 Zara A. Ioannides1,2,3*
Zara A. Ioannides1,2,3* Peter A. Csurhes1,3Nanette L. Douglas1,2,3Gem Mackenroth1,2,3Andrew Swayne1,4,5
Peter A. Csurhes1,3Nanette L. Douglas1,2,3Gem Mackenroth1,2,3Andrew Swayne1,4,5 Kate M. Thompson6,7Tracey J. Hopkins8Kerryn A. Green1,2Stefan Blum1,4,5Kaye D. Hooper1,2,3Kerstin H. Wyssusek1,9Alan Coulthard1,10
Kate M. Thompson6,7Tracey J. Hopkins8Kerryn A. Green1,2Stefan Blum1,4,5Kaye D. Hooper1,2,3Kerstin H. Wyssusek1,9Alan Coulthard1,10 Michael P. Pender1,2
Michael P. Pender1,2- 1Faculty of Medicine, The University of Queensland, Brisbane, QLD, Australia
- 2Department of Neurology, Royal Brisbane and Women's Hospital, Herston, QLD, Australia
- 3The University of Queensland Centre for Clinical Research, Herston, QLD, Australia
- 4Mater Centre for Neurosciences, Mater Hospital, Brisbane, QLD, Australia
- 5Neurology Department, Princess Alexandra Hospital, Woolloongabba, QLD, Australia
- 6Department of Psychology, Royal Brisbane and Women's Hospital, Herston, QLD, Australia
- 7School of Psychology, The University of Queensland, Brisbane, QLD, Australia
- 8Internal Medicine Day Treatment Unit, Royal Brisbane and Women's Hospital, Herston, QLD, Australia
- 9Department of Anesthesia and Perioperative Medicine, Royal Brisbane and Women's Hospital, Herston, QLD, Australia
- 10Department of Medical Imaging, Royal Brisbane and Women's Hospital, Herston, QLD, Australia
Background: Increasing evidence indicates a role for Epstein–Barr virus (EBV) in the pathogenesis of multiple sclerosis (MS). EBV-infected autoreactive B cells might accumulate in the central nervous system because of defective cytotoxic CD8+ T cell immunity. We have previously reported results of a phase I clinical trial of autologous EBV-specific T cell therapy in MS 6 months after treatment.
Objective: To investigate longer-term outcomes in MS patients who received autologous EBV-specific T cell therapy.
Methods: We assessed participants 2 and 3 years after completion of T cell therapy.
Results: We collected data from all 10 treated participants at year 2 and from 9 participants at year 3. No serious treatment-related adverse events were observed. Four participants had at least some sustained clinical improvement at year 2, including reduced fatigue in three participants, and reduced Expanded Disability Status Scale score in two participants. Three participants experienced a sustained improvement in at least some symptoms at year 3. More sustained improvement was associated with higher EBV-specific CD8+ T cell reactivity in the administered T cell product.
Conclusion: Autologous EBV-specific T cell therapy is well-tolerated, and some degree of clinical improvement can be sustained for up to 3 years after treatment.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system (CNS) characterized by progressive neurological disability. Although the primary cause of MS is unknown, genetic and environmental factors contribute to the risk of developing MS. A mounting body of evidence indicates that infection with Epstein–Barr virus (EBV) has an important role in the pathogenesis of MS (1, 2). In particular, the universality of EBV infection in people with MS (3) including those with early MS (4), and the presence of high titres of anti-EBV antibodies in the sera of patients prior to the onset of MS (5–8) and during the course of MS (9) indicate that infection with EBV has a critical role in the development of MS.
In our 2003 hypothesis, we postulated that defective elimination of EBV-infected B cells by cytotoxic CD8+ T cells might predispose to the development of MS by enabling the accumulation of EBV-infected autoreactive B cells in CNS (10). Our hypothesis is supported by observations of an accumulation of EBV-infected B cells and plasma cells in the brain in MS (11–14), although some studies have not found EBV infection in the brain (15, 16). A key prediction from the hypothesis is that EBV-specific T cell therapy should kill EBV-infected autoreactive B cells in the CNS, thereby halting disease progression and resulting in clinical improvement (10, 17).
AdE1-LMPpoly is an adenoviral vector encoding CD8+ T cell epitopes from EBV nuclear antigen 1 (EBNA1), latent membrane protein 1 (LMP1), and LMP2A which has been used to stimulate and expand EBV-specific CD8+ T cells in people with nasopharyngeal carcinoma, where the carcinoma cells express these EBV proteins (18, 19). Given that EBV-infected B cells in the brain in MS express the same three EBV proteins (11, 20), we postulated that T cells expanded with AdE1-LMPpoly might kill EBV-infected B cells in the CNS and thus halt disease progression and lead to clinical improvement in people with MS. In 2013, we first treated an MS patient with EBV-specific T cell therapy (21). The patient was a 42-year-old man with secondary progressive MS (SPMS); following autologous EBV-specific T cell therapy there was sustained clinical improvement and decreased cerebrospinal fluid (CSF) immunoglobulin G (IgG) production for 3.5 years after T cell therapy.
Following this, in 2015, we commenced an investigator-led open-label phase 1 clinical trial of autologous EBV-specific T cell therapy in patients with progressive MS (22). There were no treatment-related serious adverse effects and seven of the ten patients who received T cell therapy experienced clinical improvement. Of these, three patients showed an improvement in the Expanded Disability Status Scale (EDSS) score (23). Clinical improvement after T cell therapy was related to the level of EBV-specific CD8+ T cell reactivity in the T cell product. All six participants receiving a T cell product with strong EBV reactivity (>5% of CD8+ T cells reacting to EBV) showed clinical improvement whereas only one of the four participants receiving a T cell product with weak EBV reactivity (<5% of CD8+ T cell reacting to EBV) showed improvement (p = 0.033, Fisher's exact test) (22).
The objective of the current study was to investigate the longer-term clinical course of progressive MS in patients treated with autologous EBV-specific T cell therapy by performing comprehensive assessments at 2 and 3 years after treatment.
Materials and Methods
Study Approval
Our original phase I clinical trial of EBV-specific T cell therapy is registered with the Australian New Zealand Clinical Trials Registry (ACTRN12615000422527). The current 3-year follow-up study was approved by the Royal Brisbane and Women's Hospital Human Research Ethics Committee and The University of Queensland Medical Research Ethics Committee. All participants provided written informed consent.
