 Albert Frank Magnusen1†
Albert Frank Magnusen1† Manoj Kumar Pandey
Manoj Kumar Pandey- 1Division of Human Genetics, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
- 2Department of Paediatrics of University of Cincinnati College of Medicine, Cincinnati, OH, United States
Parkinson's disease (PD) is a movement disorder attributed to the loss of dopaminergic (DA) neurons mainly in the substantia nigra pars compacta. Motor symptoms include resting tremor, rigidity, and bradykinesias, while non-motor symptoms include autonomic dysfunction, anxiety, and sleeping problems. Genetic mutations in a number of genes (e.g., LRRK2, GBA, SNCA, PARK2, PARK6, and PARK7) and the resultant abnormal activation of microglial cells are assumed to be the main reasons for the loss of DA neurons in PD with genetic causes. Additionally, immune cell infiltration and their participation in major histocompatibility complex I (MHCI) and/or MHCII-mediated processing and presentation of cytosolic or mitochondrial antigens activate the microglial cells and cause the massive generation of pro-inflammatory cytokines and chemokines, which are all critical for the propagation of brain inflammation and the neurodegeneration in PD with genetic and idiopathic causes. Despite knowing the involvement of several of such immune devices that trigger neuroinflammation and neurodegeneration in PD, the exact disease mechanism or the innovative biomarker that could detect disease severity in PD linked to LRRK2, GBA, SNCA, PARK2, PARK6, and PARK7 defects is largely unknown. The current review has explored data from genetics, immunology, and in vivo and ex vivo functional studies that demonstrate that certain genetic defects might contribute to microglial cell activation and massive generation of a number of pro-inflammatory cytokines and chemokines, which ultimately drive the brain inflammation and lead to neurodegeneration in PD. Understanding the detailed involvement of a variety of immune mediators, their source, and the target could provide a better understanding of the disease process. This information might be helpful in clinical diagnosis, monitoring of disease progression, and early identification of affected individuals.
Introduction
Parkinson's disease (PD) is a neurodegenerative brain disorder that mainly happens due to progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNPC) and its impact on impairment of motor function that includes static tremor, bradykinesia, muscle stiffness, postural instability, balance difficulty, and walking problem (1, 2). Pro-inflammatory cytokines and chemokines have been linked to disease manifestations of Alzheimer's disease, multiple sclerosis, Huntington's disease, amyotrophic lateral sclerosis, prion disease, systemic lupus erythematosus, depression, migraine, and schizophrenia as reviewed in refs. (3–12). Microglial cells (MGCs) are residential macrophages (Mϕs) of the central nervous system (CNS), which are exquisitely sensitive to the pathophysiological insults and the resultant alteration in their morphology and phenotype to activated state (13). Such MGCs cause massive generation of pro-inflammatory cytokines, chemokines, reactive oxygen species (ROS), and nitric oxide (NO), which all contribute to the clearance of infectious agents (14). However, prolonged or excessive activation of MGCs results in pathological forms of inflammation that contribute to the progression of neurodegenerative and neoplastic diseases (15–17). Activated MGCs express major histocompatibility complex II (MHC class II), which is required for activation of naive CD4+ T cells and the production of numerous pro-inflammatory cytokines and chemokines that modulate the differentiation of effector T cells (18).
Effector T cells, i.e., T helper 1 (Th1), Th2, Th17, T regulatory (Treg), and T follicular helper (Tfh) cells as well as their signature cytokines, i.e., interferon gamma (IFNγ; TH1), interleukin 4 (IL-4; TH2) (19, 20), IL-17 (TH17) (21, 22), transforming growth factor beta (TGFβ; Treg), and IL-6 (Tfh), drive tissue inflammation in several visceral and brain diseases (23–28). The T helper cell subsets can produce IL-10, a cytokine with broad immunoregulatory properties (29). Th1 cells produce IFNγ, IL-2, and tumor necrosis factor alpha (TNFα) to clear intracellular pathogens and evoke cell-mediated immunity, whereas Th2 cells produce IL-4, IL-5, and IL-13 to clear extracellular organisms and evoke strong allergic responses (19, 30–33). In contrast to Th1 and Th2 cell differentiation, which depend on their respective effector cytokines (IFNγ and IL-4), Th17 cell differentiation does not require IL-17 but has a critical need for TGFβ and IL-6 (34–36). Treg cells produce IL-10 and TGFβ to cause immune tolerance and inhibit IFNγ synthesis (37) as well as block T helper cell differentiation of naive T cells into effector T cells (38).
The MGCs' interaction to effector T cells and the resulting production of pro-inflammatory cytokines, chemokines, and the neurodegeneration have been observed in Alzheimer's disease, amyotrophic lateral sclerosis, multiple sclerosis (MS), and prion diseases (17, 39, 40). The SNPC of PD patients have shown CD4+ T cells, CD8+ T cells, human leukocyte antigen DR isotype (HLA-DR) expressing inflammatory subset of MGCs, and increased incidence of pro-inflammatory cytokines, i.e., IFNγ, TNF, IL-1β expressing glial cells (41–43). Additionally, the striatal dopaminergic (DA) regions and cerebrospinal fluid (CSF) of PD patients have shown elevated levels of IL-1β, IL-2, IL-6, TNF, and TGFβ1 (44, 45). Peripheral blood analyses of PD patients have shown marked increases of innate and adaptive immune cells that include monocytes (MOs), IFNγ, IL-4, and IL-17 producing memory and effector T cells as well as their association to severity of the disease (43, 46–51). Elevated serum levels of TNF (52, 53), IL-1β (52, 54, 55), and IL-6 (52–54) have been observed in PD patients as reviewed in Qin et al. (56). PD patients have also shown increased serum level of cytokine receptors such as TNF receptors (e.g., TNFRs) and their link to late disease onset (57, 58). MO differentiation into the tissue-specific MGCs, Mϕs, and dendritic cells (DCs) as well as the trafficking of CD4+ and CD8+ T cells to sites of inflammation requires growth factors, i.e., granulocyte colony-stimulating factor (GCSF), granulocyte Mϕ colony-stimulating factor (GMCSF), and the Mϕ colony-stimulating factor (MCSF), as well as the number of C-C motif ligand (CCL) and the C-X-C motif ligand (CXCL) chemokines (59–69). However, the exact mechanism by which such immune inflammation occurs in PD is unknown. It is speculated that abnormal brain or circulatory level of several proteins and enzymes has been associated with the development of neuroinflammation in PD. Indeed, several of such proteins have been associated with activation of residential MGCs and the infiltrated lymphocytes and their combined impact on the generation of pro-inflammatory cytokines (e.g., IFNβ, IFNγ, TNFα, IL-1β, IL-6, IL-18, and TGFβ1), which lead to the loss of DA neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 2,4,5-trihydroxyphenethylamine or 6-hydroxydopamin (6-OHDA)-induced mouse models of idiopathic PD (Table 1A). Additionally, human patients with idiopathic PD have also suggested elevated brain or circulatory level of proteins or enzymes linked to MGC activation, pro-inflammatory cytokine and chemokine (e.g., IFNβ, IFNγ, TNFα, IL-1β, IL-2, IL-4, IL-6, IL-10, IL-12, IL-13, CCL2, CXCL1) production, loss of DA neurons, and the development of motor symptoms (Table 1B). The current review is an update on the involvement of a variety of innate and adaptive immune mediators as well as their source and targets involved in the propagation of disease manifestations in mouse and human PD associated with LRRK2, GBA, SNCA, PARK2, PARK6, and PARK7 defects. These results will likely provide much needed insights into the disease mechanism and will be useful for the identification of potential biomarkers at the level of distinguished cytokines and chemokines in different forms of PD.
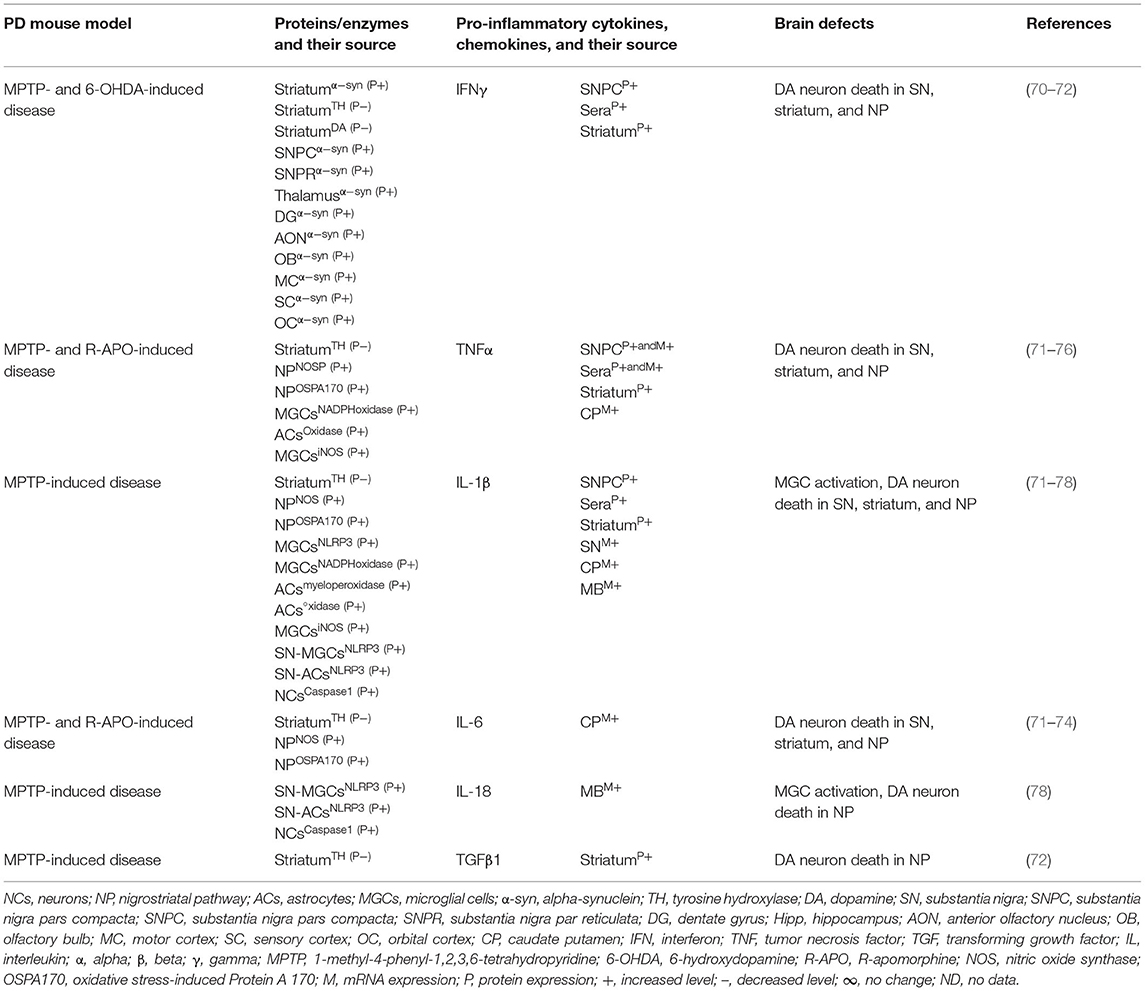
Table 1A. Cytokines and their source in the mouse model of idiopathic PD.
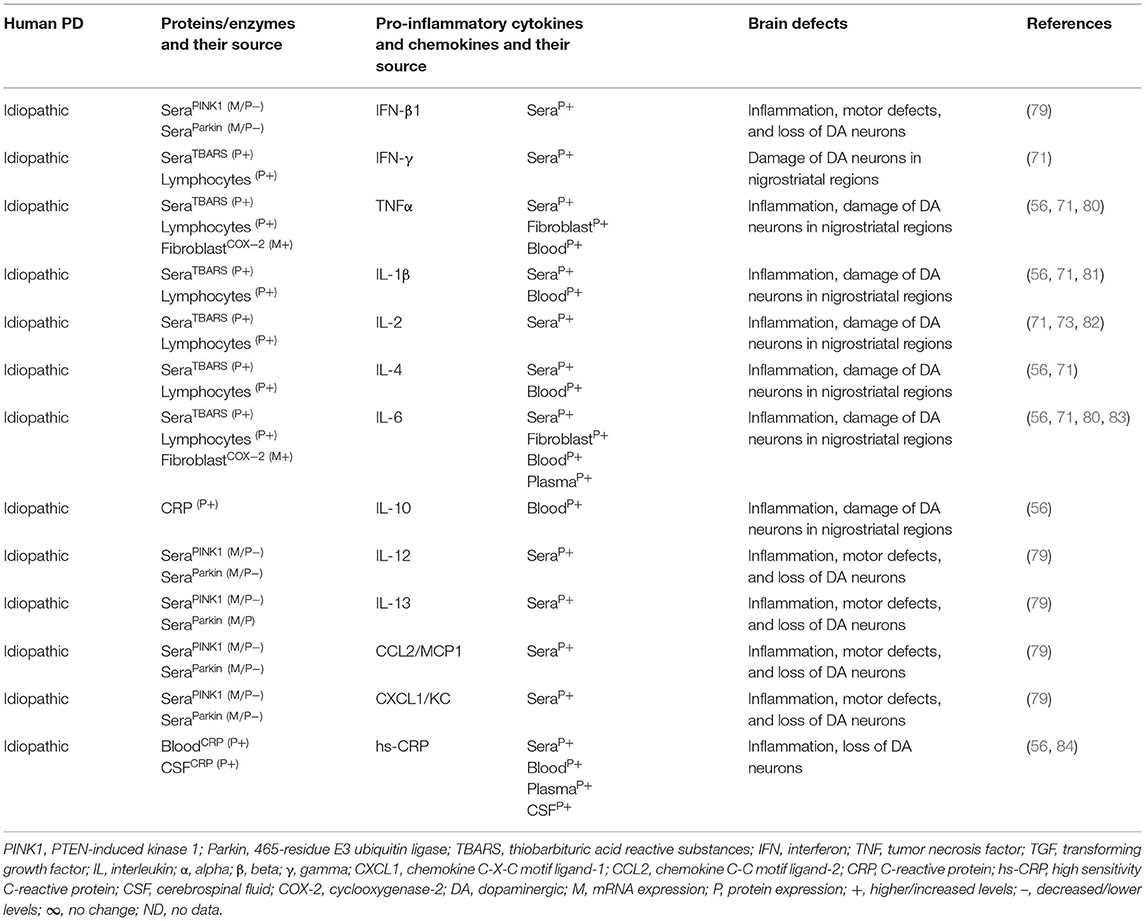
Table 1B. Cytokines and their source in idiopathic human PD.
LRRK2 Gene Defects and Pro-inflammatory Immune Mediators in PD
The leucine-rich-repeat kinase 2 (LRRK2) gene encodes a large, multidomain LRRK2 protein comprised of a GTPase and a kinase domain (85). Although the precise physiological function of LRRK2 remains largely unknown, recent studies have indicated that LRRK2 is involved in cellular functions such as neurite outgrowth, cytoskeletal maintenance, vesicle trafficking, autophagic protein degradation, and the regulation of signaling pathways, including the Wingless-INT (WNT), Fas-Fas ligand (FasL or CD95L or CD178)-associated protein with death domain (FADD), mitogen-activated protein kinase (MAPK), and nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) (86–88).
The resting neuronal cells, i.e., neurons (NCs), MGCs, and astrocytes (ACs), expressed a low level of LRRK2 (89, 90). However, several of the pro-inflammatory mediators (e.g., IFNβ, IFNγ, TNFα, IL-6, and LPS) cause upregulation of LRRK2 in immune cells, i.e., monocytes (MOs), Mϕs, and T and B cells, and in neuronal cells, i.e., MGCs and NCs (88, 91–95). LRRK2 is critical for the propagation of Crohn's disease (96, 97), leprosy (98), and neuronal toxicity (99–102).
