 Teodor M. Svedung Wettervik
Teodor M. Svedung Wettervik Anders Lewén
Anders Lewén Per Enblad
Per Enblad- Department of Neuroscience, Section of Neurosurgery, Uppsala University, Uppsala, Sweden
Neurointensive care (NIC) has contributed to great improvements in clinical outcomes for patients with severe traumatic brain injury (TBI) by preventing, detecting, and treating secondary insults and thereby reducing secondary brain injury. Traditional NIC management has mainly focused on generally applicable escalated treatment protocols to avoid high intracranial pressure (ICP) and to keep the cerebral perfusion pressure (CPP) at sufficiently high levels. However, TBI is a very heterogeneous disease regarding the type of injury, age, comorbidity, secondary injury mechanisms, etc. In recent years, the introduction of multimodality monitoring, including, e.g., pressure autoregulation, brain tissue oxygenation, and cerebral energy metabolism, in addition to ICP and CPP, has increased the understanding of the complex pathophysiology and the physiological effects of treatments in this condition. In this article, we will present some potential future approaches for more individualized patient management and fine-tuning of NIC, taking advantage of multimodal monitoring to further improve outcome after severe TBI.
Introduction
In the later decades of the twentieth century, the understanding of the critical factors responsible for neurological deterioration after traumatic brain injury (TBI) increased (1). It became clear that although the primary brain injury after TBI cannot be cured, secondary insults leading to further brain injury can be avoided by vigilant monitoring with attentive prevention and early treatment of such insults. This led to the development of standardized management protocols for severe TBI at specialized neurointensive care (NIC) units, resulting in significant improvements in the clinical outcome for these patients (2, 3).
The concept of NIC, to avoid secondary brain injury by intensive monitoring and aggressive treatment of emerging secondary insults, is still the fundamental basis for modern NIC. The purpose of this presentation was to give an overview of potential directions toward the NIC of the future. The structure of this review is based on the critical steps to achieve sufficient cerebral energy metabolism for neuronal survival in relation to multimodality monitoring (Figure 1).
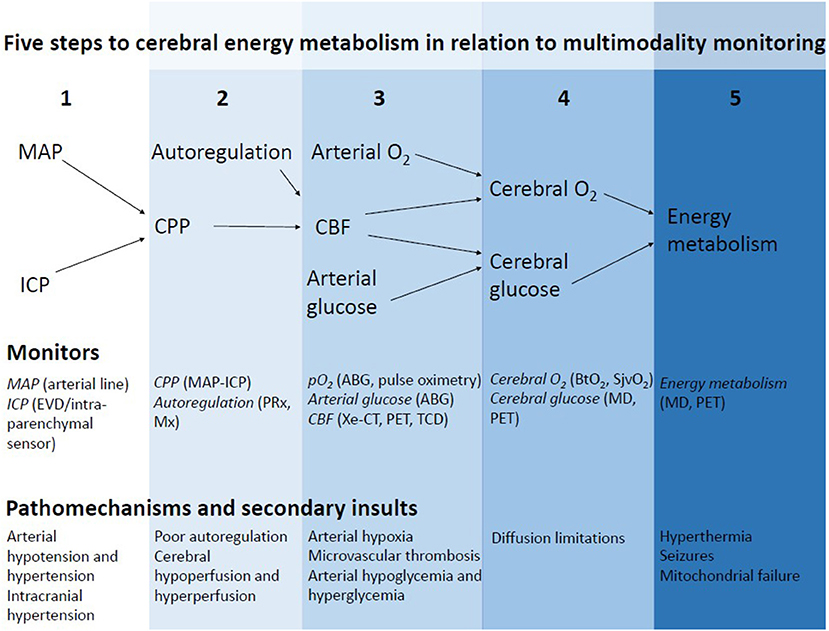
Figure 1. Five critical steps in the pathway to cerebral energy metabolism in relation to multimodality monitoring. By monitoring all the steps in this pathway to achieve sufficient cerebral energy metabolism, energy failure may be detected in time and the correct diagnosis can be made. Treatments should aim at counteracting the specific mechanism that causes cerebral energy metabolic disturbances.
Traditional Treatment Targets in Neurointensive Care
Intracranial Pressure—Step 1
In the early NIC, the main focus was to avoid high intracranial pressure (ICP). The basic understanding of ICP dynamics was derived from the Monro–Kellie hypothesis, indicating that ICP will increase due to the progression of posttraumatic intracranial hemorrhages or brain edema when there is no more compensatory reserve to reduce any other intracranial volume (4). Severe intracranial hypertension leads to brain herniation syndromes, cerebral hypoperfusion, and, ultimately, fatal outcome if untreated. In awake TBI patients, progressing intracranial hypertension may be detected by clinical evaluation as neurological deterioration, but it may be much more difficult to detect this in time in already comatose patients who are intubated and sedated.
Invasive ICP monitoring was introduced in the 1950s by Guillaume (5), further developed in the 1960s by Lundberg (6), and has ever since been used in NIC (Tables 1, 2, Figure 1). Over the years, the definition of what ICP threshold dichotomizes acceptable and dangerous ICP elevation has been extensively debated (7, 13, 14), and the value of ICP monitoring has also been questioned (15). However, the current Brain Trauma Foundation (BTF) guidelines recommend ICP monitoring in unconscious TBI patients and suggest an upper threshold at 22 mmHg (7). In the case of higher ICP, lowering therapies such as evacuation of significant intracranial hematomas, mild hyperventilation, cerebrospinal fluid (CSF) drainage, barbiturates, and decompressive craniectomy may be used in tiered protocols (7, 16, 17).
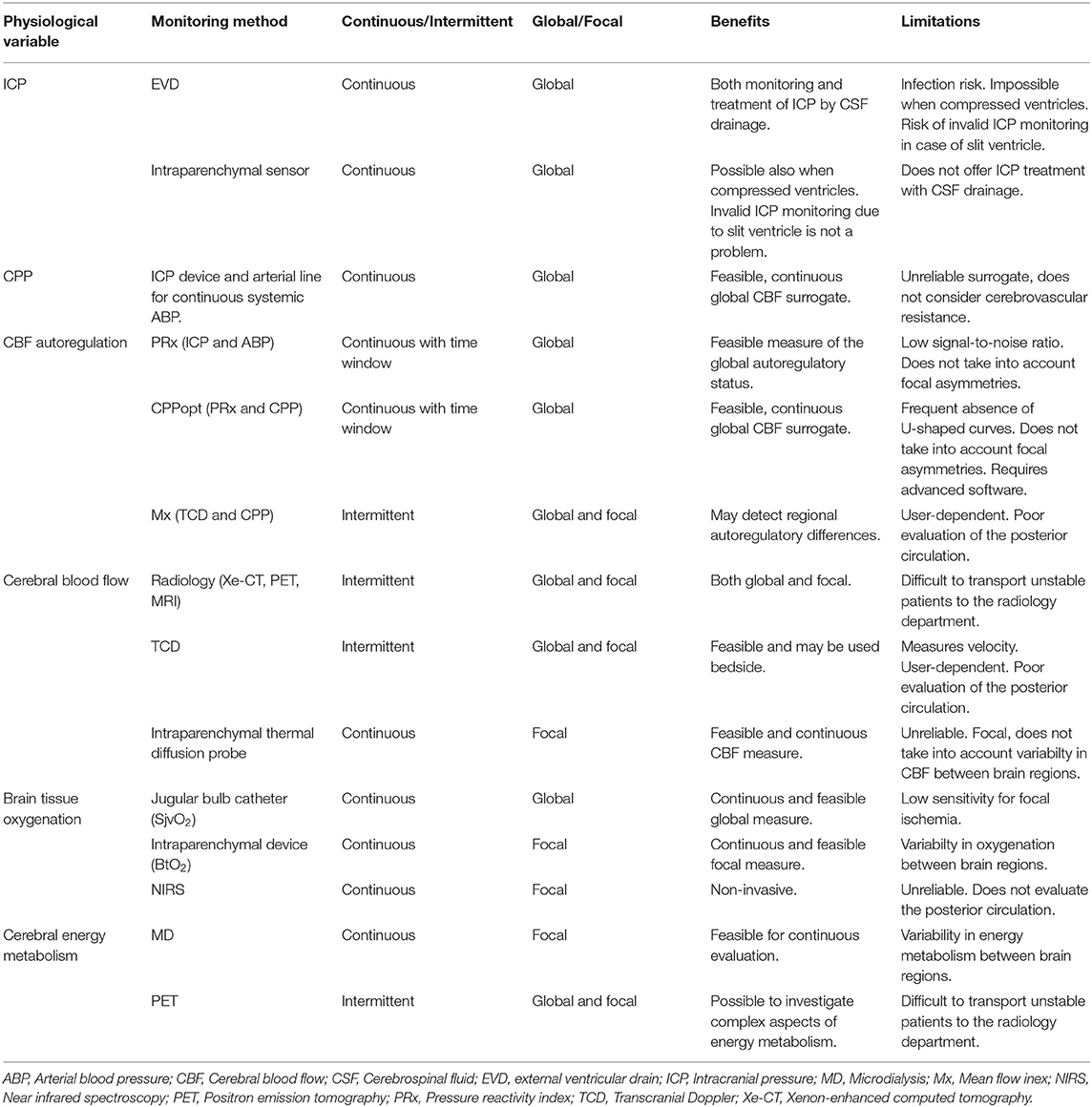
Table 1. Multimodality monitoring—a selection of methods and their benefits and limitations.
