Neuroscience. (2009) 163:55–72. doi: 10.1016/j.neuroscience.2009.05.071
67. Potter H, Woodcock JH, Boyd TD, Coughlan CM, O'Shaughnessy JR, Borges MT, et al. Safety and efficacy of sargramostim (GM-CSF) in the treatment of Alzheimer's disease. Alzheimers Dement. (2021) 7:e12158. doi: 10.1002/trc2.12158
68. Gendelman HE, Zhang Y, Santamaria P, Olson KE, Schutt CR, Bhatti D, et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson's disease trial. NPJ Parkinson Dis. (2017) 3:10. doi: 10.1038/s41531-017-0013-5
69. Kosloski LM, Kosmacek EA, Olson KE, Mosley RL, Gendelman HE. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J Neuroimmunol. (2013) 265:1–10. doi: 10.1016/j.jneuroim.2013.10.009
70. Amirzagar N, Nafissi S, Tafakhori A, Modabbernia A, Amirzargar A, Ghaffarpour M, et al. Granulocyte colony-stimulating factor for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled study of Iranian patients. J Clin Neurol. (2015) 11:164–71. doi: 10.3988/jcn.2015.11.2.164
71. Nefussy B, Artamonov I, Deutsch V, Naparstek E, Nagler A, Drory VE. Recombinant human granulocyte-colony stimulating factor administration for treating amyotrophic lateral sclerosis: A pilot study. Amyotroph Later Scler. (2010) 11:187–93. doi: 10.3109/17482960902933809
72. Pollari E, Savchenko E, Jaronen M, Kanninen K, Malm T, Wojciechowski S, et al. Granulocyte colony stimulating factor attenuates inflammation in a mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. (2011) 8:74. doi: 10.1186/1742-2094-8-74
73. Yamasaki R, Tanaka M, Fukunaga M, Tateishi T, Kikuchi H, Motomura K, et al. Restoration of microglial function by granulocyte-colony stimulating factor in ALS model mice. J Neuroimmunol. (2010) 229:51–62. doi: 10.1016/j.jneuroim.2010.07.002
74. Cashman N, Tan LY, Krieger C, Mädler B, Mackay A, Mackenzie I, et al. Pilot study of granulocyte colony stimulating factor (G-CSF)-mobilized peripheral blood stem cells in amyotrophic lateral sclerosis (ALS). Muscle Nerve. (2008) 37:620–5. doi: 10.1002/mus.20951
75. Tarella C, Rutella S, Gualandi F, Melazzini M, Scimè R, Petrini M, et al. Consistent bone marrow-derived cell mobilization following repeated short courses of granulocyte–colony-stimulating factor in patients with amyotrophic lateral sclerosis: results from a multicenter prospective trial. Cytotherapy. (2010) 12:50–9. doi: 10.3109/14653240903300682
76. Chiò A, Mora G, La Bella V, Caponnetto C, Mancardi G, Sabatelli M, et al. Repeated courses of granulocyte colony-stimulating factor in amyotrophic lateral sclerosis: clinical and biological results from a prospective multicenter study. Muscle Nerve. (2011) 43:189–95. doi: 10.1002/mus.21851
77. Johannesen S, Budeus B, Peters S, Iberl S, Meyer AL, Kammermaier T, et al. Biomarker supervised G-CSF (filgrastim) response in ALS patients. Front Neurol. (2018) 9:971. doi: 10.3389/fneur.2018.00971
78. Zhang Y, Wang L, Fu Y, Song H, Zhao H, Deng M, et al. Preliminary investigation of effect of granulocyte colony stimulating factor on amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2009) 10:430–431. doi: 10.3109/17482960802588059
79. Bernardo A, Minghetti L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr Pharm Des. (2006) 12:93–109. doi: 10.2174/138161206780574579
80. Bright JJ, Kanakasabai S, Chearwae W, Chakraborty S. PPAR Regulation of Inflammatory Signaling in CNS Diseases. PPAR Res. (2008) 2008:658520. doi: 10.1155/2008/658520
81. de la Monte SM, Tong M, Schiano I, Didsbury J. Improved brain insulin/IGF signaling and reduced neuroinflammation with T3D-959 in an experimental model of sporadic alzheimer's disease. J Alzheimers Dis. (2017) 55:849–64. doi: 10.3233/JAD-160656
82. Grieco M, Giorgi A, Gentile MC, d'Erme M, Morano S, Maras B, et al. Glucagon-Like Peptide-1: A Focus on Neurodegenerative Diseases. Frontiers in neuroscience. (2019) 13:1112–1112. doi: 10.3389/fnins.2019.01112
83. Guo J, Yang G, He Y, Xu H, Fan H, An J, et al. Involvement of α7nAChR in the protective effects of genistein against β-amyloid-induced oxidative stress in neurons via a PI3K/Akt/Nrf2 pathway-related mechanism. Cell Mol Neurobiol. (2021) 41:377–93. doi: 10.1007/s10571-020-01009-8
84. Paladugu L, Gharaibeh A, Kolli N, Learman C, Hall TC, Li L, et al. Liraglutide has anti-inflammatory and anti-amyloid properties in streptozotocin-induced and 5xFAD mouse models of alzheimer's disease. Int J Mol Sci. (2021) 22:860. doi: 10.3390/ijms22020860