Indeed, LRRK2 gene mutations have been linked to increased LRRK2 kinase substrate phosphorylation and the formation of intracellular alpha-synuclein (α-syn)-positive inclusions in Lewy bodies (LBs) and preferential loss of DA neurons and the development of motor symptoms, including tremor, rigidity, postural instability, and bradykinesia in late-onset familial and idiopathic PD (100, 103–119). The brain regions, blood, and cells of LRRK2-associated mouse models of PD have shown abnormal expression of LRRK2 kinase and their association with elevated brain and circulatory level of pro-inflammatory cytokines (e.g., IFNγ, TNFα, IL-1α, IL-1β, IL-6, IL-8, IL-10, and IL-12), chemokines (e.g., CCL2, CCL3, CCL4, CCL5, CXCL1, and CXCL10), and growth factors (e.g., GCSF and MCSF), as well as their link to the loss of NCs and the development of cognitive defects (Table 2A). The blood cells, sera, and CSF of LRRK2-associated human patients with PD have also shown abnormal expression of LRRK2 kinase and their link to elevated levels of pro-inflammatory cytokines and growth factors (e.g., IFNγ, TNFα, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-13, GCSF, PDGF, and VEGF), loss of NCs, and the development of cognitive defects in PD (Table 2B). These data suggest that LRKK2 defects and the resultant higher expression of LRRK2 kinases cause cellular activation and the higher generation of pro-inflammatory cytokines and chemokines (Tables 2A,B) that lead to DA neuron damage in LRRK2-associated PD (Figure 1A).
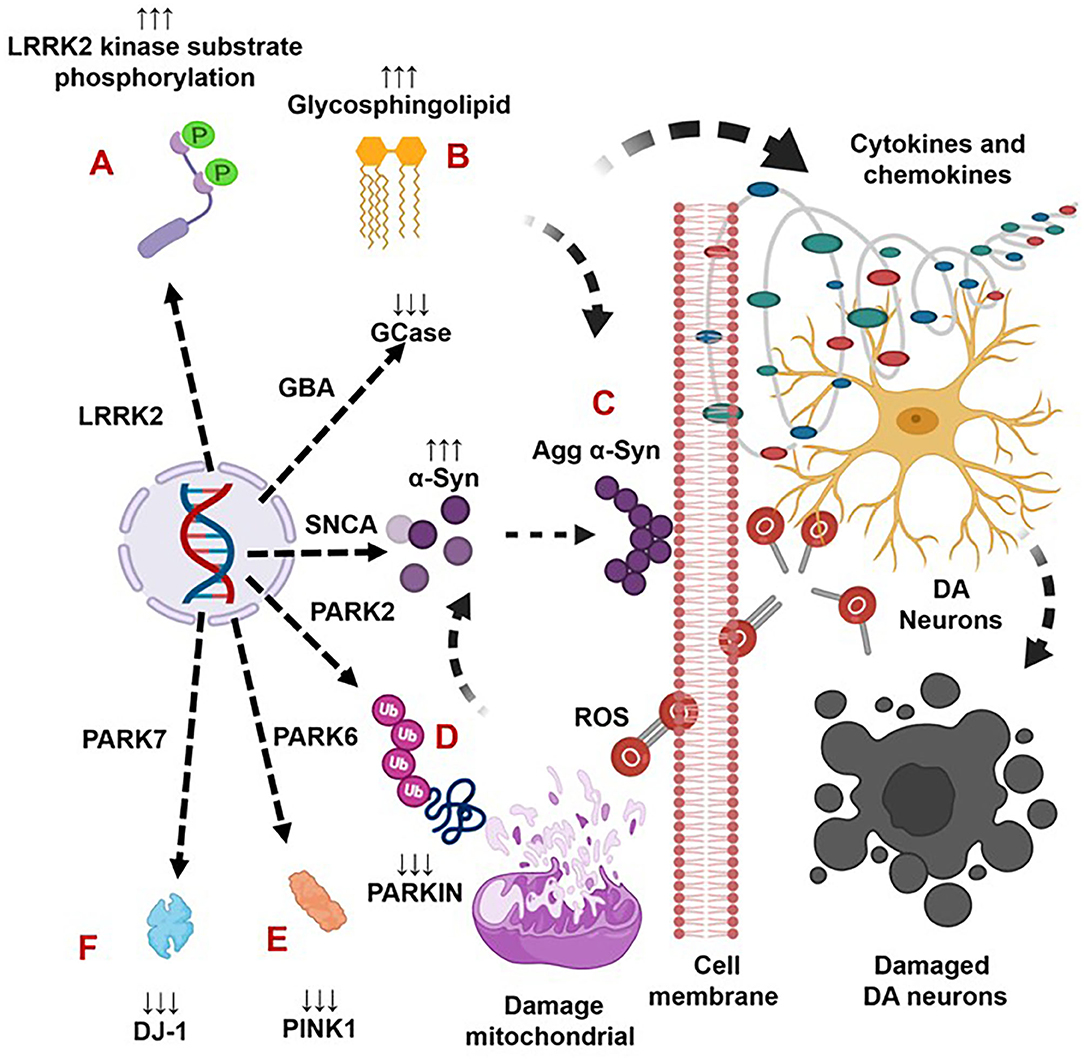
Figure 1. The genetic mutation-induced inflammatory immune reactions develop neurodegeneration in Parkinson's disease. The LRRK2 defects cause over activation of LRRK2 kinases. This defect triggers the formation of aggregated alpha-synuclein (Agg α-syn) and increased generation of pro-inflammatory cytokines and chemokines that lead to the loss of dopaminergic (DA) neurons in LRRK2-associated PD (A). The GBA mutations and the resultant deficiency of glucocerebrosidase (GCase) trigger the formation of glycosphingolipids and Agg α-syn, which trigger increased generation of pro-inflammatory cytokines and chemokines and lead to the loss of DA neurons in GBA-associated PD (B). The SNCA defects and the resultant overproduction of normal/Agg α-syn activate the brain production of inflammatory cytokines and chemokines that cause death of DA neurons in SNCA-associated PD (C). The PARK2, PARK6, and PARK7 defects and the subsequent deficiency of PARKIN, PINK, and DJ-1 proteins cause mitochondrial damage and the formation of Agg α-syn. These abnormalities trigger cellular activation and massive generation of ROS, pro-inflammatory cytokines, and chemokines that lead to the loss of DA neurons in PARK2-, PARK6-, and PARK7-associated PD (D–F).
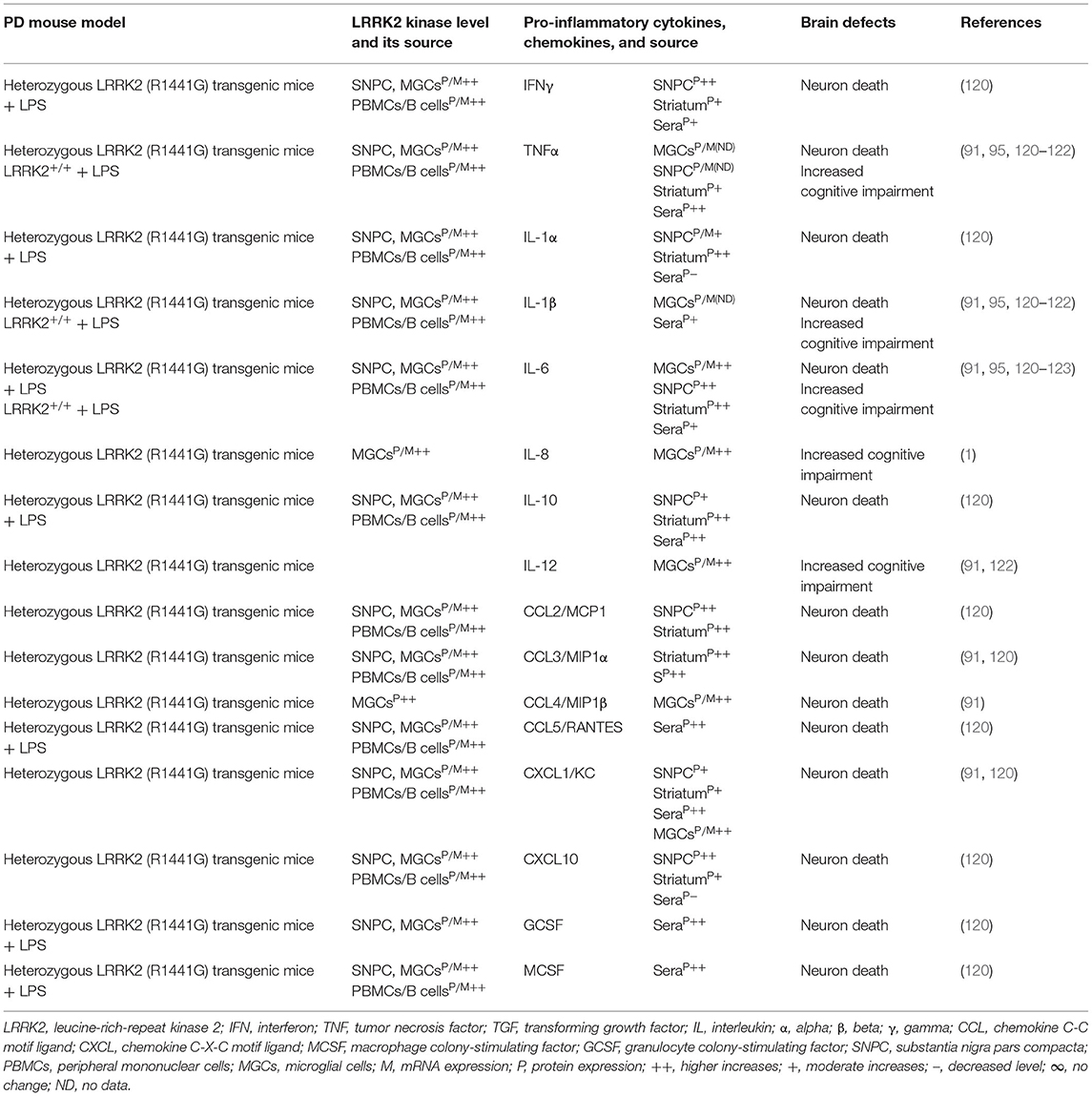
Table 2A. Cytokines and their source in the LRRK2 mouse model of PD.
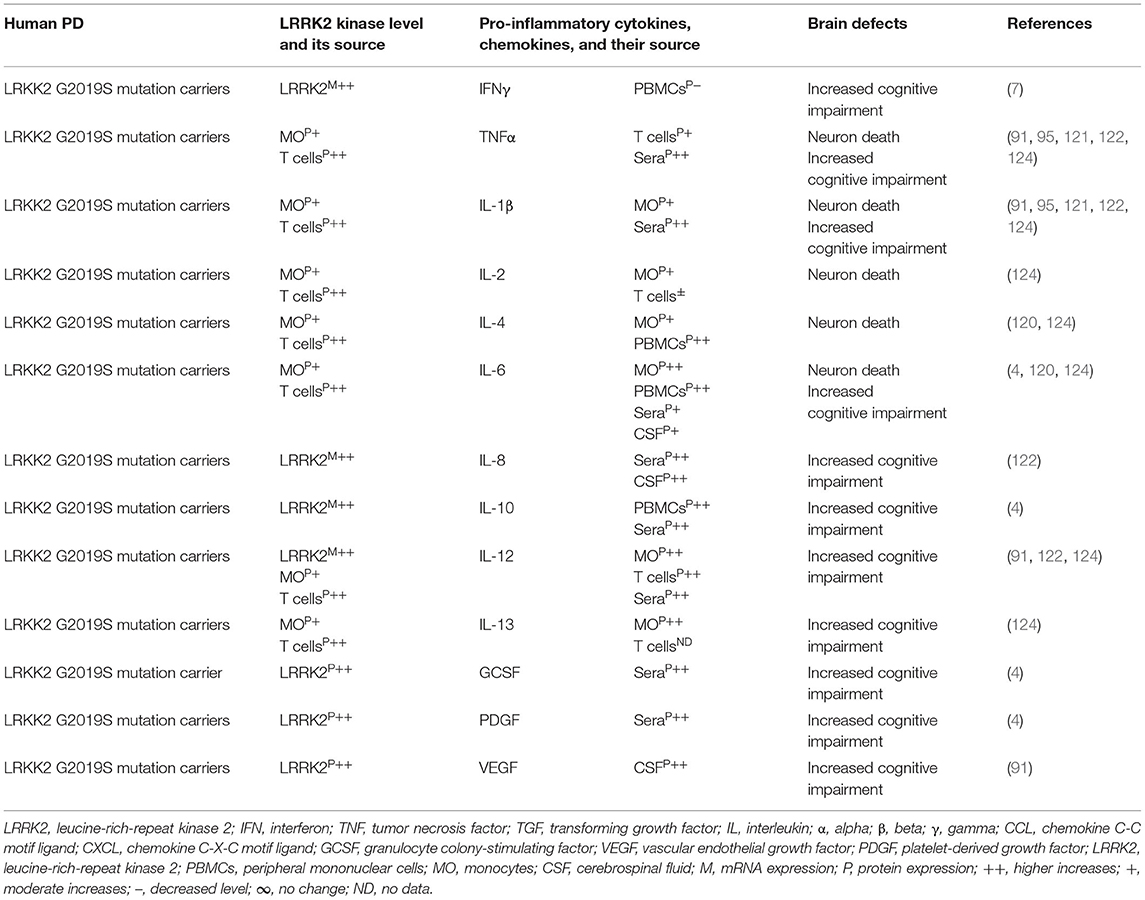
Table 2B. Cytokines and their source in the LRRK2-associated human PD.
GBA1 Gene Defects and Pro-inflammatory Immune Mediators in PD
The GBA1 gene encodes the lysosomal enzyme, acid β-glucosidase (glucocerebrosidase, GCase). This later enzyme cleaves the β-D-glucosidic bond from the glycosphingolipid substrates (glucosylceramide; GC), yielding β-D-glucose and ceramide, and its deacylated product, glucosylsphingosine (GS), resulting in the formation of β-D-glucose and sphingosine (125, 126). The three types of Gaucher disease (GD), i.e., types 1, 2, and 3, have been characterized by recessive mutations in the GBA1 gene. Pathogenic mutations in GBA1 and the resultant GCase deficiency cause excess tissue accumulation of GC and chronic tissue inflammation in type 1 GD (59, 125, 127–133). We have identified immune complexes of GC-specific immunoglobulin G (IgG) antibodies in experimental and clinical Gaucher disease, which induce massive generation of complement C5a (C5a) and the activation of C5a receptor (e.g., C5aR1). Such C5a–C5aR1 activation is what tips the balance between GC formation and its degradation through the control of an enzyme termed as glucosylceramide synthase (GCS) that produces the GC and fuels inflammation in visceral tissues (e.g., blood, bone marrow, lung, liver, spleen, and lymph node) in type 1 experimental and clinical GD (131).
Excess brain accumulation of GC has been linked to the formation of abnormal species of α-syn, microglial cell activation, generation of pro-inflammatory cytokines (e.g., TNFα, IL-1β, and IL-6), and the loss of neurons in patients with GD types 2 and 3 (134–139). Heterozygous mutations in the GBA1 gene are implicated in dementia with LBs (DLB) in idiopathic PD (140, 141). Similarly, the heterozygous GBA1 mutations have emerged as the major genetic risk for developing PD (133, 138, 142–159).
Brains of the GBA1 mouse model of PD have shown partial GCase deficiency and its impact on increased production of TNFα, IL-1β, TGFβ1, CCL2, CCL3, CCL5, VCAM-1, ICAM-1, and MCSF as well as their link to the neuronal cell death (Table 3A). Plasma, sera, CSF, and blood-derived MOs of PD patients with GBA mutations have shown partial GCase deficiency and its impact on the higher production of pro-inflammatory cytokines (e.g., IFNγ, TNFα, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-13, CCL2, CCL3, CCL18, and SF), midbrain damage, and cognitive defects (Table 3B). These studies suggest that GBA defects and the resultant GCase deficiency cause excess tissue storage of glycosphingolipids and/or the formation of abnormal species of α-syn. These abnormal proteins and/or lipids trigger residential and infiltrated immune cell (e.g., MOs and MGCs) activation and massive brain generation of pro-inflammatory cytokines and chemokines (Tables 3A,B), which are all critical for the development of brain inflammation and neurodegeneration in GBA-associated PD (Figure 1B).
