
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 22 February 2021
Sec. Neuroinfectious Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.634827
 Caleb R. S. McEntire1
Caleb R. S. McEntire1 Kun-Wei Song1Robert P. McInnis1John Y. Rhee1Michael Young1Erika Williams1Leah L. Wibecan2
Kun-Wei Song1Robert P. McInnis1John Y. Rhee1Michael Young1Erika Williams1Leah L. Wibecan2 Neal Nolan1Amanda M. Nagy2Jeffrey Gluckstein1
Neal Nolan1Amanda M. Nagy2Jeffrey Gluckstein1 Shibani S. Mukerji3
Shibani S. Mukerji3 Farrah J. Mateen3*
Farrah J. Mateen3*The World Health Organization (WHO) monitors the spread of diseases globally and maintains a list of diseases with epidemic or pandemic potential. Currently listed diseases include Chikungunya, cholera, Crimean-Congo hemorrhagic fever, Ebola virus disease, Hendra virus infection, influenza, Lassa fever, Marburg virus disease, Neisseria meningitis, MERS-CoV, monkeypox, Nipah virus infection, novel coronavirus (COVID-19), plague, Rift Valley fever, SARS, smallpox, tularemia, yellow fever, and Zika virus disease. The associated pathogens are increasingly important on the global stage. The majority of these diseases have neurological manifestations. Those with less frequent neurological manifestations may also have important consequences. This is highlighted now in particular through the ongoing COVID-19 pandemic and reinforces that pathogens with the potential to spread rapidly and widely, in spite of concerted global efforts, may affect the nervous system. We searched the scientific literature, dating from 1934 to August 2020, to compile data on the cause, epidemiology, clinical presentation, neuroimaging features, and treatment of each of the diseases of epidemic or pandemic potential as viewed through a neurologist's lens. We included articles with an abstract or full text in English in this topical and scoping review. Diseases with epidemic and pandemic potential can be spread directly from human to human, animal to human, via mosquitoes or other insects, or via environmental contamination. Manifestations include central neurologic conditions (meningitis, encephalitis, intraparenchymal hemorrhage, seizures), peripheral and cranial nerve syndromes (sensory neuropathy, sensorineural hearing loss, ophthalmoplegia), post-infectious syndromes (acute inflammatory polyneuropathy), and congenital syndromes (fetal microcephaly), among others. Some diseases have not been well-characterized from a neurological standpoint, but all have at least scattered case reports of neurological features. Some of the diseases have curative treatments available while in other cases, supportive care remains the only management option. Regardless of the pathogen, prompt, and aggressive measures to control the spread of these agents are the most important factors in lowering the overall morbidity and mortality they can cause.
The World Health Organization (WHO) provides guidance on diseases that are international threats to populations with a focus on diseases of epidemic and pandemic potential (1). Close observation of these infectious diseases is required to ensure prevention, foster early detection, mitigate further spread, and provide supportive care to those affected. To date, of all of the millions of pathogens globally, only ~20 infections are listed as having epidemic or pandemic potential by the WHO. While some may be familiar to the neurologist, others may be only known to practitioners in tropical settings or locations where few neurologists practice. Here we synthesize the available knowledge on the neurological manifestations of the WHO's listed diseases of epidemic and pandemic potential by pathogen.
Searches were performed using the keywords of the disease and infectious organism (if different) as well as the terms “neurology,” “brain,” “nervous system,” and related terms for each of the listed disease of epidemic or pandemic potential on the WHO website as of April 1, 2020. Search engines included PubMed, Google Scholar, and the reference lists of articles found through those searches. All searches took place in spring 2020 with updates continuing as more information became available, particularly on evolving pandemics, such as SARS-CoV-2, influenza, Ebola Virus disease, and Neisseria meningitidis meningitis until September 2020. Articles in English or with an abstract in English were included, using data sources dating from 1934 until present.