Subjects
The study population consisted of the patients with progressive forms of MS, as defined by the 2010 Revised McDonald Criteria (24), who received autologous EBV-specific T cell therapy in our previously published phase I clinical trial (22). These were 5 patients with primary progressive MS (PPMS, 2 males, and 3 females) and 5 patients with SPMS (2 males and 3 females) who were between the ages of 42 and 61 at the time of receiving T cell therapy (Table 1). Patients were assessed at 2 years and at 3 years after completion of T cell therapy.
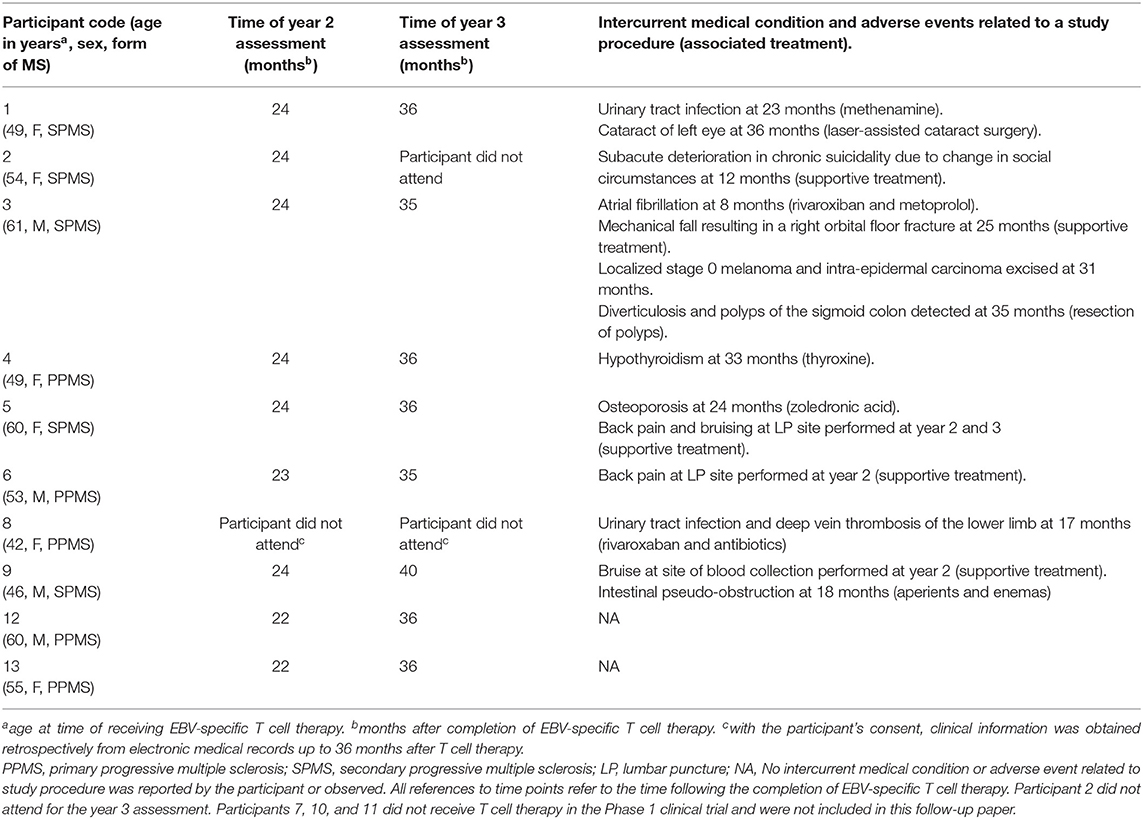
Table 1. Time of clinical assessments, intercurrent medical conditions, and adverse events during the observation period.
Clinical Assessments
At each assessment neurological symptoms were assessed in a face-to-face interview with a neurologist. At each visit we obtained a detailed medical history, including the temporal profile of any changes in neurological symptoms; performed a clinical examination, including a detailed neurological examination; and assessed the EDSS score (24). To assess fatigue, we administered the self-reported Fatigue Severity Scale (25). To assess quality of life (QOL) a single question from the MS QOL Instrument was used; this question asks “Overall, how would you rate your own quality of life?” on a scale from 0 (worst—as bad or worse than being dead) to 10 (best), as used by Cosio et al. (26).
To evaluate cognitive function, a clinical neuropsychologist administered a comprehensive neuropsychological test battery at 2 and 3 years following T cell therapy. The Minimal Assessment of Cognitive Function in Multiple Sclerosis (MACFIMS) formed the basis of the comprehensive cognitive assessment and is a validated battery for the assessment of cognitive function in patients with MS (27). The MACFIMS was supplemented with a measure of reaction time, the Computerized Test of Information Processing, and a screen of depressive symptoms, the Beck Depression Inventory (BDI)—Fast Screen for Medical Patients (28). To minimize the impact of practice effects due to repeated assessment, alternate forms of three measures (the Brief Visual Memory Test, the California Verbal Learning Test—Second Edition, and the Delis-Kaplan Executive Function System Sorting Test) were administered in an alternating fashion according to 10 computer-generated randomization schedules established prior to participant recruitment. Pairwise comparisons of change over time were conducted for each of the 15 tests used in the comprehensive neuropsychological test battery. Scores measured at 2 and 3 years were compared to baseline. Paired t-tests or Wilcoxon matched-pairs signed rank test were used according to data distribution as assessed by the Kolmogorov-Smirnov test. After application of statistical corrections for multiple comparisons a p-value threshold for significance was set at p < 0.0033.
As a mood disorder could affect cognition, in addition to administering the BDI, we also screened patients for depression by asking two questions used by Mohr et al. (29), namely, (a) “During the past 2 weeks, have you often been bothered by feeling down, depressed, or hopeless?” and (b) “During the past 2 weeks, have you often been bothered by little interest or pleasure in doing things.”
At years 2 and 3, brain and spinal cord MRI acquired at 3 Tesla before and after intravenous injection of gadolinium-containing contrast material (including 3-dimensional T1 and 3-dimensional T2 fluid-attenuated inversion recovery sequences for assessment of enhancing lesions and T2 lesions) were performed. Previously published MRI brain scan data from baseline and 6 months following T cell therapy was used to assess the presence of new T2 lesions compared to the prior MRI scans. MRI scans were also assessed for the number of gadolinium-enhancing lesions. Lumbar puncture for CSF analysis of intrathecal IgG production with matched serum analysis was performed at years 2 and 3.