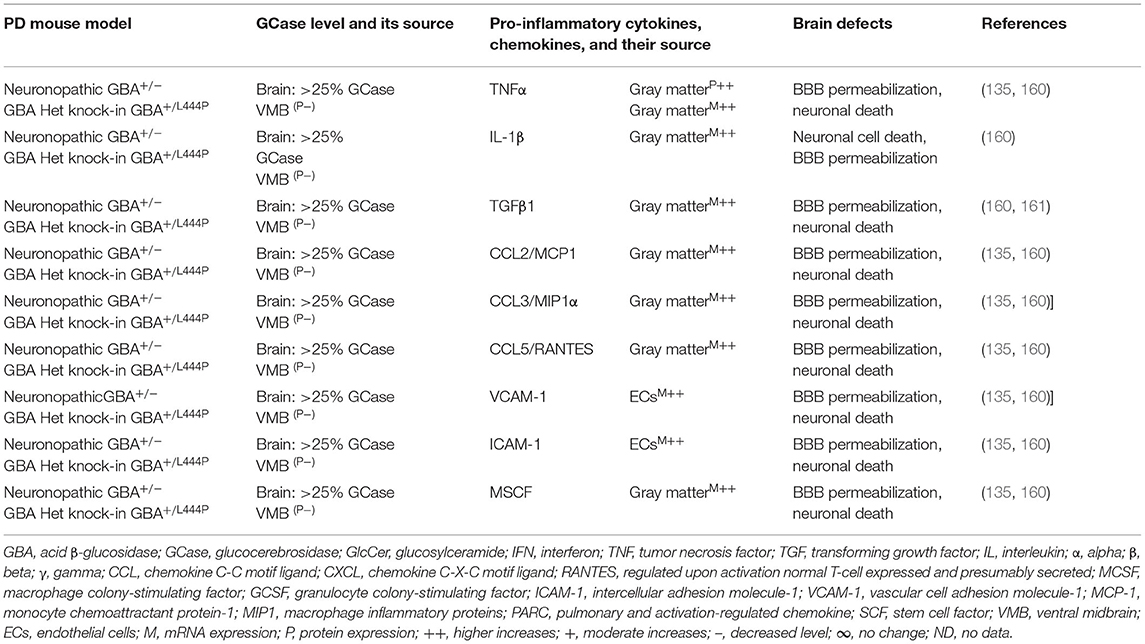
Table 3A. Cytokines and their source in the mouse model of GBA1 PD.
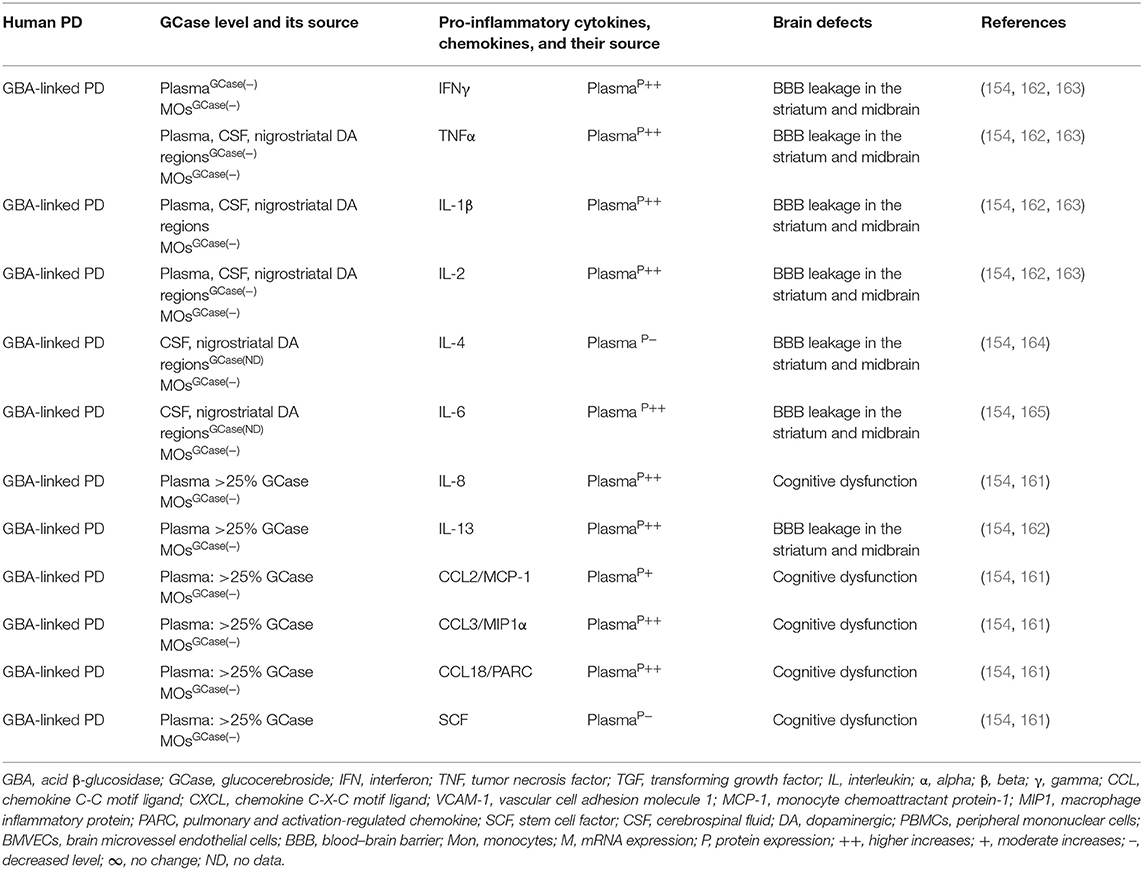
Table 3B. Cytokines and their source in the GBA-associated human PD.
SNCA gENE Defects and Pro-inflammatory Immune Mediators in PD
SNCA encodes the α-syn, which is an 18-kDa protein composed of 140 amino acids and expressed in presynaptic terminals of the neocortex, hippocampus, substantia nigra (SN), NCs, ACs, and oligodendrocytes as well as CSF, serum, plasma, and hematopoietic cells (166–173). The brain α-syn interacts with proteins and lipids and controls the synaptic vesicle recycling and neurotransmitter release (174–177). However, the SNCA defect and the resultant excess generation and/or formation of normal endogenous or aggregated Agg α-syn in cytoplasmic inclusions of NCs termed as LBs and Lewy neurites (LNs) lead to neuronal toxicity and neurodegeneration in early- and late-onset PD (166, 178–185). Strikingly, LBs and LNs of the idiopathic forms of PD have also shown excess of α-syn and the Agg α-syn without any SNCA mutation (183, 186–188). In contrast, overexpression of wild-type SNCA and the resultant higher production of WT α-syn show their link to neurotoxicity in Drosophila melanogaster (189) and rodent models (190). Normal and Agg α-syn have shown TLR2- or TLR4-mediated MGC activation and neuronal loss in PD and mouse models (70, 191–198). PD genome-wide association studies (GWAS) identified the risk variants in certain loci associated to disease risk such as HLA-DR locus, which encodes for the major histocompatibility complex I (MHC class II) known for triggering the antigen presentation to CD4+ T cells (199–202). Two classical pathways of antigen presentation have been described for the presentation of endogenous antigens on MHC I molecules and the presentation of exogenous antigens, such as intracellular pathogens, on MHC class II molecules [reviewed by Blum et al. (203)]. The MHCII pathway is performed by specialized antigen-presenting cells, i.e., Mϕs, DCs, and DA neurons, which present peptides on MHCII molecules, ensuring its efficient recognition by CD4+ T cells (204). In addition to the increased brain infiltration of effector T-cell subsets in PD patients (42, 43), MHCII-mediated presentation of α-syn to CD4+ T cells has been linked to neuroinflammation in a mouse model and human PD (205–207). α-Syn peptide-stimulated T cells have shown development of activated subsets of helper and cytotoxic T cells and increased production of IFNγ, IL-2, and IL-5 (205). In addition, one of the peptide regions strongly binds to MHC encoded by HLA (DRB1*15:01, DRB5*01:01) linked to PD by GWAS (201, 208–210).
The sera, MGCs, and brain regions of the SNCA mouse model of PD have shown overexpression of different species of α-syn and pro-inflammatory cytokines (e.g., IFNγ, TNFα, IL-1α, IL-1β, IL-6, IL-10, TGFβ, CCL2, CCL3, CCL5, CXCL10, and ICAM-1) as well as their link to neuronal cell death and cognitive defects (Table 4A). The blood-derived immune cells, sera, and brain regions of PD patients with SNCA defect have also shown overexpression of α-syn and their association with cellular activation and increased generation of pro-inflammatory mediators (e.g., IFNγ, TNFα, IL-1β, IL-4, IL-5, IL-6, IL-18, and CCL2) as well as their link to neuronal cell damage (Table 4B). Hence, SNCA defects and the resultant increased making of normal and/or Agg α-syn promote the activation of peripheral immune cells and the brain MGCs. Such cells cause massive generation of NO, ROS, and pro-inflammatory cytokines and chemokines (Tables 4A,B), which are all critical for promoting brain inflammation and neurodegeneration in SNCA-associated PD (Figure 1C).
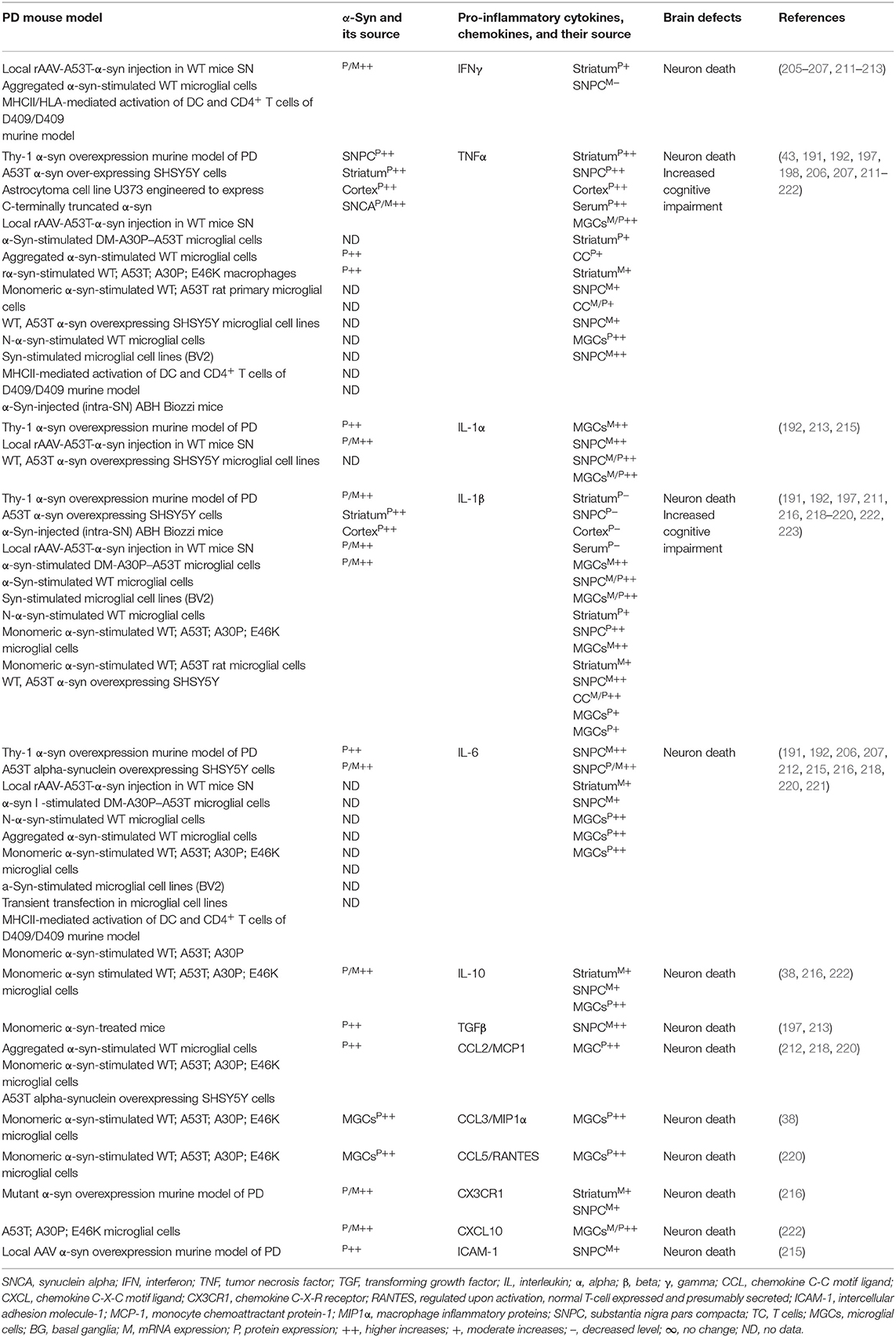
Table 4A. Cytokines and their source in the mouse model of SNCA PD.

Table 4B. Cytokines and their source in the SNCA-associated human PD.
PARK2 Gene Defects and Pro-inflammatory Immune Mediators in PD
The PARK2 gene encodes cytosolic ubiquitin E3 ligase termed as parkin protein, which is critical for the targeting, breakdown, and recycling of damaged proteins as well as the regulation of mitophagy and survival of DA neurons (224). PARK2 mutations cause a loss of parkin function that leads to the excess accumulation of dysfunctional mitochondria and the resultant massive generation of oxidative stress and death of DA neurons in autosomal recessive and idiopathic PD (225–235). CD4+ and CD8+ cell infiltration, MGC activation, increased generation of pro-inflammatory cytokines, and the loss of DA neurons have been observed in mouse model and human PD (43, 236).
Parkin plays a protective role during bacterial and viral infection and chemically induced oxidative and ER stress by altering the mitochondrial ROS and pro-inflammatory cytokine-mediated downstream signaling cascades (237–247). Biochemical and genetic studies reveal that parkin also acts in tandem with phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1), which is accountable for controlling the mitochondrial quality (248). Indeed, mutations in the genes that encode PINK1 and Parkin showed massive mitochondrial damage and the development of familial PD (229). It has been shown that autophagy, the recycling of self-components through lysosomal degradation, is involved in the presentation of endogenous antigens on both MHC class I and class II molecules (249, 250), highlighting that vacuolar content can also be presented on MHC class I/II molecules. The mitochondrial MHCI-mediated antigen processing and presentation to CD8+ T cells have been valued for induction of neuroinflammation in mouse models and human PD (42, 43, 205, 251, 252). To understand the exact role of parkin and PINK1 in the development of brain inflammation in PD, Matheoud et al. (252) have discovered a pathway for mitochondrial antigen presentation, in which mitochondria-derived vesicles targeted endolysosomes for processing and presentation by MHC class I molecules. Using both in vitro and in vivo experiments, this study has demonstrated that parkin and PINK1 inhibit mitochondria-derived vesicle formation and mitochondrial antigen presentation, and therefore, in the absence of PINK1 or parkin, mitochondrial antigen presentation triggers DC and CD8+ T-cell activation and increased generation of pro-inflammatory cytokines. These data suggest that PINK1 and/or parkin has a key role in the activation of innate and adaptive immune cells by repressing the presentation of mitochondrial antigens, which suggests the involvement of autoimmune reactions in PD (252). PARK2 mutations and their link to α-syn inclusions and LB formation have also been observed in exceptional cases of PARK2-associated PD (253–255). The exact mechanism by which PARK2 defects propagate brain inflammation and neurodegeneration in PD is poorly defined.
The MGCs, Mϕs, and sera of the PARK2 mouse model displayed decreased expression of parkin and its link to the increased generation of pro-inflammatory cytokines and chemokines (e.g., IFNβ1, TNFα, IL-1β, IL-12, IL-13, IL-17, CCL2, and CXCL1), loss of DA neurons, and cognitive defects in PD (Table 5A). The sera, MGCs, Mϕs, and midbrain regions of PARK2-associated human PD also displayed decreased expression of parkin and its link to increased generation of pro-inflammatory cytokines (e.g., IFNβ1, TNFα, IL-1β, IL-6, IL-12, IL-13, CCL2, CCL4, and CXCL1), loss of DA neurons, and cognitive defects in PD (Table 5B). These findings suggest that PARK2 and the resultant deficiency of parkin are associated with mitochondrial damage and/or the formation of Agg α-syn. These defects cause cellular activation and massive generation of pro-inflammatory cytokines and chemokines (Tables 5A,B), which lead to the loss of DA neurons in PARK2-associated PD (Figure 1D).