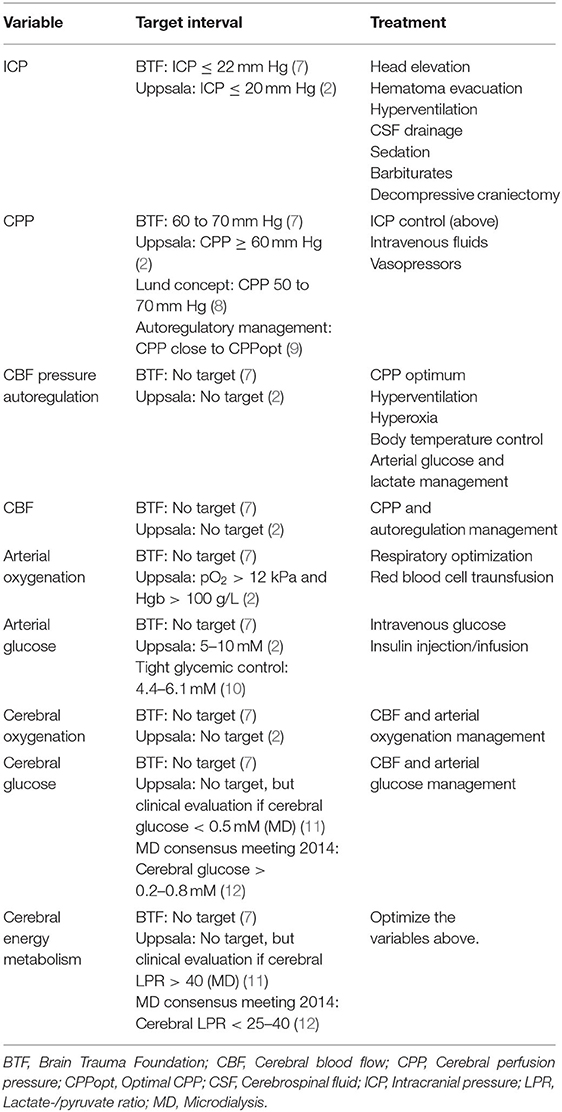
Table 2. Monitoring variables, target intervals, and treatments.
Cerebral Perfusion Pressure—Step 2
Cerebral perfusion pressure (CPP), defined as the pressure gradient between the mean arterial blood pressure (MAP) and ICP, is another important treatment target in TBI during NIC (Tables 1, 2, Figure 1). CPP is considered a surrogate measure for global cerebral blood flow (CBF). Early studies stressed the importance of keeping CPP at high levels above 70 mmHg (18) to avoid cerebral ischemia and counteract the vasodilatory cascade. However, the Lund concept suggested that CPP values as low as 50 mmHg can usually be tolerated, whereas values above 70 mmHg may induce brain edema (8). Today, the current guidelines recommend a CPP between 60 and 70 mmHg as a balance between the risk of cerebral hypo- and hyperperfusion (7).
Systemic Monitoring Variables
In traditional NIC of TBI, ICP and CPP have been the two main treatment targets, but other variables have also been considered, e.g., to maintain sufficient arterial oxygenation (pO2 ≥ 12 kPa), to avoid systemic hyperthermia (T < 38°C), and to keep arterial glucose within normal limits (5–10 mM) (2, 19). Figure 2 demonstrates the Uppsala standardized TBI management protocol as an example of a local escalated ICP-oriented management protocol with a focus on avoiding secondary insults (2, 11, 19).
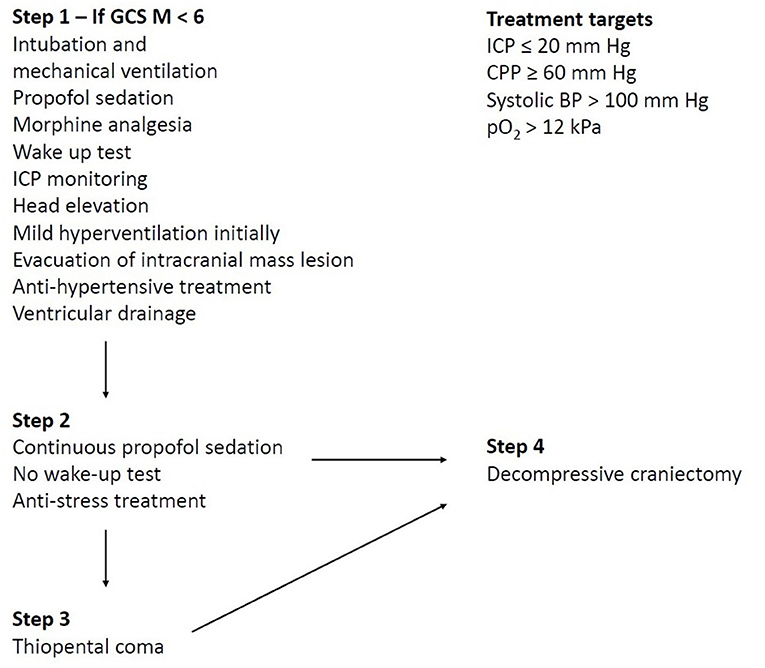
Figure 2. The Uppsala standardized traumatic brain injury (TBI) management protocol as an example of a local escalated intracranial pressure (ICP)-oriented management protocol with a focus on avoiding secondary insults. The figure demonstrates an example of a local ICP-oriented management protocol (2). The therapeutic intensity is gradually increased by starting with ICP-lowering treatments that carry a lower risk of complications and escalating to treatments with greater risks if the ICP target cannot otherwise be controlled. Decompressive craniectomy may be performed both on a primary indication (first surgical procedure, i.e., the fourth step is used as the first step) in the case of severe brain edema in the early course, but chiefly on a secondary indication (when all other treatments are exhausted).
Indicative Observations and Future Perspectives
Multimodality Monitoring of the Complex Pathophysiology in Traumatic Brain Injury
The traditional NIC treatment targets in TBI, ICP, and CPP, are two important surrogate measures of the cerebral environment, but many other variables are also crucial steps in the pathway to achieve an optimal cerebral energy metabolic state (Figure 1). CBF delivers oxygen and glucose to the injured brain. The autoregulation of CBF by the cerebral vessels is often disturbed after TBI and focal CBF disturbances may occur despite normal ICP and CPP (20–24). In addition, systemic traumatic injuries could precipitate for arterial hypoxia with secondary brain tissue hypoxia despite normal CBF in the large cerebral vessels. Furthermore, microvascular thrombosis and diffusion limitations from brain edema may limit oxygen delivery in the microvascular circulation despite normal macrovascular CBF and arterial oxygenation (25, 26). However, even if brain tissue oxygenation and the level of energy metabolites are at adequate levels, cerebral energy failure may still occur due to mitochondrial dysfunction (26–28). In addition, mechanisms such as seizures and hyperthermia increase energy consumption and may be detrimental if the compensatory cerebral energy reserve is compromised (29–33).
Multimodality monitoring makes it possible to continuously surveil the different steps in the pathway to cerebral energy metabolism (Tables 1, 2 and Figure 1). The methods used are both global and focal, as well as continuous and intermittent. Some methods give direct information about the crucial parameters for the energy metabolism and others provide indirect measures. The complete picture is obtained by an integrated analysis of all measures. The future direction of NIC is a more detailed analysis of each of the crucial pathways for cerebral energy metabolism using multimodality monitoring in order to identify the correct cause of energy metabolic disturbances (Table 3) (34). This may in turn lead to a more timely and cause-specific treatment. The following sections will review the background and potential future applications of some monitoring techniques for the fine-tuning of next-generation NIC.
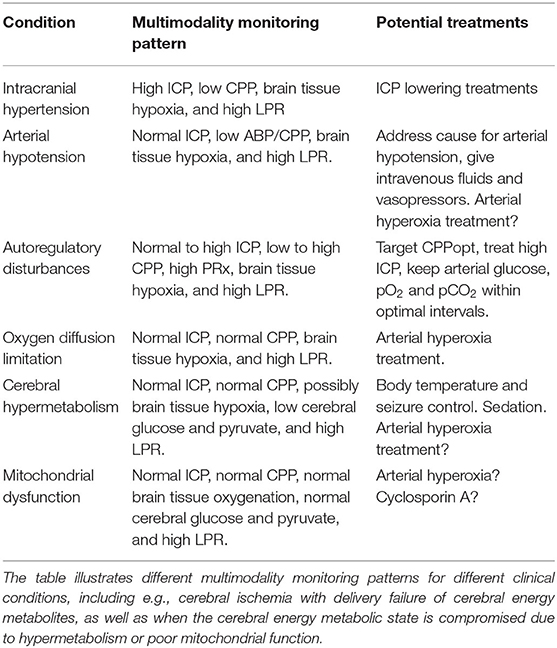
Table 3. Secondary brain injury conditions, multimodality monitoring patterns and potential cause-specific treatments.