Results
Subjects
Nine of the 10 patients who received EBV-specific T cell therapy in the original phase I trial consented to participate in the current follow-up study. Participant 8 was unable to participate due to social circumstances; however, she provided consent for us to access her Queensland Health electronic medical record for up to 3 years after she received T cell therapy and to include relevant data in study reports and/or publication. Participant 2 did not consent to the year 3 assessment. Lumbar puncture was contraindicated in participant 3 at years 2 and 3 because of anti-coagulation. Owing to social circumstances, participant 3 was unable to attend for the MRI scans and neuropsychological assessment at year 3. Participants 7, 10 and 11 from the original phase I clinical trial did not receive T cell therapy and were therefore not included in this follow-up study.
Intercurrent Medical Conditions, Adverse Events, and New MS Therapy
No treatment-related serious adverse events were observed during the study. Intercurrent medical conditions and adverse events related to a study procedure during the follow-up period are presented in Table 1. Participant 1 commenced ocrelizumab at 33 months after T cell therapy because of an MS relapse (Table 2). Participant 13 commenced ocrelizumab at 34 months after T cell therapy because of a deterioration in walking ability (Table 2).
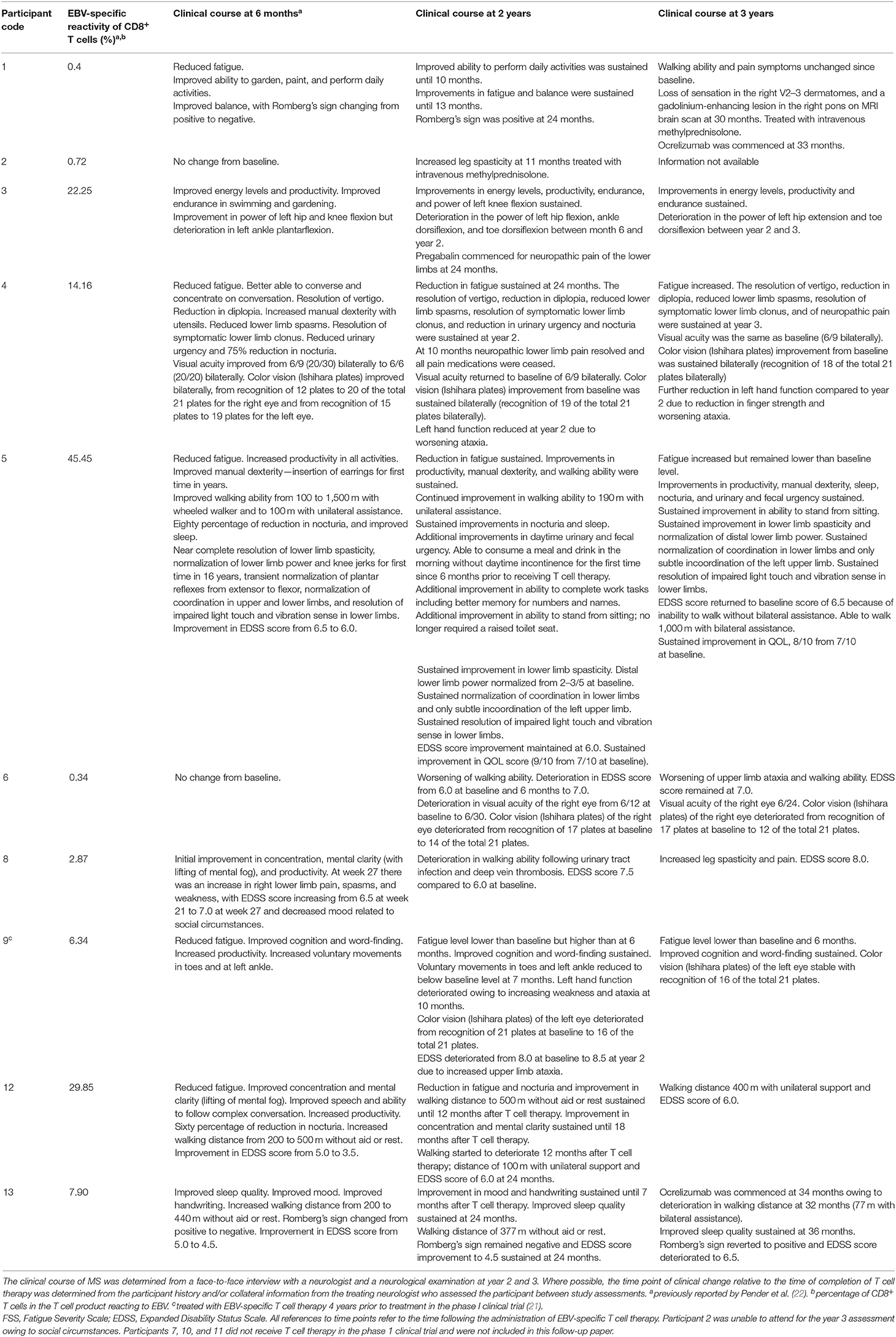
Table 2. Clinical course of MS following EBV-specific T cell therapy.
Clinical Outcomes Following T Cell Therapy
The year 2 assessment
Four of the 7 participants who experienced clinical improvement at the 6-month assessment (published previously) (22) were found to have some degree of sustained improvement at the 2-year assessment (participants 3, 4, 5, and 13) (Table 2; Figures 1A-F). Interestingly, the T cell therapy received by all of these 4 participants had strong EBV reactivity [>5% of CD8+ T cells reacting to EBV, (22) as presented in Table 2]. In participants 4, 5 and 13 there was a reduction in the Fatigue Severity Scale score at year 2 compared to baseline (Figure 1D). The improvement in QOL score that had been observed at 6 months was sustained at year 2 in participants 3, 4, and 5, but not in participant 13 (Figure 1F). In participant 3, despite the sustained improvements in energy levels, productivity, endurance and power of left knee flexion, lower limb neuropathic pain developed and there was deterioration in the power of other left lower limb movements at year 2. In participant 4, despite the sustained improvement in many symptoms, there was deterioration in left hand function due to worsening ataxia at year 2. In participants 5 and 13 the clinical improvements were accompanied by a sustained improvement in EDSS score at year 2 compared to baseline (Table 2 and Figure 1B). Indeed, participant 5 showed further improvements in memory, ability to complete work tasks, daytime urinary and fecal urgency, and walking ability at year 2, compared to month 6 after T cell therapy. The T cell therapy received by participants 5 and 13 not only had strong EBV reactivity but also reactivity to all 3 EBV proteins, namely EBNA1, LMP1, and LMP2A (22). A deterioration in MS symptoms and/or neurological examination findings between the 6-month and the 2-year assessments was observed in 6 participants (participants 1, 2, 6, 8, 9, and 12), of whom 4 (participants 1, 2, 6, and 8) received T cell therapy with weak EBV reactivity (<5% of CD8+ T cells reacting to EBV) (Table 2). The duration of clinical improvement in participants 1, 9, and 12, who experienced clinical improvement at the 6-month assessment that was not sustained at the 2-year assessment, was 13 months in participant 1, 10 months in participant 9, and 12 months in participant 12 (Table 2). The EDSS in participant 9 deteriorated from 8.0 at baseline and month 6 to 8.5 at year 2 owing to worsening of upper limb ataxia; however, the improvements in cognition and fatigue which were noted by the participant at month 6 were sustained at year 2 and at year 3. In this participant the duration of sustained clinical improvement (10 months) after administration of the T cell product in this clinical trial (6.34% of CD8+ T cells reacted to EBV) was substantially less than after the participant's first course of EBV-specific T cell therapy in 2013 (3.5 years as reported in 2018) (22) after the patient received a T cell product with considerably higher EBV reactivity (38.46% of CD8+ T cells reacted to EBV) (21). Although participant 12 had received T cell therapy with very strong EBV reactivity (29.85% of CD8+ T cells reacted to EBV), the reactivity of the CD8+ T cells was directed at only EBNA1 (22).