85. Glotfelty EJ, Olson L, Karlsson TE, Li Y, Greig NH. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson's disease. Expert Opin Investig Drugs. (2020) 29:595–602. doi: 10.1080/13543784.2020.1764534
86. Ntetsika T, Papathoma PE, Markaki I. Novel targeted therapies for Parkinson's disease. Mol Med. (2021) 27:17. doi: 10.1186/s10020-021-00279-2
87. Yaribeygi H, Rashidy-Pour A, Atkin SL, Jamialahmadi T, Sahebkar A. GLP-1 mimetics and cognition. Life Sci. (2021) 264:118645. doi: 10.1016/j.lfs.2020.118645
88. Gejl M, Gjedde A, Egefjord L, Møller A, Hansen SB, Vang K, et al. In alzheimer's disease, 6-month treatment with glp-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front Aging Neurosci. (2016) 8:108. doi: 10.3389/fnagi.2016.00108
89. Mulvaney CA, Duarte GS, Handley J, Evans DJ, Menon S, Wyse R, et al. GLP-1 receptor agonists for Parkinson's disease. Cochrane Database Syst Rev. (2020) 7:Cd012990. doi: 10.1002/14651858.CD012990.pub2
90. Wang SY, Wu SL, Chen TC, Chuang CS. Antidiabetic agents for treatment of parkinson's disease: a meta-analysis. Int J Environ Res Public Health. (2020) 17:4805. doi: 10.3390/ijerph17134805
91. Watson KT, Wroolie TE, Tong G, Foland-Ross LC, Frangou S, Singh M, et al. Neural correlates of liraglutide effects in persons at risk for Alzheimer's disease. Behav Brain Res. (2019) 356:271–8. doi: 10.1016/j.bbr.2018.08.006
92. Petry FDS, Hoppe JB, Klein CP, Dos Santos BG, Hözer RM, Bifi F, et al. Genistein attenuates amyloid-beta-induced cognitive impairment in rats by modulation of hippocampal synaptotoxicity and hyperphosphorylation of Tau. J Nutr Biochem. (2021) 87:108525. doi: 10.1016/j.jnutbio.2020.108525
93. Tong M, Deochand C, Didsbury J, de la Monte SM. T3D-959: a multi-faceted disease remedial drug candidate for the treatment of alzheimer's disease. J Alzheimers Dis. (2016) 51:123–38. doi: 10.3233/JAD-151013
94. Chamberlain S, Gabriel H, Strittmatter W, Didsbury J. An exploratory phase iia study of the PPAR delta/gamma agonist T3D-959 assessing metabolic and cognitive function in subjects with mild to moderate alzheimer's disease. J Alzheimers Dis. (2020) 73:1085–103. doi: 10.3233/JAD-190864
95. Geldmacher DS, Fritsch T, McClendon MJ, Landreth G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch Neurol. (2011) 68:45–50. doi: 10.1001/archneurol.2010.229
96. Machado MMF, Bassani TB, Cóppola-Segovia V, Moura ELR, Zanata SM, Andreatini R, et al. PPAR-γ agonist pioglitazone reduces microglial proliferation and NF-κB activation in the substantia nigra in the 6-hydroxydopamine model of Parkinson's disease. Pharmacol Rep. (2019) 71:556–64. doi: 10.1016/j.pharep.2018.11.005
97. Pinto M, Nissanka N, Peralta S, Brambilla R, Diaz F, Moraes CT. Pioglitazone ameliorates the phenotype of a novel Parkinson's disease mouse model by reducing neuroinflammation. Mol Neurodegener. (2016) 11:25. doi: 10.1186/s13024-016-0090-7
98. Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S, et al. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson's disease. Neurobiol Dis. (2014) 71:280–91. doi: 10.1016/j.nbd.2014.08.011
99. Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer's disease. Alzheimer Dis Associ Disord. (2011) 25:206. doi: 10.1097/WAD.0b013e318204b550
100. Bachmann MF, Whitehead P. Active immunotherapy for chronic diseases. Vaccine. (2013) 31:1777–1784. doi: 10.1016/j.vaccine.2013.02.001
101. Shahaduzzaman M, Nash K, Hudson C, Sharif M, Grimmig B, Lin X, et al. Anti-human α-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-α-synuclein rat model of Parkinson's disease. PLoS ONE. (2015) 10:e0116841. doi: 10.1371/journal.pone.0116841
102. Gustafsson G, Eriksson F, Möller C, da Fonseca TL, Outeiro TF, Lannfelt L, et al. Cellular uptake of α-synuclein oligomer-selective antibodies is enhanced by the extracellular presence of α-synuclein and mediated via Fcγ receptors. Cell Mol Neurobiol. (2017) 37:121–31. doi: 10.1007/s10571-016-0352-5
103. Lindström V, Fagerqvist T, Nordström E, Eriksson F, Lord A, Tucker S, et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h [A30P] α-synuclein mice. Neurobiol Dis. (2014) 69:134–43. doi: 10.1016/j.nbd.2014.05.009
104. Huang YR, Xie XX, Ji M, Yu X, Zhu J, Zhang LX, et al. Naturally occurring autoantibodies against α-synuclein rescues memory and motor deficits and attenuates α-synuclein pathology in mouse model of Parkinson's disease. Neurobiol Dis. (2019) 124:202–17. doi: 10.1016/j.nbd.2018.11.024
105. Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti–α-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol. (2018) 75:1206–14. doi: 10.1001/jamaneurol.2018.1487
106. Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of lewy body disease. PLoS ONE. (2011) 6:e19338. doi: 10.1371/journal.pone.0019338
107. Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, Zago W, et al. First-in-human assessment of PRX002, an anti–α-synuclein monoclonal antibody, in healthy volunteers. Move Disord. (2017) 32:211–8. doi: 10.1002/mds.26878
108. Weihofen A, Liu Y, Arndt JW, Huy C, Quan C, Smith BA, et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson's disease models. Neurobiol Dis. (2019) 124:276–88. doi: 10.1016/j.nbd.2018.10.016
109. Brys M, Fanning L, Hung S, Ellenbogen A, Penner N, Yang M, et al. Randomized phase I clinical trial of anti–α-synuclein antibody BIIB054. Move Disord. (2019) 34:1154–63. doi: 10.1002/mds.27738
110. Valera E, Masliah E. Immunotherapy for neurodegenerative diseases: focus on α-synucleinopathies. Pharmacol Ther. (2013) 138:311–22. doi: 10.1016/j.pharmthera.2013.01.013
111. Sanchez-Guajardo V, Annibali A, Jensen PH, Romero-Ramos M. α-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol. (2013) 72:624–45. doi: 10.1097/NEN.0b013e31829768d2
112. Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener. (2015) 10:10. doi: 10.1186/s13024-015-0008-9
113. Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, et al. Next-generation active immunization approach for synucleinopathies: implications for Parkinson's disease clinical trials. Acta Neuropathol. (2014) 127:861–79. doi: 10.1007/s00401-014-1256-4
114. McFarthing K, Simuni T. Clinical trial highlights: targetting alpha-synuclein. J Parkinson Dis. (2019) 9:5–16. doi: 10.3233/JPD-189004
115. Cheng L, Zhang J, Li XY, Yuan L, Pan YF, Chen XR, et al. A novel antibody targeting sequence 31–35 in amyloid β protein attenuates Alzheimer's disease-related neuronal damage. Hippocampus. (2017) 27:122–33. doi: 10.1002/hipo.22676
116. Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, et al. 11C-PiB PET assessment of change in fibrillar amyloid-β load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. (2010) 9:363–72. doi: 10.1016/S1474-4422(10)70043-0
117. Salloway S, Sperling R, Gilman S, Fox N, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. (2009) 73:2061–70. doi: 10.1212/WNL.0b013e3181c67808
118. Abushouk AI, Elmaraezy A, Aglan A, Salama R, Fouda S, Fouda R, et al. Bapineuzumab for mild to moderate Alzheimer's disease: a meta-analysis of randomized controlled trials. BMC Neurol. (2017) 17:66. doi: 10.1186/s12883-017-0850-1
119. Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, et al. Epitope and isotype specificities of antibodies to β-amyloid peptide for protection against Alzheimer's disease-like neuropathology. Proc Natl Acad Sci USA. (2003) 100:2023–8. doi: 10.1073/pnas.0436286100
120. Farlow MR, Brosch JR. Immunotherapy for Alzheimer's disease. Neurol Clin. (2013) 31:869–78. doi: 10.1016/j.ncl.2013.03.012
121. Lu M, Brashear HR. Pharmacokinetics, pharmacodynamics, and safety of subcutaneous bapineuzumab: a single-ascending-dose study in patients with mild to moderate Alzheimer disease. Clin Pharmacol Drug Dev. (2019) 8:326–35. doi: 10.1002/cpdd.584
122. Salloway SP, Sperling R, Fox NC, Sabbagh MN, Honig LS, Porsteinsson AP, et al. Long-term follow up of patients with mild-to-moderate Alzheimer's disease treated with bapineuzumab in a phase III, open-label, extension study. J Alzheimers Dis. (2018) 64:689–707. doi: 10.3233/JAD-171157
123. Siemers ER, Friedrich S, Dean RA, Sethuraman G, DeMattos R, Jennings D, et al. P4-346: Safety, tolerability and biomarker effects of an Abeta monoclonal antibody administered to patients with Alzheimer's disease. Alzheimers Dement. (2008) 4:T774. doi: 10.1016/j.jalz.2008.05.2416
124. DeMattos RB, Racke MM, Gelfanova V, Forster B, Knierman MD, Bryan MT, et al. Identification, characterization, and comparison of amino-terminally truncated Aβ42 peptides in Alzheimer's disease brain tissue and in plasma from Alzheimer's patients receiving solanezumab immunotherapy treatment. Alzheimers Dement. (2009) 5:P156–7. doi: 10.1016/j.jalz.2009.05.541