CBF Pressure Autoregulation and CPP Management—Step 2
Global and regional CBF can be measured intermittently with different imaging techniques, and regional CBF can also be measured continuously by means of an intraparenchymal probe (Table 1). It would be advantageous to continuously monitor global CBF, but there is, at present, no feasible way. CPP is currently the best surrogate measure for global CBF, but this concept leaves the cerebrovascular reactivity out of the equation, and there is an interest to find better surrogate measures of CBF that takes both CPP and the cerebrovascular status into account.
Lassen described the cerebral autoregulation in 1959 as he demonstrated that CBF is maintained over a wide range of MAPs (35, 36). The autoregulatory capacity may become deranged following TBI, which is strongly associated with poor outcome (14, 21, 37). Several methods to monitor the cerebral autoregulation in the NIC have been introduced (Table 1), such as the mean flow index (Mx) and the pressure reactivity index (PRx). PRx has received the greatest interest and is continuously measured as the correlation coefficient between MAP and ICP over 5 min (37). Negative index values indicate intact pressure autoregulation, such as when an increase in MAP leads to cerebral vasoconstriction to maintain normal CBF with a corresponding reduction in CBV and ICP. Positive index values indicate disturbed pressure autoregulation, such as when an increase in MAP leads to passive cerebral vasodilation with corresponding increases in CBF, CBV, and ICP (37–39). The clinical utility of PRx is still under development. However, as low PRx values are associated with better clinical outcome, autoregulatory-oriented management that aims at improving PRx has been suggested, as outlined below.
Autoregulatory management aims at giving the patient treatments that improve the autoregulatory capacity. Particularly, the association between pressure autoregulation and CPP has received interest. In a first attempt to consider autoregulation in CPP management, patients were classified as either pressure active or pressure passive, and the absolute autoregulatory status was suggested to determine whether the patient would benefit from high or low CPP (40). It was then demonstrated in an observational study including two centers with different CPP philosophy (Uppsala and Edinburgh) that patients with pressure-passive cerebral vessels (high PRx) had better outcomes if treated with ICP-oriented therapy with relatively lower CPP targets, whereas pressure-active patients (low PRx) benefitted from CPP-oriented treatment with relatively higher CPP targets (41). In the next attempt, it was found that PRx varies with CPP in a U-shaped way and that the CPP with the concurrently lowest PRx could be targeted where autoregulation works best (22), as demonstrated in Figure 3. Several studies have supported that deviation of the absolute CPP above and below the optimal (CPPopt) is associated with poor outcomes (24, 42–45). Furthermore, brain tissue oxygenation reaches a plateau when CPP approaches CPPopt, indicating optimal CBF (46). These findings support that CPPopt may be a better CBF surrogate that takes into account both the absolute CPP and the cerebrovascular status for the individual patient.
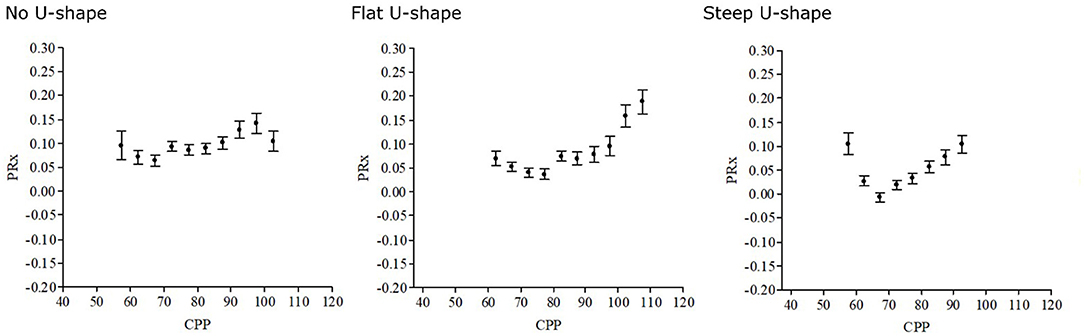
Figure 3. Optimal cerebral perfusion pressure (CPPopt)—differences in curve shape. The CPPopt curves may vary over time and between patients, as illustrated in these three different curves. There are questions on how the curve shape and the absolute pressure reactivity index (PRx) are relevant in CPPopt management. Patients with a steeper CPPopt curve may, in theory, be more vulnerable to changes in CPP.
However, despite these promising findings, several concerns have been raised regarding CPPopt that need to be addressed in future trials. The U-shaped association between CPP and PRx is absent during ~40% of the monitoring time (47), which limits the feasibility of CPPopt as a target (48). Secondly, the appearance of the U-shaped curve may vary from flat to steep and the absolute PRx at CPPopt may vary from negative to positive values (Figure 3). The necessity to keeping CPP close to CPPopt probably depends on the curve shape and the absolute PRx (42). For example, if CPPopt is high, the absolute PRx is negative, and the U-shaped curve is relatively flat, the Lassen plateau phase of autoregulation should be acceptably wide and a much lower absolute CPP below CPPopt could then be allowed since higher targets are otherwise associated with complications such as acute respiratory distress syndrome (49). On the contrary, it could be more critical to keep CPP close to CPPopt if the curve is steep and PRx is high. Thirdly, although patients that spontaneously had CPP values close to CPPopt had better clinical outcome in retrospective studies (43), CPPopt as an active treatment target has so far not been sufficiently investigated prospectively (50). Particularly, CPPopt is often above 70 mmHg, i.e., above the upper fixed threshold according to the BTF guidelines (7), but using vasopressors to achieve such high targets could carry adverse effects. The safety and feasibility of CPPopt as a treatment target is investigated in a multicenter randomized controlled trial (COGiTATE) (9). The results of this trial will determine the future directions of CPP research and management in TBI care.
Autoregulatory Management and Systemic Physiology—Step 2
In addition to CPP, several other variables that control the cerebrovascular resistance are associated with pressure autoregulation and could potentially be targeted in an integrated autoregulatory protocol. Arterial hyperglycemia may disturb the endothelial and myogenic function of the cerebral vessels (51, 52). In line with these findings, we and others have demonstrated that arterial hyperglycemia is independently associated with a higher PRx (53, 54), indicating that a tighter glycemic control at least below 10 mM could be beneficial from an autoregulatory point of view. Furthermore, lactate is a cerebral vasodilator and higher systemic levels may increase CBF (55). However, we have found that a higher arterial lactate is independently associated with a higher PRx (33), and it is possible that the corresponding increase in CBF (55) represents dysregulated hyperemia. This calls for caution in clinical TBI trials on lactate-based fluids as a way of increasing the delivery of an alternative energy fuel to the brain due to the possible negative effect of lactate on pressure autoregulation. Hyperthermia could induce cerebral vasodilation, and some findings support an association between a higher body temperature and a higher PRx (56), whereas other studies have found no such association (33). Arterial hypoxia also leads to cerebral vasodilation in order to increase CBF to maintain normal brain tissue oxygenation (57), whereas higher oxygen levels may increase the cerebrovascular tone. We have found that higher arterial oxygen levels above 12 kPa are associated with lower PRx, i.e., better pressure autoregulation (58). Similarly, increased cerebrovascular tone by hyperventilation is associated with better pressure autoregulation (56, 59, 60).
Hence, several systemic variables, in addition to CPP, could potentially be targeted in an integrated autoregulatory protocol including, e.g., temperature management, glycemic control, and respiratory targets to optimize pO2 and pCO2.
Arterial and Brain Oxygenation—Steps 3 and 4
Ischemic and hypoxic secondary brain injuries are common after TBI (61), and treatment aims at maintaining adequate CBF and arterial oxygen content for aerobic energy metabolism. Arterial oxygen content is monitored with repeated arterial blood gas analyses of hemoglobin (Hgb) and pO2 and continuously with pulse oximetry (SpO2). The arterial oxygen content is, to a great extent, dependent on the level of oxygen-binding Hgb, whereas dissolved oxygen only constitutes a fraction of the total arterial oxygen content under normal conditions (62). There is great controversy regarding the definition of clinically relevant anemia for cerebral oxygen delivery, but an Hgb below 9 g/dl may discriminate when the risk of cerebral hypoxia is significantly increased (63). Although red blood cell transfusion (RBCT) could improve cerebral oxygen delivery (64), adverse effects such as a worsening in pressure autoregulation (65) and an increased risk of thromboembolic events have been described (66). Clinical studies on liberal vs. restricted RBCT based on Hgb thresholds (e.g., 10 vs. 7 g/dl) indicate a neutral to negative effect on clinical outcomes with the liberal approach (64, 66–69). The observations that both anemia and RBCT are independently associated with poor outcomes make the clinical management difficult, but it is possible that the indication for RBCT could be guided by means of multimodal monitoring. Despite, or because, the controversy in this matter, there is currently no BTF guidelines on RBCT in severe TBI (7).