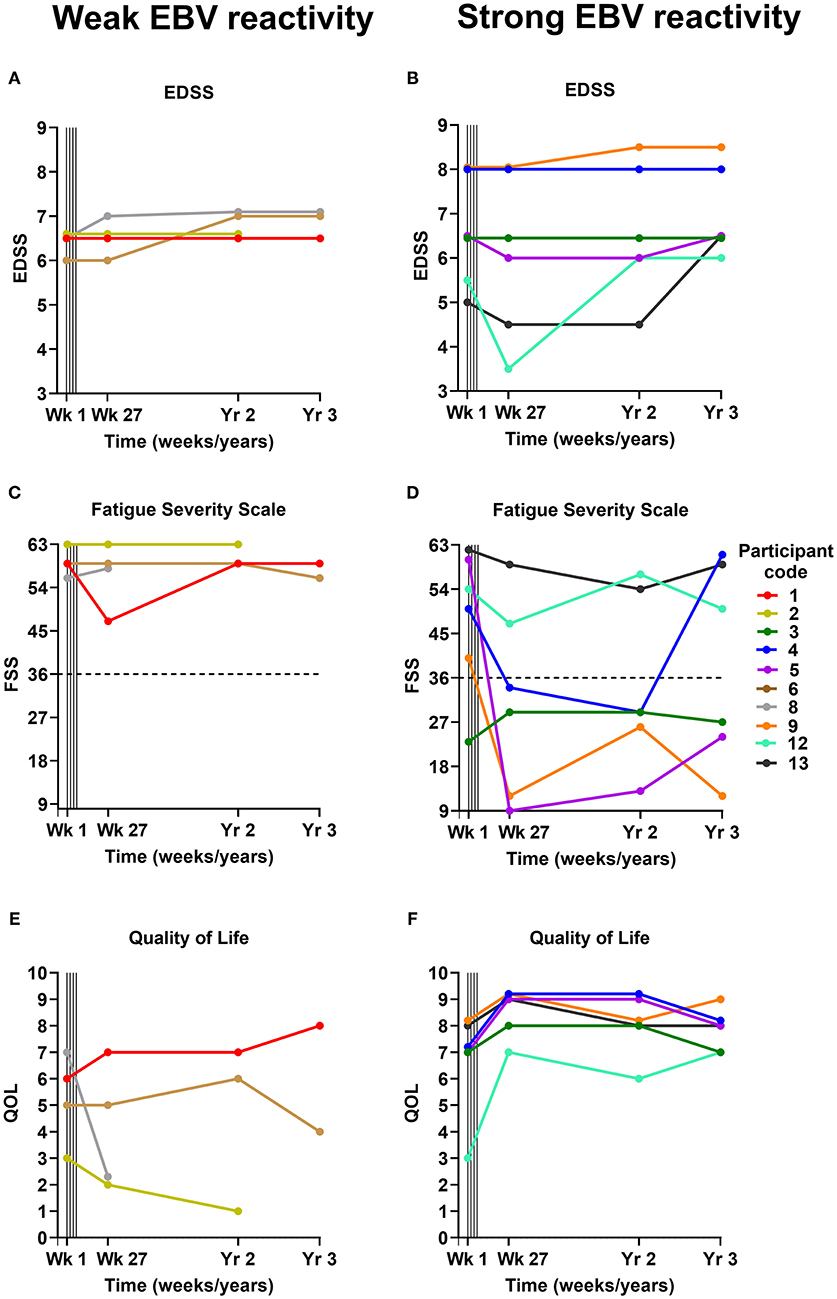
Figure 1. EDSS (A,B), Fatigue Severity Scale (C,D), and Quality of Life (E,F) scores before and after EBV-specific T cell therapy. The participants are grouped according to whether they received a T cell product with weak EBV reactivity (<5% of CD8+ T cells reacting to EBV) (A,C,E) or strong EBV reactivity (>5% of CD8+ T cells reacting to EBV) (B,D,F). For the Fatigue Severity Scale (C,D) a total score of 36 (indicated by dotted horizontal lines) or more suggests that a person is suffering from fatigue; the maximum score is 63 and the minimum score is 9. Vertical lines indicate successive T cell infusions of 5 × 106, 1 × 107, 1.5 × 107, and 2 × 107 cells at 2-week intervals from week 1 to 7. EDSS, Expanded Disability Status Scale; FSS, Fatigue Severity Scale; QOL, Quality of Life; EBV, Epstein–Barr virus; Wk, week; Yr, year.
The year 3 assessment
Participant 2 did not consent to the year 3 assessment. Three of the 6 participants who received T cell therapy with strong EBV reactivity (participants 3, 4, and 5) experienced an improvement in one or more of their MS symptoms at the year 3 assessment compared to baseline (Table 2). Energy levels and productivity were improved in participant 3, although there was an increase in left lower limb weakness. In participant 4, improvements in diplopia and lower limb spams, and resolution of longstanding vertigo, symptomatic lower limb clonus and neuropathic pain, which occurred at 6 months, were sustained at 3 years. Remarkably, all pain medications (paracetamol and amitriptyline) were ceased in this participant at 10 months as they were no longer required, and she remained pain-free at year 3 with an increase in QOL score (Figure 1F) and color vision at 3 years compared to baseline. However, her Fatigue Severity Scale score, which was reduced at year 2, increased to above the baseline level (Figure 1D) and there was further deterioration in left hand function due to weakness and ataxia. In participant 5 there were sustained improvements in productivity, manual dexterity, sleep quality, nocturia, urinary and fecal urgency, lower limb spasticity, distal lower limb power, coordination in the lower limbs, and lower limb sensation at year 3, with an associated reduction in the Fatigue Severity Scale score (Figure 1D) and an increase in QOL score (Figure 1F) compared to baseline. Despite these sustained improvements her walking deteriorated between year 2 and year 3 so that her EDSS score increased from 6.0 at year 2 to 6.5 (the baseline score) at year 3 (Figure 1B), although she was still able to walk 1,000 m with bilateral assistance, compared to 100 m at baseline. In participant 13 the EDSS score, which had been decreased at year 2 compared to baseline, increased from 4.5 to 6.5 at year 3 which was higher than her baseline score of 5.0 (Figure 1B). Of all the participants in the study, participant 5 received T cells with the highest EBV reactivity [45.45% of CD8+ T cells reacted to EBV (22)] and had the greatest degree of sustained clinical improvement at year 3.
