
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurol., 07 July 2021
Sec. Applied Neuroimaging
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.645625
This article is part of the Research TopicApplied Neuroimaging - Case Report Collection 2021View all 5 articles
 Masaki Ikeda1,2,3*
Masaki Ikeda1,2,3* Koichi Okamoto2
Koichi Okamoto2 Keiji Suzuki4
Keiji Suzuki4 Eriko Takai3Hiroo Kasahara3Natsumi Furuta3Minori Furuta3Yuichi Tashiro5Chisato Shimizu2Shin Takatama6Isao Naito6Mie Sato7Yasujiro Sakai2Manabu Takahashi8
Eriko Takai3Hiroo Kasahara3Natsumi Furuta3Minori Furuta3Yuichi Tashiro5Chisato Shimizu2Shin Takatama6Isao Naito6Mie Sato7Yasujiro Sakai2Manabu Takahashi8 Masakuni Amari2Masamitsu Takatama2
Masakuni Amari2Masamitsu Takatama2 Tetsuya Higuchi9
Tetsuya Higuchi9 Yoshito Tsushima9Hideaki Yokoo10Masahiko Kurabayashi11Shun Ishibashi10
Yoshito Tsushima9Hideaki Yokoo10Masahiko Kurabayashi11Shun Ishibashi10 Kenji Ishii12
Kenji Ishii12 Yoshio Ikeda3
Yoshio Ikeda3In Alzheimer's disease, the apolipoprotein E gene (APOE) ε2 allele is a protective genetic factor, whereas the APOE ε4 allele is a genetic risk factor. However, both the APOE ε2 and the APOE ε4 alleles are genetic risk factors for lobar intracerebral hemorrhage. The reasons for the high prevalence of lobar intracerebral hemorrhage and the low prevalence of Alzheimer's disease with the APOE ε2 allele remains unknown. Here, we describe the case of a 79-year-old Japanese female with Alzheimer's disease, homozygous for the APOE ε2 allele. This patient presented with recurrent lobar hemorrhages and multiple cortical superficial siderosis. The findings on the 11C-labeled Pittsburgh Compound B-positron emission tomography (PET) were characteristic of Alzheimer's disease. 18F-THK5351 PET revealed that the accumulation of 18F-THK 5351 in the right pyramidal tract at the pontine level, the cerebral peduncle of the midbrain, and the internal capsule, reflecting the lesions of the previous lobar intracerebral hemorrhage in the right frontal lobe. Moreover, 18F-THK5351 accumulated in the bilateral globus pallidum, amygdala, caudate nuclei, and the substantia nigra of the midbrain, which were probably off-target reaction, by binding to monoamine oxidase B (MAO-B). 18F-THK5351 were also detected in the periphery of prior lobar hemorrhages and a cortical subarachnoid hemorrhage, as well as in some, but not all, areas affected by cortical siderosis. Besides, 18F-THK5351 retentions were observed in the bilateral medial temporal cortices and several cortical areas without cerebral amyloid angiopathy or prior hemorrhages, possibly where tau might accumulate. This is the first report of a patient with Alzheimer's disease, carrying homozygous APOE ε2 allele and presenting with recurrent lobar hemorrhages, multiple cortical superficial siderosis, and immunohistochemically vascular amyloid β. The 18F-THK5351 PET findings suggested MAO-B concentrated regions, astroglial activation, Waller degeneration of the pyramidal tract, neuroinflammation due to CAA related hemorrhages, and possible tau accumulation.
The apolipoprotein E gene (APOE) ε2 allele has an exceptionally low likelihood of causing Alzheimer's dementia in APOE2 homozygotes (1) and, it protects against Alzheimer's disease (AD) (2). In contrast, the APOE ε4 allele is a genetic risk factor of AD (3). However, the APOE ε2 and the APOE ε4 alleles, responsible for causing severe alterations in the vascular wall structure causing cerebral amyloid angiopathy (CAA), are common genetic risk factors for lobar intracerebral hemorrhage (ICH) (4–7). The reason for the high prevalence of lobar ICH despite the very low prevalence of AD in individuals with the APOE ε2 allele remains unknown. 18F-THK5351, which was developed as a tau binding tracer (8, 9), has been shown to strongly bind to monoamine oxidase B (MAO-B) (10). Recently, in a patient of cerebral infarction, the off-target retentions of 18F-THK5351 revealed Wallerian degeneration of the pyramidal tract, basal ganglia, thalamus, and cerebral cortices (11). In this report, we describe a patient with AD who carried a homozygous APOE ε2 allele. This patient presented with recurrent lobar hemorrhages and multiple cortical superficial siderosis (cSS) with hypobetalipoproteinemia. Additionally, several 18F-THK5351 PET findings reflected an increase in the MAO-B, reactive astrogliosis, and Wallerian degeneration of the pyramidal tract due to lobar ICH, as well as the possible accumulation of tau, related to AD pathology, in cerebral cortical areas.
11C-labeled Pittsburgh Compound B positron emission tomography (11C-PiB PET) was performed at the Gunma University Hospital as described previously by the authors (12, 13). 18F-THK5351 PET examinations were performed at the Tokyo Metropolitan Institute of Gerontology using the Discovery PET/CT 710 scanner (GE Healthcare, Milwaukee, WI, USA). Emission data were acquired for 20 min, starting 40 min after intravenous injection of 18F-THK5351 at a dose of approximately 190 MBq (5.1 mCi). In total, 47 slice images, with a voxel size of 2 × 2 × 3.27 mm and a matrix size of 128 × 128, were obtained. The data, collected in three-dimensional mode, were reconstructed after correcting for decay, attenuation, and scatter. 18F-THK5351 PET images were then normalized using the cerebellum as a reference region; cerebellar uptake was set to the value of one, as in the previously reported method (11, 14).
A Cerebrospinal fluid was obtained by a lumbar puncture of the L3/L4 or L4/L5 intervertebral space, and the CSF samples were centrifuged for 10 min at 1,800 × g at 4°C within 3 h of collection. Samples were divided into aliquots of 0.5 mL and stored at −80°C in polypropylene tubes until they were analyzed using enzyme-linked immunosorbent assay (ELISA) kits for human CSF amyloid β (Aβ) Aβ1-42 and Aβ1-40 (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) (12). Phosphorylated tau 181 (P-Tau) and human total tau (T-Tau) levels in the CSF were measured using the sandwich using ELISA INNOTESTⓇ PHOSPHO-TAU (181P) (15) and the sandwich ELISA INNOTESTⓇ T-Tau-Ag (both FUJIREBIO, Ghent, Belgium) (16), respectively, as described previously (12, 17).