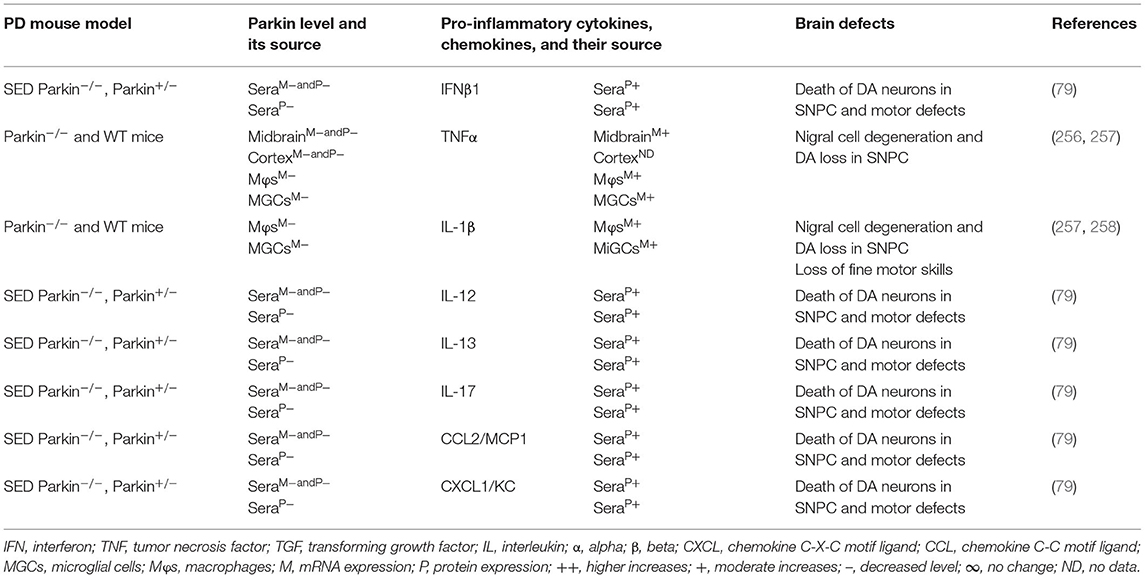
Table 5A. Cytokines and their source in the mouse model of PARK2 PD.
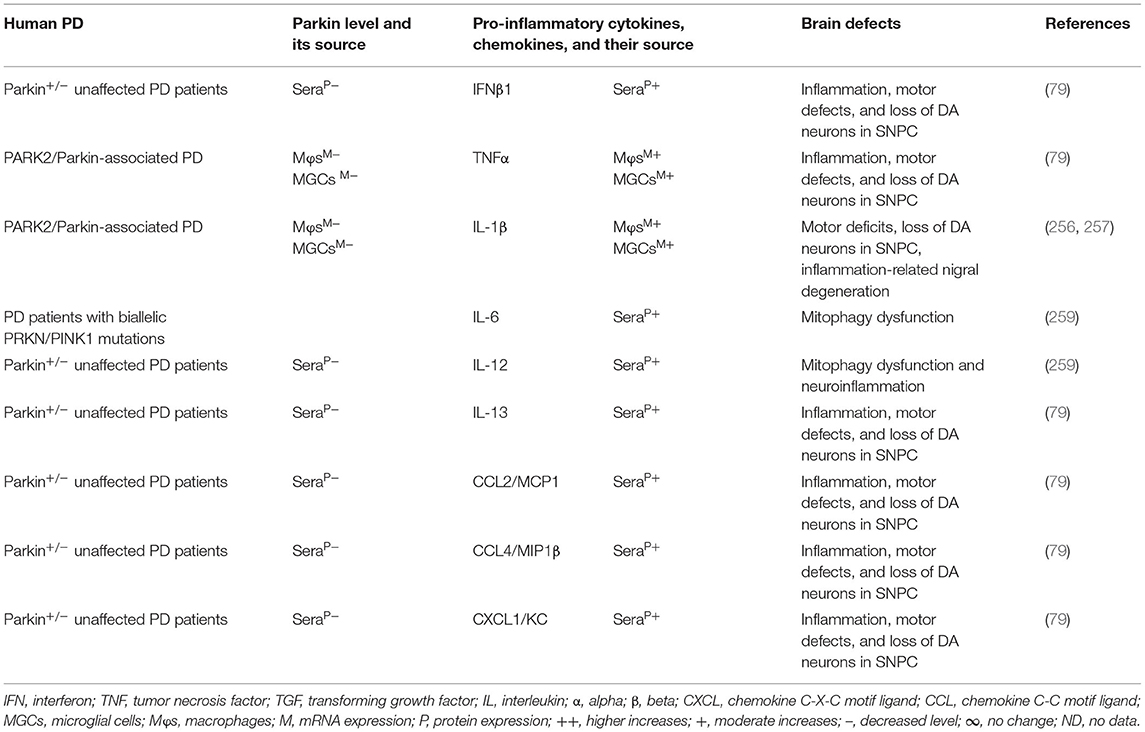
Table 5B. Cytokines and their source in the PARK2-associated human PD.
PARK6 Gene Defects and Pro-inflammatory Immune Mediators in PD
The PARK6 gene encodes PINK1, which is a universally expressed serine/threonine kinase with a mitochondrial targeting sequence that directs the import of PINK1 as well as the activation and recruitment of parkin into the mitochondria for clearance of damaged mitochondria (260–267). PINK1-deficient cells, including NCs, are more susceptible to various insults (268, 269). PINK1 and parkin control the degradation of dysfunctional mitochondria (270, 271). PARK6 defects and the resultant deficiency of PINK1 lead to mitochondrial dysfunctions and the development of autosomal recessive and early-onset PD (261, 272–274). Pink1-deficient Drosophila displayed mitochondrial damage associated with apoptotic muscle degeneration and DA neuron loss, whereas Parkin overexpression protected such PINK1-induced defects (248, 275, 276). Several studies have shown that PINK1, like parkin, modulates NF-κB activity and brain generation of pro-inflammatory cytokines (277). PINK1-deficient T cells have reduced protein kinase B (PKB or Akt) activity, which is critical for inducible regulatory T cells (iTreg) development (278). PINK1-deficient iTreg cells showed reduced capacity to suppress lymphocyte proliferation (278). Importantly, the autologous transfer of Treg cells to MPTP-treated mice attenuated MGC activation and provides neuroprotection (279).
Strikingly, Treg cells from PD patients also have impaired suppressor function (47). T-cell subset infiltration and their interaction with MGCs and DA neurons are critical for the development of neuroinflammation and neurodegeneration in MPTP-induced mouse model and human patients with PD (43, 47, 48, 280, 281). Gram-negative bacteria-induced intestinal infection in Pink1−/− mice showed mitochondrial antigen presentation to CD8+ T cells in the periphery and in the brain and their link to loss of DA axonal varicosities in the striatum and the motor impairment. These data suggest the relevance of the gut–brain axis that could develop brain inflammation and neurodegeneration in PD (282, 283).
The blood, brain regions, and cells of the mouse model of PARK6-associated PD have shown PINK1 deficiency and its impact on increased blood or brain generation of pro-inflammatory cytokines and chemokines (e.g., IFNγ, IFNβ1, TNFα, IL-1β, IL-2, IL-6, IL-10, IL-12, IL-13, IL-17, TGFβ, CCL2, CCL4, and CXCL1), loss of neuronal cells, and the development of cognitive defects in PD (Table 6A). Additionally, PARK6-associated PD patients have also shown PINK1 deficiency and its impact on increased generation of pro-inflammatory cytokines and chemokines (e.g., IFNβ1, IL-6, IL-12, IL-13, CCL2, CCL4, and CXCL1), loss of NCs, and the development of cognitive defects (Table 6B). These findings suggest that PARK6 and the resultant PINK1 defects trigger residential and infiltrated immune cell activation and increased production of pro-inflammatory cytokines and chemokines (Tables 6A,B), which ultimately lead to the loss of DA neurons in PARK6-associated PD (Figure 1E).
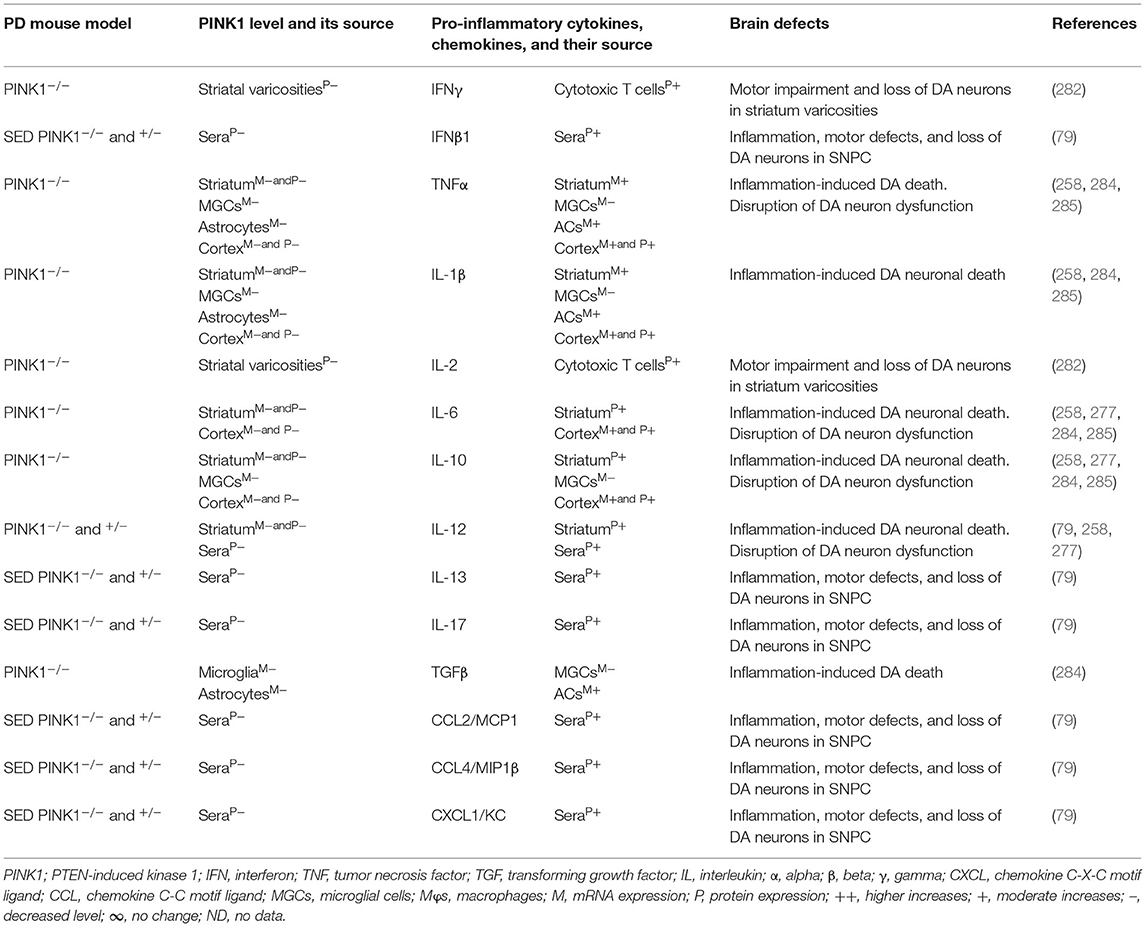
Table 6A. Cytokines and their source in the mouse model of PARK6 PD.
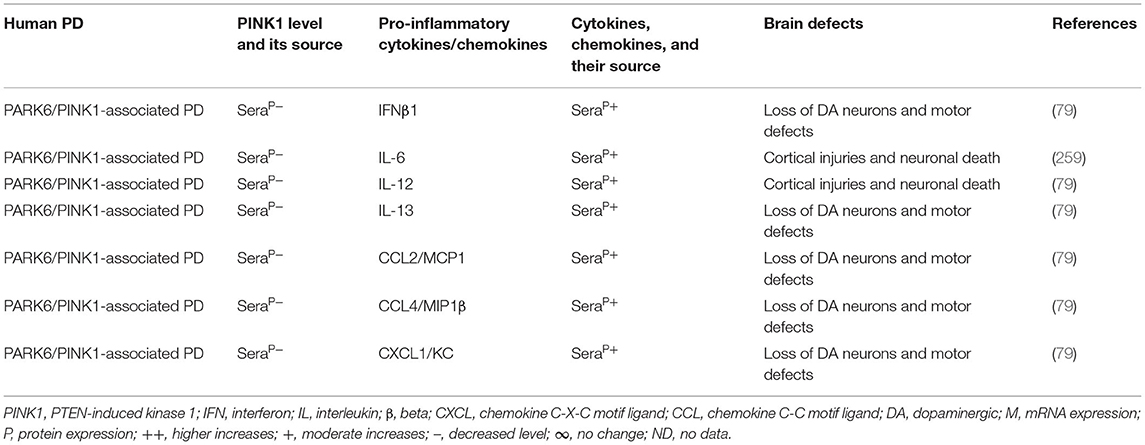
Table 6B. Cytokines and their source in the PARK6-associated human PD.
PARK7 gENE Defects and Pro-inflammatory Immune Mediators in PD
PARK7 encodes a protein deglycase DJ-1, which belongs to the peptidase C56 family of proteins and ubiquitously expressed under physiological conditions (286). Like PINK1 and parkin, DJ-1 is required for controlling mitochondrial damage and production of oxidative stress (287–289). Several chemicals and physiological factors trigger the upregulation of DJ-1, which protects the oxidative and endoplasmic reticulum stress-induced damage of endothelial cells, Mϕs, fibroblast, NCs, and islet β cells (290–296), and therefore, DJ-1 deficiency has been associated with the development of several diseases (e.g., stroke, male infertility, cancers, diabetes, and neurodegenerative illnesses) (290, 297, 298). Escherichia coli- or Pseudomonas aeruginosa-mediated excess activation of MAPK signaling and the resultant induction of brain inflammation have been observed in DJ-1-deficient Caenorhabditis elegans (299). Mutations in PARK7 and the resultant deficiency or the oxidized form of DJ-1 protein cause autosomal recessive early-onset and idiopathic PD as reviewed in ref. (300).
Brain regions and their cells of the mouse model of PARK7-associated PD have shown DJ-1 deficiency and its effect on increased production of IFNγ, IL-1β, IL-1Ra, IL-6, IL-16, IL-17, CXCL11, and NGF as well as on the damage of ACs and DA neurons (Table 7A). Furthermore, abnormal cellular and brain region expression of DJ-1 has been associated with the formation of α-syn and Tau containing LBs, mitochondrial damage, increased production of ROS, and their link to the loss of NCs in PD patients with PARK7 mutation (Table 7B). These data suggest that PARK7 and the resultant DJ-1 deficiency induced mitochondrial damage and/or the formation of Agg α-syn and Tau comprising LB. These abnormal proteins cause massive generation of pro-inflammatory cytokines and chemokines (Tables 7A,B), which ultimately lead to the death of DA neurons in PARK7-associated PD (Figure 1F).
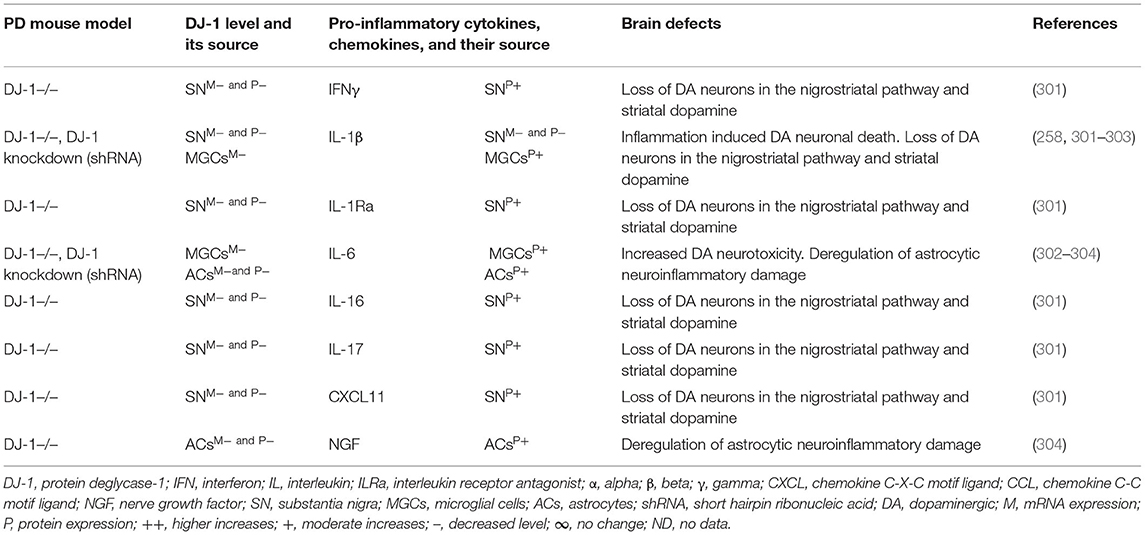
Table 7A. Cytokines and their source in the mouse model of PARK7 PD.