At the year 3 assessment, 4 participants had the same EDSS score as at baseline (participants 1, 3, 4, and 5). All of these participants had shown clinical improvement at 6 months. Participant 1 had received a T cell product with weak EBV reactivity, and participants 3, 4, and 5 had received a T cell product with strong EBV reactivity. Five participants (participants 6, 8, 9, 12, and 13) had an increased EDSS score at year 3 compared to baseline (Figures 1A,B). Participants 6 and 8 had received a weak T cell product and had not improved at 6 months, whereas participants 9, 12, and 13 had received a strong T cell product and had shown clinical improvement at 6 months.
Neuropsychological Assessment
Neuropsychological data are presented in Table 3. After correction for multiple comparisons there were no significant differences between the year 2 or 3 assessments and the baseline assessments for any of the 15 neuropsychological tests. The increase in verbal generativity that was observed on the Controlled Oral Word Association Test (COWAT) 6 months after T cell therapy (22) was maintained at both the year 2 and 3 assessments, whereas the increased attainment in the Paced Auditory Serial Addition Test (PASAT3), a measure of working memory/processing speed, observed at the 6-month assessment, (22) trended back toward baseline attainment at years 2 and 3. Depressive symptomatology was stable across the 3 year follow-up period, although participant 2 who experienced decreased mood at 6 months and 2 years after T cell therapy did not consent to the year 3 assessment, and participant 8 who also experienced decreased mood 6 months after T cell therapy did not consent to either the year 2 or the year 3 assessment.
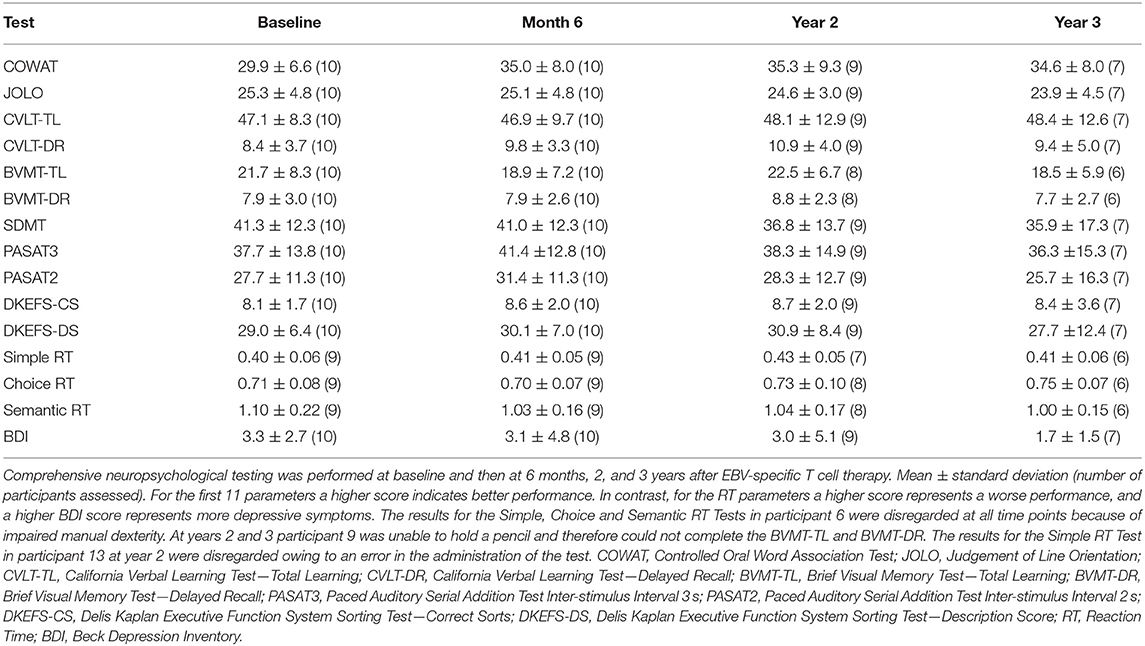
Table 3. Comprehensive neuropsychological assessment data.
MRI Brain and Spinal Cord Scans
The MRI brain scan findings are presented in Table 4. Five participants (participants 1, 4, 6, 8, and 9) had one gadolinium-enhancing MRI brain lesion at baseline. In participant 6, the enhancing lesion did not change throughout the observation period and was thought to be unrelated to MS. Compared with the 6-month scan, the number of enhancing lesions at year 2 increased in 3 participants (participants 1, 2, and 9) and decreased in participant 4. Participants 1 and 2 experienced a clinical decline between the 6-month and 2-year assessments whereas participants 4 and 9 experienced some degree of sustained clinical improvement; therefore the change in number of enhancing brain lesions did not appear to be related to the clinical course. Participants 1, 2, 4, 6, 9, and 13 had 1–3 new lesions at year 2, compared to month 6, whereas participants 3, 5, and 12 had no new lesions. Participants 4 and 9 had one enhancing brain lesion at year 3. Participants 1, 4, 5, 9, and 13 had one new lesion at year 3 compared to year 2, whereas participants 6 and 12 had no new lesions at year 3.