There is also limited knowledge and lack of guidelines regarding specific arterial oxygen thresholds (7), but pO2 levels above 12 kPa and SaO2 above 95% may be targeted (2). Additionally, brain tissue oxygenation can be monitored globally with a jugular vein bulb (SjvO2) and focally with an intraparenchymal probe (BtO2) (Table 1). BTF currently recommends SjvO2 monitoring and avoiding ischemic SjvO2 levels below 50% (7). There is no BTF recommendation on BtO2 monitoring (7), but focal brain tissue hypoxia below 20 mmHg has been associated with poor clinical outcomes (70).
Although there is only a small fraction of free and dissolved pO2 in the blood (62), an increase in pO2 to hyperoxic levels could still be beneficial in TBI. Increasing the fraction of the inspired oxygen (FiO2), i.e., normobaric hyperoxia (NBO), has been proven to increase arterial and brain tissue oxygenation (71). NBO may then compensate for ischemic hypoxia, overcome diffusion barriers, and improve mitochondrial function (62). Hyperoxia reduces cerebral glycolytic enzymes and thereby decreases pyruvate and lactate (57, 71, 72), but there are questions whether cerebral oxidative energy metabolism improves. A PET study found that hyperoxia improves energy metabolism in ischemic brain regions (73), and microdialysis (MD) studies support true energy metabolic improvements in the case of poor substrate supply (58) or anaerobic energy metabolism (74). On the other end, some studies suggest that hyperoxia induces detrimental reactive oxygen species that negatively affect cerebral tissue survival, although this may only occur to a small extent in a limited number of patients (75). Brain tissue oxygen-guided treatment protocols have been introduced, in which low brain tissue oxygenation may be treated with an increase in FiO2. However, there is still clinical equipoise of this approach due to small positive to neutral effects on clinical outcomes in prospective trials (76–78).
Delivery of Cerebral Energy Metabolites, Cerebral Energy Metabolism, Neurochemical Monitoring, and Treatments—Steps 3, 4, and 5
Cerebral nutrients, such as glucose and lactate, are delivered by the CBF. The arterial levels may be monitored with repeated arterial blood gases to ensure sufficient arterial content. The cerebral energy metabolites, glucose, pyruvate, and lactate, as well as the rate of oxidative energy metabolism (lactate/pyruvate ratio, LPR) may be evaluated with hourly measures by means of cerebral MD, i.e., a double-lumen, semi-permeable catheter that is perfused with artificial CSF (79, 80). The catheter can either be placed in macroscopically normal-appearing brain tissue to estimate global cerebral energy metabolism or in peri-contusional areas to estimate focal cerebral energy metabolism in “tissue-at-risk” (81). Recently, the new MD system (Loke) has been introduced, which measures cerebral glucose, pyruvate, and lactate minute by minute. This may reveal the complex explanatory variables for cerebral energy metabolism in higher resolution. The cerebral energy metabolism may also be evaluated with radiological snapshots using PET (Table 1).
Glucose is the main cerebral energy fuel. The arterial glucose level and CBF determine cerebral glucose delivery, but the cerebral glucose level also depends on the cerebral energy metabolic rate. Arterial and cerebral glucose are normally correlated, but this association may be disturbed after TBI (33, 54, 82–84) as the CBF and cerebral energy metabolic rate could have greater influence on cerebral glucose levels. The immediate effects of TBI include a sympathetic stress response that, among other things, gives rise to a surge in arterial glucose (85). This is beneficial since it is necessary to avoid arterial hypoglycemia and neuroglycopenia for the vulnerable brain, but arterial hyperglycemia is also associated with worse outcomes after TBI (86–88). Although the latter association could reflect more severe underlying traumatic injuries, hyperglycemia could, per se, induce secondary brain injury by causing disturbances in cerebral autoregulation and mitochondrial function (54, 89). Since both too low and too high arterial glucose could exert a negative effect on the brain, tight glycemic control with intensive insulin therapy (IIT) management has been suggested in TBI care. IIT showed promising results in general ICU patients at an early stage (10, 90, 91), but recent studies show that IIT causes an increased burden of arterial hypoglycemia that outweighs the benefits of avoiding hyperglycemia, resulting in a neutral to negative net effect on outcomes in various ICU populations as well as in TBI (92–97). Specifically, TBI patients treated with IIT developed more severe energy metabolic disturbances with reductions in cerebral glucose and oxidative energy metabolism than those treated with conventional glycemic control (95, 98). Hence, a narrow arterial glucose interval is appealing in theory, but is currently not feasible due to the risk of overtreatment. BTF does not have any recommendation on optimal glucose levels or management in TBI (7). Due to the lack of clear evidence of benefits for IIT in the NIC setting, a looser glycemic control is mostly applied, e.g., between 5 and 10 mM (99). Cerebral MD glucose below 0.2–0.8 mM is considered dangerously low, and such thresholds could aid in guiding treatment for when the termination of insulin treatment or infusion of glucose is warranted (12).
The understanding of arterial lactate as an additional cerebral energy substrate (100) has increased in the last years. However, the arterial contribution to cerebral lactate is only 10% when the arterial lactate concentration is 1 mM, but increases to 60% at 5 mM (101). An increased consumption of lactate as an energy fuel may, in turn, spare cerebral glucose (55, 102, 103). Higher lactate decreases the cerebrovascular tone, induces vasodilation, and increases CBF (55, 104), but possibly at the expense of a worsened pressure autoregulation (33). Exogenous, hypertonic lactate may exert anti-edematous effects and alleviate intracranial hypertension (102). Animal TBI studies support a reduction in lesion volume after exogenous lactate infusion (105). Clinical trials also indicate that exogenous lactate improves outcomes (106), although a higher endogenous arterial lactate is independently associated with worse outcomes after TBI (33). Future studies are needed to determine the role of intravenous lactate supplement in TBI care. Nevertheless, high endogenous levels of cerebral lactate above 4 mM indicate anaerobic energy metabolism and are associated with unfavorable clinical outcomes (12, 107).
Furthermore, high cerebral MD LPRs above 25–40 indicate disturbed cerebral energy metabolism (12, 107). The etiology for LPR elevations may differ. A high LPR with concurrently low cerebral glucose and pyruvate has been suggested to reflect cerebral ischemia/substrate delivery failure, whereas a high LPR with concurrently normal/high cerebral glucose and pyruvate has been suggested to reflect mitochondrial dysfunction (26). Substrate delivery could, in theory, be improved by a higher CPP, improved pressure autoregulation, and higher arterial substrate content (Table 3). The importance of mitochondrial dysfunction has gained increased interest and understanding in recent years. There is currently no treatment for this condition, but cyclosporine A, which is an immunosuppressant, improves the mitochondrial function in animal TBI models and has demonstrated some promise in preliminary human trials, but larger studies are needed to evaluate its efficacy (108, 109).
MD may also aid in monitoring more complex dynamic pathophysiological processes in the brain following TBI. Neuroinflammation after TBI may exert both beneficial and negative effects on the brain, depending on the time window after injury and the specific mechanisms (110). MD can be used to evaluate neuroinflammatory biomarkers bedside in order to better understand the neuroinflammatory mechanisms, their relation to the dynamic clinical course, secondary insults, and clinical outcomes in TBI (111, 112). This may aid in the development of optimal neuroprotective agents, as outlined below, and the cerebral MD biomarkers could potentially be used to improve the indication for such treatments.
Neuroprotective Agents
In addition to the secondary physiological insults, there is a cascade of injury mechanisms on the cellular and molecular levels after the primary brain injury, e.g., toxic release of excitatory neurotransmitters, apoptotic pathways, neuroinflammation, and mitochondrial dysfunction (113). Many neuroprotective agents have been developed to counteract these pathomechanisms, but there has so far been limited success in translation from animal to clinical studies (113). For example, N-methyl-D-aspartate (NMDA) receptor antagonists and calcium channel antagonists both reduce excitotoxicity in animal TBI models, but their efficacy in human TBI trials has been limited (114, 115). Human TBI trials on immunomodulating agents have also been disappointing. Progesterone had a neutral effect (116), whereas corticosteroids even worsened clinical outcomes (117). As outlined above, cyclosporine A has demonstrated some promise in preliminary human trials, but larger studies are needed to evaluate its efficacy (108, 109).
One reason for the so far disappointing results of neuroprotective drugs in humans may be that the therapeutic time window is limited, and it may be difficult to administer the treatments within the right time in the clinical setting. Furthermore, TBI in humans is very heterogeneous, and the subtype may be of importance for the therapeutic effect. Animal trials are often based on relatively homogenous TBI models, while the clinical trials often have included all types of TBI patients. Many pharmacological trials have also been done in the dawn of NIC, when secondary insults were not yet as efficiently surveyed and managed as today, and the relative importance of these agents might have been less significant in that setting. In conclusion, despite the limited effect of the neuroprotective agents shown in human TBI trials, it is possible that neuroprotective agents with a more optimal treatment timing, better understanding of the injury processes, and an improved patient selection in a high-quality NIC setting could yield improved outcomes. Future studies that take these aspects into account are needed.