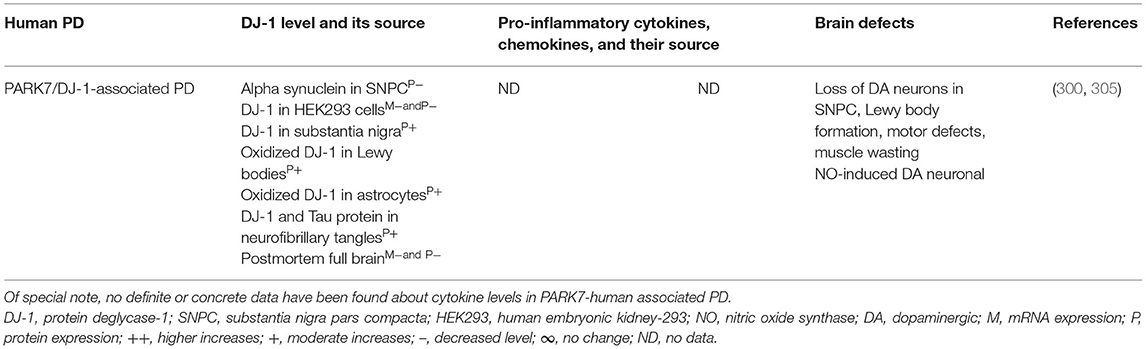
Table 7B. Cytokines and their source in the PARK7-associated human PD.
Conclusion
The molecular mechanisms by which LRRK2, GBA, SNCA, PARK2, PARK6, and PARK7 defects trigger neuroinflammation and neurodegeneration in PD are poorly defined and need more studies. However, the abnormal function of LRRK2, GBA, SNCA, PARK2, PARK6, and PARK7 genes has been linked to alteration in innate and adaptive immune responses in cancer, stroke, diabetes, male infertility, Crohn's disease, and infectious diseases (59, 96–98, 125, 127–133, 237–245, 290, 297, 298, 306–309). Findings from mouse models, cell system, and human specimens have shown that the abnormal expressions of LRRK2, GBA, SNCA, PARK2, PARK6, and PARK7 genes and their corresponding proteins or enzymes (e.g., LRRK2, GCase, α-syn, parkin, PINK1, and DJ-1) are linked to the activation of MGCs, ACs, and NCs and the massive production of growth factors (e.g., GCSF, GMCSF, MCSF) and CCL and CXCL chemokines (i.e., CCL2/MCP1, CCL3/MIP1α, CCL4/MIP1β, CCL5/RANTES, CXCL1, and CXCL10), which are all accountable for the development and trafficking of immunological cells from the peripheral blood and bone marrow to the sites of inflammation for the generation of pro-inflammatory cytokines that lead to tissue destruction (61–69). The CCL2/MCP1, CCL3/MIP1α, CCL4/MIP1β, CCL5/RANTES, CXCL1, and CXCL10 chemokines are specific chemoattractants for tissue recruitment of several inflammatory subsets of MOs, Mφs, DCs, and CD4+ and CD8+ T cells (59, 60). Certain inflammatory conditions cause accelerated migration of immunological cell precursors out of the bone marrow and into the circulation (310–312). A similar condition is thought to occur in PD due to genetic defects in LRRK2, GBA, SNCA, PARK2, PARK6, and PARK7 genes and the resultant alteration in the expression of their corresponding proteins or enzymes, i.e., LRRK2, GCase, α-Syn, parkin, PINK1, and DJ-1, which leads to the establishment of a network of several of the innate and adaptive immune cells, i.e., MOs and memory and effector T cells (43, 46–51). Hence, it is possible that immune cell integration and the resultant generation of pro-inflammatory cytokines at the periphery alter the blood–brain barrier integrity. This situation permits the recruitment of immune cells, to the specific region of the brain where infiltrated (e.g., MOs, DCs, CD4+ T cells, and CD8+ T cells) and residential immune cells (e.g., MGCs) meet and amplify their activation, and the resultant massive generation of pro-inflammatory cytokines (e.g., IFNγ, TNFα, IL-1β, IL-6, IL-8, IL-12, and IL-17), which are all lethal to DA neurons, and this condition develops neurodegeneration in PD.
Author Contributions
AFM and SLH prepared and designed the tables. RR designed the figures and assisted in the writing and critical review of the text. MKP conceptualized, designed, wrote, reviewed, edited, and approved the submitted version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Division of Human Genetics, Cincinnati Children's Hospital Medical Center; Cincinnati, OH, USA (31-40327-584000-143032 to MKP), and Michael J. Fox Foundation; New York, NY, USA; (31-90000-584000-305961 to MKP). This study was supported in part by Cincinnati Children's Research Innovation/Pilot Research Award (31-41642-584000-143032 to MKP), and University of Cincinnati Center for Clinical and Translational Science Just-in-Time Core Research Award (31-41268-584000 to MKP).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with the author MKP.
Acknowledgments
We would like to acknowledge BioRender tools for developing the figures and Mr. Charles D. Loftice for office and laboratory support.
References
1. Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. (2008) 79:368. doi: 10.1136/jnnp.2007.131045
2. Bonnet AM, Jutras MF, Czernecki V, Corvol JC, Vidailhet M. Nonmotor symptoms in Parkinson's disease in 2012: relevant clinical aspects. Parkinson Dis. (2012) 2012:198316. doi: 10.1155/2012/198316
3. Subhramanyam CS, Wang C, Hu Q, Dheen ST. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol. (2019) 94:112–20. doi: 10.1016/j.semcdb.2019.05.004
4. Talaat RM, Mohamed SF, Bassyouni IH, Raouf AA. Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: correlation with disease activity. Cytokine. (2015) 72:146–53. doi: 10.1016/j.cyto.2014.12.027
5. Taipa R, das Neves SP, Sousa AL, Fernandes J, Pinto C, Correia AP, et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer's disease and their correlation with cognitive decline. Neurobiol Aging. (2019) 76:125–32. doi: 10.1016/j.neurobiolaging.2018.12.019
6. Mrugacz M, Ostrowska L, Bryl A, Szulc A, Zelazowska-Rutkowska B, Mrugacz G. Pro-inflammatory cytokines associated with clinical severity of dry eye disease of patients with depression. Adv Med Sci. (2017) 62:338–44. doi: 10.1016/j.advms.2017.03.003
7. He W, Long T, Pan Q, Zhang S, Zhang Y, Zhang D, et al. Microglial NLRP3 inflammasome activation mediates IL-1β release and contributes to central sensitization in a recurrent nitroglycerin-induced migraine model. J Neuroinflamm. (2019) 16:78. doi: 10.1186/s12974-019-1459-7
8. Sirivichayakul S, Kanchanatawan B, Thika S, Carvalho AF, Maes M. A new schizophrenia model: immune activation is associated with the induction of different neurotoxic products which together determine memory impairments and schizophrenia symptom dimensions. CNS Neurol Disord Drug Targets. (2019) 18:124–40. doi: 10.2174/1871527317666181119115532
9. Decourt B, Lahiri DK, Sabbagh MN. Targeting tumor necrosis factor alpha for Alzheimer's disease. Curr Alzheimer Res. (2017) 14:412–25. doi: 10.2174/1567205013666160930110551
10. Zhang L, Zheng H, Wu R, Kosten TR, Zhang XY, Zhao J. The effect of minocycline on amelioration of cognitive deficits and pro-inflammatory cytokines levels in patients with schizophrenia. Schizophrenia Res. (2019) 212:92–8. doi: 10.1016/j.schres.2019.08.005
11. Moaaz M, Youssry S, Elfatatry A, El Rahman MA. Th17/Treg cells imbalance and their related cytokines (IL-17, IL-10 and TGF-β) in children with autism spectrum disorder. J Neuroimmunol. (2019) 337:577071. doi: 10.1016/j.jneuroim.2019.577071
12. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. (2018) 21:1359–69. doi: 10.1038/s41593-018-0242-x
13. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
14. Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. (2011) 11:775–87. doi: 10.1038/nri3086
15. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
16. Cameron B, Landreth GE. Inflammation, microglia, alzheimer's disease. Neurobiol Dis. (2010) 37:503–9. doi: 10.1016/j.nbd.2009.10.006
17. Perry VH, Nicoll JAR, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. (2010) 6:193–201. doi: 10.1038/nrneurol.2010.17
18. O'Keefe GM, Nguyen VT, Benveniste EN. Regulation and function of class II major histocompatibility complex, CD40, and B7 expression in macrophages and microglia: implications in neurological diseases. J Neurovirol. (2002) 8:496–512. doi: 10.1080/13550280290100941
19. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. (1989) 7:145–73. doi: 10.1146/annurev.iy.07.040189.001045
20. Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. (2001) 15:985–95. doi: 10.1016/S1074-7613(01)00243-6
21. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
22. Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. (2004) 21:467–76. doi: 10.1016/j.immuni.2004.08.018
23. Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. (2009) 324:1569–72. doi: 10.1126/science.1174182
24. Harker JA, Lewis GM, Mack L, Zuniga EI. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science. (2011) 334:825–9. doi: 10.1126/science.1208421
25. O'Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. (2010) 327:1098–102. doi: 10.1126/science.1178334
26. Machhi J, Kevadiya BD, Muhammad IK, Herskovitz J, Olson KE, Mosley RL, et al. Harnessing regulatory T cell neuroprotective activities for treatment of neurodegenerative disorders. Mol Neurodegener. (2020) 15:32. doi: 10.1186/s13024-020-00375-7
27. Sommer A, Winner B, Prots I. The Trojan horse - neuroinflammatory impact of T cells in neurodegenerative diseases. Mol Neurodegener. (2017) 12:78. doi: 10.1186/s13024-017-0222-8
28. Solleiro-Villavicencio H, Rivas-Arancibia S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4+T cells in neurodegenerative diseases. Front Cell Neurosci. (2018) 12:114. doi: 10.3389/fncel.2018.00114
29. Jankovic D, Kugler DG, Sher A. IL-10 production by CD4+ effector T cells: a mechanism for self-regulation. Mucosal Immunol. (2010) 3:239–46. doi: 10.1038/mi.2010.8
30. O'Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. (1998) 8:275–83. doi: 10.1016/S1074-7613(00)80533-6
31. Sher A, Coffman RL. Regulation of immunity to parasites by T cells and T cell-derived cytokines. Annu Rev Immunol. (1992) 10:385–409. doi: 10.1146/annurev.iy.10.040192.002125
32. Romagnani S. Lymphokine production by human T cells in disease states. Annu Rev Immunol. (1994) 12:227–57. doi: 10.1146/annurev.iy.12.040194.001303
33. Bottomly K. A functional dichotomy in CD4+ T lymphocytes. Immunol Today. (1988) 9:268–74. doi: 10.1016/0167-5699(88)91308-4
34. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. (2003) 198:1951–7. doi: 10.1084/jem.20030896
35. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
36. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
37. Sojka DK, Fowell DJ. Regulatory T cells inhibit acute IFN-gamma synthesis without blocking T-helper cell type 1 (Th1) differentiation via a compartmentalized requirement for IL-10. Proc Natl Acad Sci USA. (2011) 108:18336–41. doi: 10.1073/pnas.1110566108
38. Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med. (2011) 3:111ra120. doi: 10.1126/scitranslmed.3003130
39. Ellwardt E, Walsh JT, Kipnis J, Zipp F. Understanding the role of T cells in CNS homeostasis. Trends Immunol. (2016) 37:154–65. doi: 10.1016/j.it.2015.12.008
40. Korn T, Kallies A. T cell responses in the central nervous system. Nat Rev Immunol. (2017) 17:179–94. doi: 10.1038/nri.2016.144
41. Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debré P, et al. FcεRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-α in glial cells. J Neurosci. (1999) 19:3440. doi: 10.1523/JNEUROSCI.19-09-03440.1999
42. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. (1988) 38:1285–91. doi: 10.1212/WNL.38.8.1285
43. Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Investig. (2009) 119:182–92. doi: 10.1172/JCI36470
44. Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci Lett. (1994) 180:147–50. doi: 10.1016/0304-3940(94)90508-8
45. Chen X, Hu Y, Cao Z, Liu Q, Cheng Y. Cerebrospinal fluid inflammatory cytokine aberrations in Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis: a systematic review and meta-analysis. Front Immunol. (2018) 9:2122. doi: 10.3389/fimmu.2018.02122
46. Grozdanov V, Bliederhaeuser C, Ruf WP, Roth V, Fundel-Clemens K, Zondler L, et al. Inflammatory dysregulation of blood monocytes in Parkinson's disease patients. Acta Neuropathol. (2014) 128:651–63. doi: 10.1007/s00401-014-1345-4
47. Saunders JAH, Estes KA, Kosloski LM, Allen HE, Dempsey KM, Torres-Russotto DR, et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson's disease. J Neuroimmune Pharmacol. (2012) 7:927–38. doi: 10.1007/s11481-012-9402-z
48. Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Related Disord. (2005) 11:493–8. doi: 10.1016/j.parkreldis.2005.07.005
49. Chen X, Liu Z, Cao B-B, Qiu Y-H, Peng Y-P. TGF-β1 neuroprotection via inhibition of microglial activation in a rat model of Parkinson's disease. J Neuroimmune Pharmacol. (2017) 12:433–46. doi: 10.1007/s11481-017-9732-y
50. Yang L, Guo C, Zhu J, Feng Y, Chen W, Feng Z, et al. Increased levels of pro-inflammatory and anti-inflammatory cellular responses in parkinson's disease patients: search for a disease indicator. Med Sci Monit. (2017) 23:2972–8. doi: 10.12659/MSM.904240
51. Kustrimovic N, Rasini E, Legnaro M, Bombelli R, Aleksic I, Blandini F, et al. Dopaminergic receptors on CD4+ T naive and memory lymphocytes correlate with motor impairment in patients with Parkinson's disease. Sci Rep. (2016) 6:33738. doi: 10.1038/srep33738
52. Bu X-L, Wang X, Xiang Y, Shen L-L, Wang Q-H, Liu Y-H, et al. The association between infectious burden and Parkinson's disease: a case-control study. Parkinsonism Related Disord. (2015) 21:877–81. doi: 10.1016/j.parkreldis.2015.05.015
53. Williams-Gray CH, Wijeyekoon R, Yarnall AJ, Lawson RA, Breen DP, Evans JR, et al. Serum immune markers and disease progression in an incident Parkinson's disease cohort (ICICLE-PD). Movement Disord. (2016) 31:995–1003. doi: 10.1002/mds.26563
54. Dursun E, Gezen-Ak D, Hanagasi H, Bilgiç B, Lohmann E, Ertan S, et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer's disease, mild cognitive impairment or Parkinson's disease. J Neuroimmunol. (2015) 283:50–7. doi: 10.1016/j.jneuroim.2015.04.014
55. Hu Y, Yu S-Y, Zuo L-J, Cao C-J, Wang F, Chen Z-J, et al. Parkinson disease with REM sleep behavior disorder. Neurology. (2015) 84:888. doi: 10.1212/WNL.0000000000001308
56. Qin X-Y, Zhang S-P, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: a systematic review and meta-analysis. JAMA Neurol. (2016) 73:1316–24. doi: 10.1001/jamaneurol.2016.2742
57. McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR, et al. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson's disease. J Neurosci. (2006) 26:9365. doi: 10.1523/JNEUROSCI.1504-06.2006
58. Scalzo P, Kümmer A, Cardoso F, Teixeira AL. Serum levels of interleukin-6 are elevated in patients with Parkinson's disease and correlate with physical performance. Neurosci Lett. (2010) 468:56–8. doi: 10.1016/j.neulet.2009.10.062
59. Pandey MK, Grabowski GA. Immunological cells and functions in Gaucher disease. Crit Rev Oncog. (2013) 18:197–220. doi: 10.1615/CritRevOncog.2013004503
60. Pandey MK, Grabowski GA. Cytology of Gaucher disease in advances in Gaucher disease: basic and clinical perspectives. Future Med. (2013) 78–93. doi: 10.2217/ebo.12.209
61. Ripa RS. Granulocyte-colony stimulating factor therapy to induce neovascularization in ischemic heart disease. Dan Med J. (2012) 59:B4411.