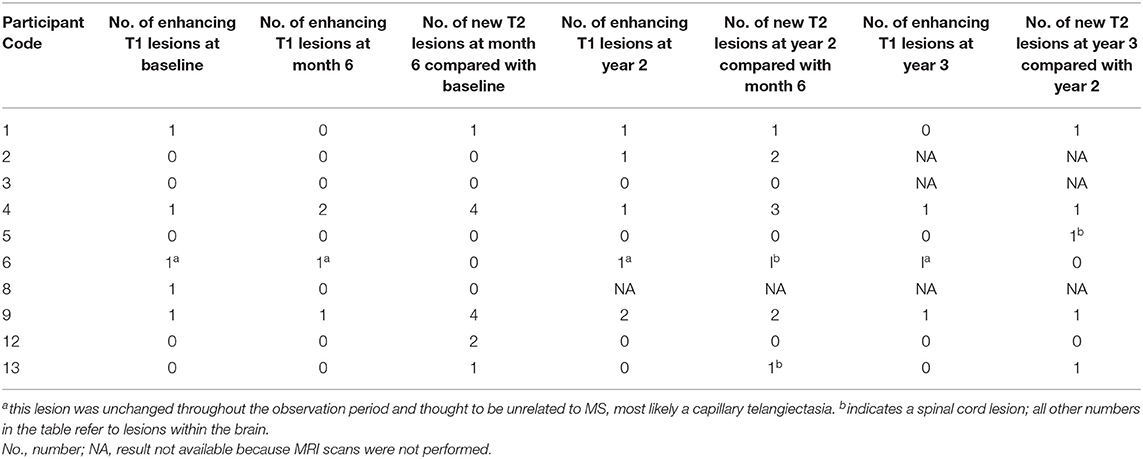
Table 4. Brain MRI findings after T cell therapy.
Intrathecal IgG Synthesis
CSF IgG production and leukocyte count were assessed in 8 participants at year 2, and 6 participants at year 3 (Table 5). Of the four participants with decreased intrathecal IgG synthesis 6 months after T cell therapy (participants 1, 6, 9, and 13), two of these (participants 6 and 13) still had decreased intrathecal IgG production at year 3 compared to baseline. In participant 13, ocrelizumab, which was commenced 2 months before the year 3 lumbar puncture, might have contributed to the decreased intrathecal IgG synthesis at year 3, but not at year 2 (Table 5). In participants 1 and 9, who had shown decreased intrathecal IgG synthesis 6 months after T cell therapy, the level of intrathecal IgG production had increased to above the baseline level by year 2. In participant 4, intrathecal IgG synthesis was increased at year 2, but decreased at year 3, compared to baseline.
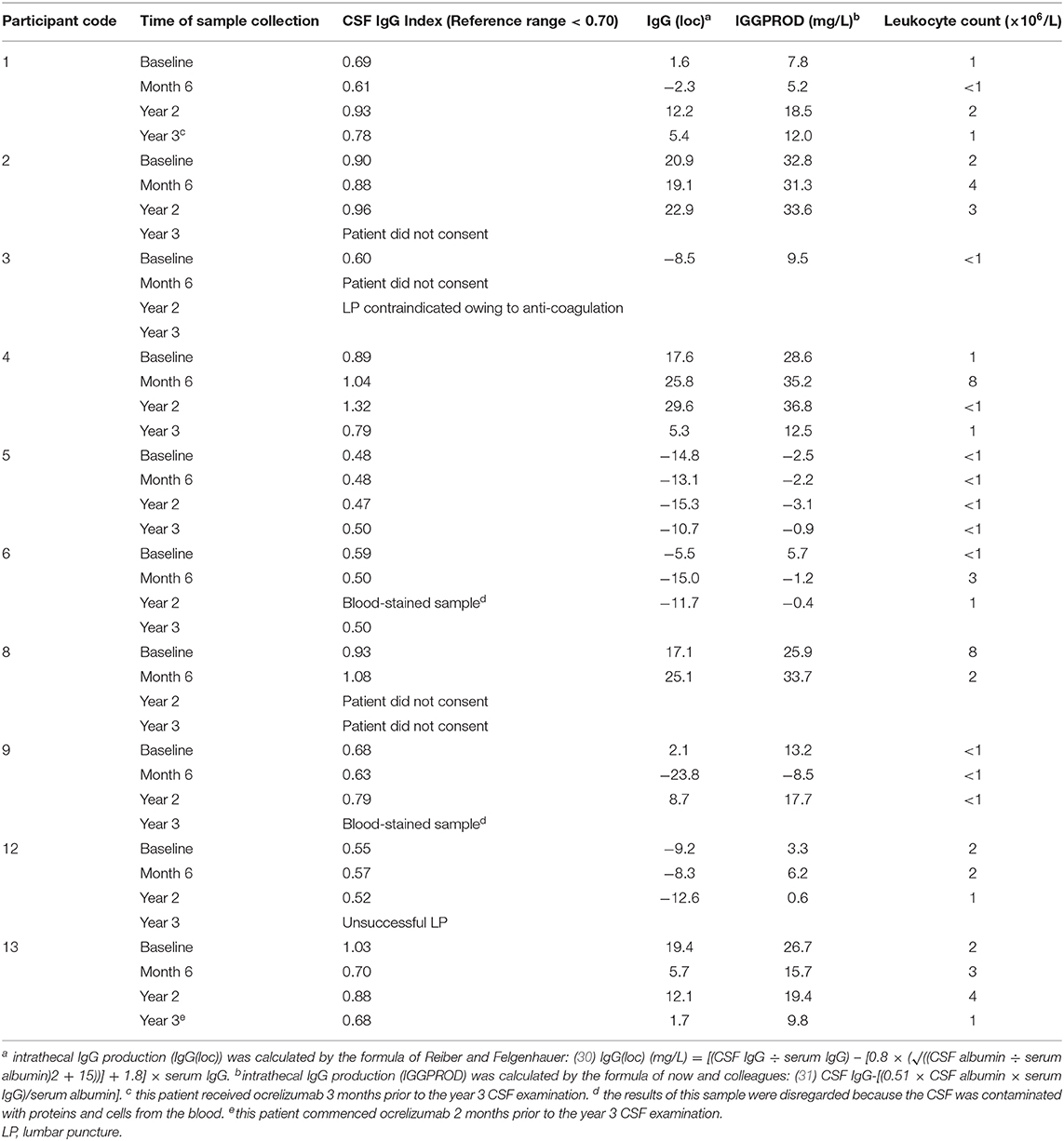
Table 5. CSF IgG analysis and leukocyte count after T cell therapy.
Discussion
In the current study we followed up ten patients with progressive MS for 2 years after autologous EBV-specific T cell therapy and nine of these patients for 3 years after T cell therapy. There were no serious treatment-related adverse events. Of the seven patients who had shown clinical improvement 6 months after T cell therapy (22), four still had at least some degree of sustained clinical improvement at year 2, with two of these still having a reduced EDSS score compared to baseline. At year 3, three of the participants who had clinical improvement at month 6 still showed some evidence of sustained clinical improvement, although the EDSS score that had still been reduced at year 2 had increased to baseline in one patient and above baseline in the other. We also observed a reduction in the Fatigue Severity Scale score compared to baseline in three of the participants with sustained clinical improvement at year 2, as we did in the participants with clinical improvement 6 months after T cell therapy (22). These clinical improvements lasting for up to 3 years after EBV-specific T cell therapy are contrary to the expected decline of patients with progressive forms of MS (32).