125. Seubert P, Barbour R, Khan K, Motter R, Tang P, Kholodenko D, et al. Antibody capture of soluble Aβ does not reduce cortical Aβ amyloidosis in the PDAPP mouse. Neurodegenerat Dis. (2008) 5:65–71. doi: 10.1159/000112834
126. Siemers ER, Friedrich S, Dean RA, Gonzales CR, Farlow MR, Paul SM, et al. Safety and changes in plasma and cerebrospinal fluid amyloid β after a single administration of an amyloid β monoclonal antibody in subjects with Alzheimer disease. Clin Neuropharmacol. (2010) 33:67–73. doi: 10.1097/WNF.0b013e3181cb577a
127. Doggrell SA. Grasping at straws: the failure of solanezumab to modify mild Alzheimer's disease. Exp Opin Biol Ther. (2018) 18:1189–92. doi: 10.1080/14712598.2018.1543397
128. Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. (2018) 378:321–30. doi: 10.1056/NEJMoa1705971
129. Jia Q, Deng Y, Qing H. Potential therapeutic strategies for Alzheimer's disease targeting or beyond β-amyloid: insights from clinical trials. BioMed Res Int. (2014) 2014:837157. doi: 10.1155/2014/837157
130. Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, et al. Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer's disease: a PET substudy interim analysis. Alzheimers Res Ther. (2019) 11:101. doi: 10.1186/s13195-019-0559-z
131. Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease. Alzheimers Res Ther. (2017) 9:95. doi: 10.1186/s13195-017-0318-y
132. Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, et al. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci. (2012) 32:9677–89. doi: 10.1523/JNEUROSCI.4742-11.2012
133. Garber K. Genentech's Alzheimer's antibody trial to study disease prevention. Nat Biotechnol. (2012) 30:731–2. doi: 10.1038/nbt0812-731
134. Ultsch M, Li B, Maurer T, Mathieu M, Adolfsson O, Muhs A, et al. Structure of crenezumab complex with Aβ shows loss of β-hairpin. Sci Rep. (2016) 6:39374. doi: 10.1038/srep39374
135. Yang T, Dang Y, Ostaszewski B, Mengel D, Steffen V, Rabe C, et al. Target engagement in an alzheimer trial: crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann Neurol. (2019) 86:215–24. doi: 10.1002/ana.25513
136. Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, Cho W, et al. ABBY: a phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. (2018) 90:e1889–97. doi: 10.1212/WNL.0000000000005550
137. Salloway S, Honigberg LA, Cho W, Ward M, Friesenhahn M, Brunstein F, et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer's disease (BLAZE). Alzheimers Res Ther. (2018) 10:96. doi: 10.1186/s13195-018-0424-5
138. Tariot PN, Lopera F, Langbaum JB, Thomas RG, Hendrix S, Schneider LS, et al. The alzheimer's prevention initiative autosomal-dominant alzheimer's disease trial: a study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer's disease, including a placebo-treated noncarrier cohort. Alzheimers Dement. (2018) 4:150–60. doi: 10.1016/j.trci.2018.02.002
139. Arndt JW, Qian F, Smith BA, Quan C, Kilambi KP, Bush MW, et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci Rep. (2018) 8:6412. doi: 10.1038/s41598-018-24501-0
140. Budd SH, O'Gorman J, Chiao P, Bussiere T, Tian Y, Zhu Y, et al. Clinical development of aducanumab, an anti-Aβ human monoclonal antibody being investigated for the treatment of early alzheimer's disease. J Prevent Alzheimers Dis. (2017) 4:255–63. doi: 10.14283/jpad.2017.39
141. Ferrero J, Williams L, Stella H, Leitermann K, Mikulskis A, O'Gorman J, et al. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer's disease. Alzheimers Dement. (2016) 2:169–76. doi: 10.1016/j.trci.2016.06.002
142. Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. (2016) 537:50–6. doi: 10.1038/nature19323
143. Armour KL, van de Winkel JG, Williamson LM, Clark MR. Differential binding to human FcγRIIa and FcγRIIb receptors by human IgG wildtype and mutant antibodies. Mol Immunol. (2003) 40:585–93. doi: 10.1016/j.molimm.2003.08.004
144. Liu YH, Wang YR, Xiang Y, Zhou HD, Giunta B, Mañucat-Tan NB, et al. Clearance of amyloid-beta in Alzheimer's disease: shifting the action site from center to periphery. Mol Neurobiol. (2015) 51:1–7. doi: 10.1007/s12035-014-8694-9
145. Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, et al. Passive immunotherapy against Aβ in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. (2004) 1:24. doi: 10.1186/1742-2094-1-24
146. Landen JW, Andreasen N, Cronenberger CL, Schwartz PF, Börjesson-Hanson A, Östlund H, et al. Ponezumab in mild-to-moderate Alzheimer's disease: randomized phase II PET-PIB study. Alzheimers Dement. (2017) 3:393–401. doi: 10.1016/j.trci.2017.05.003