Multimodality Monitoring and Precision Medicine
The multimodality monitoring pattern may reveal the underlying etiology of cerebral energy failure following TBI so that cause-specific treatments may be initiated (Table 3). For example, in cases of intracranial hypertension, low CPP, brain tissue hypoxia, and cerebral energy failure, treatments should aim at lowering ICP to improve the cerebral environment. In other cases with normal ICP, CPP within optimal targets, and normal arterial oxygenation, but poor brain tissue oxygenation and concurrent cerebral energy metabolic failure (Figure 4), arterial hyperoxia could be used to possibly overcome cerebral diffusion barriers. In cases where all variables such as ICP, CPP, and brain tissue oxygenation are within the targeted intervals, except for the energy metabolic state (LPR), mitochondrial dysfunction is a plausible explanation. There is currently no treatment for this condition, but there are ongoing trials for, e.g., neuroprotective agents that may relieve mitochondrial dysfunction.
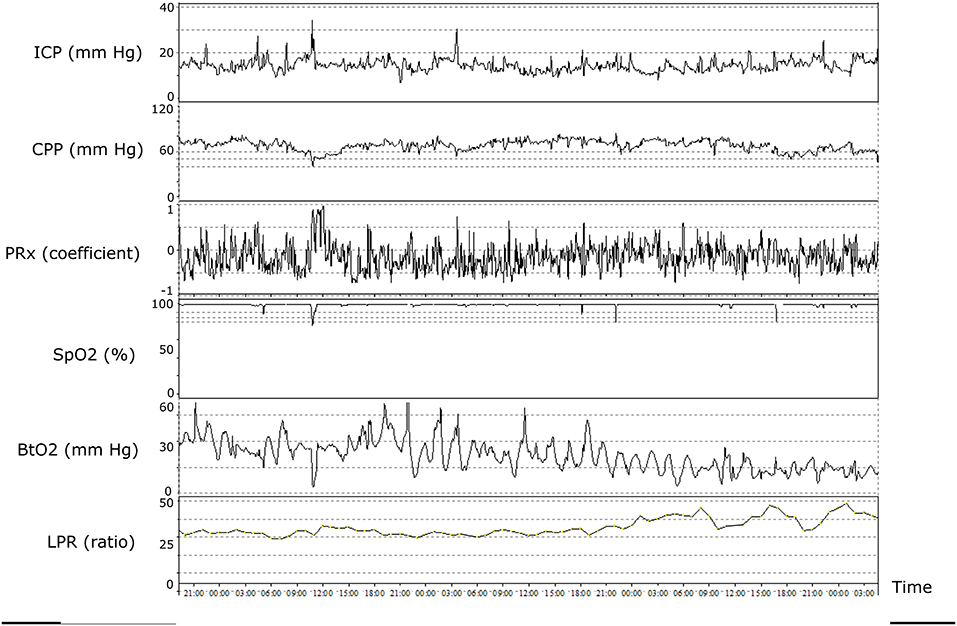
Figure 4. Illustrative patient case with multimodal monitoring. The figure demonstrates the temporal evolution of cerebral physiology over ~2 days. Although intracranial pressure (ICP), cerebral perfusion pressure (CPP), pressure reactivity index (PRx), and oxygen saturation (SpO2) were within adequate intervals, the patient developed brain tissue hypoxia and cerebral energy failure (high lactate/pyruvate ratio). The pathophysiology may include microvascular thrombosis and/or increased diffusion limitations from cerebral edema.
Cerebral Monitoring in a Resource-Limited Setting
High-resolution multimodal monitoring offers the best method to fully evaluate the cerebral environment after TBI, but such tools may be limited to a few research-minded academic centers. There is, hence, also an interest in more feasible monitoring tools. PRx and CPPopt both require high-resolution data and advanced software. However, low-frequency autoregulation index (LAx) and the corresponding LAx-derived CPPopt are based on minute-by-minute data, require less advance software, and may be a fair substitute for centers without access to PRx and CPPopt (118). Transcranial Doppler is also a feasible noninvasive and accessible method to assess cerebral blood flow velocity, particularly in the absence of CPP and advanced radiological imaging, and could be used to guide blood pressure management.
Concluding Remarks
Traditional NIC has focused on reducing secondary brain injury by treating elevated ICP and maintaining the CPP sufficiently high to avoid cerebral ischemia. However, recent findings support that the TBI pathophysiology is much more complex and cerebral energy failure frequently occurs in the absence of intracranial hypertension and low CPP. Particularly, the roles of pathomechanisms such as disturbances in pressure autoregulation, microvascular thrombosis, oxygen diffusion limitations, and mitochondrial dysfunction have gained increased understanding. By multimodal monitoring, cerebral energy metabolic failure may be detected earlier and its etiology could be better diagnosed. This may, in turn, lead to precision medicine with more cause-specific treatments to avoid secondary brain injury for these patients. Future studies are needed to elaborate on the strengths and limitations of certain monitoring tools and their role in guiding cause-specific treatments in NIC of severe TBI.
Author Contributions
TS: conceptualization, writing–original draft. AL: conceptualization, writing–review and editing. PE: conceptualization, supervision, writing–review and editing. All authors contributed to the article and approved the submitted version.
Funding
The research was supported by the Department of Neuroscience/Neurosurgery, Uppsala University, Sweden.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rose J, Valtonen S, Jennett B. Avoidable factors contributing to death after head injury. Br Med J. (1977) 2:615–8. doi: 10.1136/bmj.2.6087.615
2. Elf K, Nilsson P, Enblad P. Outcome after traumatic brain injury improved by an organized secondary insult program and standardized neurointensive care. Crit Care Med. (2002) 30:2129–34. doi: 10.1097/00003246-200209000-00029
3. Patel HC, Menon DK, Tebbs S, Hawker R, Hutchinson PJ, Kirkpatrick PJ. Specialist neurocritical care and outcome from head injury. Int Care Med. (2002) 28:547–53. doi: 10.1007/s00134-002-1235-4
4. Mokri B. The Monro-Kellie hypothesis applications in CSF volume depletion. Neurology. (2001) 56:1746–8. doi: 10.1212/WNL.56.12.1746
5. Guillaume J, Janny P. Continuous intracranial manometry; physiopathologic and clinical significance of the method. La Presse medicale. (1951) 59:953–5.
6. Lundberg N, Troupp H, Lorin H. Continuous recording of the ventricular-fluid pressure in patients with severe acute traumatic brain injury. A preliminary report. J Neurosurg. (1965) 22:581–90. doi: 10.3171/jns.1965.22.6.0581
7. Carney N, Totten AM, O'Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery. (2017) 80:6–15. doi: 10.1227/NEU.0000000000001432
8. Grande PO. Critical evaluation of the lund concept for treatment of severe traumatic head injury, 25 years after its introduction. Front Neurol. (2017) 8:315. doi: 10.3389/fneur.2017.00315
9. Beqiri E, Smielewski P, Robba C, Czosnyka M, Cabeleira MT, Tas J, et al. Feasibility of individualised severe traumatic brain injury management using an automated assessment of optimal cerebral perfusion pressure: the COGiTATE phase II study protocol. BMJ Open. (2019) 9:e030727. doi: 10.1136/bmjopen-2019-030727
10. van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, et al. Intensive insulin therapy in critically ill patients. N Engl J Med. (2001) 345:1359–67. doi: 10.1056/NEJMoa011300
11. Svedung Wettervik T. Aspects on Mechanisms, Treatment and Outcome in Severe Traumatic Brain Injury. Uppsala: Acta Universitatis Upsaliensis (2020).
12. Hutchinson PJ, Jalloh I, Helmy A, Carpenter KL, Rostami E, Bellander BM, et al. Consensus statement from the 2014 International Microdialysis Forum. Int Care Med. (2015) 41:1517–28. doi: 10.1007/s00134-015-3930-y
13. Bratton SL, Chestnut RM, Ghajar J, McConnell Hammond FF, Harris OA, Hartl R, et al. Guidelines for the management of severe traumatic brain injury. IX. Cerebral perfusion thresholds. J Neurotrauma. (2007) 24 Suppl 1:S59–64. doi: 10.1089/neu.2007.9987
14. Sorrentino E, Diedler J, Kasprowicz M, Budohoski KP, Haubrich C, Smielewski P, et al. Critical thresholds for cerebrovascular reactivity after traumatic brain injury. Neurocritical Care. (2012) 16:258–66. doi: 10.1007/s12028-011-9630-8
15. Chesnut RM, Temkin N, Carney N, Dikmen S, Rondina C, Videtta W, et al. A trial of intracranial-pressure monitoring in traumatic brain injury. N Engl J Med. (2012) 367:2471–81. doi: 10.1056/NEJMoa1207363
16. Hawryluk GWJ, Aguilera S, Buki A, Bulger E, Citerio G, Cooper DJ, et al. A management algorithm for patients with intracranial pressure monitoring: the Seattle International Severe Traumatic Brain Injury Consensus Conference (SIBICC). Int Care Med. (2019) 45:1783–94. doi: 10.1007/s00134-019-05805-9
17. Hawryluk GWJ, Rubiano AM, Totten AM, O'Reilly C, Ullman JS, Bratton SL, et al. Guidelines for the Management of Severe Traumatic Brain Injury: 2020 update of the decompressive craniectomy recommendations. Neurosurgery. (2020) 87:427–34. doi: 10.1093/neuros/nyaa278
18. Rosner MJ, Rosner SD, Johnson AH. Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg. (1995) 83:949–62. doi: 10.3171/jns.1995.83.6.0949
19. Wettervik TS, Lenell S, Nyholm L, Howells T, Lewen A, Enblad P. Decompressive craniectomy in traumatic brain injury: usage and clinical outcome in a single centre. Acta Neurochirurg. (2018) 160:229–37. doi: 10.1007/s00701-017-3418-3