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Chikungunya virus is a positive-sense alphavirus most often spread by Aedes mosquitos as a direct infection to humans, although it can rarely pass through maternal-fetal transmission or blood products. Chikungunya presents with a systemic course that is relatively uniform across individuals, but neurologic manifestations can be more heterogeneous. Its systemic course begins with an incubation period from 1 to 12 days and then progresses to fever, malaise, and headache, as well as a characteristic symmetric arthralgia in as many as 90% of patients (2). Chikungunya means “that which bends up” in Kimakonde, the language of the ethnic group in whom the disease was first identified (3), referring to the stooped posture individuals with the disease adopt due to joint pain (Tables 1, 2).
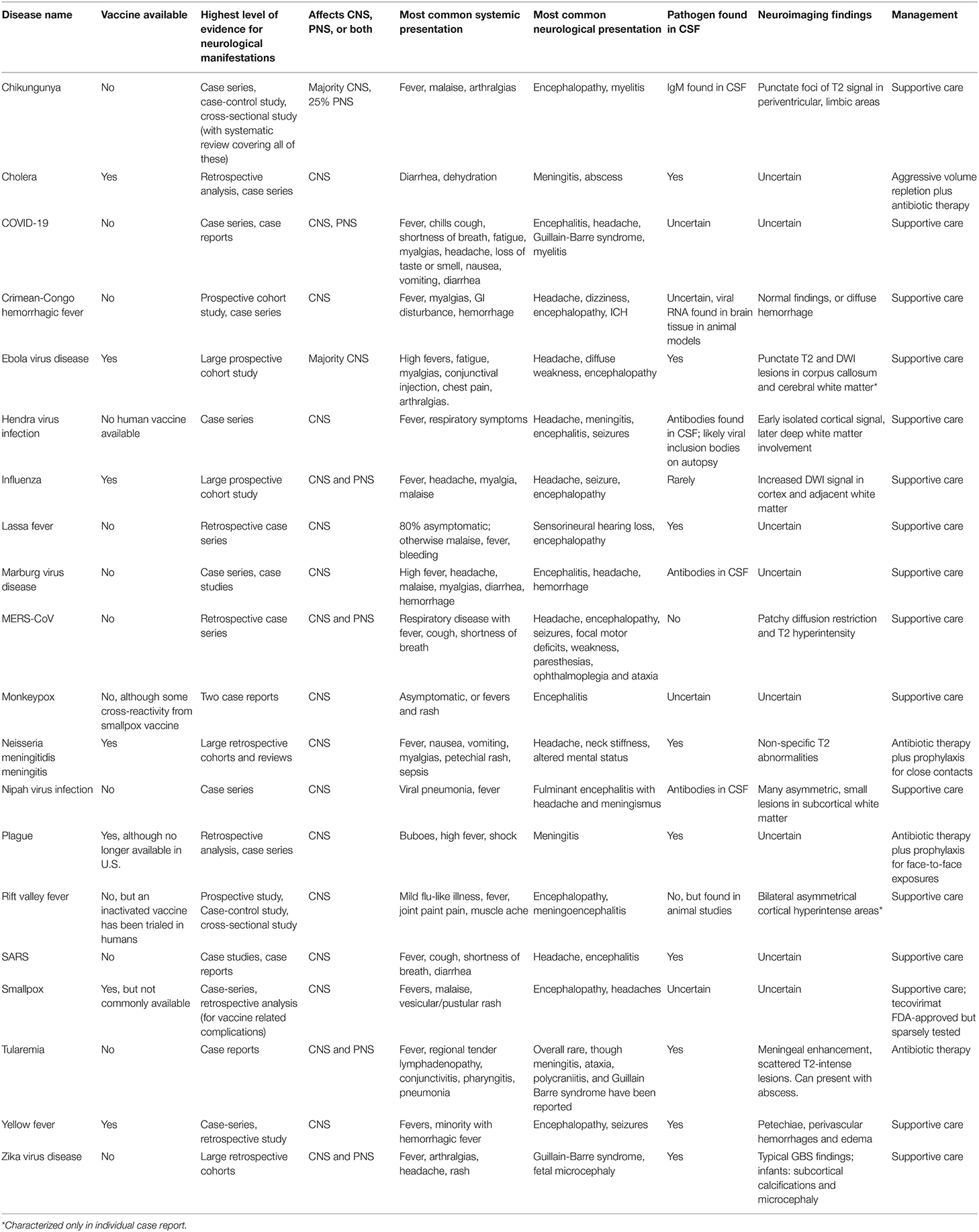
Table 1. Overview of presentation, diagnosis, and management of the WHO Diseases of Epidemic & Pandemic Potential.