Brain tissue samples were obtained when the patient underwent surgery for hematoma. Immunohistochemical studies were performed on using an anti-Aβ1-42 antibody and an anti-Aβ1-40 antibody (both IBL, Fujioka, Gunma, Japan).
After obtaining informed consent for the genetic test of the APOE gene, we purified genomic DNA from lymphocytes in the peripheral blood of the patient. For the analysis of APOE allele polymorphism, purified genomic DNA samples were examined as previously described (12). The Apolipoprotein E protein was confirmed by the LSI Medience Corporation (Tokyo, Japan).
This report describes the case of a 79-year-old Japanese woman diagnosed with AD, presenting with recurrent hemorrhages despite the absence of vascular risk factors such as hypertension, diabetes mellitus, hypercholesterolemia, coronary heart disease, and smoking. However, fatty liver and hypocholesterolemia were observed on medical examination. Although her mother had died of a subarachnoid hemorrhage (SAH). At age 70 years, the patient experienced a loss of recent memory, and since then, the amnesia has gradually progressed. At the age of 72 years, she was admitted to the Gunma University Hospital for a headache caused by a brain hemorrhage; she felt a little headache and dullness without nausea, vomiting or loss of consciousness, a state that were quite different from a typical acute SAH. At the time of her first admission, cerebral magnetic resonance imaging (MRI) revealed a hemorrhage in the left parietal subcortical area (Figure 1A) and multiple regions of cSS (Figures 1A,B). Although there were no white matter hyperintensities, perivascular spaces, chronic ischemic infarcts, or lacunae, there were multiple regions of cSS and a few lobar microbleeds. On neuropsychological examinations, the scores were 23/30 in the Mini-Mental State Examination (MMSE) (disorientation of time, delayed recall deficits, acalculia, and agraphia), 19/30 in the Montreal Cognitive Assessment (MoCA) (visual-spatial disorientation, clock drawing, delayed recall, and disorientation of time), and 9/18 in the Frontal Assessment Battery (FAB) (deficits of similarities, motor series, conflicting instructions, and prehension behavior). The Instrumental Activities of Daily Living scales (IADL) (18) was 4/8 (Abilities required for shopping, food preparation, responsibility for own medications, and handling finances were disturbed). She could do activities of daily living (ADL) to some extent but needed some assistance from her family. According to gradual amnestic progression and executive dysfunction of ADL, she was diagnosed with mild dementia with AD at the first admission; the findings were also suggestive of possible CAA according to the modified Boston criteria (19). At age 73 years, the second cerebral hemorrhage occurred. This time she was admitted to the Geriatric Research Institute and Hospital for a left hemiparalysis with a lobar ICH at the right frontal lobe (Figure 1C). The hematoma was removed by a neurosurgical operation. The score of MMSE at the time of the second admission had decreased to 18/30, implying a cognitive decline, when compared to the first admission. The score of the revised Hasegawa Dementia Scale (HDS-R) (20) was 17/30, suggesting moderate dementia. At age 77 years, she was admitted for a headache for the third time; this time she had left hemispatial neglect, caused by a lobar ICH in the right parieto-occipital lobes (Figure 1D). Currently, she has no additional hemorrhages but is still affected by dementia and left hemiparalysis. The patient has not on any anti-platelet, anti-coagulation, or antihyperlipidemic drugs for the clinical course.
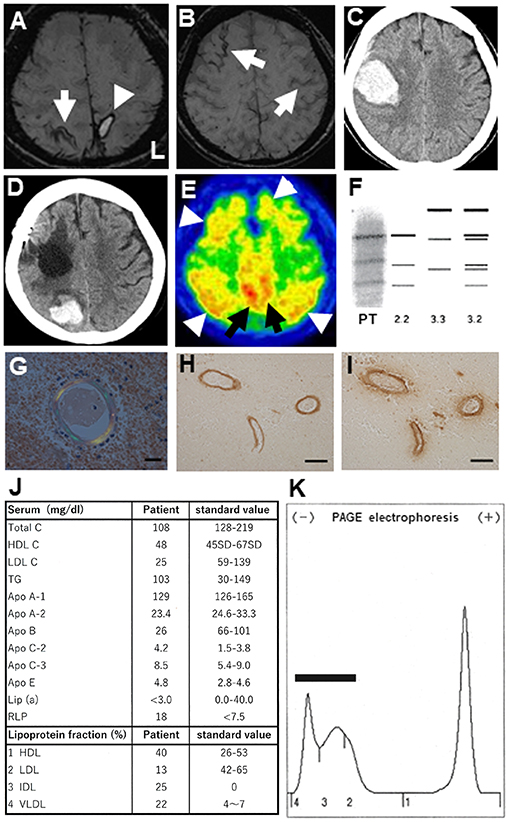
Figure 1. (A) Brain magnetic resonance imaging (MRI; 3T System, Skyra, Siemens, Germany) at the first admission. Susceptibility weighted images (SWI) showing a cortical hemorrhage (arrowhead) at the left parietal lobe cortex and cortical superficial siderosis (cSS) in the right parietal cortex (arrow). (B) In the same MRI, cSS was additionally observed in the left and right frontal cortices (arrows). (C) Brain computed tomography (CT) showing a second lobar hemorrhage at the right frontal subcortical area concurrent with left hemiparesis. (D) Brain CT showing a third lobar hemorrhage at the right parieto-occipital subcortical region. (E) 11C-PiB PET revealing widespread 11C-PiB retentions in the cerebral cortices (arrowheads) and precuneus (arrows). (F) Electrophoresis revealing a 2/2 pattern in the apolipoprotein E phenotype for this patient (PT). (G) Congophilic amyloid deposits shown in blood vessels of the resected brain tissue, as shown using polarizing microscopy (Scale bar: 20 μm). (H,I) Aβ-reactive structures in blood vessel walls identified by immunohistochemistry using anti-Aβ1-42 (H) and anti-Aβ1-40 (I) antibodies. Scale bars: 100 μm. (J) Laboratory serum results from the patient's first admission. (K) Serum lipoprotein electrophoresis reveals a broad-β pattern (bar), where intermediate-density lipoprotein (fraction 3) and very-low-density lipoprotein (fraction 4) are aberrantly elevated.