62. Zohlnhofer D, Ott I, Mehilli J, Schomig K, Michalk F, Ibrahim T, et al. Stem cell mobilization by granulocyte colony-stimulating factor in patients with acute myocardial infarction: a randomized controlled trial. JAMA. (2006) 295:1003. doi: 10.1001/jama.296.16.1968-b
63. Zhang H, Bai H, Yi Z, He X, Mo S. Effect of stem cell factor and granulocyte-macrophage colony-stimulating factor-induced bone marrow stem cell mobilization on recovery from acute tubular necrosis in rats. Ren Fail. (2012) 34:350–7. doi: 10.3109/0886022X.2011.647340
64. Rapoport AP, Abboud CN, DiPersio JF. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF): receptor biology, signal transduction, neutrophil activation. Blood Rev. (1992) 6:43–57. doi: 10.1016/0268-960X(92)90007-D
65. Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. (1994) 84:1737–46. doi: 10.1182/blood.V84.6.1737.1737
66. Avigan D, Wu Z, Gong J, Joyce R, Levine J, Elias A, et al. Selective in vivo mobilization with granulocyte macrophage colony-stimulating factor (GM-CSF)/granulocyte-CSF as compared to G-CSF alone of dendritic cell progenitors from peripheral blood progenitor cells in patients with advanced breast cancer undergoing autologous transplantation. Clin Cancer Res. (1999) 5:2735–41.
67. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. (2006) 354:610–21. doi: 10.1056/NEJMra052723
68. Mantovani A, Bonecchi R, Locati M. Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol. (2006) 6:907–18. doi: 10.1038/nri1964
69. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, dendritic cells. Science. (2010) 327:656–61. doi: 10.1126/science.1178331
70. Chesselet M-F, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR. A progressive mouse model of Parkinson's disease: the Thy1-aSyn (“line 61”) mice. Neurotherapeutics. (2012) 9:297–314. doi: 10.1007/s13311-012-0104-2
71. Brodacki B, Staszewski J, Toczyłowska B, Kozłowska E, Drela N, Chalimoniuk M, et al. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci Lett. (2008) 441:158–62. doi: 10.1016/j.neulet.2008.06.040
72. Ciesielska A, Joniec I, Kurkowska-Jastrzebska I, Przybyłkowski A, Gromadzka G, Członkowska A, et al. Influence of age and gender on cytokine expression in a murine model of Parkinson's disease. Neuroimmunomodulation. (2007) 14:255–65. doi: 10.1159/000113432
73. Mandel S, Grünblatt E, Youdim M. cDNA microarray to study gene expression of dopaminergic neurodegeneration and neuroprotection in MPTP and 6-hydroxydopamine models: implications for idiopathic Parkinson's disease. Neuroimmunomodulation. (2000) 117–24. doi: 10.1007/978-3-7091-6301-6_7
74. Lofrumento DD, Saponaro C, Cianciulli A, De Nuccio F, Mitolo V, Nicolardi G, et al. MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. Neuroimmunomodulation. (2011) 18:79–88. doi: 10.1159/000320027
75. Chung YC, Bok E, Huh SH, Park J-Y, Yoon S-H, Kim SR, et al. Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J Immunol. (2011) 187:6508. doi: 10.4049/jimmunol.1102435
76. Chung YC, Kim SR, Jin BK. Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson's disease. J Immunol. (2010) 185:1230–7. doi: 10.4049/jimmunol.1000208
77. Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, et al. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Different. (2019) 26:213–28. doi: 10.1038/s41418-018-0124-5
78. Javed H, Thangavel R, Selvakumar GP, Dubova I, Schwartz N, Ahmed ME, et al. NLRP3 inflammasome and glia maturation factor coordinately regulate neuroinflammation and neuronal loss in MPTP mouse model of Parkinson's disease. Int Immunopharmacol. (2020) 83:106441. doi: 10.1016/j.intimp.2020.106441
79. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. (2018) 561:258–62. doi: 10.1038/s41586-018-0448-9
80. Lopez de Maturana R, Aguila JC, Sousa A, Vazquez N, Del Rio P, Aiastui A, et al. Leucine-rich repeat kinase 2 modulates cyclooxygenase 2 and the inflammatory response in idiopathic and genetic Parkinson's disease. Neurobiol Aging. (2014) 35:1116–24. doi: 10.1016/j.neurobiolaging.2013.11.018
81. McGeer PL, Yasojima K, McGeer EG. Association of interleukin-1β polymorphisms with idiopathic Parkinson's disease. Neurosci Lett. (2002) 326:67–9. doi: 10.1016/S0304-3940(02)00300-2
82. Klüter H, Vieregge P, Stolze H, Kirchner H. Defective production of interleukin-2 in patients with idiopathic Parkinson's disease. J Neurol Sci. (1995) 133:134–9. doi: 10.1016/0022-510X(95)00180-A
83. Dufek M, Rektorova I, Thon V, Lokaj J, Rektor I. Interleukin-6 may contribute to mortality in Parkinson's disease patients: a 4-year prospective study. Parkinson's Dis. (2015) 2015:898192. doi: 10.1155/2015/898192
84. Qiu X, Xiao Y, Wu J, Gan L, Huang Y, Wang J. C-reactive protein and risk of Parkinson's disease: a systematic review and meta-analysis. Front Neurol. (2019) 10:384. doi: 10.3389/fneur.2019.00384
85. Rudenko IN, Chia R, Cookson MR. Is inhibition of kinase activity the only therapeutic strategy for LRRK2-associated Parkinson's disease? BMC Med. (2012) 10:20. doi: 10.1186/1741-7015-10-20
86. Rideout HJ, Stefanis L. The neurobiology of LRRK2 and its role in the pathogenesis of Parkinson's disease. Neurochem Res. (2014) 39:576–92. doi: 10.1007/s11064-013-1073-5
87. Wallings R, Manzoni C, Bandopadhyay R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. (2015) 282:2806–26. doi: 10.1111/febs.13305
88. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. (2010) 185:5577–85. doi: 10.4049/jimmunol.1000548
89. Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet. (2014) 23:4201–14. doi: 10.1093/hmg/ddu138
90. Miklossy J, Arai T, Guo JP, Klegeris A, Yu S, McGeer EG, et al. LRRK2 expression in normal and pathologic human brain and in human cell lines. J Neuropathol Exp Neurol. (2006) 65:953–63. doi: 10.1097/01.jnen.0000235121.98052.54
91. Gillardon F, Schmid R, Draheim H. Parkinson's disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience. (2012) 208:41–8. doi: 10.1016/j.neuroscience.2012.02.001
92. Kuss M, Adamopoulou E, Kahle PJ. Interferon-γ induces leucine-rich repeat kinase LRRK2 via extracellular signal-regulated kinase ERK5 in macrophages. J Neurochem. (2014) 129:980–7. doi: 10.1111/jnc.12668
93. Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer. (1980) 26:171–6. doi: 10.1002/ijc.2910260208
94. Thévenet J, Pescini Gobert R, van Huijsduijnen RH, Wiessner C, Sagot YJ. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS ONE. (2011) 6:e21519. doi: 10.1371/journal.pone.0021519
95. Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, et al. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. (2012) 32:1602–11. doi: 10.1523/JNEUROSCI.5601-11.2012
96. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. (2010) 42:1118–25. doi: 10.1038/ng.717
97. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
98. Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, et al. Genomewide association study of leprosy. N Engl J Med. (2009) 361:2609–18. doi: 10.1056/NEJMoa0903753
99. Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. (2010) 7:243–9. doi: 10.1038/cmi.2010.10
100. West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, et al. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. (2005) 102:16842. doi: 10.1073/pnas.0507360102
101. Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. (2006) 23:329–41. doi: 10.1016/j.nbd.2006.04.001
102. Luzón-Toro B, Rubio de la Torre E, Delgado A, Pérez-Tur J, Hilfiker S. Mechanistic insight into the dominant mode of the Parkinson's disease-associated G2019S LRRK2 mutation. Hum Mol Genet. (2007) 16:2031–9. doi: 10.1093/hmg/ddm151
103. Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, et al. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. eLife. (2016) 5:1–28. doi: 10.7554/eLife.12813
104. Greggio E. Role of LRRK2 kinase activity in the pathogenesis of Parkinson's disease. Biochem Soc Trans. (2012) 40:1058–62. doi: 10.1042/BST20120054
105. Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. (2008) 7:583–90. doi: 10.1016/S1474-4422(08)70117-0
106. Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. (2009) 41:1303–7. doi: 10.1038/ng.485
107. Haugarvoll K, Rademakers R, Kachergus JM, Nuytemans K, Ross OA, Gibson JM, et al. Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology. (2008) 70:1456–60. doi: 10.1212/01.wnl.0000304044.22253.03
108. Haugarvoll K, Wszolek ZK. Clinical features of LRRK2 parkinsonism. Parkinsonism Relat Disord. (2009) 3(15 Suppl):S205–8. doi: 10.1016/S1353-8020(09)70815-6
109. Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. (2009) 41:1308–12. doi: 10.1038/ng.487
110. Wider C, Dickson DW, Wszolek ZK. Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegener Dis. (2010) 7:175–9. doi: 10.1159/000289232
111. Sheng Z, Zhang S, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med. (2012) 4:164ra161. doi: 10.1126/scitranslmed.3004485
112. Jaleel M, Nichols RJ, Deak M, David Campbell G, Gillardon F, Knebel A, et al. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. (2007) 405:307–17. doi: 10.1042/BJ20070209
113. Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med. (2010) 16:998–1000. doi: 10.1038/nm.2199
114. Daher JP, Volpicelli-Daley LA, Blackburn JP, Moehle MS, West AB. Abrogation of α-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc Natl Acad Sci USA. (2014) 111:9289–94. doi: 10.1073/pnas.1403215111
115. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. (2004) 44:601–7. doi: 10.1016/j.neuron.2004.11.005
116. Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. (2004) 44:595–600. doi: 10.1016/j.neuron.2004.10.023
117. Dächsel JC, Farrer MJ. LRRK2 and Parkinson disease. Arch Neurol. (2010) 67:542–7. doi: 10.1001/archneurol.2010.79
118. Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson's disease: progress and therapeutic implications. Movement Disord. (2013) 28:14–23. doi: 10.1002/mds.25249
119. Lesage S, Dürr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, et al. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N Engl J Med. (2006) 354:422–3. doi: 10.1056/NEJMc055540
120. Kozina E, Sadasivan S, Jiao Y, Dou Y, Ma Z, Tan H, et al. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain. (2018) 141:1753–69. doi: 10.1093/brain/awy077
121. Russo I, Berti G, Plotegher N, Bernardo G, Filograna R, Bubacco L, et al. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J Neuroinflamm. (2015) 12:230. doi: 10.1186/s12974-015-0449-7
122. Dzamko N, Rowe DB, Halliday GM. Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Movement Disord. (2016) 31:889–97. doi: 10.1002/mds.26529
123. Kim B, Yang M-S, Choi D, Kim J-H, Kim H-S, Seol W, et al. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS ONE. (2012) 7: e34693. doi: 10.1371/journal.pone.0034693
124. Cook DA, Kannarkat GT, Cintron AF, Butkovich LM, Fraser KB, Chang J, et al. LRRK2 levels in immune cells are increased in Parkinson's disease. NPJ Parkinson's Dis. (2017) 3:11. doi: 10.1038/s41531-017-0010-8
125. Grabowski GA, Petsko GA, Kolodny EH. Gaucher disease. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, editors. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: Mc Graw Hill (2010) 1–13.
126. Brady RO, Kanfer J, Shapiro D. The metabolism of glucocerebrosides. Purification I, and properties of a glucocerebroside-cleaving enzyme from spleen tissue. J Biol Chem. (1965) 240:39–43. doi: 10.1016/S0021-9258(18)97611-8
127. Xu YH, Quinn B, Witte D, Grabowski GA. Viable mouse models of acid beta-glucosidase deficiency: the defect in Gaucher disease. Am J Pathol. (2003) 163:2093–101. doi: 10.1016/S0002-9440(10)63566-3
128. Pandey MK, Rani R, Zhang W, Setchell K, Grabowski GA. Immunological cell type characterization and Th1-Th17 cytokine production in a mouse model of Gaucher disease. Mol Genet Metab. (2012) 106:310–22. doi: 10.1016/j.ymgme.2012.04.020
129. Pandey MK, Grabowski GA. Cytology of Gaucher disease. In: Grabowski GA, editor. Advances in Gaucher Disease: Basic and Clinical Perspectives. London: Future Medicine Ltd. (2013). p. 78–93.