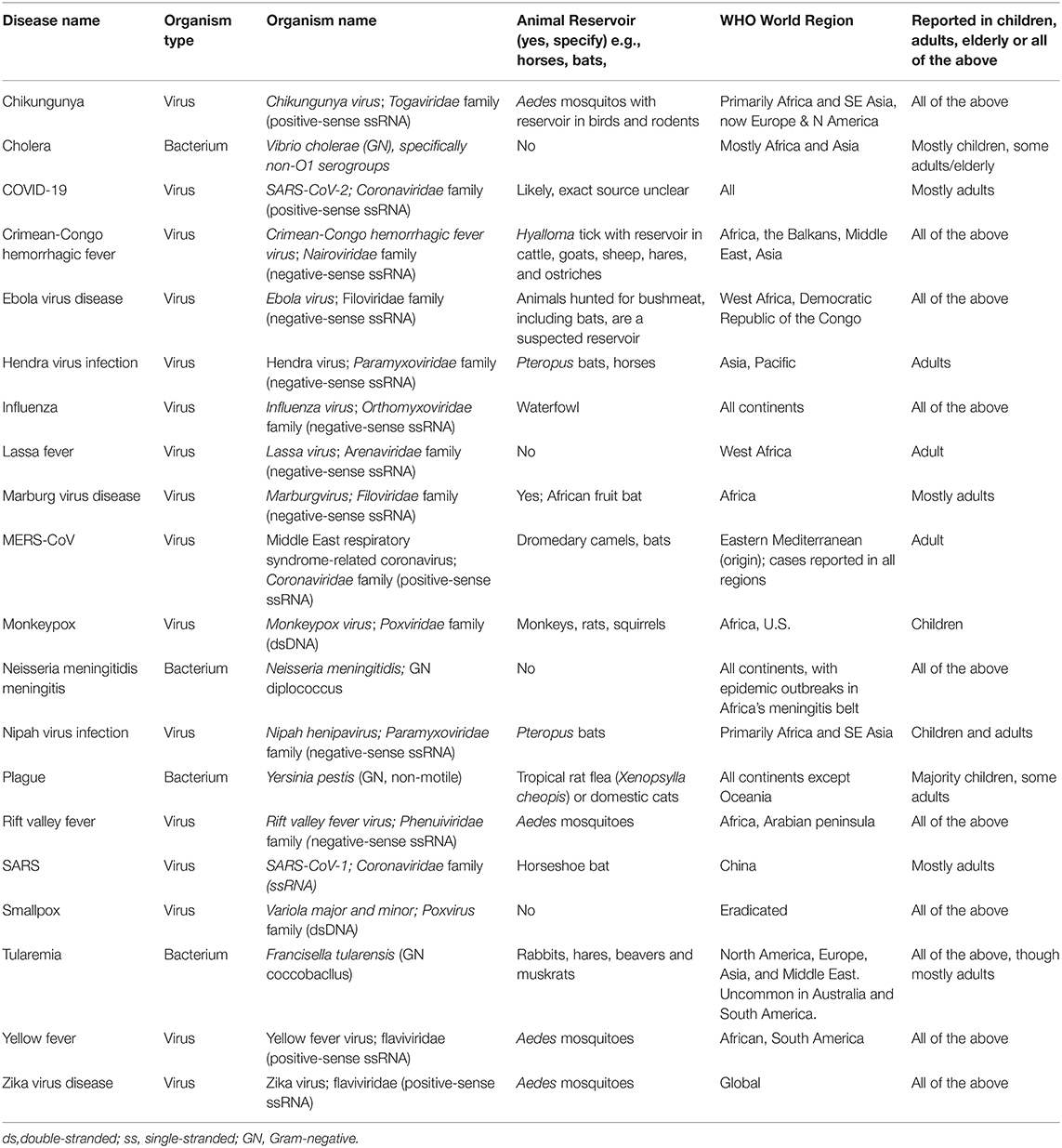
Table 2. Overview of epidemiology of the WHO Diseases of Epidemic & Pandemic Potential.