At the time of the first hospitalization, 11C-PiB PET revealed widespread 11C-PiB retentions in the cerebral cortices and precuneus, which were compatible with those of AD (Figure 1E).
The APOE genotyping revealed a homozygous ε2 allele (not shown). The apolipoprotein E (ApoE) was confirmed as a 2/2 protein by the western blot (Figure 1F). Congophilic amyloid deposits in blood vessels were confirmed on polarizing microscopy, undertaken using tissue resected during the second hospitalization (Figure 1G). Aβ-immunoreactive structures in blood vessels walls were revealed by an anti-Aβ1-42 antibody (Figure 1H) and an anti-Aβ1-40 antibody (Figure 1I) on immunohistochemical studies. Based on the pathological results, this case was suggestive of “probable CAA” with supporting pathological evidence according to the modified Boston criteria (19). Laboratory examination of the serum at the first admission demonstrated hypolipidemia, defined by low serum levels of total cholesterol (Total C), low-density lipoprotein cholesterol (LDL C), apolipoprotein A-2 (ApoA-2), and apolipoprotein B (Apo B). Chylomicrons were scarcely detectable (data not shown). The serum levels of apolipoprotein C-2 (ApoC-2), apolipoprotein E (ApoE), remnant-like lipoprotein (RLP), intermediate-density lipoprotein (IDL), and very-low-density lipoprotein (VLDL) were increased (Figure 1J). Furthermore, polyacrylamide gel (PAGE) electrophoresis of her serum revealed a broad-β band which seemed to be mimicking type III dysliplidemia (Figure 1K) (BML Co. Tokyo, Japan). There was no detection of aberrantly truncated apolipoprotein B on the western blot (data not shown). In the case, the CSF level of Aβ1-42 was 144.49 pg/mL, which was lower than that in non-dementia subjects (mean ± standard deviation, 431.55 ± 103.46 pg/mL, n = 20) and comparable to that in age-matched AD patients (183.32 ± 14.68 pg/mL, n = 25); the Aβ1-40 was 3037.22 pg/mL, which was lower than that in non-dementia subjects (mean ± standard deviation, 5118.79 ± 1882.47 pg/mL, n = 20) and age-matched AD patients (5358.47 ± 2376.80 pg/mL, n = 25); the P-Tau level was 42.94 pg/mL, which was higher than that in non-dementia subjects (29.33 ± 11.36 pg/mL, n = 20) and comparable to those of age-matched AD patients (75.40 ± 7.5 pg/mL, n = 25); and the T-Tau level was 424.50 pg/mL, which was higher than that in non-dementia subjects (143.55 ± 14.63 pg/mL, n = 20) and comparable to that in age-matched AD patients (503.30 ± 53.03 pg/mL, n = 25). These findings of CSF biomarkers and 11C-PiB PET of the patient were compatible with those of AD. Post-discharge the second hemorrhage at the age of 73 years, the 76-year-old patient was examined using 18F-THK5351 PET in a stable condition.
18F-THK5351 retentions areas were observed at the right side of the pyramidal tract at the pontine level, the cerebral peduncle of the midbrain, and the internal capsule (red circles in Figure 2 A-G-M-S, B-H-N-T, C-I-O-U, and D-J-P-V). The off-target 18F-THK5351 retentions, binding to MAO-B derived from astrocytes, located in the pyramidal tract, were considered a result of a previous right frontal lobar hemorrhage.
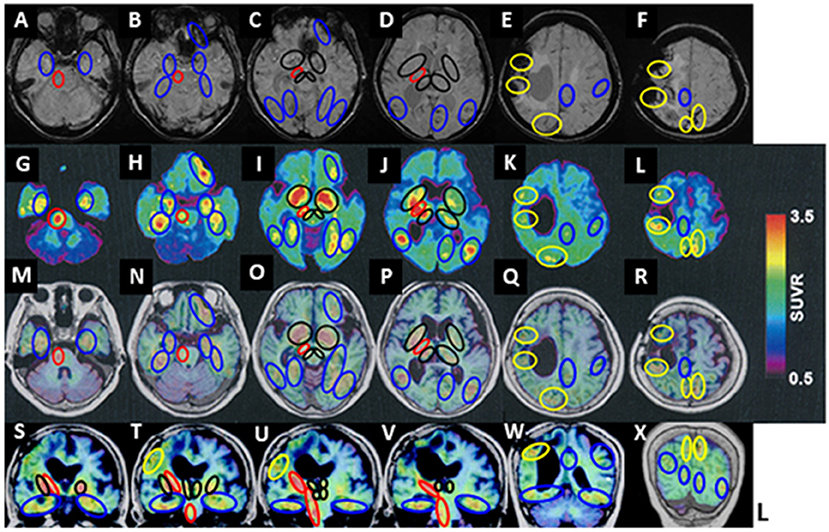
Figure 2. 18F-THK5351 imaging and MRI of the patient's brain. (A–F) Susceptibility-weighted MRI. (G–L) 18F-THK5351. (M–R) 18F-THK 5351 on transverse MRI (T1-weighted images). (S–X) 18F-THK 5351 on coronal MRI (T1-weighted images). 18F-THK5351 retentions areas were observed in the pyramidal tract at the pontine level, the cerebral peduncle of the midbrain, and the internal capsule, all at the right side (red circles: A-G-M-S, B-H-N-T, C-I-O-U, and D-J-P-V). The bilateral substantia nigra of the midbrain, the globus pallidum, the anterior caudate nuclei, amygdala, and the medial areas of thalamus (black circles: C-I-O-U and D-J-P-V) were considered the off-target retentions of 18F-THK5351 binding to MAO-B. They were also visible along the previous lobar hemorrhagic lesions in the right frontal lobe and the left parietal cortex, as well as an asymptomatic old cortical SAH lesion in the right parietal cortex (yellow circles: E-K-Q-W and F-L-R-X). Additional 18F-THK5351 retentions were observed in the bilateral medial temporal areas (blue circles: A-G-M-S and B-H-N-T), the left anterior frontal cortical areas (blue circles: B-H-N-T and C-I-O-U), the posterior areas in bilateral temporal cortices, and the bilateral fusiform, lingual, and parahippocampal gyruses (blue circles: C-I-O and D-J-P).
In addition, they were detected at the bilateral substantia nigra of the midbrain, medial areas of the thalamus, the amygdala, the globus pallidum (black circles in Figure 2 T, C-I-O-U, D-J-P-V), and the anterior caudate nuclei (black circles in Figure 2 D-J-P), in which originally contained a high concentration of MAO-B.