20. Paulson OB, Strandgaard S, Edvinsson L. Cerebral autoregulation. Cerebrovasc Brain Metab Rev. (1990) 2:161–92.
21. Czosnyka M, Smielewski P, Kirkpatrick P, Menon DK, Pickard JD. Monitoring of cerebral autoregulation in head-injured patients. Stroke. (1996) 27:1829–34. doi: 10.1161/01.STR.27.10.1829
22. Czosnyka M, Smielewski P, Piechnik S, Steiner LA, Pickard JD. Cerebral autoregulation following head injury. J Neurosurg. (2001) 95:756–63. doi: 10.3171/jns.2001.95.5.0756
23. Zeiler FA, Ercole A, Czosnyka M, Smielewski P, Hawryluk G, Hutchinson PJA, et al. Continuous cerebrovascular reactivity monitoring in moderate/severe traumatic brain injury: a narrative review of advances in neurocritical care. Br J Anaesth. (2020) 124:440–53. doi: 10.1016/j.bja.2019.11.031
24. Svedung Wettervik TM, Howells T, Enblad P, Lewén A. Temporal neurophysiological dynamics in traumatic brain injury-the role of pressure reactivity and optimal cerebral perfusion pressure for predicting outcome. J Neurotr. (2019) 36:1818–27. doi: 10.1089/neu.2018.6157
25. Veenith TV, Carter EL, Geeraerts T, Grossac J, Newcombe VF, Outtrim J, et al. Pathophysiologic mechanisms of cerebral ischemia and diffusion hypoxia in traumatic brain injury. JAMA Neurol. (2016) 73:542–50. doi: 10.1001/jamaneurol.2016.0091
26. Lazaridis C. Cerebral oxidative metabolism failure in traumatic brain injury: “Brain shock”. J Crit Care. (2017) 37:230–3. doi: 10.1016/j.jcrc.2016.09.027
27. Vespa P, Bergsneider M, Hattori N, Wu HM, Huang SC, Martin NA, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. (2005) 25:763–74. doi: 10.1038/sj.jcbfm.9600073
28. Verweij BH, Muizelaar JP, Vinas FC, Peterson PL, Xiong Y, Lee CP. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg. (2000) 93:815–20. doi: 10.3171/jns.2000.93.5.0815
29. Mrozek S, Vardon F, Geeraerts T. Brain temperature: physiology and pathophysiology after brain injury. Anesthesiol Res Prac. (2012) 2012:989487. doi: 10.1155/2012/989487
30. Stocchetti N, Rossi S, Zanier ER, Colombo A, Beretta L, Citerio G. Pyrexia in head-injured patients admitted to intensive care. Int Care Med. (2002) 28:1555–62. doi: 10.1007/s00134-002-1513-1
31. Rossi S, Zanier ER, Mauri I, Columbo A, Stocchetti N. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J Neurol Neurosurg Psychiatry. (2001) 71:448–54. doi: 10.1136/jnnp.71.4.448
32. Wang CX, Stroink A, Casto JM, Kattner K. Hyperthermia exacerbates ischaemic brain injury. Int J Stroke. (2009) 4:274–84. doi: 10.1111/j.1747-4949.2009.00317.x
33. Svedung Wettervik TM, Engquist H, Lenell S, Howells T, Hillered L, Rostami E, et al. Systemic hyperthermia in traumatic brain injury-relation to intracranial pressure dynamics, cerebral energy metabolism, and clinical outcome. J Neurosurg Anesthesiol. (2020). doi: 10.1097/ANA.0000000000000695. [Epub ahead of print].
34. Le Roux P, Menon DK, Citerio G, Vespa P, Bader MK, Brophy GM, et al. Consensus summary statement of the international multidisciplinary consensus conference on multimodality monitoring in neurocritical care. Neurocritical care. (2014) 21:1–26. doi: 10.1007/s12028-014-0041-5
36. Lassen NA. Cerebral blood flow and oxygen consumption in man. Physiol Rev. (1959) 39:183–238. doi: 10.1152/physrev.1959.39.2.183
37. Czosnyka M, Smielewski P, Kirkpatrick P, Laing RJ, Menon D, Pickard JD. Continuous assessment of the cerebral vasomotor reactivity in head injury. Neurosurgery. (1997) 41:11–9. doi: 10.1097/00006123-199707000-00005
38. Brady KM, Lee JK, Kibler KK, Easley RB, Koehler RC, Shaffner DH. Continuous measurement of autoregulation by spontaneous fluctuations in cerebral perfusion pressure: comparison of 3 methods. Stroke. (2008) 39:2531–7. doi: 10.1161/STROKEAHA.108.514877
39. Zeiler FA, Donnelly J, Calviello L, Lee JK, Smielewski P, Brady K, et al. Validation of pressure reactivity and pulse amplitude indices against the lower limit of autoregulation, part I: experimental intracranial hypertension. J Neurotrauma. (2018) 35:2803–11. doi: 10.1089/neu.2017.5603
40. Lang EW, Chesnut RM. A bedside method for investigating the integrity and critical thresholds of cerebral pressure autoregulation in severe traumatic brain injury patients. Br J Neurosurg. (2000) 14:117–26. doi: 10.1080/02688690050004534
41. Howells T, Elf K, Jones PA, Ronne-Engstrom E, Piper I, Nilsson P, et al. Pressure reactivity as a guide in the treatment of cerebral perfusion pressure in patients with brain trauma. J Neurosurg. (2005) 102:311–7. doi: 10.3171/jns.2005.102.2.0311
42. Donnelly J, Czosnyka M, Adams H, Robba C, Steiner LA, Cardim D, et al. Individualizing thresholds of cerebral perfusion pressure using estimated limits of autoregulation. Crit Care Med. (2017) 45:1464–71. doi: 10.1097/CCM.0000000000002575
43. Steiner LA, Czosnyka M, Piechnik SK, Smielewski P, Chatfield D, Menon DK, et al. Continuous monitoring of cerebrovascular pressure reactivity allows determination of optimal cerebral perfusion pressure in patients with traumatic brain injury. Crit Care Med. (2002) 30:733–8. doi: 10.1097/00003246-200204000-00002
44. Aries MJ, Czosnyka M, Budohoski KP, Steiner LA, Lavinio A, Kolias AG, et al. Continuous determination of optimal cerebral perfusion pressure in traumatic brain injury. Crit Care Med. (2012) 40:2456–63. doi: 10.1097/CCM.0b013e3182514eb6
45. Svedung Wettervik T, Howells T, Lewén A, Enblad P. Blood pressure variability and optimal cerebral perfusion pressure-new therapeutic targets in traumatic brain injury. Neurosurgery. (2020) 86:E300–e9. doi: 10.1093/neuros/nyz515
46. Jaeger M, Dengl M, Meixensberger J, Schuhmann MU. Effects of cerebrovascular pressure reactivity-guided optimization of cerebral perfusion pressure on brain tissue oxygenation after traumatic brain injury. Crit Care Med. (2010) 38:1343–7. doi: 10.1097/CCM.0b013e3181d45530
47. Howells T, Smielewski P, Donnelly J, Czosnyka M, Hutchinson PJA, Menon DK, et al. Optimal cerebral perfusion pressure in centers with different treatment protocols. Crit Care Med. (2018) 46:e235–e41. doi: 10.1097/CCM.0000000000002930
48. Weersink CS, Aries MJ, Dias C, Liu MX, Kolias AG, Donnelly J, et al. Clinical and physiological events that contribute to the success rate of finding “optimal” cerebral perfusion pressure in severe brain trauma patients. Crit Care Med. (2015) 43:1952–63. doi: 10.1097/CCM.0000000000001165
49. Robertson CS, Valadka AB, Hannay HJ, Contant CF, Gopinath SP, Cormio M, et al. Prevention of secondary ischemic insults after severe head injury. Crit Care Med. (1999) 27:2086–95. doi: 10.1097/00003246-199910000-00002
50. Dias C, Silva MJ, Pereira E, Monteiro E, Maia I, Barbosa S, et al. Optimal cerebral perfusion pressure management at bedside: a single-center pilot study. Neurocritical Care. (2015) 23:92–102. doi: 10.1007/s12028-014-0103-8
51. Lott ME, Hogeman C, Herr M, Gabbay R, Sinoway LI. Effects of an oral glucose tolerance test on the myogenic response in healthy individuals. Am J Physiol Heart Circ Physiol. (2007) 292:H304–H10. doi: 10.1152/ajpheart.00940.2005
52. Ward ME, Yan L, Angle MR. Modulation of rat pial arteriolar responses to flow by glucose. Anesthesiology. (2002) 97:471–7. doi: 10.1097/00000542-200208000-00026
53. Donnelly J, Czosnyka M, Sudhan N, Varsos GV, Nasr N, Jalloh I, et al. Increased blood glucose is related to disturbed cerebrovascular pressure reactivity after traumatic brain injury. Neurocritical Care. (2015) 22:20–5. doi: 10.1007/s12028-014-0042-4
54. Svedung Wettervik T, Howells T, Ronne-Engstrom E, Hillered L, Lewen A, Enblad P, et al. High arterial glucose is associated with poor pressure autoregulation, high cerebral lactate/pyruvate ratio and poor outcome following traumatic brain injury. Neurocritical Care. (2019) 31:526–33. doi: 10.1007/s12028-019-00743-2
55. Carteron L, Solari D, Patet C, Quintard H, Miroz JP, Bloch J, et al. Hypertonic lactate to improve cerebral perfusion and glucose availability after acute brain injury. Crit Care Med. (2018) 46:1649–55. doi: 10.1097/CCM.0000000000003274
56. Zeiler FA, Ercole A, Beqiri E, Cabeleira M, Aries M, Zoerle T, et al. Cerebrovascular reactivity is not associated with therapeutic intensity in adult traumatic brain injury: a CENTER-TBI analysis. Acta Neuroch. (2019) 161:1955–64. doi: 10.1007/s00701-019-03980-8
57. Menzel M, Doppenberg EM, Zauner A, Soukup J, Reinert MM, Bullock R. Increased inspired oxygen concentration as a factor in improved brain tissue oxygenation and tissue lactate levels after severe human head injury. J Neurosurg. (1999) 91:1–10. doi: 10.3171/jns.1999.91.1.0001
58. Wettervik TS, Engquist H, Howells T, Lenell S, Rostami E, Hillered L, et al. Arterial oxygenation in traumatic brain injury-relation to cerebral energy metabolism, autoregulation, and clinical outcome. J Inten Care Med. (2020). doi: 10.1177/0885066620944097. [Epub ahead of print].