The most common clinical presentation of neurologic Chikungunya is encephalopathy, which can be present in up to 40% of individuals with neurologic involvement. Case series of numerous epidemics have also reported meningoencephalitis, Guillain-Barré Syndrome (GBS), myeloradiculitis, facial palsy, sensorineural deafness, optic neuritis, and more (4, 5). Approximately 75% of adult and pediatric cases have a pure central nervous system (CNS) phenotype, while ~13% have a pure peripheral nervous system (PNS) disorder. Approximately 9% have disorders of both the CNS and PNS.
The time course of neurologic signs varies by the syndrome; encephalopathy often appears concurrently with initial systemic symptoms, occasionally lagging by 1 to 2 days. Spinal disease, with weakness and occasional paresthesias of multiple limbs, can appear at symptomatic onset or with a delay of up to 3 weeks. GBS appears weeks after the initial illness, consistent with its usual presentation (5).
The exact proportion of individuals infected with Chikungunya who exhibit neurologic involvement is unclear, but it appears to be the most common “atypical” involvement in severe disease. Case series during epidemics have reported neurologic involvement ranging from 0.1% (5) to 16.3% (6). A neurological disorder was the primary issue in 61% (7) and 79% (8) of more severely affected patients in intensive care units.
Diagnosis of chikungunya is with real-time reverse-transcription polymerase chain reaction (RT-PCR) or enzyme-linked immunosorbent assay (ELISA) of the serum depending on the timing of testing. Within 5 days of initial symptom onset, serum RT-PCR for viral RNA has up to 100% sensitivity and 98% specificity, while ELISA for immunoglobulin M (IgM) is appropriate for patients who have shown clinical symptoms for at least 6–8 days (9, 10). Viral RNA has not been shown in the CSF of affected individuals, although viral particles have been found in glial cells of infected mice (11). Treatment of the disease is supportive care, including rest and fluids, as well as acetaminophen or non-steroidal anti-inflammatory drugs to alleviate arthralgias. Corticosteroids have been used in some published case reports but with no data for their efficacy (5). GBS should be treated as per its standard protocols with intravenous immunoglobulins (IVIg) or plasmapheresis.
Cholera was classified as a pandemic by the WHO in 2010 in light of the disease causing an estimated 28,800–130,000 deaths per year and affecting 3–5 million individuals globally (12). The causative organism Vibrio cholerae is a gram-negative, comma shaped bacterium most commonly found in seawater and known to have a number of serogroups. Specifically, serogroups O1 and O139 have been implicated in the majority of intestinal infections that are most common worldwide (13). Non-O1 serotypes have long been associated with more invasive disease phenotype and extraintestinal involvement (14).
Infants and neonates make up the majority of cholera cases with neurologic sequelae, presenting with V. cholerae meningitis, abscess, or both. In some cases there appears to have been a specific causative risk factor, as in one case where an individual had eviscerated fish from a nearby reservoir in the family sink 2 weeks prior to presentation (15), or a case where a neonate developed V. cholerae O139 septicemia and lethargy in the context of hospital workers managing an adult with bacteremia of the same organism in a nearby ward (16). In other cases no specific source of infection was found, but environmental factors in a resource-limited setting could be implicated. One 10-day-old boy with V. cholerae meningitis had no discrete risk factor identified, but recent studies of water in the rural area of India in which he resided had isolated non-O1 non-0139 V. cholerae from 41% of drinking water samples as well as 100% of water, sediment, and plankton samples from three nearby lakes (17).