We have previously reported that clinical improvement after EBV-specific T cell therapy is associated with a higher level of EBV-specific CD8+ T cell reactivity in the administered T cell product (22). In the present study all four of the participants with some degree of sustained clinical improvement at year 2 and all three of the participants with some sustained improvement at year 3 had received T cells with strong EBV reactivity. Remarkably, participant 5 who had received T cells with the strongest EBV reactivity and who had shown pronounced neurological improvement and reduction in fatigue at 6 months had the greatest degree of sustained improvement in neurological function and fatigue 3 years after T cell therapy. These findings suggest that EBV-specific T cell therapy has a beneficial effect in progressive MS.
Previously we have postulated that clinical improvement following EBV-specific T cell therapy results from the killing of EBV-infected B cells in the CNS by the adoptively transferred CD8+ T cells and that this prevents further autoimmune attack on the CNS and allows neurological recovery through remyelination, dendritic and axonal sprouting, and synaptic remodeling (22). Similarly, this might also account for the sustained clinical improvements we observed in four participants at year 2 and three participants at year 3 after T cell therapy. An important goal of future studies should be to determine whether EBV-specific T cell therapy decreases the number of EBV-infected B cells in the CNS and whether this correlates with clinical improvement in MS. However, at present there is no safe and reliable way of quantifying EBV-infected B cells and plasma cells in the CNS.
A key question is why the clinical improvement in responders waned over time. With increasing duration of MS there is a progressive decrease in the frequency of EBV-specific T cells in the CD8+ population, indicating T cell exhaustion (33, 34). We suggest that CD8+ T cell exhaustion induced by a persistently high EBV latent antigen load in MS might gradually decrease the cytotoxic effect of the adoptively transferred CD8+ T cells on EBV-infected B cells in the CNS and thereby allow further autoimmune attack on the CNS. Repeated administration of EBV-specific T cell therapy every 12 months should help sustain clinical improvement in MS.
Limitations of our study include the small cohort of participants and the relatively high level of disability prior to treatment. These factors could impact upon the ability to detect clinical or radiological change over the follow-up period. The fact that some participants displayed clinical improvement is remarkable but should be interpreted with some caution. For example, participant 9 had an improved Fatigue Severity Scale score 2 and 3 years after T cell therapy when the EDSS score had increased. In this case, the reduction in reported fatigue might have been due to reduced energy expenditure as a result of decreased physical activity. Future studies incorporating activity monitoring devices could provide more insight.
This study adds to the accumulating evidence for a pathogenic role of EBV infection in MS. Our data suggests that T cell therapy targeting EBV-infected B cells in MS patients has a favorable safety profile and the potential for long-term efficacy. A phase I clinical trial of allogeneic EBV-specific T cell therapy is currently in progress (35).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
This follow-up study was approved by the Royal Brisbane and Women's Hospital Human Research Ethics Committee and The University of Queensland Medical Research Ethics Committee. All participants provided written informed consent. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MP conceived and designed the research. ZI, PC, ND, GM, AS, KT, TH, KH, KW, AC, and MP collected and/or analyzed the data. ZI wrote the manuscript with critical input from all authors. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by MS Queensland.
Conflict of Interest
ZI: Received clinical trial support from Atara Biotherapeutics. MP: Received research grants from Atara Biotherapeutics and MS Queensland, and consultancy fees, clinical trial support, and travel support from Atara Biotherapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the patients who participated in this study. The authors are also grateful to the staff members of the following departments who assisted with the collection of data for the study: Internal Medicine Day Treatment Unit, Royal Brisbane and Women's Hospital; Level 3, The University of Queensland Centre for Clinical Research; the Herston Imaging Research Facility; and Pathology Queensland at the Royal Brisbane and Women's Hospital.
References
1. Ascherio A, Munger KL, Lünemann JD. The initiation and prevention of multiple sclerosis. Nat Rev Neurol. (2012) 8:602–12. doi: 10.1038/nrneurol.2012.198
2. Pender MP, Burrows SR. Epstein-Barr virus and multiple sclerosis: potential opportunities for immunotherapy. Clin Transl Immunology. (2014) 3:e27. doi: 10.1038/cti.2014.25
3. Pakpoor J, Disanto G, Gerber JE, Dobson R, Meier UC, Giovannoni G, et al. The risk of developing multiple sclerosis in individuals seronegative for Epstein-Barr virus: a meta-analysis. Mult Scler. (2013) 19:162–6. doi: 10.1177/1352458512449682
4. Abrahamyan S, Eberspächer B, Hoshi MM, Aly L, Luessi F, Groppa S, et al. Complete Epstein-Barr virus seropositivity in a large cohort of patients with early multiple sclerosis. J Neurol Neurosurg Psychiatry. (2020) 91:681–6. doi: 10.1136/jnnp-2020-322941
5. Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernan MA, Olek MJ, et al. Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospective study. JAMA. (2001) 286:3083–8. doi: 10.1001/jama.286.24.3083
6. Sundström P, Juto P, Wadell G, Hallmans G, Svenningsson A, Nystrom L, et al. An altered immune response to Epstein-Barr virus in multiple sclerosis: a prospective study. Neurology. (2004) 62:2277–82. doi: 10.1212/01.WNL.0000130496.51156.D7
7. Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Temporal relationship between elevation of epstein-barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA. (2005) 293:2496–500. doi: 10.1001/jama.293.20.2496
8. Levin LI, Munger KL, O'Reilly EJ, Falk KI, Ascherio A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol. (2010) 67:824–30. doi: 10.1002/ana.21978