59. Svedung Wettervik T, Howells T, Hillered L, Nilsson P, Engquist H, Lewen A, et al. Mild hyperventilation in traumatic brain injury-relation to cerebral energy metabolism, pressure autoregulation, and clinical outcome. World Neurosurg. (2020) 133:e567–e75. doi: 10.1016/j.wneu.2019.09.099
60. Steiner LA, Balestreri M, Johnston AJ, Coles JP, Chatfield DA, Pickard JD, et al. Effects of moderate hyperventilation on cerebrovascular pressure-reactivity after head injury. Acta Neuroch. (2005) 95:17–20. doi: 10.1007/3-211-32318-X_4
61. Graham DI, Adams JH. Ischaemic brain damage in fatal head injuries. Lancet. (1971) 1:265–6. doi: 10.1016/S0140-6736(71)91003-8
62. Diringer MN, Zazulia AR, Powers WJ. Does ischemia contribute to energy failure in severe TBI? Transl Stroke Res. (2011) 2:517–23. doi: 10.1007/s12975-011-0119-8
63. Oddo M, Levine JM, Kumar M, Iglesias K, Frangos S, Maloney-Wilensky E, et al. Anemia and brain oxygen after severe traumatic brain injury. Int Care Med. (2012) 38:1497–504. doi: 10.1007/s00134-012-2593-1
64. Yamal JM, Rubin ML, Benoit JS, Tilley BC, Gopinath S, Hannay HJ, et al. Effect of hemoglobin transfusion threshold on cerebral hemodynamics and oxygenation. J Neurotrauma. (2015) 32:1239–45. doi: 10.1089/neu.2014.3752
65. Sekhon MS, Griesdale DE, Czosnyka M, Donnelly J, Liu X, Aries MJ, et al. The effect of red blood cell transfusion on cerebral autoregulation in patients with severe traumatic brain injury. Neurocritical care. (2015) 23:210–6. doi: 10.1007/s12028-015-0141-x
66. Robertson CS, Hannay HJ, Yamal JM, Gopinath S, Goodman JC, Tilley BC, et al. Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: a randomized clinical trial. JAMA. (2014) 312:36–47. doi: 10.1001/jama.2014.6490
67. Utter GH, Shahlaie K, Zwienenberg-Lee M, Muizelaar JP. Anemia in the setting of traumatic brain injury: the arguments for and against liberal transfusion. J Neurotrauma. (2011) 28:155–65. doi: 10.1089/neu.2010.1451
68. Gobatto ALN, Link MA, Solla DJ, Bassi E, Tierno PF, Paiva W, et al. Transfusion requirements after head trauma: a randomized feasibility controlled trial. Crit Care. (2019) 23:89. doi: 10.1186/s13054-018-2273-9
69. Salim A, Hadjizacharia P, DuBose J, Brown C, Inaba K, Chan L, et al. Role of anemia in traumatic brain injury. J Am Coll Surg. (2008) 207:398–406. doi: 10.1016/j.jamcollsurg.2008.03.013
70. Chang JJ, Youn TS, Benson D, Mattick H, Andrade N, Harper CR, et al. Physiologic and functional outcome correlates of brain tissue hypoxia in traumatic brain injury. Crit Care Med. (2009) 37:283–90. doi: 10.1097/CCM.0b013e318192fbd7
71. Tolias CM, Reinert M, Seiler R, Gilman C, Scharf A, Bullock MR. Normobaric hyperoxia–induced improvement in cerebral metabolism and reduction in intracranial pressure in patients with severe head injury: a prospective historical cohort-matched study. J Neurosurg. (2004) 101:435–44. doi: 10.3171/jns.2004.101.3.0435
72. Reinert M, Schaller B, Widmer HR, Seiler R, Bullock R. Influence of oxygen therapy on glucose-lactate metabolism after diffuse brain injury. J Neurosurg. (2004) 101:323–9. doi: 10.3171/jns.2004.101.2.0323
73. Nortje J, Coles JP, Timofeev I, Fryer TD, Aigbirhio FI, Smielewski P, et al. Effect of hyperoxia on regional oxygenation and metabolism after severe traumatic brain injury: preliminary findings. Crit Care Med. (2008) 36:273–81. doi: 10.1097/01.CCM.0000292014.60835.15
74. Vilalta A, Sahuquillo J, Merino MA, Poca MA, Garnacho A, Martinez-Valverde T, et al. Normobaric hyperoxia in traumatic brain injury: does brain metabolic state influence the response to hyperoxic challenge? J Neurotrauma. (2011) 28:1139–48. doi: 10.1089/neu.2010.1720
75. Vidal-Jorge M, Sánchez-Guerrero A, Mur-Bonet G, Castro L, Rădoi A, Riveiro M, et al. Does normobaric hyperoxia cause oxidative stress in the injured brain? A microdialysis study using 8-iso-prostaglandin F2α as a biomarker. J Neurotrauma. (2017) 34:2731–42. doi: 10.1089/neu.2017.4992
76. Spiotta AM, Stiefel MF, Gracias VH, Garuffe AM, Kofke WA, Maloney-Wilensky E, et al. Brain tissue oxygen-directed management and outcome in patients with severe traumatic brain injury. J Neurosurg. (2010) 113:571–80. doi: 10.3171/2010.1.JNS09506
77. Martini RP, Deem S, Yanez ND, Chesnut RM, Weiss NS, Daniel S, et al. Management guided by brain tissue oxygen monitoring and outcome following severe traumatic brain injury. J Neurosurg. (2009) 111:644–9. doi: 10.3171/2009.2.JNS08998
78. Okonkwo DO, Shutter LA, Moore C, Temkin NR, Puccio AM, Madden CJ, et al. Brain oxygen optimization in severe traumatic brain injury phase-II: a phase II randomized trial. Crit Care Med. (2017) 45:1907–14. doi: 10.1097/CCM.0000000000002619