Interestingly, the infant cases reported of cholera meningitis have presented with lethargy, fever, and refusal to feed, but none have presented with the diarrhea typical of intestinal cholera. Prognosis of these infants appears poor; 2 of 6 cases we have been able to identify in the literature have resulted in death, while 3 of the remaining patients have had lasting neurologic sequelae (18).
Neurological consequences of any cholera serotype have also been described in adults, but often with more concrete risk factors for intracranial infection. For instance, one 58-year-old fisherman developed a left-sided paresis and a Babinski sign and was found to have intracerebral abscess and bacteremia with non-cholera toxin-producing, non-O1, non-O139 V. cholerae. While he had no recently identified risk factor for the disease beyond seawater contact, he had craniotomy for a subdural hematoma 5 years before presentation that could have allowed the organism intracranial access (19).
The gold standard for diagnosis of cholera is stool culture for V. cholerae, although several rapid antigen-based tests are available depending on the resource availability of the area where testing occurs (20). CSF culture has also successfully grown the organism in most cases (17, 18, 21, 22), although in at least one case report, there was no yield (23). Given the rarity of cholera meningitis no large-scale studies have examined antibiotic use, but it is prudent to treat the disease with antibiotics given its poor prognosis. The available literature indicates that most organisms have been susceptible to third-generation cephalosporins, piperacillin/tazobactam, fluoroquinolones, and tetracyclines (18). Supportive care should also be offered.
Crimean-Congo Hemorrhagic Fever (CCHF) is a hemorrhagic illness caused by infection with the Crimean-Congo hemorrhagic fever virus (CCHFV), a negative-sense RNA virus of the Nairoviridae genus of the Bunyaviridae family with a genome comprised of three segments (24). CCHF was first recognized in Crimea in 1944 during an outbreak involving Soviet troops and was subsequently named Crimean Hemorrhagic Fever (25). In 1956, an identical pathogen was identified in the former Belgian Congo (current Democratic Republic of the Congo) and named the “Congo Virus”; the names were later consolidated to CCHFV (25). As the most widespread tickborne virus, CCHFV is endemic throughout areas of Africa, southeastern Europe, Asia, and the Middle East (24). Although typically contained, outbreaks have been increasing and the geographic distribution expanding in recent years due to a number of factors including climate change (26, 27).
Infection with CCHFV can occur directly from a bite from an infected Hyalomma tick, through the butchering of livestock that act as amplifying hosts for the virus, or via contact with infected bodily fluids (27). The highest risk for infection occurs in the spring and summer, when tick bites are most likely to occur, and often is limited to an isolated rural case. The clinical presentation of CCHF begins with an incubation period of 1–13 days (25). This is followed by a pre-hemorrhagic phase, characterized by the sudden onset of a non-specific febrile illness with myalgias, diarrhea, nausea, and vomiting (27). The hemorrhagic phase follows in 3 to 6 days and can evolve from petechiae to widespread hemorrhage in severe cases, with mortality ranging from 9 to 50% (24). Survivors typically will enter the convalescent phase 15 to 20 days after symptomatic onset (24).
Common neurologic manifestations in CCHF include headache, seen in 68% of patients (28), dizziness, photophobia, neck pain and stiffness, and encephalopathy. Given the extensive hemorrhage often associated with the disease, intracranial hemorrhage (ICH) is an expected finding. There have been reported cases of intraparenchymal, subarachnoid, and subdural hemorrhages diagnosed on imaging (29–31) and on autopsy (32), in addition to fatal cerebellar tonsillar herniation secondary to cerebral edema with associated frontal intraparenchymal hemorrhage (33), While ICH may be found in more severe and often fatal cases, there are also milder forms of illness that are not associated with intracranial abnormalities in spite of clinical findings that implicate CNS involvement (fever, headache, alteration of consciousness, agitation, and myoclonic jerks) (34). Little is reported on the long-term neurologic sequelae of survivors of CCHF.