9. Sumaya CV, Myers LW, Ellison GW, Ench Y. Increased prevalence and titer of Epstein-Barr virus antibodies in patients with multiple sclerosis. Ann Neurol. (1985) 17:371–7. doi: 10.1002/ana.410170412
10. Pender MP. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol. (2003) 24:584–8. doi: 10.1016/j.it.2003.09.005
11. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. (2007) 204:2899–912. doi: 10.1084/jem.20071030
12. Tzartos JS, Khan G, Vossenkamper A, Cruz-Sadaba M, Lonardi S, Sefia E, et al. Association of innate immune activation with latent Epstein-Barr virus in active MS lesions. Neurology. (2012) 78:15–23. doi: 10.1212/WNL.0b013e31823ed057
13. Hassani A, Corboy JR, Al-Salam S, Khan G. Epstein-Barr virus is present in the brain of most cases of multiple sclerosis and may engage more than just B cells. PLoS ONE. (2018) 13:e0192109. doi: 10.1371/journal.pone.0192109
14. Moreno MA, Or-Geva N, Aftab BT, Khanna R, Croze E, Steinman L. Molecular signature of Epstein-Barr virus infection in MS brain lesions. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e466. doi: 10.1212/NXI.0000000000000466
15. Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, et al. Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain. (2009) 132:3318–28. doi: 10.1093/brain/awp200
16. Peferoen LA, Lamers F, Lodder LN, Gerritsen WH, Huitinga I, Melief J, et al. Epstein Barr virus is not a characteristic feature in the central nervous system in established multiple sclerosis. Brain. (2010) 133:e137. doi: 10.1093/brain/awp296
17. Pender MP. The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis. Neuroscientist. (2011) 17:351–67. doi: 10.1177/1073858410381531
18. Smith C, Cooper L, Burgess M, Rist M, Webb N, Lambley E, et al. Functional reversion of antigen-specific CD8+ T cells from patients with Hodgkin lymphoma following in vitro stimulation with recombinant polyepitope. J Immunol. (2006) 177:4897–906. doi: 10.4049/jimmunol.177.7.4897
19. Smith C, Tsang J, Beagley L, Chua D, Lee V, Li V, et al. Effective treatment of metastatic forms of Epstein-Barr virus-associated nasopharyngeal carcinoma with a novel adenovirus-based adoptive immunotherapy. Cancer Res. (2012) 72:1116–25. doi: 10.1158/0008-5472.CAN-11-3399
20. Serafini B, Severa M, Columba-Cabezas S, Rosicarelli B, Veroni C, Chiappetta G, et al. Epstein-Barr virus latent infection and BAFF expression in B cells in the multiple sclerosis brain: implications for viral persistence and intrathecal B-cell activation. J Neuropathol Exp Neurol. (2010) 69:677–93. doi: 10.1097/NEN.0b013e3181e332ec
21. Pender MP, Csurhes PA, Smith C, Beagley L, Hooper KD, Raj M, et al. Epstein-Barr virus-specific adoptive immunotherapy for progressive multiple sclerosis. Mult Scler. (2014) 20:1541–4. doi: 10.1177/1352458514521888
22. Pender MP, Csurhes PA, Smith C, Douglas NL, Neller MA, Matthews KK, et al. Epstein-Barr virus-specific T cell therapy for progressive multiple sclerosis. JCI Insight. (2018) 3:e124714. doi: 10.1172/jci.insight.124714
23. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. (1983) 33:1444–52. doi: 10.1212/WNL.33.11.1444
24. Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. (2011) 69:292–302. doi: 10.1002/ana.22366
25. Krupp LB, LaRocca NG, Muir-Nash J, Steinberg AD. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol. (1989) 46:1121–3. doi: 10.1001/archneur.1989.00520460115022
26. Cosio D, Jin L, Siddique J, Mohr DC. The effect of telephone-administered cognitive-behavioral therapy on quality of life among patients with multiple sclerosis. Ann Behav Med. (2011) 41:227–34. doi: 10.1007/s12160-010-9236-y
27. Benedict RH, Cookfair D, Gavett R, Gunther M, Munschauer F, Garg N, et al. Validity of the minimal assessment of cognitive function in multiple sclerosis (MACFIMS). J Int Neuropsychol Soc. (2006) 12:549–58. doi: 10.1017/S1355617706060723
28. Beck A, Steer R, Brown G. BDI-Fast Screen for Medical Patients: Manual. Pearson Clinical. Available online at: https://www.pearsonclinical.com/psychology/products/100000173/bdi-fastscreen-for-medical-patients-bdi.html (accessed November 10, 2020).
29. Mohr DC, Hart SL, Julian L, Tasch ES. Screening for depression among patients with multiple sclerosis: two questions may be enough. Mult Scler. (2007) 13:215–9. doi: 10.1177/1352458506070926
30. Reiber H, Felgenhauer K. Protein transfer at the blood cerebrospinal fluid barrier and the quantitation of the humoral immune response within the central nervous system. Clin Chim Acta. (1987) 163:319–28. doi: 10.1016/0009-8981(87)90250-6
31. Blennow K, Fredman P, Wallin A, Gottfries CG, Karlsson I, Langstrom G, et al. Protein analysis in cerebrospinal fluid. II. Reference values derived from healthy individuals 18-88 years of age. Eur Neurol. (1993) 33:129–33. doi: 10.1159/000116919
32. Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. N Engl J Med. (2000) 343:1430–8. doi: 10.1056/NEJM200011163432001
33. Jilek S, Schluep M, Meylan P, Vingerhoets F, Guignard L, Monney A, et al. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain. (2008) 131:1712–21. doi: 10.1093/brain/awn108
34. Pender MP, Csurhes PA, Burrows JM, Burrows SR. Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin Transl Immunology. (2017) 6:e126. doi: 10.1038/cti.2016.87
35. United States National Library of Medicine. Phase 1 Study to Evaluate the Safety and Efficacy of ATA188 in Subjects With Progressive Multiple Sclerosis. Available online at: https://clinicaltrials.gov/ct2/show/NCT03283826 (accessed February 16, 2021).
Keywords: B cell, CD8+ T cell, disease-modifying therapy, Epstein-Barr virus, progressive multiple sclerosis, T cell therapy
Citation: Ioannides ZA, Csurhes PA, Douglas NL, Mackenroth G, Swayne A, Thompson KM, Hopkins TJ, Green KA, Blum S, Hooper KD, Wyssusek KH, Coulthard A and Pender MP (2021) Sustained Clinical Improvement in a Subset of Patients With Progressive Multiple Sclerosis Treated With Epstein–Barr Virus-Specific T Cell Therapy. Front. Neurol. 12:652811. doi: 10.3389/fneur.2021.652811
Received: 13 January 2021; Accepted: 22 February 2021;
Published: 15 March 2021.
Edited by:
Bert A. 'T. Hart, University Medical Center Groningen, NetherlandsReviewed by:
Cris S. Constantinescu, University of Nottingham, United KingdomRachele Bigi, Sapienza University of Rome, Italy
Copyright © 2021 Ioannides, Csurhes, Douglas, Mackenroth, Swayne, Thompson, Hopkins, Green, Blum, Hooper, Wyssusek, Coulthard and Pender. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zara A. Ioannides, ei5pb2FubmlkZXNAdXEuZWR1LmF1