79. Ungerstedt U, Pycock C. Functional correlates of dopamine neurotransmission. Bull Schw Akad Med Wis. (1974) 30:44.
80. Persson L, Hillered L. Chemical monitoring of neurosurgical intensive care patients using intracerebral microdialysis. J Neurosurg. (1992) 76:72–80. doi: 10.3171/jns.1992.76.1.0072
81. Engstrom M, Polito A, Reinstrup P, Romner B, Ryding E, Ungerstedt U, et al. Intracerebral microdialysis in severe brain trauma: the importance of catheter location. J Neurosurg. (2005) 102:460–9. doi: 10.3171/jns.2005.102.3.0460
82. Rostami E, Bellander BM. Monitoring of glucose in brain, adipose tissue, and peripheral blood in patients with traumatic brain injury: a microdialysis study. J Diab Sci Tech. (2011) 5:596–604. doi: 10.1177/193229681100500314
83. Diaz-Parejo P, Ståhl N, Xu W, Reinstrup P, Ungerstedt U, Nordström CH. Cerebral energy metabolism during transient hyperglycemia in patients with severe brain trauma. Int Care Med. (2003) 29:544–50. doi: 10.1007/s00134-003-1669-3
84. Magnoni S, Tedesco C, Carbonara M, Pluderi M, Colombo A, Stocchetti N. Relationship between systemic glucose and cerebral glucose is preserved in patients with severe traumatic brain injury, but glucose delivery to the brain may become limited when oxidative metabolism is impaired: implications for glycemic control. Crit Care Med. (2012) 40:1785–91. doi: 10.1097/CCM.0b013e318246bd45
85. Rosner MJ, Newsome HH, Becker DP. Mechanical brain injury: the sympathoadrenal response. J Neurosurg. (1984) 61:76–86. doi: 10.3171/jns.1984.61.1.0076
86. Jeremitsky E, Omert LA, Dunham CM, Wilberger J, Rodriguez A. The impact of hyperglycemia on patients with severe brain injury. J Trauma. (2005) 58:47–50. doi: 10.1097/01.TA.0000135158.42242.B1
87. Lam AM, Winn HR, Cullen BF, Sundling N. Hyperglycemia and neurological outcome in patients with head injury. J Neurosurg. (1991) 75:545–51. doi: 10.3171/jns.1991.75.4.0545
88. Young B, Ott L, Dempsey R, Haack D, Tibbs P. Relationship between admission hyperglycemia and neurologic outcome of severely brain-injured patients. Ann Surg. (1989) 210:466–72. doi: 10.1097/00000658-198910000-00007
89. Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, et al. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. (2002) 16:1738–48. doi: 10.1096/fj.01-1027com
90. Krinsley JS. Effect of an intensive glucose management protocol on the mortality of critically ill adult patients. Mayo Clinic Proc. (2004) 79:992–1000. doi: 10.4065/79.8.992
91. Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. (2006) 354:449–61. doi: 10.1056/NEJMoa052521
92. Bilotta F, Caramia R, Cernak I, Paoloni FP, Doronzio A, Cuzzone V, et al. Intensive insulin therapy after severe traumatic brain injury: a randomized clinical trial. Neurocrit Care. (2008) 9:159–66. doi: 10.1007/s12028-008-9084-9
93. Coester A, Neumann CR, Schmidt MI. Intensive insulin therapy in severe traumatic brain injury: a randomized trial. J Traum Acute Care Surg. (2010) 68:904–11. doi: 10.1097/TA.0b013e3181c9afc2
94. Meier R, Béchir M, Ludwig S, Sommerfeld J, Keel M, Steiger P, et al. Differential temporal profile of lowered blood glucose levels (3.5 to 6.5 mmol/l versus 5 to 8 mmol/l) in patients with severe traumatic brain injury. Crit Care. (2008) 12:R98. doi: 10.1186/cc6974
95. Vespa P, Boonyaputthikul R, McArthur DL, Miller C, Etchepare M, Bergsneider M, et al. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit Care Med. (2006) 34:850–6. doi: 10.1097/01.CCM.0000201875.12245.6F
96. Finfer S, Liu B, Chittock DR, Norton R, Myburgh JA, McArthur C, et al. Hypoglycemia and risk of death in critically ill patients. N Engl J Med. (2012) 367:1108–18. doi: 10.1056/NEJMoa1204942
97. Finfer S, Chittock DR, Su SY, Blair D, Foster D, Dhingra V, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. (2009) 360:1283–97. doi: 10.1056/NEJMoa0810625
98. Oddo M, Schmidt JM, Carrera E, Badjatia N, Connolly ES, Presciutti M, et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med. (2008) 36:3233–8. doi: 10.1097/CCM.0b013e31818f4026
99. Rostami E. Glucose and the injured brain-monitored in the neurointensive care unit. Front Neurol. (2014) 5:91. doi: 10.3389/fneur.2014.00091
100. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. (1994) 91:10625–9. doi: 10.1073/pnas.91.22.10625
101. Boumezbeur F, Petersen KF, Cline GW, Mason GF, Behar KL, Shulman GI, et al. The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J Neurosci. (2010) 30:13983–91. doi: 10.1523/JNEUROSCI.2040-10.2010
102. Bouzat P, Sala N, Suys T, Zerlauth JB, Marques-Vidal P, Feihl F, et al. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Int Care Med. (2014) 40:412–21. doi: 10.1007/s00134-013-3203-6
103. Quintard H, Patet C, Zerlauth JB, Suys T, Bouzat P, Pellerin L, et al. Improvement of neuroenergetics by hypertonic lactate therapy in patients with traumatic brain injury is dependent on baseline cerebral lactate/pyruvate ratio. J Neurotrauma. (2016) 33:681–7. doi: 10.1089/neu.2015.4057
104. Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. (2010) 468:232–43. doi: 10.1038/nature09613
105. Alessandri B, Schwandt E, Kamada Y, Nagata M, Heimann A, Kempski O. The neuroprotective effect of lactate is not due to improved glutamate uptake after controlled cortical impact in rats. J Neurotrauma. (2012) 29:2181–91. doi: 10.1089/neu.2011.2067
106. Patet C, Suys T, Carteron L, Oddo M. Cerebral lactate metabolism after traumatic brain injury. Curr Neurol Neurosci Rep. (2016) 16:31. doi: 10.1007/s11910-016-0638-5
107. Timofeev I, Carpenter KL, Nortje J, Al-Rawi PG, O'Connell MT, Czosnyka M, et al. Cerebral extracellular chemistry and outcome following traumatic brain injury: a microdialysis study of 223 patients. Brain. (2011) 134(Pt 2):484–94. doi: 10.1093/brain/awq353
108. Sánchez-Aguilar M, Tapia-Pérez JH, Sánchez-Rodríguez JJ, Viñas-Ríos JM, Martínez-Pérez P, de la Cruz-Mendoza E, et al. Effect of rosuvastatin on cytokines after traumatic head injury. J Neurosurg. (2013) 118:669–75. doi: 10.3171/2012.12.JNS121084
109. Kelsen J, Karlsson M, Hansson MJ, Yang Z, Fischer W, Hugerth M, et al. Copenhagen head injury ciclosporin study: a phase IIA safety, pharmacokinetics, and biomarker study of ciclosporin in severe traumatic brain injury patients. J Neurotrauma. (2019) 36:3253–63. doi: 10.1089/neu.2018.6369
110. Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. (2017) 13:171. doi: 10.1038/nrneurol.2017.13
111. Dyhrfort P, Shen Q, Clausen F, Thulin M, Enblad P, Kamali-Moghaddam M, et al. Monitoring of protein biomarkers of inflammation in human traumatic brain injury using microdialysis and proximity extension assay technology in neurointensive care. J Neurotrauma. (2019) 36:2872–85. doi: 10.1089/neu.2018.6320
112. Hillered L, Dahlin AP, Clausen F, Chu J, Bergquist J, Hjort K, et al. Cerebral microdialysis for protein biomarker monitoring in the neurointensive care setting-a technical approach. Front Neurol. (2014) 5:245. doi: 10.3389/fneur.2014.00245
113. McConeghy KW, Hatton J, Hughes L, Cook AM. A review of neuroprotection pharmacology and therapies in patients with acute traumatic brain injury. CNS Drugs. (2012) 26:613–36. doi: 10.2165/11634020-000000000-00000
114. Murray GD, Teasdale GM, Schmitz H. Nimodipine in traumatic subarachnoid haemorrhage: a re-analysis of the HIT I and HIT II trials. Acta Neuroch. (1996) 138:1163–7. doi: 10.1007/BF01809745
115. Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. (2002) 1:383–6. doi: 10.1016/S1474-4422(02)00164-3
116. Skolnick BE, Maas AI, Narayan RK, van der Hoop RG, MacAllister T, Ward JD, et al. A clinical trial of progesterone for severe traumatic brain injury. N Engl J Med. (2014) 371:2467–76. doi: 10.1056/NEJMoa1411090
117. Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet. (2004) 364:1321–8. doi: 10.1016/S0140-6736(04)17188-2
118. Depreitere B, Güiza F, Van den Berghe G, Schuhmann MU, Maier G, Piper I, et al. Pressure autoregulation monitoring and cerebral perfusion pressure target recommendation in patients with severe traumatic brain injury based on minute-by-minute monitoring data. J Neurosurg. (2014) 120:1451–7. doi: 10.3171/2014.3.JNS131500
Keywords: multimodality monitoring, secondary brain injury, secondary insults, neurointensive care, traumatic brain injury
Citation: Svedung Wettervik TM, Lewén A and Enblad P (2021) Fine Tuning of Traumatic Brain Injury Management in Neurointensive Care—Indicative Observations and Future Perspectives. Front. Neurol. 12:638132. doi: 10.3389/fneur.2021.638132
Received: 05 December 2020; Accepted: 20 January 2021;
Published: 24 February 2021.
Edited by:
Jian Hai, Tongji Hospital Affiliated to Tongji University, ChinaReviewed by:
Marek Czosnyka, University of Cambridge, United KingdomDanilo Cardim, University of Texas Southwestern Medical Center, United States
Copyright © 2021 Svedung Wettervik, Lewén and Enblad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teodor M. Svedung Wettervik, dGVvZG9yLnN2ZWR1bmctd2V0dGVydmlrQG5ldXJvLnV1LnNl