130. Pandey MK, Tinch SL, Jabre NA, Grabowski GA. Gaucher disease: glucosylceramide-mediated TLR4-MyD88 induction causes enhanced CXCL-13 secretion and increased B cell trafficking in a mouse model. Mol Genet Metab. (2014) 111:S84. doi: 10.1016/j.ymgme.2013.12.199
131. Pandey MK, Burrow TA, Rani R, Martin LJ, Witte D, Setchell KD, et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature. (2017) 543:108–12. doi: 10.1038/nature21368
132. Pandey MK, Grabowski GA, Kohl J. An unexpected player in Gaucher disease: the multiple roles of complement in disease development. Semin Immunol. (2018) 37:30–42. doi: 10.1016/j.smim.2018.02.006
133. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. New Engl J Med. (2009) 361:1651–61. doi: 10.1056/NEJMoa0901281
134. Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. (2004) 82:192–207. doi: 10.1016/j.ymgme.2004.04.011
135. Vitner EB, Farfel-Becker T, Eilam R, Biton I, Futerman AH. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher's disease. Brain. (2012) 135:1724–35. doi: 10.1093/brain/aws095
136. Vitner EB, Dekel H, Zigdon H, Shachar T, Farfel-Becker T, Eilam R, et al. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Human Mol Genet. (2010) 19:3583–90. doi: 10.1093/hmg/ddq273
137. Dasgupta N, Xu YH, Li R, Peng Y, Pandey MK, Tinch SL, et al. Neuronopathic Gaucher disease: dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Human Mol Genet. (2015) 24:7031–48. doi: 10.1093/hmg/ddv404
138. Gegg ME, Burke D, Heales SJR, Cooper JM, Hardy J, Wood NW, et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. (2012) 72:455–63. doi: 10.1002/ana.23614
139. Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, et al. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson's disease. Brain. (2014) 137:834–48. doi: 10.1093/brain/awt367
140. Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology. (2012) 79:1944–50. doi: 10.1212/WNL.0b013e3182735e9a
141. Moors TE, Paciotti S, Ingrassia A, Quadri M, Breedveld G, Tasegian A, et al. Characterization of brain lysosomal activities in GBA-related and sporadic Parkinson's disease and dementia with lewy bodies. Mol Neurobiol. (2019) 56:1344–55. doi: 10.1007/s12035-018-1090-0
142. Halperin A, Elstein D, Zimran A. Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol Dis. (2006) 36:426–8. doi: 10.1016/j.bcmd.2006.02.004
143. Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. (2004) 41:937. doi: 10.1136/jmg.2004.024455
144. Rosenbloom B, Balwani M, Bronstein JM, Kolodny E, Sathe S, Gwosdow AR, et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher registry. Blood Cells Mol Dis. (2011) 46:95–102. doi: 10.1016/j.bcmd.2010.10.006
145. Lopez G, Kim J, Wiggs E, Cintron D, Groden C, Tayebi N, et al. Clinical course and prognosis in patients with Gaucher disease and parkinsonism. Neurol Genet. (2016) 2:e57. doi: 10.1212/NXG.0000000000000057
146. Zhang Y, Shu L, Zhou X, Pan H, Xu Q, Guo J, et al. A meta-analysis of GBA-related clinical symptoms in Parkinson's disease. Parkinson's Dis. (2018) 2018:3136415. doi: 10.1155/2018/3136415
147. Velez-Pardo C, Lorenzo-Betancor O, Jimenez-Del-Rio M, Moreno S, Lopera F, Cornejo-Olivas M, et al. The distribution and risk effect of GBA variants in a large cohort of PD patients from Colombia and Peru. Parkinsonism Related Disord. (2019) 63:204–8. doi: 10.1016/j.parkreldis.2019.01.030
148. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson's disease: the mutation matters. Ann Neurol. (2016) 80:662–73. doi: 10.1002/ana.24777
149. Thaler A, Gurevich T, Bar Shira A, Gana Weisz M, Ash E, Shiner T, et al. A “dose” effect of mutations in the GBA gene on Parkinson's disease phenotype. Parkinsonism Related Disord. (2017) 36:47–51. doi: 10.1016/j.parkreldis.2016.12.014
150. Neumann J, Bras J, Deas E, O'Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain. (2009) 132:1783–94. doi: 10.1093/brain/awp044
151. Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. (2015) 84:880–7. doi: 10.1212/WNL.0000000000001315
152. Jesús S, Huertas I, Bernal-Bernal I, Bonilla-Toribio M, Cáceres-Redondo MT, Vargas-González L, et al. GBA variants influence motor and non-motor features of Parkinson's disease. PLoS ONE. (2016) 11:e0167749. doi: 10.1371/journal.pone.0167749
153. Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, et al. Features of GBA-associated Parkinson's disease at presentation in the UK Tracking Parkinson's study. J Neurol Neurosurg Psychiatry. (2018) 89:702–9. doi: 10.1136/jnnp-2017-317348
154. Atashrazm F, Hammond D, Perera G, Dobson-Stone C, Mueller N, Pickford R, et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson's disease. Sci Rep. (2018) 8:15446. doi: 10.1038/s41598-018-33921-x
155. Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med. (2011) 17:485–93. doi: 10.1016/j.molmed.2011.05.003
156. Clarke E, Jantrachotechatchawan C, Buhidma Y, Broadstock M, Yu L, Howlett D, et al. Age-related neurochemical and behavioural changes in D409V/WT GBA1 mouse: relevance to lewy body dementia. Neurochem Int. (2019) 129:104502. doi: 10.1016/j.neuint.2019.104502
157. Alcalay RN, Dinur T, Quinn T, Sakanaka K, Levy O, Waters C, et al. Comparison of parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. (2014) 71:752–7. doi: 10.1001/jamaneurol.2014.313
158. Hugo Fernandes JR, Elizabeth Hartfield M, Helen Christian C, Emmanoulidou E, Zheng Y, Booth H, et al. ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson's iPSC-derived dopamine neurons. Stem Cell Rep. (2016) 6:342–56. doi: 10.1016/j.stemcr.2016.01.013
159. Schöndorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, et al. iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. (2014) 5:4028. doi: 10.1038/ncomms5028
160. Yun SP, Kim D, Kim S, Kim S, Karuppagounder SS, Kwon S-H, et al. alpha-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol Neurodegen. (2018) 13:1 doi: 10.1186/s13024-017-0233-5
161. Chahine LM, Qiang J, Ashbridge E, Minger J, Yearout D, Horn S, et al. Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol. (2013) 70:852–8. doi: 10.1001/jamaneurol.2013.1274
162. Miliukhina IV, Usenko TS, Senkevich KA, Nikolaev MA, Timofeeva AA, Agapova EA, et al. Plasma cytokines profile in patients with Parkinson's disease associated with mutations in GBA gene. Bull Exp Biol Med. (2020) 168:423–6. doi: 10.1007/s10517-020-04723-x
163. Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson's disease. J Neural Transm Suppl. (2000) 277–90. doi: 10.1007/978-3-7091-6301-6_19
164. Álvarez-Luquín DD, Arce-Sillas A, Leyva-Hernández J, et al. Regulatory impairment in untreated Parkinson's disease is not restricted to Tregs: other regulatory populations are also involved. J Neuroinflammation. (2019) 16:212. doi: 10.1186/s12974-019-1606-1
165. Hofmann KW, Schuh AFS, Saute J, Townsend R, Fricke D, Leke R, et al. Interleukin-6 Serum Levels in Patients with Parkinson's Disease. Neurochem Res. (2009) 34:1401–4. doi: 10.1007/s11064-009-9921-
166. Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, et al. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology. (2004) 62:1835–8. doi: 10.1212/01.WNL.0000127517.33208.F4
167. Scherzer CR, Grass JA, Liao Z, Pepivani I, Zheng B, Eklund AC, et al. GATA transcription factors directly regulate the Parkinson's disease-linked gene alpha-synuclein. Proc Natl Acad Sci USA. (2008) 105:10907–12. doi: 10.1073/pnas.0802437105
168. El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, et al. Alpha-synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. FASEB J. (2003) 17:1945–7. doi: 10.1096/fj.03-0098fje
169. Borghi R, Marchese R, Negro A, Marinelli L, Forloni G, Zaccheo D, et al. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neurosci Lett. (2000) 287:65–7. doi: 10.1016/S0304-3940(00)01153-8
170. Barbour R, Kling K, Anderson JP, Banducci K, Cole T, Diep L, et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener Dis. (2008) 5:55–9. doi: 10.1159/000112832
171. Kim S, Seo JH, Suh YH. Alpha-synuclein Parkinson's disease, and Alzheimer's disease. Parkinsonism Relat Disord. (2004) 1(10 Suppl.): S9–13. doi: 10.1016/j.parkreldis.2003.11.005
172. Richter-Landsberg C, Gorath M, Trojanowski JQ, Lee VM. alpha-synuclein is developmentally expressed in cultured rat brain oligodendrocytes. J Neurosci Res. (2000) 62:9–14. doi: 10.1002/1097-4547(20001001)62:1<9::AID-JNR2>3.0.CO;2-U
173. Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K. Demonstration of alpha-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp Neurol. (2002) 176:98–104. doi: 10.1006/exnr.2002.7929
174. Chandra S, Chen X, Rizo J, Jahn R, Südhof TC. A broken alpha -helix in folded alpha -synuclein. J Biol Chem. (2003) 278:15313–8. doi: 10.1074/jbc.M213128200
175. Atias M, Tevet Y, Sun J, Stavsky A, Tal S, Kahn J, et al. Synapsins regulate α-synuclein functions. Proc Natl Acad Sci USA. (2019) 116:11116. doi: 10.1073/pnas.1903054116
176. Sun J, Wang L, Bao H, Premi S, Das U, Chapman ER, et al. Functional cooperation of α-synuclein and VAMP2 in synaptic vesicle recycling. Proc Natl Acad Sci USA. (2019) 116:11113. doi: 10.1073/pnas.1903049116
177. Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci. (2014) 34:9364. doi: 10.1523/JNEUROSCI.4787-13.2014
178. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045.
179. Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann Neurol. (2004) 55:164–73. doi: 10.1002/ana.10795
180. Farrer M, Kachergus J, Forno L, Lincoln S, Wang D-S, Hulihan M, et al. Comparison of kindreds with parkinsonism and α-synuclein genomic multiplications. Ann Neurol. (2004) 55:174–9. doi: 10.1002/ana.10846
181. Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. (2004) 364:1167–9. doi: 10.1016/S0140-6736(04)17103-1
182. Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Agid Y, et al. Causal relation between α-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. (2004) 364:1169–71. doi: 10.1016/S0140-6736(04)17104-3
183. Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. (1997) 388:839–40. doi: 10.1038/42166
184. Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
185. Olanow CW, Brundin P. Parkinson's disease and alpha synuclein: is Parkinson's disease a prion-like disorder? Movement Disord. (2013) 28:31–40. doi: 10.1002/mds.25373
186. Golbe LI. Alpha-synuclein and Parkinson's disease. Movement Disord. (1999) 14:6–9. doi: 10.1002/1531-8257(199901)14:1<6::AID-MDS1004>3.0.CO;2-L
187. Johnson J, Hague SM, Hanson M, Gibson A, Wilson KE, Evans EW, et al. SNCA multiplication is not a common cause of Parkinson disease or dementia with Lewy bodies. Neurology. (2004) 63:554. doi: 10.1212/01.WNL.0000133401.09043.44
188. Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. (2005) 74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400
189. Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. (2000) 404:394–8. doi: 10.1038/35006074
190. Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K. The role of α-synuclein in Parkinson's disease: insights from animal models. Nat Rev Neurosci. (2003) 4:727–38. doi: 10.1038/nrn1199
191. Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson's disease. Neurobiol Aging. (2008) 29:1690–701. doi: 10.1016/j.neurobiolaging.2007.04.006
192. Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJA. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res. (2011) 69:337–42. doi: 10.1016/j.neures.2010.12.020
193. Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, et al. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE. (2013) 8:e55375. doi: 10.1371/journal.pone.0055375
194. Kim C, Ho D-H, Suk J-E, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. (2013) 4:1562. doi: 10.1038/ncomms2534
195. Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Lachaud CC, Guilliams T, Fernandez-Montesinos R, et al. Preconditioning of microglia by α-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS ONE. (2013) 8:e79160. doi: 10.1371/journal.pone.0079160
196. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. (2013) 61:349–60. doi: 10.1002/glia.22437
197. Couch Y, Alvarez-Erviti L, Sibson NR, Wood MJA, Anthony DC. The acute inflammatory response to intranigral α-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflamm. (2011) 8:166. doi: 10.1186/1742-2094-8-166
198. Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E, et al. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp Neurol. (2012) 237:318–34. doi: 10.1016/j.expneurol.2012.06.025
199. Ferreira M, Massano J. An updated review of Parkinson's disease genetics and clinicopathological correlations. Acta Neurol Scand. (2017) 135:273–84. doi: 10.1111/ane.12616
200. Holmans P, Moskvina V, Jones L, Sharma M, Vedernikov A, Buchel F, et al. A pathway-based analysis provides additional support for an immune-related genetic susceptibility to Parkinson's disease. Hum Mol Genet. (2013) 22:1039–49. doi: 10.1093/hmg/ddt554
201. Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet. (2010) 42:781–5. doi: 10.1038/ng.642
202. Witoelar A, Jansen IE, Wang Y, Desikan RS, Gibbs JR, Blauwendraat C, et al. Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol. (2017) 74:780–92. doi: 10.1001/jamaneurol.2017.0469
203. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
204. Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. (2011) 11:823–36. doi: 10.1038/nri3084
205. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson's disease recognize α-synuclein peptides. Nature. (2017) 546:656–61. doi: 10.1038/nature22815
206. Pandey MK, Gross C, Caballero M, Mckay MA, Tiwari D, Schroeder LM, et al. Immune cells encounter with α-synuclein fuels neurodegeneration in Parkinson's disease. J Immunol. (2016) 196:51–5.
207. Magnusen D, McKay M, Magnusen A, Pandey M. Dendritic cells, CD4+ T cells, and α synuclein triangle fuels neuroinflammation in Parkinson's disease (P3.053). Neurology. (2018) 90(15 Supplement).
208. William Wissemann T, Erin Hill-Burns M, Cyrus Zabetian P, Stewart Factor A, Patsopoulos N, Hoglund B, et al. Association of Parkinson disease with structural and regulatory variants in the HLA region. Am J Human Genetics. (2013) 93:984–93. doi: 10.1016/j.ajhg.2013.10.009
209. Hill-Burns EM, Wissemann WT, Hamza TH, Factor SA, Zabetian CP, Payami H. Identification of a novel Parkinson's disease locus via stratified genome-wide association study. BMC Genomics. (2014) 15:118. doi: 10.1186/1471-2164-15-118
210. Kannarkat GT, Cook DA, Lee JK, Chang J, Chung J, Sandy E, et al. Common genetic variant association with altered HLA expression, synergy with pyrethroid exposure, and risk for Parkinson's disease: an observational and case–control study. NPJ Parkinson's Dis. (2015) 1:15002. doi: 10.1038/npjparkd.2015.2
211. Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV α-synucleinopathy. J Neurosci. (2009) 29:3365. doi: 10.1523/JNEUROSCI.5427-08.2009
212. Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, et al. Nitrated alpha-synuclein-activated microglial profiling for Parkinson's disease. J Neurochem. (2008) 104:1504–25. doi: 10.1111/j.1471-4159.2007.05087.x
213. Sznejder-Pachołek A, Joniec-Maciejak I, Wawer A, Ciesielska A, Mirowska-Guzel D. The effect of α-synuclein on gliosis and IL-1α, TNFα, IFNγ, TGFβ expression in murine brain. Pharmacol Rep. (2017) 69:242–51. doi: 10.1016/j.pharep.2016.11.003
214. Stefanova N, Schanda K, Klimaschewski L, Poewe W, Wenning GK, Reindl M. Tumor necrosis factor-α-induced cell death in U373 cells overexpressing α-synuclein. J Neurosci Res. (2003) 73:334–40. doi: 10.1002/jnr.10662
215. Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol. (2008) 67:1149–58. doi: 10.1097/NEN.0b013e31818e5e99
216. Su X, Federoff HJ, Maguire-Zeiss KA. Mutant α-synuclein overexpression mediates early proinflammatory activity. Neurotoxicity Res. (2009) 16:238–54. doi: 10.1007/s12640-009-9053-x
217. Lee S-b, Park SM, Ahn KJ, Chung KC, Paik SR, Kim J. Identification of the amino acid sequence motif of α-synuclein responsible for macrophage activation. Biochem Biophys Res Commun. (2009) 381:39–43. doi: 10.1016/j.bbrc.2009.02.002
218. Lee E-J, Woo M-S, Moon P-G, Baek M-C, Choi I-Y, Kim W-K, et al. α-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J Immunol. (2010) 185:615. doi: 10.4049/jimmunol.0903480
219. Beraud D, Twomey M, Bloom B, Mittereder A, Neitzke K, Ton V, et al. α-synuclein alters toll-like receptor expression. Front Neurosci. (2011) 5:80. doi: 10.3389/fnins.2011.00080
220. Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Fernandez-Montesinos R, Caro M, Lachaud CC, et al. Glial innate immunity generated by non-aggregated alpha-synuclein in mouse: differences between wild-type and Parkinson's disease-linked mutants. PLoS ONE. (2010) 5:e13481. doi: 10.1371/journal.pone.0013481
221. Rojanathammanee L, Murphy EJ, Combs CK. Expression of mutant alpha-synuclein modulates microglial phenotype in vitro. J Neuroinflamm. (2011) 8:44. doi: 10.1186/1742-2094-8-44
222. Hoenen C, Gustin A, Birck C, Kirchmeyer M, Beaume N, Felten P, et al. Alpha-synuclein proteins promote pro-inflammatory cascades in microglia: stronger effects of the A53T mutant. PLoS ONE. (2016) 11:e0162717. doi: 10.1371/journal.pone.0162717