Besides, 18F-THK5351 retentions were visible along with previous lobar hemorrhagic lesions in the left parietal cortex (at the first hemorrhage) and the right frontal lobe (at the second hemorrhage), and an asymptomatic old cortical SAH lesion in the right parietal cortex (yellow circles in Figure 2 E-K-Q-W and F-L-R-X).
Additional 18F-THK5351 retentions were observed in the bilateral medial temporal areas (blue circles in Figure 2 A-G-M-S and B-H-N-T), the bilateral areas of entorhinal and parahippocampal gyruses, and hippocampus (blue circles in Figure 2 B-H-N-T, C-I-O), the bilateral areas of parahippocampal, fusiform, and lingual gyruses (blue circles in Figure 2 C-I-O), the left anterior frontal cortical areas (blue circles in Figure 2 B-H-N and C-I-O), the bilateral posterior areas in the temporal cortices (blue circles in Figure 2 C-I-O and D-J-P), bilateral basis areas in the temporal lobes (blue circles in Figure 2 S, T, U, V, and W), the left calcarine sulcus (blue circles in Figure 2 D-J-P), the left precuneus area and the left frontal cortex (blue circles in Figure 2 E-K-Q-W).
To our knowledge, this is the first report of an AD patient, homozygous for the APOE ε2 allele, presenting with recurrent lobar hemorrhage, vascular Aβ amyloid deposits, hypobetalipoproteinemia, and pathological 18F-THK5351 findings. The vasculopathologic changes associated with the APOE ε2 allele might have a crucial role in the severity and clinical course of lobar ICH, and the screening of patients with ICH for the APOE ε2 may identify those at increased risk of mortality and poor functional outcomes (21). The APOE ε2, as well as the APOE ε4, allele was reported to be significantly associated with lobar ICH and cSS (21, 22), and their presence may predict the recurrence of anti-coagulation-associated ICH (23). Clinically, some patients with the APOE ε2 allele present with hypocholesterolemia (24). In animal study, APOE ε2 transgenic mice showed a decreased serum level of low-density lipoprotein cholesterol (25). These biochemical aberrations may lead to the fragility in the structure of cerebral blood vessel walls, resulting in recurrent ICH in AD patients with the APOE ε2 allele (26, 27). APOE ε2/ε3 carriers have larger total volumes of white matter hyperintensities compared to APOE ε4 carriers and a higher prevalence of microbleeds compared to APOE ε3 homozygotes (28). Because the homozygous APOE ε2 allele has an extremely low frequency (1), no prior publication has confirmed a proven vascular amyloid pathology, in a homozygous APOE ε2 allele carrying AD patient with recurrent lobar ICH, using 18F-THK5351 PET and 11C-PiB PET. In vivo study revealed that the levels of Aβ40 and Aβ42 in both soluble and insoluble fractions of the brain lysate were reduced in APOE ε2, but not in APOE ε3 or APOE ε4 gene integrated in human amyloid precursor protein/human presenilin 1 (APP/PS1) double transgenic mice (29). The CAA patients had a distinctive CSF biomarker profile, with significantly lower concentrations of Aβ40 and Aβ42 than those of non-demented controls and AD patients, which were found in APOE ε2, ε4, and ε3 genotypes (30–33). Lower CSF levels of Aβ1-40 and Aβ1-42 in an AD patient with CAA carrying APOE ε2 homozygote had not been reported before this case. So far, pathological reports of autopsy studies showed reduced neurofibrillary tangles (NFTs) in postmortem AD brains of APOE ε2 carriers (2, 34, 35). Although the mechanism underlying this reduction of NFTs is poorly understood; as APOE ε2 was significantly correlated with tau pathology only in Aβ positive AD patients but not with those in Aβ negative individuals, the protective effect of APOE ε2 against AD tau may be partially mediated through its effect on Aβ deposition (36). It remains still controversial why APOE ε2 may protect against tau pathology independently of Aβ in AD (37–39). 18F-THK5351 was initially developed to target tau aggregates in NFTs (8, 9), however, 18F-THK5351 has been shown to strongly bind off-target to MAO-B in the basal ganglia and thalamus (10), as well as the melanin-containing cells of the substantia nigra (40). 18F-THK5351 is also highly concentrated in astrocytes, compared to other types of glial cells (41, 42). The MAO-B concentration is also increased in reactive astrogliosis (43), which describes a spectrum of astrocytic changes that occur in response to brain injury (44). On the other hand, Wallerian degeneration, the progressive anterograde demyelination and disintegration of distal axons, develops following a brain injury to neuronal soma or proximal axons (45). In the early stage of brain injury, axonal swelling, and breakdown of myelin sheath occur, followed by loss of myelinated fibers and infiltration of macrophages. In the later stage, the degenerated axons are replaced by gliosis, which is primarily the proliferation of astrocytes to form a scar. A previous case report demonstrated ipsilateral myelin loss and gliosis in the pyramidal tract lesion 5 years after a left middle cerebral artery infarction (45). As for the other tau PET tracers and CAA, two reports of Tau PET have been published (46, 47). In the former report, paired helical filament (PHF) tau burden overlapped cerebral microbleeds and/or cSS on 18F-AV1451 PET, raising the possibility that the characteristic markers of vascular amyloid were associated with local production of PHF tau (46). In the latter report, the amyloid deposition, evaluated by 18F-florbetaben PET, was not different in vicinity of cSS, but tau depositions were elevated in vicinity of cSS-affected regions compared to non-cSS-affected brain regions. Their case of probable CAA suggested that cSS may be associated with an elevation in local tau accumulation but not with an increase in fibrillary amyloid deposition (47). 18F-THK5351 PET has been reported to strongly bind to MAO-B derived from astrocytes and possibly tau, probably reflecting astrogliosis related CAA hematoma and cSS, also possibly tau accumulation in AD pathology (48). In our case, 3 year have passed since the 18F-THK5351 PET examination for the previous ICH in the right frontal lobe presenting as contralateral hemiparalysis. In this patient, Wallerian degeneration may have developed in the ipsilateral pyramidal tract followed by astrogliosis, and the marker 18F-THK5351 PET, targeting MAO-B containing tissues, may have identified this Wallerian degeneration. The 18F-THK5351 is now recognized as a dual-purpose compound that binds to both MAO-B and tau aggregates, thus tau isoforms might have accumulated in the damaged fasciculus. MAO-B is highly concentrated in astrocytes and serotonergic and histaminergic neurons (43), astrocytes proliferate in response to inflammation caused by brain injury (44). Therefore, regional changes in MAO-B concentration can be an index of astrogliosis and be detected on 18F-THK5351 PET imaging, although tau accumulation may co-exist in the AD brain (48). These findings suggest that 18F-THK5351 accumulates in lesions where astrogliosis occurs and that 18F-THK5351 PET can be an imaging modality to visualize and quantify astrogliosis, as recently demonstrated in patients with a cerebral infarction or neurological disorders (49–51). In our case, 18F-THK5351 retentions were observed in some, but not all, regions with cSS, presumably because this effect might depend on severity of cSS and the degree of astrogliosis that is correlated with the extent of hemosiderin deposition (52). 