147. Landen JW, Cohen S, Billing CB Jr, Cronenberger C, Styren S, Burstein AH, et al. Multiple-dose ponezumab for mild-to-moderate Alzheimer's disease: Safety and efficacy. Alzheimers Dement. (2017) 3:339–47. doi: 10.1016/j.trci.2017.04.003
148. Landen JW, Zhao Q, Cohen S, Borrie M, Woodward M, Billing CB Jr, et al. Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease: a phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin Neuropharmacol. (2013) 36:14–23. doi: 10.1097/WNF.0b013e31827db49b
149. van Ameijde J, Crespo R, Janson R, Juraszek J, Siregar B, Verveen H, et al. Enhancement of therapeutic potential of a naturally occurring human antibody targeting a phosphorylated Ser 422 containing epitope on pathological tau. Acta Neuropathol Commun. (2018) 6:59. doi: 10.1186/s40478-018-0562-9
150. Dai C, l, Hu W, Tung YC, Liu F, Gong CX, et al. Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3 × Tg-AD mice. Alzheimers Res Ther. (2018) 10:13. doi: 10.1186/s13195-018-0341-7
151. Dai CL, Tung YC, Liu F, Gong CX, Iqbal K. Tau passive immunization inhibits not only tau but also Aβ pathology. Alzheimers Res Ther. (2017) 9:1. doi: 10.1186/s13195-016-0227-5
152. Albert M, Mairet-Coello G, Danis C, Lieger S, Caillierez R, Carrier S, et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain. (2019) 142:1736–50. doi: 10.1093/brain/awz100
153. d'Abramo C, Acker CM, Jimenez H, Davies P. Passive immunization in JNPL3 transgenic mice using an array of phospho-tau specific antibodies. PLoS ONE. (2015) 10:e0135774. doi: 10.1371/journal.pone.0135774
154. Gerson JE, Farmer KM, Henson N, Castillo-Carranza DL, Murillo MC, Sengupta U, et al. Tau oligomers mediate α-synuclein toxicity and can be targeted by immunotherapy. Mol Neurodegener. (2018) 13:13. doi: 10.1186/s13024-018-0245-9
155. Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Kayed R. Specific targeting of tau oligomers in htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. (2014) 40:S97–111. doi: 10.3233/JAD-132477
156. Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Hernandez C, Barrett AD, Dineley K, et al. Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer's disease mouse model. J Neurosci. (2015) 35:4857–68. doi: 10.1523/JNEUROSCI.4989-14.2015
157. Castillo-Carranza DL, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Gerson JE, Singh G, et al. Passive immunization with tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci. (2014) 34:4260–72. doi: 10.1523/JNEUROSCI.3192-13.2014
158. Alzforum. Therputics, C2N 8E12. (2018). Retrieved from https://www.alzforum.org/therapeutics/c2n-8e12
159. Alzforum. Therpeutics, Gosuranemab (2020). Retrieved from https://www.alzforum.org/therapeutics/gosuranemab
160. Alzforum. Therapeutics, Zagotenemab. (2018). Retrieved from https://www.alzforum.org/therapeutics/zagotenemab
161. Alzforum. Theraputics, Semorinemab. (2020). Retrieved from https://www.alzforum.org/therapeutics/semorinemab
162. Qureshi IA, Tirucherai G, Ahlijanian MK, Kolaitis G, Bechtold C, Grundman M. A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement. (2018) 4:746–55. doi: 10.1016/j.trci.2018.10.007
163. Jäkel L, Boche D, Nicoll JA, Verbeek MM. Aβ43 in human Alzheimer's disease: effects of active Aβ42 immunization. Acta Neuropathol Communi. (2019) 7:141. doi: 10.1186/s40478-019-0791-6
164. Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson K, Jenkins L, et al. Evaluation of the safety and immunogenicity of synthetic Aβ42 (AN1792) in patients with AD. Neurology. (2005) 64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67
165. Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, et al. Aβ species removal after Aβ42 immunization. J Neuropathol Exp Neurol. (2006) 65:1040–8. doi: 10.1097/01.jnen.0000240466.10758.ce
166. Nicoll JA, Buckland GR, Harrison CH, Page A, Harris S, Love S, et al. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer's disease. Brain. (2019) 142:2113–26. doi: 10.1093/brain/awz142
167. Sakai K, Boche D, Carare R, Johnston D, Holmes C, Love S, et al. Aβ immunotherapy for Alzheimer's disease: effects on apoE and cerebral vasculopathy. Acta Neuropathol. (2014) 128:777–89. doi: 10.1007/s00401-014-1340-9
168. Muhs A, Hickman DT, Pihlgren M, Chuard N, Giriens V, Meerschman C, et al. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc Nat Acad Sci USA. (2007) 104:9810–5. doi: 10.1073/pnas.0703137104