223. Chatterjee K, Roy A, Banerjee R, Choudhury S, Mondal B, Halder S, et al. Inflammasome and α-synuclein in Parkinson's disease: a cross-sectional study. J Neuroimmunol. (2020) 338:577089. doi: 10.1016/j.jneuroim.2019.577089
224. Lim KL, Ng XH, Grace LG, Yao TP. Mitochondrial dynamics and Parkinson's disease: focus on parkin. Antioxid Redox Signal. (2012) 16:935–49. doi: 10.1089/ars.2011.4105
225. Dawson TM, Dawson VL. The role of parkin in familial and sporadic Parkinson's disease. Movement Disord. (2010) 25:S32–9. doi: 10.1002/mds.22798
226. Ishikawa A, Takahashi H. Clinical and neuropathological aspects of autosomal recessive juvenile parkinsonism. J Neurol. (1998) 245:P4–9. doi: 10.1007/PL00007745
227. Periquet M, Lücking CB, Vaughan JR, Bonifati V, Dürr A, De Michele G, et al. Origin of the mutations in the parkin gene in Europe: exon rearrangements are independent recurrent events, whereas point mutations may result from founder effects. Am J Human Genet. (2001) 68:617–26. doi: 10.1086/318791
228. Kilarski LL, Pearson JP, Newsway V, Majounie E, Knipe MD, Misbahuddin A, et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson's disease. Movement Disord. (2012) 27:1522–9. doi: 10.1002/mds.25132
229. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. (2015) 85:257–73. doi: 10.1016/j.neuron.2014.12.007
230. Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, et al. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology. (1998) 51:890. doi: 10.1212/WNL.51.3.890
231. van de Warrenburg BPC, Lammens M, Lücking CB, Denèfle P, Wesseling P, Booij J, et al. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology. (2001) 56:555. doi: 10.1212/WNL.56.4.555
232. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. (1998) 392:605–8. doi: 10.1038/33416
233. Klein C, Lohmann K. Parkinson disease(s). Neurology. (2009) 72:106. doi: 10.1212/01.wnl.0000333666.65522.8d
234. Klein C, Schlossmacher MG. Parkinson disease, 10 years after its genetic revolution: multiple clues to a complex disorder. Neurology. (2007) 69:2093. doi: 10.1212/01.wnl.0000271880.27321.a7
235. Pilcher H. Parkin implicated in sporadic Parkinson's disease. Lancet Neurol. (2005) 4:798. doi: 10.1016/S1474-4422(05)70237-4
236. González H, Contreras F, Prado C, Elgueta D, Franz D, Bernales S, et al. Dopamine receptor D3 expressed on CD4+ T cells favors neurodegeneration of dopaminergic neurons during Parkinson's disease. J Immunol. (2013) 190:5048–56. doi: 10.4049/jimmunol.1203121
237. de Léséleuc L, Orlova M, Cobat A, Girard M, Huong NT, Ba NN, et al. PARK2 mediates interleukin 6 and monocyte chemoattractant protein 1 production by human macrophages. PLoS Negl Trop Dis. (2013) 7:e2015. doi: 10.1371/journal.pntd.0002015
238. Chopra R, Kalaiarasan P, Ali S, Srivastava AK, Aggarwal S, Garg VK, et al. PARK2 and proinflammatory/anti-inflammatory cytokine gene interactions contribute to the susceptibility to leprosy: a case–control study of North Indian population. BMJ Open. (2014) 4:e004239. doi: 10.1136/bmjopen-2013-004239
239. Mira MT, Alcaïs A, Van Thuc N, Moraes MO, Di Flumeri C, Hong Thai V, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature. (2004) 427:636–40. doi: 10.1038/nature02326
240. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. (2013) 501:512–6. doi: 10.1038/nature12566
241. Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ. Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Human Mol Genet. (2005) 14:799–811. doi: 10.1093/hmg/ddi074
242. Dupré-Crochet S, Erard M, Nüβe O. ROS production in phagocytes: why, when, and where? J Leukocyte Biol. (2013) 94:657–70. doi: 10.1189/jlb.1012544
243. Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. (2011) 208:417–20. doi: 10.1084/jem.20110367
244. Pelletier M, Lepow TS, Billingham LK, Murphy MP, Siegel RM. New tricks from an old dog: mitochondrial redox signaling in cellular inflammation. Semin Immunol. (2012) 24:384–92. doi: 10.1016/j.smim.2013.01.002
245. Li J, Ma C, Long F, Yang D, Liu X, Hu Y, et al. Parkin impairs antiviral immunity by suppressing the mitochondrial reactive oxygen species-Nlrp3 axis and antiviral inflammation. iScience. (2019) 16:468–84. doi: 10.1016/j.isci.2019.06.008
246. Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, et al. Parkin mediates neuroprotection through activation of IκB kinase/nuclear factor-κB signaling. J Neurosci. (2007) 27:1868. doi: 10.1523/JNEUROSCI.5537-06.2007
247. Anne Müller-Rischart K, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. (2013) 49:908–21. doi: 10.1016/j.molcel.2013.01.036
248. Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. (2006) 441:1157–61. doi: 10.1038/nature04788
249. English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. (2009) 10:480–7. doi: 10.1038/ni.1720
250. Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. (2005) 307:593. doi: 10.1126/science.1104904
251. Forno LS, DeLanney LE, Irwin I, Di Monte D, Langston JW. Astrocytes and Parkinson's disease. Prog Brain Res. (1992) 94:429–36. doi: 10.1016/S0079-6123(08)61770-7
252. Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson's disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell. (2016) 166:314–27. doi: 10.1016/j.cell.2016.05.039
253. Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. (2001) 50:293–300. doi: 10.1002/ana.1132
254. Sasaki S, Shirata A, Yamane K, Iwata M. Parkin-positive autosomal recessive juvenile parkinsonism with α-synuclein-positive inclusions. Neurology. (2004) 63:678. doi: 10.1212/01.WNL.0000134657.25904.0B
255. Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, et al. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. (2005) 58:411–22. doi: 10.1002/ana.20587
256. Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. (2008) 28:10825. doi: 10.1523/JNEUROSCI.3001-08.2008
257. Tran TA, Nguyen AD, Chang J, Goldberg MS, Lee JK, Tansey MG. Lipopolysaccharide and tumor necrosis factor regulate Parkin expression via nuclear factor-kappa B. PLoS ONE. (2011) 6:e23660. doi: 10.1371/journal.pone.0023660
258. Stojkovska I, Wagner BM, Morrison BE. Parkinson's disease and enhanced inflammatory response. Exp Biol Med. (2015) 240:1387–95. doi: 10.1177/1535370215576313
259. Borsche M, König IR, Delcambre S, Petrucci S, Balck A, Brüggemann N, et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain. (2020) 143:3041–51. doi: 10.1093/brain/awaa246
260. Bonifati V, Rohé CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C, et al. Early-onset parkinsonism associated with PINK1 mutations. Neurology. (2005) 65:87. doi: 10.1212/01.wnl.0000167546.39375.82
261. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. (2004) 304:1158. doi: 10.1126/science.1096284
262. Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, et al. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Human Mol Genet. (2005) 14:3477–92. doi: 10.1093/hmg/ddi377
263. Kim Y, Park J, Kim S, Song S, Kwon S-K, Lee S-H, et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. (2008) 377:975–80. doi: 10.1016/j.bbrc.2008.10.104
264. Narendra DP, Jin SM, Tanaka A, Suen D-F, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. (2010) 8:e1000298. doi: 10.1371/journal.pbio.1000298
265. Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RLA, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. (2010) 107:378. doi: 10.1073/pnas.0911187107
266. Pridgeon JW, Olzmann JA, Chin L-S, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. (2007) 5:e172. doi: 10.1371/journal.pbio.0050172
267. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. (2010) 189:211–21. doi: 10.1083/jcb.200910140
268. Deng H, Jankovic J, Guo Y, Xie W, Le W. Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y. Biochem Biophys Res Commun. (2005) 337:1133–8. doi: 10.1016/j.bbrc.2005.09.178
269. Youn J, Lee C, Oh E, Park J, Kim JS, Kim H-T, et al. Genetic variants of PARK genes in Korean patients with early-onset Parkinson's disease. Neurobiol Aging. (2019) 75:224.e9–15. doi: 10.1016/j.neurobiolaging.2018.10.030
270. Büeler H. Mitochondrial dynamics, cell death and the pathogenesis of Parkinson's disease. Apoptosis. (2010) 15:1336–53. doi: 10.1007/s10495-010-0465-0
271. Dagda RK, Chu CT. Mitochondrial quality control: insights on how Parkinson's disease related genes PINK1, parkin, and Omi/HtrA2 interact to maintain mitochondrial homeostasis. J Bioenerget Biomembr. (2009) 41:473–9. doi: 10.1007/s10863-009-9255-1
272. Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, et al. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol. (2004) 56:336–41. doi: 10.1002/ana.20256
273. Gustavsson EK, Trinh J, McKenzie M, Bortnick S, Petersen MS, Farrer MJ, et al. Genetic identification in early onset parkinsonism among Norwegian patients. Movement Disord Clin Prac. (2017) 4:499–508. doi: 10.1002/mdc3.12501
274. Kawajiri S, Saiki S, Sato S, Hattori N. Genetic mutations and functions of PINK1. Trends Pharmacol Sci. (2011) 32:573–80. doi: 10.1016/j.tips.2011.06.001
275. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. (2006) 441:1162–6. doi: 10.1038/nature04779
276. Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang J-W, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. (2006) 103:10793. doi: 10.1073/pnas.0602493103
277. Akundi RS, Huang Z, Eason J, Pandya JD, Zhi L, Cass WA, et al. Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS ONE. (2011) 6:e16038. doi: 10.1371/journal.pone.0016038
278. Ellis GI, Zhi L, Akundi R, Büeler H, Marti F. Mitochondrial and cytosolic roles of PINK1 shape induced regulatory T-cell development and function. Euro J Immunol. (2013) 43:3355–60. doi: 10.1002/eji.201343571
279. Reynolds AD, Stone DK, Hutter JAL, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal Dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol. (2010) 184:2261. doi: 10.4049/jimmunol.0901852
280. Fiszer U, Mix E, Fredrikson S, Kostulas V, Olsson T, Link H. γδ+ T cells are increased in patients with Parkinson's disease. J Neurol Sci. (1994) 121:39–45. doi: 10.1016/0022-510X(94)90154-6
281. Stevens CH, Rowe D, Morel-Kopp M-C, Orr C, Russell T, Ranola M, et al. Reduced T helper and B lymphocytes in Parkinson's disease. J Neuroimmunol. (2012) 252:95–9. doi: 10.1016/j.jneuroim.2012.07.015
282. Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, et al. Intestinal infection triggers Parkinson's disease-like symptoms in Pink1(-/-) mice. Nature. (2019) 571:565–9. doi: 10.1038/s41586-019-1405-y
283. Houser MC, Tansey MG. The gut-brain axis: is intestinal inflammation a silent driver of Parkinson's disease pathogenesis? NPJ Parkinson's Dis. (2017) 3:3. doi: 10.1038/s41531-016-0002-0
284. Sun L, Shen R, Agnihotri SK, Chen Y, Huang Z, Büeler H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci Rep. (2018) 8:383. doi: 10.1038/s41598-017-18786-w
285. Kim J, Byun J-W, Choi I, Kim B, Jeong H-K, Jou I, et al. PINK1 deficiency enhances inflammatory cytokine release from acutely prepared brain slices. Exp Neurobiol. (2013) 22:38–44. doi: 10.5607/en.2013.22.1.38
286. Honbou K, Suzuki NN, Horiuchi M, Niki T, Taira T, Ariga H, et al. The crystal structure of DJ-1, a protein related to male fertility and Parkinson's disease. J Biol Chem. (2003) 278:31380–4. doi: 10.1074/jbc.M305878200
287. Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxidants Redox Signal. (2011) 15:111–22. doi: 10.1089/ars.2010.3481
288. Kahle PJ, Waak J, Gasser T. DJ-1 and prevention of oxidative stress in Parkinson's disease and other age-related disorders. Free Radical Biol Med. (2009) 47:1354–61. doi: 10.1016/j.freeradbiomed.2009.08.003
289. Girotto S, Cendron L, Bisaglia M, Tessari I, Mammi S, Zanotti G, et al. DJ-1 is a copper chaperone acting on SOD1 activation. J Biol Chem. (2014) 289:10887–99. doi: 10.1074/jbc.M113.535112
290. Jain D, Jain R, Eberhard D, Eglinger J, Bugliani M, Piemonti L, et al. Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J Mol Cell Biol. (2012) 4:221–30. doi: 10.1093/jmcb/mjs025
291. Mitsumoto A, Nakagawa Y, Takeuchi A, Okawa K, Iwamatsu A, Takanezawa Y. Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat. Free Radical Res. (2001) 35:301–10. doi: 10.1080/10715760100300831
292. Mitsumoto A, Nakagawa Y. DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radical Res. (2001) 35:885–93. doi: 10.1080/10715760100301381
293. Yokota T, Sugawara K, Ito K, Takahashi R, Ariga H, Mizusawa H. Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, proteasome inhibition. Biochem Biophys Res Commun. (2003) 312:1342–8. doi: 10.1016/j.bbrc.2003.11.056
294. Taira T, Saito Y, Niki T, Iguchi-Ariga SMM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. (2004) 5:213–8. doi: 10.1038/sj.embor.7400074
295. Shinbo Y, Niki T, Taira T, Ooe H, Takahashi-Niki K, Maita C, et al. Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death Different. (2006) 13:96–108. doi: 10.1038/sj.cdd.4401704
296. Cao J, Lou S, Ying M, Yang B. DJ-1 as a human oncogene and potential therapeutic target. Biochem Pharmacol. (2015) 93:241–50. doi: 10.1016/j.bcp.2014.11.012
297. Yanagisawa D, Kitamura Y, Inden M, Takata K, Taniguchi T, Morikawa S, et al. DJ-1 protects against neurodegeneration caused by focal cerebral ischemia and reperfusion in rats. J Cereb Blood Flow Metab. (2008) 28:563–78. doi: 10.1038/sj.jcbfm.9600553
298. An C-N, Jiang H, Wang Q, Yuan R-P, Liu J-M, Shi W-L, et al. Down-regulation of DJ-1 protein in the ejaculated spermatozoa from Chinese asthenozoospermia patients. Fertility Sterility. (2011) 96:19–23.e2. doi: 10.1016/j.fertnstert.2011.04.048
299. Cornejo Castro EM, Waak J, Weber SS, Fiesel FC, Oberhettinger P, Schütz M, et al. Parkinson's disease-associated DJ-1 modulates innate immunity signaling in Caenorhabditis elegans. J Neural Transm. (2010) 117:599–604. doi: 10.1007/s00702-010-0397-4
300. Repici M, Giorgini F. DJ-1 in Parkinson's disease: clinical insights and therapeutic perspectives. J Clin Med. (2019) 8:1377. doi: 10.3390/jcm8091377
301. Chien CH, Lee MJ, Liou HC, Liou HH, Fu WM. Microglia-derived cytokines/chemokines are involved in the enhancement of LPS-induced loss of nigrostriatal dopaminergic neurons in DJ-1 knockout mice. PLoS ONE. (2016) 11:e0151569. doi: 10.1371/journal.pone.0151569
302. Nash Y, Schmukler E, Trudler D, Pinkas-Kramarski R, Frenkel D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J Neurochem. (2017) 143:584–94. doi: 10.1111/jnc.14222
303. Trudler D, Weinreb O, Mandel SA, Youdim MBH, Frenkel D. DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J Neurochem. (2014) 129:434–47. doi: 10.1111/jnc.12633
304. Waak J, Weber SS, Waldenmaier A, Görner K, Alunni-Fabbroni M, Schell H, et al. Regulation of astrocyte inflammatory responses by the Parkinson's disease-associated gene DJ−1. FASEB J. (2009) 23:2478–89. doi: 10.1096/fj.08-125153
305. Taipa R, Pereira C, Reis I, Alonso I, Bastos-Lima A, Melo-Pires M, et al. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain. (2016) 139:1680–7. doi: 10.1093/brain/aww080
306. Sturchio A, Dwivedi AK, Vizcarra JA, Chirra M, Keeling EG, Mata IF, et al. Genetic parkinsonisms and cancer: a systematic review and meta-analysis. Rev Neurosci. (2020) 32:159–67. doi: 10.1515/revneuro-2020-0083
307. Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci USA. (2010) 107:15145–50. doi: 10.1073/pnas.1009941107
308. Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, et al. Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet. (2010) 42:77–82. doi: 10.1038/ng.491
309. Lee S, She J, Deng B, Kim J, de Andrade M, Na J, et al. Multiple-level validation identifies PARK2 in the development of lung cancer and chronic obstructive pulmonary disease. Oncotarget. (2016) 7:44211–23. doi: 10.18632/oncotarget.9954
310. Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J Exp Med. (2004) 199:47–58. doi: 10.1084/jem.20031104
311. Nagaoka H, Gonzalez-Aseguinolaza G, Tsuji M, Nussenzweig MC. Immunization and infection change the number of recombination activating gene (RAG)-expressing B cells in the periphery by altering immature lymphocyte production. J Exp Med. (2000) 191:2113–20. doi: 10.1084/jem.191.12.2113
Keywords: neuroimmunology, immunogenetics, innate and adaptive immunity, glycosphingolipid, aggregated proteins, brain disease, neuroinflammation, mitochondrial disease
Citation: Magnusen AF, Hatton SL, Rani R and Pandey MK (2021) Genetic Defects and Pro-inflammatory Cytokines in Parkinson's Disease. Front. Neurol. 12:636139. doi: 10.3389/fneur.2021.636139
Received: 30 November 2020; Accepted: 06 May 2021;
Published: 22 June 2021.
Edited by:
Marcelo A. Kauffman, University of Buenos Aires, ArgentinaReviewed by:
Luca Magistrelli, University of Eastern Piedmont, ItalyQiying Sun, Xiangya Hospital, Central South University, China
Copyright © 2021 Magnusen, Hatton, Rani and Pandey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manoj Kumar Pandey, bWFub2oucGFuZGV5QGNjaG1jLm9yZw==
†These authors have contributed equally to this work