18F-THK5351 retentions were observed only in some areas rather than all peripheral regions at the lobar hemorrhage in this patient. Several cSS lesions reflected 18F-THK5351 PET retentions, however, the other cSS lesions did not reflect, which might probably depend upon severity of cSS or neuroinflammation of CAA. It seems that old cSS showed less or no THK5351 retention [such as in bilateral prefrontal lobes cortices/subcortices (Figure 2 D-J-P and E-K-Q)], while the recent lesions of hematoma or siderosis showed THK5351 retentions [such as in left parietal cortical area (hematoma at the first admission) and right frontal cortico-subcortical areas (hematoma at the second admission)]. This case signifies that both AD and CAA pathologies could co-occur and contribute to cognitive decline (53), indicating that CAA, especially, cSS strongly increases the risk of hemorrhages, even when the clinical changes are gradual. We found that CSF Aβ1-40 of this patient was lower than those of AD patients and non-demented subjects, as, in the recent publication, the levels of CSF Aβ1-40 in AD with disseminated cSS were lower than that in AD patients with focal cSS and those without cSS (54, 55). This result is thought be in accordance with neuropathological reports demonstrating deposition of Aβ40 within the vessel walls of CAA patients (56). CSF P-Tau levels, in this case, were not significantly higher than those in AD patients, because CSF P-Tau levels in AD patients with CAA pathology have been reported to be either slightly elevated or within the normal levels (55, 57, 58). This could be because of the change in permeability of the cerebral blood vessels due to CAA; decreased ability of parenchymal P-Tau to flow out into the lumen of vessels or less amount of tau phosphorylation in CAA than in AD patients may have led to lower P-Tau levels in CSF. On the other hand, Aβ 40 is thought to deposit in the blood vessels affected by CAA, rather than flowing into the lumen of vessels (59). Although the fact that the pathophysiologic mechanisms are not completely understood, recent studies show that APOE2 allele is strongly associated with cSS and increased frequency and severity of lobar ICH (22). Furthermore, while APOE ε2 promotes so-called CAA-related vasculopathic changes (vessel cracking, detachment, and delamination of the outermost layer of the tunica media, and fibrinoid necrosis), this can lead to vessel rupture (5, 22), another evidence of increased ICH risk in APOE ε2 carriers was recently reported, with or without AD (60). Hypobetalipoproteinemia may not be a direct cause for the ICH in this case, but might be an additional contributing factor, because hypocholesterolemia was reported to be a risk factor of ICH (61–63). This AD case carried the homozygous APOE ε2 allele and severe hypocholesterolemia, which may have exacerbated vascular changes in the CAA, thereby leading to more severe CAA, multiple cSS, and recurrent lobar ICH. The mechanism underlying the risk for ICH because of an APOE ε2 genotype is still unclear and perhaps complex (60), which should be elucidated hereafter. In future, a large-scale investigation of cholesterol levels in AD patients with a homozygous APOE ε2 allele will be required to define its clinical significance and etiologies. This AD case with multiple cSS regions and lobar hemorrhages is also worth reporting; it demonstrates that 18F-THK5351 retentions reflected MAO-B, reactive astrogliosis, and Wallerian degeneration of the pyramidal tract due to CAA related lobar ICH, and possibly tau accumulation.
Finally, the therapies targeting APOE ε2 and APOE2 for AD have been extensively developed; a viral-mediated APOE ε2 overexpression, converting APOE ε4 to APOE ε2, and plasma APOE-based therapy were described detail in a recent review (64). Although these trials have some effect, unfortunately, they have not been effective, regardless of their successful experimental results. Besides severe CAA pathology, this case presented with hypobetalipoproteinemia, however, we could not elucidate the clear molecular and pathological relationships between them. To validate the mechanisms of effective therapies targeting APOE2/APOE ε2, more evidence from humans and animal models is required. Further, more AD patients carrying the APOE ε2 allele are required to analyze pathological and biochemical findings. Furthermore, improved understanding of the roles of APOE2/APOE ε2 in other diseases, CAA/Stroke and different proteinopathies, including tau, TDP-43, and α-synuclein pathologies, may allow a comprehensive assessment of the safety of APOE2 targeted therapeutics for AD.
A limitation of this study is that it includes only one AD patient carrying a homozygous APOE ε2 allele with recurrent hemorrhages and multiple cSS regions. This impedes statistical analyses for cholesterol levels in comparison to other APOE alleles or healthy control groups. The other limitation is that we could not confirm how histopathological association between 18F-THK5351 retention and astrogliosis pathology since we histologically observed only a part of the brain. To better understand the pathological mechanism, a detailed investigation using neuroradiological, biochemical, genetical, and pathological methods is required in CAA and AD patients.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
The studies involving human participants were reviewed and approved by the ethics committee of the Gunma University Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written consent to publish the clinical information was obtained from the patient's family.
MI collected the clinical data, interpreted the data, and wrote the manuscript. ET analyzed genomic DNA from the patient's blood samples and CSF biomarkers from the patient. KO, KS, and HY performed pathological examinations and evaluation of the results. TH and YTs performed the neuroimaging examination for 11C PiB-PET, and KI performed and evaluated the neuroimaging examination for 18F THK5351-PET. MF, NF, YTa, CS, MS, YS, MA, and MTakat collected the clinical information. SI, MK, and MTakah performed and evaluated the western blot of ApoB protein, also discussed the hypobetalipoproteinemia in this patient. ST and IN performed the neurosurgical operation to removal the hematoma at the second admission. MI performed the clinical data analysis and evaluated the specificities and neurological significances. All authors contributed to the article and approved the submitted version.