169. Immune A. ACI 24 anti Abeta Vaccine for Prevention and Therapy in Alzheimer's Disease and Down Syndrome. (2019). Retrieved from https://ir.acimmune.com/static-files/276dac58-d882-48a6-8cb4-d47ccad53b69
170. Farlow MR, Andreasen N, Riviere ME, Vostiar I, Vitaliti A, Sovago J, et al. Long-term treatment with active Aβ immunotherapy with CAD106 in mild Alzheimer's disease. Alzheimers Res Ther. (2015) 7:23. doi: 10.1186/s13195-015-0108-3
171. Vandenberghe R, Riviere ME, Caputo A, Sovago J, Maguire RP, Farlow M, et al. Active Aβ immunotherapy CAD106 in alzheimer's disease: a phase 2b study. Alzheimers Dement. (2017) 3:10–22. doi: 10.1016/j.trci.2016.12.003
172. Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer's disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. (2012) 11:597–604. doi: 10.1016/S1474-4422(12)70140-0
173. Hull M, Sadowsky C, Arai H, Le Prince L, Holstein A, Booth K, et al. Long-Term extensions of randomized vaccination trials of ACC-001 and QS-21 in mild to moderate alzheimer's disease. Curr Alzheimer Res. (2017) 14:696–708. doi: 10.2174/1567205014666170117101537
174. Ketter N, Liu E, Di J, Honig LS, Lu M, Novak G, et al. A randomized, double-blind, phase 2 study of the effects of the vaccine vanutide cridificar with QS-21 adjuvant on immunogenicity, safety and amyloid imaging in patients with mild to moderate alzheimer's disease. J Prev Alzheimers Dis. (2016) 3:192–201. doi: 10.14283/jpad.2016.118
175. Pasquier F, Sadowsky C, Holstein A, Leterme Gle P, Peng Y, Jackson N, et al. Two phase 2 multiple ascending-dose studies of vanutide cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate alzheimer's disease. J Alzheimers Dis. (2016) 51:1131–43. doi: 10.3233/JAD-150376
176. van Dyck CH, Sadowsky C, Le Prince Leterme G, Booth K, Peng Y, Marek K, et al. Vanutide cridificar (ACC-001) and QS-21 adjuvant in individuals with early alzheimer's disease: amyloid imaging positron emission tomography and safety results from a phase 2 study. J Prev Alzheimers Dis. (2016) 3:75–84. doi: 10.14283/jpad.2016.91
177. Lacosta AM, Pascual-Lucas M, Pesini P, Casabona D, Pérez-Grijalba V, Marcos-Campos I, et al. Safety, tolerability and immunogenicity of an active anti-Aβ 40 vaccine (ABvac40) in patients with Alzheimer's disease: a randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res Ther. (2018) 10:12. doi: 10.1186/s13195-018-0340-8
178. Rajamohamedsait H, Rasool S, Rajamohamedsait W, Lin Y, Sigurdsson EM. Prophylactic active tau immunization leads to sustained reduction in both tau and amyloid-β pathologies in 3xTg mice. Sci Rep. (2017) 7:17034. doi: 10.1038/s41598-017-17313-1
179. Kontsekova E, Zilka N, Kovacech B, Novak P, Novak M. First-in-man tau vaccine targeting structural determinants essential for pathological tau–tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer's disease model. Alzheimers Res Ther. (2014) 6:44. doi: 10.1186/alzrt278
180. Novak P, Schmidt R, Kontsekova E, Kovacech B, Smolek T, Katina S, et al. Fundamant: an interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer's disease. Alzheimers Res Ther. (2018) 10:108. doi: 10.1186/s13195-018-0436-1
181. Novak P, Schmidt R, Kontsekova E, Zilka N, Kovacech B, Skrabana R, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer's disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. (2017) 16:123–34. doi: 10.1016/S1474-4422(16)30331-3
182. Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, López-Deber MP, Reis P, et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau. P301L mice that model tauopathy. PLoS ONE. (2013) 8:e72301. doi: 10.1371/journal.pone.0072301
183. Ayalon G, Lee SH, Adolfsson O, Foo-Atkins C, Atwal JK, Blendstrup M, et al. Antibody semorinemab reduces tau pathology in a transgenic mouse model and engages tau in patients with Alzheimer's disease. Sci Transl Med. (2021) 13:eabb2639. doi: 10.1126/scitranslmed.abb2639
184. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. (1993) 362:59–62. doi: 10.1038/364362c0
185. Ido A, Fukuyama H, Urushitani M. Protein misdirection inside and outside motor neurons in amyotrophic lateral sclerosis (ALS): a possible clue for therapeutic strategies. Int J Mol Sci. (2011) 12:6980–7003. doi: 10.3390/ijms12106980
186. Khare SD, Dokholyan NV. Common dynamical signatures of familial amyotrophic lateral sclerosis-associated structurally diverse Cu, Zn superoxide dismutase mutants. Proc Natl Acad Sci USA. (2006) 103:3147–52. doi: 10.1073/pnas.0511266103