This study was supported by a Grant-in-Aid for Scientific Research (C) (MI: 18K07491, MTakah: 20K08870, TH: 19K08220, YTs: 18K07627, HY: 19K07411, KI: 17K09813, YI: 19K07813), (B) (SI: 19H03712, MK: 20H03671) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, MI also received a Donation of love from the JOMO-SHINBUN newspaper company (Maebashi, Gunma, Japan).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the patient and his family for providing clinical data and allowing the publication of this case report. We especially thank Dr. Hirofumi Kawakami, an Advanced Application Specialist who belongs to Headquarter of Academy, GE Healthcare Japan, and Yoshikatsu Tsunemi (Inspection Specialist), Department of Clinical Laboratory Medicine, Geriatrics Research Institute and Hospital, for their excellent technical assistance. We thank Dr. Mikio Shoji for academic discussion about this manuscript. We would like to thank Editage (www.editage.com) for American English language editing.
APOE, apolipoprotein E gene; AD, Alzheimer's disease; ICH, intracerebral hemorrhage; CAA, cerebral amyloid angiopathy; MAO-B, monoamine oxidase B; cSS, cortical superficial siderosis; 11C PiB-PET, 11C-labeled Pittsburgh Compound B-positron emission tomography; Aβ, amyloid β; P-Tau, phosphorylated tau 181; T-Tau, human total tau; CSF, cerebrospinal fluid; SAH, subarachnoid hemorrhage; MRI, magnetic resonance imaging; WMH, white matter hyperintensities; PVC, perivascular spaces; ADL, activities of daily living.
1. Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, et al. Exceptionally low likelihood of Alzheimer's dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. (2020) 11:667. doi: 10.1038/s41467-019-14279-8
2. Serrano-Pozo A, Qian J, Monsell S, Betensky R, Hyman B. APOEε2 is associated with milder clinical and pathological Alzheimer's disease. Ann Neurol. (2015) 77:917–29. doi: 10.1002/ana.24369
3. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small G, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. (1993) 261:921–3. doi: 10.1126/science.8346443
4. Nicoll JA, Burnett C, Love S, Graham DI, Dewar D, Ironside JW, et al. High frequency of apolipoprotein E ε2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol. (1997) 41:716–21. doi: 10.1002/ana.410410607
5. Greenberg SM, Vonsattel JP, Segal AZ, Chiu RI, Clatworthy AE, Liao A, et al. Association of apolipoprotein E ε2 and vasculopathy in cerebral amyloid angiopathy. Neurology. (1998) 50:961–5. doi: 10.1212/wnl.50.4.961
6. O'Donnell HC, Rosand J, Knudsen KA, Furie KL, Segal AZ, Chiu RI, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Eng J Med. (2000) 342:240–5. doi: 10.1056/NEJM200001273420403
7. Rannikmäe K, Kalaria RN, Greenberg SM, Chui HC, Schmitt FA, Samarasekera N, et al. APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J Neurol Neurosurg Psychiatry. (2014) 85:300–5. doi: 10.1136/jnnp-2013-306485
8. Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N, et al. 18F-THK5351: a novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer disease. J Nucl Med. (2016) 57:208–14. doi: 10.2967/jnumed.115.164848
9. Tago T, Furumoto S, Okamura N, Harada R, Adachi H, Ishikawa Y, et al. Structure-activity relationship of 2-arylquinolines as PET imaging tracers for tau pathology in Alzheimer disease. J Nucl Med. (2016) 57:608–14. doi: 10.2967/jnumed.115.166652
10. Ng KP, Pascoal TA, Mathotaarachchi S, Therriault J, Kang MS, Shin M, et al. Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimers Res Ther. (2017) 9:25. doi: 10.1186/s13195-017-0253-y
11. Ishibashi K, Kameyama M, Tago T, Toyohara J, Ishii K. Potential use of 18F-THK5351 PET to identify wallerian degeneration of the pyramidal tract caused by cerebral infarction. Clin Nucl Med. (2017) 42:e523–4. doi: 10.1097/RLU.0000000000001868
12. Ikeda M, Tashiro Y, Takai E, Kurose S, Fugami N, Tsuda K, et al. CSF levels of Aβ1-38/Aβ1-40/Aβ1-42 and 11C-PiB PET studies in three clinical variants of primary progressive aphasia and Alzheimer's disease. Amyloid. (2014) 21:238–45. doi: 10.3109/13506129.2014.949231
13. Kasahara H, Ikeda M, Nagashima K, Fujita Y, Makioka K, Tsukagoshi S, et al. Deep white matter lesions are associated with early recognition of dementia in Alzheimer's disease. J Alzheimers Dis. (2019) 68:797–808. doi: 10.3233/JAD-180939
14. Ishibashi K, Miura Y, Hirata K, Toyohara J, Ishii. Relationship between the temporal course of astrogliosis and symptom improvement in cerebral infarction: report of a case monitored using 18 F-THK5351 positron emission tomography. BMC Med Imaging. (2020) 20:81. doi: 10.1186/s12880-020-00481-4
15. Vanmechelen E, Vanderstichele H, Davidsson P, Van Kerschaver E, Van Der Perre B, Sjögren M, Andreasen N, et al. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett. (2000) 285:49–52. doi: 10.1016/s0304-3940(00)01036-3
16. Andreasen N, Minthon L, Davidsson P, Vanmechelen E, Vanderstichele H, Winblad B, et al. Evaluation of CSF-tau and CSF-Aβ42 as diagnostic markers for Alzheimer's disease in clinical practice. Arch Neurol. (2001) 58:373–9. doi: 10.1001/archneur.58.3.373
17. Ikeda M, Kuwabara T, Takai E, Kasahara H, Furuta M, Sekine A, et al. Increased neurofilament light chain and YKL-40 CSF Levels in one Japanese IBMPFD patient with VCP R155C mutation: a clinical case report with CSF biomarker analyses. Front Neurol. (2020) 11:757. doi: 10.3389/fneur.2020.00757
18. Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. (1969) 9:179–86.
19. Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Dichgans M, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology. (2010) 74:1346–50. doi: 10.1212/WNL.0b013e3181dad605
20. Katoh S, Shimogaki H, Onodera A, Ueda H, Oikawa K, Ikeda K, et al. Development of the revised version of Hasegawa's Dementia-Scale (HDS-R). Jpn J Geriatr Psychiatry. (1991) 2:1339–47. (Japanese)
21. Biffi A, Anderson CD, Jagiella JM, Schmidt H, Kissela B, Hansen BM, et al. APOE genotype and extent of bleeding and outcome in lobar intracerebral haemorrhage: a genetic association study. Lancet Neurol. (2011) 10:702–9. doi: 10.1016/S1474-4422(11)70148-X
22. Charidimou A, Zonneveld HI, Shams S, Kantarci K, Shoamanesh A, Hilal S, et al. APOE and cortical superficial siderosis in CAA: meta-analysis and potential mechanisms. Neurology. (2019) 93:e358–71. doi: 10.1212/WNL.0000000000007818
23. Biffi A, Urday S, Kubiszewski P, Gilkerson L, Sekar P, Rodriguez-Torres A, et al. Combining imaging and genetics to predict recurrence of anticoagulation-associated intracerebral hemorrhage. Stroke. (2020) 51:2153–60. doi: 10.1161/STROKEAHA.120.028310
24. Mahley RW, Huang Y, Rall SC. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia): questions, quandaries, and paradoxes. J Lipid Res. (1999) 40:1933–49.
25. Huang Y, Liu XQ, Rall SC, Mahley RW. Apolipoprotein E2 reduces the low-density lipoprotein level in transgenic mice by impairing lipoprotein lipase-mediated lipolysis of triglyceride-rich lipoproteins. J Biol Chem. (1998) 273:17483–90. doi: 10.1074/jbc.273.28.17483
26. McCarron MO, Nicoll JA, Stewart J, Ironside JW, Mann DM, Love S, et al. The apolipoprotein E epsilon2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. J Neuropathol Exp Neurol. (1999) 58:711–8. doi: 10.1097/00005072-199907000-00005
27. Nelson PT, Pious NM, Jicha GA, Wilcock DM, Fardo DW, Estus S, et al. APOE-ε2 and APOE-ε4 correlate with increased amyloid accumulation in cerebral vasculature. J Neuropathol Exp Neurol. (2013) 72:708–15. doi: 10.1097/NEN.0b013e31829a25b9
28. Groot C, Sudre CH, Barkhof F, Teunissen CE, van Berckel BN, Seo SW, et al. Clinical phenotype, atrophy, and small vessel disease in APOEε2 carriers with Alzheimer disease. Neurology. (2018) 91:e1851–9. doi: 10.1212/WNL.0000000000006503
29. Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, et al. Gene transfer of human ApoE isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. (2013) 5: 212ra161. doi: 10.1126/scitranslmed.3007000
30. Costa AS, Pinho J, Kučikiene D, Reich A, Schulz JB, Reetz K. Cerebral amyloid angiopathy in amyloid-positive patients from a memory clinic cohort. J Alzheimers Dis. (2021) 79:1661–72. doi: 10.3233/JAD-201218
31. Banerjee G, Ambler G, Keshavan A, Paterson RW, Foiani MS, Toombs J, et al. Cerebrospinal fluid biomarkers in cerebral amyloid angiopathy. J Alzheimers Dis. (2020) 74:1189–1201. doi: 10.3233/JAD-191254
32. Charidimou A, Imaizumi T, Moulin S, Biffi A, Samarasekera N, Yakushiji Y, et al. Brain hemorrhage recurrence, small vessel disease type, and cerebral microbleeds: a meta-analysis. Neurology. (2017) 89:820–29. doi: 10.1212/WNL.0000000000004259
33. Shams S, Martola J, Charidimou A, Cavallin L, Granberg T, Shams M, et al. Cortical superficial siderosis: prevalence and biomarker profile in a memory clinic population. Neurology. (2016) 87:1110–7. doi: 10.1212/WNL.0000000000003088
34. Bennett DA, De Jager PL, Leurgans SE, Schneider JA. Neuropathologic intermediate phenotypes enhance association to Alzheimer susceptibility alleles. Neurology. (2009) 72:1495–503. doi: 10.1212/WNL.0b013e3181a2e87d
35. Nagy Z, Esiri MM, Jobst KA, Johnston C, Litchfield S, Sim E, et al. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer's disease. Neuroscience. (1995) 69:757–61. doi: 10.1016/0306-4522(95)00331-c
36. Farfel JM, Yu L, De Jager PL, Schneider JA, Bennett DA. Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol Aging. (2016) 37:19–25. doi: 10.1016/j.neurobiolaging.2015.09.011
37. Goldberg TE, Huey ED, Devanand DP. Association of APOE ε2 genotype with Alzheimer's and non-Alzheimer's neurodegenerative pathologies. Nat Commun. (2020) 11:4727. doi: 10.1038/s41467-020-18198-x
38. Zhao N, Liu CC, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, et al. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat Commun. (2018) 9:4388. doi: 10.1038/s41467-018-06783-0
39. Ghebremedhin E, Schultz C, Botez G, Rüb U, Sassin I, Braak E, et al. Argyrophilic grain disease is associated with apolipoprotein E epsilon 2 allele. Acta Neuropathol. (1998) 96:222–4. doi: 10.1007/s004010050886
40. Tago T, Toyohara J, Harada R, Furumoto S, Okamura N, Kudo Y, et al. Characterization of the binding of tau imaging ligands to melanin-containing cells: putative off-target-binding site. Ann Nucl Med. (2019) 33:375–82. doi: 10.1007/s12149-019-01344-x
41. Gulyás B, Pavlova E, Kása P, Gulya K, Bakota L, Várszegi S, et al. Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-L-deprenyl using whole hemisphere autoradiography. Neurochem Int. (2011) 58:60–8. doi: 10.1016/j.neuint.2010.10.013
42. Levitt P, Pintar JE, Breakefield XO. Immunocytochemical demonstration of monoamine oxidase B in brain astrocytes and serotonergic neurons. Proc Natl Acad Sci USA. (1982) 79:6385–9. doi: 10.1073/pnas.79.20.6385
43. Saura J, Luque JM, Cesura AM, Da Prada M, Chan-Palay V, Huber G, et al. Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience. (1994) 62:15–30. doi: 10.1016/0306-4522(94)90311-5
44. Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. (2009) 32:638–47. doi: 10.1016/j.tins.2009.08.002
45. Jones KC, Hawkins C, Armstrong D, Deveber G, Macgregor D, Moharir M, et al. Association between radiographic Wallerian degeneration and neuropathological changes post childhood stroke. Dev Med Child Neurol. (2013) 55:173–7. doi: 10.1111/dmcn.12010
46. Kim HJ, Cho H, Werring DJ, Jang YK, Kim YJ, Lee JS, et al. 18F-AV-1451 PET imaging in three patients with probable cerebral amyloid angiopathy. J Alzheimers Dis. (2017) 57:711–6. doi: 10.3233/JAD-161139
47. Brendel M, Catak C, Beyer L, Linn J, Wahl H, Janowitz D, et al. Colocalization of tau but not β-amyloid with cortical superficial siderosis in a case with probable CAA. Case Rep Neurol. (2020) 12:232–7. doi: 10.1159/000506765
48. Harada R, Ishiki A, Kai H, Sato N, Furukawa K, Furumoto S, et al. Correlations of 18F-THK5351 PET with postmortem burden of tau and astrogliosis in Alzheimer disease. J Nucl Med. (2018) 59:671–4. doi: 10.2967/jnumed.117.197426
49. Ishibashi K, Miura Y, Hirata K, Toyohara J, Ishii K. 18F-THK5351 PET can identify astrogliosis in multiple sclerosis plaques. Clin Nucl Med. (2020) 45:e98–100. doi: 10.1097/RLU.0000000000002751
50. Schönecker S, Brendel M, Palleis C, Beyer L, Höglinger GU, Schuh E, et al. PET imaging of astrogliosis and tau facilitates diagnosis of Parkinsonian syndromes. Front Aging Neurosci. (2019) 11:249. doi: 10.3389/fnagi.2019.00249
51. Takami Y, Yamamoto Y, Norikane T, Mitamura K, Hatakeyama T, Nishiyama Y. 18F-THK5351 PET can identify lesions of acute traumatic brain injury. Clin Nucl Med. (2020) 45:e491–2. doi: 10.1097/RLU.0000000000003165
52. Charidimou A, Perosa V, Frosch MP, Scherlek AA, Greenberg SM, van Veluw SJ. Neuropathological correlates of cortical superficial siderosis in cerebral amyloid angiopathy. Brain. (2020) 143:3343–51. doi: 10.1093/brain/awaa266
53. Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. (2018) 83:74–83. doi: 10.1002/ana.25123
54. Martínez-Lizana E, Carmona-Iragui M, Alcolea D, Gómez-Choco M, Vilaplana E, Sánchez-Saudinós MB, et al. Cerebral amyloid angiopathy-related atraumatic convexal subarachnoid hemorrhage: an ARIA before the tsunami. J Cereb Blood Flow Metab. (2015) 35:710–7. doi: 10.1038/jcbfm.2015.25
55. Catak C, Zedde M, Malik R, Janowitz D, Soric V, Seegerer A, et al. Decreased CSF levels of ß-amyloid in patients with cortical superficial siderosis. Front Neurol. (2019) 10:439. doi: 10.3389/fneur.2019.00439
56. Vinters HV. Emerging concepts in Alzheimer's disease. Annu Rev Pathol. (2015) 10:291–319. doi: 10.1146/annurev-pathol-020712-163927
57. van Etten ES, Verbeek MM, van der Grond J, Zielman R, van Rooden S, van Zwet EW, et al. β-Amyloid in CSF: biomarker for preclinical cerebral amyloid angiopathy. Neurology. (2017) 88:169–76. doi: 10.1212/WNL.0000000000003486
58. Shams S, Granberg T, Martola J, Li X, Shams M, Fereshtehnejad SM, et al. Cerebrospinal fluid profiles with increasing number of cerebral microbleeds in a continuum of cognitive impairment. J Cereb Blood Flow Metab. (2016) 36:621–8. doi: 10.1177/0271678X15606141
59. Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol. (2011) 70:871–80. doi: 10.1002/ana.22516
60. Goldberg TE, Huey ED, Devanand DP Associations of APOE e2 genotype with cerebrovascular pathology: a postmortem study of 1275 brains. J Neurol Neurosurg Psychiatry. (2021) 92:7–11. doi: 10.1136/jnnp-2020-323746
61. Yilmaz P, Ikram MA, Ikram MK, Niessen WJ, Viswanathan A, Charidimou A, et al. Application of an imaging-based sum score for cerebral amyloid angiopathy to the general population: risk of major neurological diseases and mortality. Front Neurol. (2019) 10:1276. doi: 10.3389/fneur.2019.01276
62. An SJ, Kim TJ, Yoon BW. Epidemiology, risk factors, and clinical features of intracerebral hemorrhage: an update. J Stroke. (2017) 19:3–10. doi: 10.5853/jos.2016.00864
63. Mustanoja S, Strbian D, Putaala J, Meretoja A, Curtze S, Haapaniemi E, et al. Association of prestroke statin use and lipid levels with outcome of intracerebral hemorrhage. Stroke. (2013) 44:2330–2. doi: 10.1161/STROKEAHA.113.001829
Keywords: Alzheimer's disease, apolipoprotein ϵ allele, cerebral amyloid angiopathy, cortical superficial siderosis, recurrent lobar brain hemorrhages, hypobetalipoproteinemia, 18F-THK5351 PET, 11C-PiB PET
Citation: Ikeda M, Okamoto K, Suzuki K, Takai E, Kasahara H, Furuta N, Furuta M, Tashiro Y, Shimizu C, Takatama S, Naito I, Sato M, Sakai Y, Takahashi M, Amari M, Takatama M, Higuchi T, Tsushima Y, Yokoo H, Kurabayashi M, Ishibashi S, Ishii K and Ikeda Y (2021) Recurrent Lobar Hemorrhages and Multiple Cortical Superficial Siderosis in a Patient of Alzheimer's Disease With Homozygous APOE ε2 Allele Presenting Hypobetalipoproteinemia and Pathological Findings of 18F-THK5351 Positron Emission Tomography: A Case Report. Front. Neurol. 12:645625. doi: 10.3389/fneur.2021.645625
Received: 15 January 2021; Accepted: 05 May 2021;
Published: 07 July 2021.
Edited by:
Achim Gass, University of Heidelberg, GermanyReviewed by:
Katsutoshi Furukawa, Tohoku University, JapanCopyright © 2021 Ikeda, Okamoto, Suzuki, Takai, Kasahara, Furuta, Furuta, Tashiro, Shimizu, Takatama, Naito, Sato, Sakai, Takahashi, Amari, Takatama, Higuchi, Tsushima, Yokoo, Kurabayashi, Ishibashi, Ishii and Ikeda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masaki Ikeda, aWtlZGFtOEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.