187. Kim JM, Billington E, Reyes A, Notarianni T, Sage J, Agbas E, et al. Impaired Cu–Zn superoxide dismutase (SOD1) and calcineurin (Cn) interaction in ALS: a presumed consequence for TDP-43 and zinc aggregation in Tg SOD1 G93A rodent spinal cord tissue. Neurochem Res. (2019) 44:228–33. doi: 10.1007/s11064-017-2461-z
188. Perry J, Shin D, Getzoff E, Tainer J. The structural biochemistry of the superoxide dismutases. Biochim Biophys Acta. (2010) 1804:245–62. doi: 10.1016/j.bbapap.2009.11.004
189. Proctor EA, Fee L, Tao Y, Redler RL, Fay JM, Zhang Y, et al. Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. (2016) 113:614–9. doi: 10.1073/pnas.1516725113
190. Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. (1997) 69:1936–44. doi: 10.1046/j.1471-4159.1997.69051936.x
191. Hashimoto K, Hayashi Y, Watabe K, Inuzuka T, Hozumi I. Metallothionein-III prevents neuronal death and prolongs life span in amyotrophic lateral sclerosis model mice. Neuroscience. (2011) 189:293–8. doi: 10.1016/j.neuroscience.2011.05.034
192. Kaneko M, Noguchi T, Ikegami S, Sakurai T, Kakita A, Toyoshima Y, et al. Zinc transporters ZnT3 and ZnT6 are downregulated in the spinal cords of patients with sporadic amyotrophic lateral sclerosis. J Neurosci Res. (2015) 93:370–9. doi: 10.1002/jnr.23491
193. Nagano S, Satoh M, Sumi H, Fujimura H, Tohyama C, Yanagihara T, et al. Reduction of metallothioneins promotes the disease expression of familial amyotrophic lateral sclerosis mice in a dose-dependent manner. Eur J Neurosc. (2001) 13:1363–70. doi: 10.1046/j.0953-816x.2001.01512.x
194. Smith AP, Lee NM. Role of zinc in ALS. Amyotro Lateral Scler. (2007) 8:131–43. doi: 10.1080/17482960701249241
195. Takeuchi S, Fujiwara N, Ido A, Oono M, Takeuchi Y, Tateno M, et al. Induction of protective immunity by vaccination with wild-type apo superoxide dismutase 1 in mutant SOD1 transgenic mice. J Neuropathol Exp Neurol. (2010) 69:1044–56. doi: 10.1097/NEN.0b013e3181f4a90a
196. Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. (2006) 6:37–46. doi: 10.1007/s11910-996-0008-9
197. Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. (2004) 27:723–49. doi: 10.1146/annurev.neuro.27.070203.144244
198. Valentine JS, Doucette PA, Zittin Potter. S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. (2005) 74:563–93. doi: 10.1146/annurev.biochem.72.121801.161647
199. Urushitani M, Ezzi SA, Julien JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. (2007) 104:2495–500. doi: 10.1073/pnas.0606201104
200. Dong QX, Zhu J, Liu SY, Yu XL, Liu RT. An oligomer-specific antibody improved motor function and attenuated neuropathology in the SOD1-G93A transgenic mouse model of ALS. Int Immunopharmacol. (2018) 65:413–21. doi: 10.1016/j.intimp.2018.10.032
201. Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. (2014) 23:3579–95. doi: 10.1093/hmg/ddu068
202. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
203. Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
204. Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. (2015) 130:845–61. doi: 10.1007/s00401-015-1476-2
205. Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. (2013) 339:1335–8. doi: 10.1126/science.1232927
206. May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. (2014) 128:485–503. doi: 10.1007/s00401-014-1329-4
207. Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, et al. Aggregation-prone c9FTD/ALS poly (GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. (2014) 128:505–24. doi: 10.1007/s00401-014-1336-5
208. Zhou Q, Lehmer C, Michaelsen M, Mori K, Alterauge D, Baumjohann D, et al. Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins. EMBO Mol Med. (2017) 9:687–702. doi: 10.15252/emmm.201607054
209. Nguyen L, Montrasio F, Pattamatta A, Tusi SK, Bardhi O, Meyer KD, et al. Antibody therapy targeting RAN proteins rescues C9 ALS/FTD phenotypes in C9orf72 mouse model. Neuron. (2020) 105:645–62.e11. doi: 10.1016/j.neuron.2019.11.007
210. Zhou Q, Mareljic N, Michaelsen M, Parhizkar S, Heindl S, Nuscher B, et al. Active poly-GA vaccination prevents microglia activation and motor deficits in a C9orf72 mouse model. EMBO Mol Med. (2020) 12:e10919. doi: 10.15252/emmm.201910919
211. Miller TW, Shirley TL, Wolfgang WJ, Kang X, Messer A. DNA vaccination against mutant huntingtin ameliorates the HDR6/2 diabetic phenotype. Mol Ther. (2003) 7:572–9. doi: 10.1016/S1525-0016(03)00063-7
212. Ramsingh AI, Manley K, Rong Y, Reilly A, Messer A. Transcriptional dysregulation of inflammatory/immune pathways after active vaccination against Huntington′ s disease. Hum Mol Genet. (2015) 24:6186–97. doi: 10.1093/hmg/ddv335
213. Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G. Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease. J Clin Invest. (2003) 112:415–22. doi: 10.1172/JCI200318104
214. Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer's disease. Nat Rev Immunol. (2006) 6:404–16. doi: 10.1038/nri1843
215. Fu L, Li Y, Hu Y, Zheng Y, Yu B, Zhang H, et al. Norovirus P particle-based active Aβ immunotherapy elicits sufficient immunogenicity and improves cognitive capacity in a mouse model of Alzheimer's disease. Sci Rep. (2017) 7:41041. doi: 10.1038/srep41041
216. Yang P, Guo Y, Sun Y, Yu B, Zhang H, Wu J, et al. Active immunization with norovirus P particle-based amyloid-β chimeric protein vaccine induces high titers of anti-Aβ antibodies in mice. BMC Immunol. (2019) 20:9. doi: 10.1186/s12865-019-0289-9
217. Shinnick TM, Sutcliffe JG, Green N, Lerner RA. Synthetic peptide immunogens as vaccines. Ann Rev Microbiol. (1983) 37:425–46. doi: 10.1146/annurev.mi.37.100183.002233
218. Soto C. Constraining the loop, releasing prion infectivity. Proc Natl Acad Sci USA. (2009) 106:10–1. doi: 10.1073/pnas.0811625106
219. Thomas S, Hoxha K, Tran A, Prendergast GC. Bin1 antibody lowers the expression of phosphorylated tau in Alzheimer's disease. J Cell Biochem. (2019) 120:18320–31. doi: 10.1002/jcb.29142
220. Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen K, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. (2013) 18:1225–34. doi: 10.1038/mp.2013.1
221. Crotti A, Sait HR, McAvoy KM, Estrada K, Ergun A, Szak S, et al. BIN1 favors the spreading of tau via extracellular vesicles. Sci Rep. (2019) 9:9477. doi: 10.1038/s41598-019-45676-0
222. Lasorsa A, Malki I, Cantrelle FX, Merzougui H, Boll E, Lambert JC, et al. Structural basis of tau interaction with BIN1 and regulation by tau phosphorylation. Front Mol Neurosci. (2018) 11:421. doi: 10.3389/fnmol.2018.00421
223. Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. (2010) 303:1832–40. doi: 10.1001/jama.2010.574
224. Erny D, de Angelis ALH, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
225. Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Move Disord. (2015) 30:350–8. doi: 10.1002/mds.26069
226. Keene AC, Joiner WJ. Neurodegeneration: paying it off with sleep. Curr Biol. (2015) 25:R234–6. doi: 10.1016/j.cub.2015.02.003
227. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatics. Nature. (2015) 523:337. doi: 10.1038/nature14432
228. Dunckley T, Coon KD, Stephan DA. Discovery and development of biomarkers of neurological disease. Drug Discov Today. (2005) 10:326–34. doi: 10.1016/S1359-6446(04)03353-7
229. Dorsey ER, Johnston SC. The impact of clinical trials in neurology. In: Ravina B, Cummings J, McDermott M, and Poole RM, editors. Clinical Trials in Neurology: Design, Conduct, Analysis. Cambridge: Cambridge University Press (2012). p. 1–7. doi: 10.1017/CBO9781139032445.002
230. O'Neill G. Unique challenges in the development of therapies for neurological disorders. In: Ravina B, Cummings J, McDermott M, Poole R, editors. Clinical Trials in Neurology: Design, Conduct, Analysis. Cambridge: Cambridge University Press (2012). p. 19–27. doi: 10.1017/CBO9781139032445.004
Keywords: neurodegenerative diseases, neuropathology, neuroinflammation, anti-inflammatory agents, immunotherapy, active immunization, passive immunization, vaccination
Citation: Mortada I, Farah R, Nabha S, Ojcius DM, Fares Y, Almawi WY and Sadier NS (2021) Immunotherapies for Neurodegenerative Diseases. Front. Neurol. 12:654739. doi: 10.3389/fneur.2021.654739
Received: 17 January 2021; Accepted: 05 May 2021;
Published: 07 June 2021.
Edited by:
Jennifer S. Yokoyama, University of San Francisco, United StatesReviewed by:
Valerie Joers, University of Florida, United StatesAnthoula Charalampos Tsolaki, Aristotle University of Thessaloniki, Greece
Copyright © 2021 Mortada, Farah, Nabha, Ojcius, Fares, Almawi and Sadier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Najwane Said Sadier, am9hbm5lLnNhZGllckBhZHUuYWMuYWU=
†These authors have contributed equally to this work and share first authorship