Despite the frequent lack of CNS involvement on neuroimaging, animal studies have demonstrated the presence of CCHFV antigen in the brain (primarily glial cells and meninges) for certain strains of CCHFV along with neuropathological evidence of gliosis and meningoencephalitis. Viral RNA has also been isolated from ocular samples in mice. There is evidence of vascular congestion in the CNS, indicating a route of entry for CCHFV through the blood-brain-barrier related to the virus's known tropism for endothelial cells (35). Serum RT-PCR for CCHFV RNA is sensitive and specific for the disease, and several real-time tests exist for a rapid diagnosis (36, 37).
Ebolavirus is a virus in the Filoviridae family, whose infamy as one of the most dangerous human pathogens has earned it recognition as a potential bioweapon (38, 39). It is an RNA virus that primarily infects macrophages and endothelial cells using the C1 protein associated with Niemann-Pick disease as a receptor. Interestingly, cells derived from patients with Niemann-Pick disease are resistant to infection (39, 40). Transmission to humans occurs through infected animals, and bats are recognized as one of the most important disease reservoirs. Human to human transmission occurs through bodily fluids, and can even be transmitted from people who died from infection, where the virus can remain viable for many days (39). Ebola recently gained new attention following an epidemic of unprecedented magnitude, which spread through West Africa from 2013 to 2016. The estimated case fatality was 50% (41, 42). The size of this epidemic led to accelerated scientific discoveries about the virus, including the numerous neurologic manifestations of disease, and the possibility of disease relapse from immunologically privileged sites in the body (39).
Symptoms of Ebola virus disease (EVD) typically begin after an incubation period lasting 4 to 10 days. Early disease presents with non-specific symptoms, including high fevers, headaches, fatigue, myalgias, and body aches; there may be associated conjunctival injection, chest pain, and arthralgias. A few days later, infected individuals may develop abdominal pain with nausea, copious vomiting and watery diarrhea, which can lead to severe volume depletion and shock. Hemorrhagic disease is a late complication that affects a minority of infected people, and usually manifests with petechiae, ecchymoses, oozing from sites of venipuncture, hematemesis, or melena (39).
There is better understanding of neurologic complications from EVD in the wake of recent epidemics; however, neurologic evaluation of acutely infected patients poses significant challenges. Strict quarantining and extensive personal protective equipment can limit a comprehensive neurologic exam and has largely prohibited lumbar puncture (LP) for CSF analysis, except in rare cases. Acute neurologic manifestations vary in prevalence from very common, to quite rare. Commonly seen neurologic manifestations include headaches, diffuse weakness, and a varying degree of encephalopathy. Rarely, EVD can be complicated by hemorrhagic or ischemic strokes. Cerebellar signs and myoclonus have also been observed, but are considered rare (43). Seizures can occur, and patients can develop a syndrome concerning for meningitis or encephalitis. Neuropsychiatric signs and symptoms vary widely, with a report of ideomotor slowing, aggression, and disinhibition, which may be related to delirium, but also raise concern for CNS involvement (44). LP was performed on three patients with confirmed EVD in Guinea who exhibited worsening neuropsychiatric symptoms, but no meningeal signs. Although biochemical and cytological analysis could not be performed, quantitative reverse transcription polymerase chain reaction (RT-PCR) was positive for EVD in CSF in each case (44). Separately reported, another patient with EVD developed frank meningeal signs, and then underwent LP, which was notable for sterile CSF, no red or white cells, but positive EVD RT-PCR (45). Importantly, one case report identified a patient who was recovering from EVD, but at 2 weeks developed marked neurologic decline, with LP showing an elevated opening pressure. Interestingly, RT-PCR returned positive in CSF, but was negative in the serum (39).
A proportion of people who survive EVD may develop chronic or delayed onset neurologic complications, and cases have surfaced that demonstrate reactivation of virus within immunologically privileged sites. Survivors of a 2007 EVD outbreak in Uganda exhibited headaches, retro-orbital pain, hearing loss, memory problems, and confusion more commonly than controls, 2 years following infection (39, 46). Hearing loss and generalized fatigue have also been reported (39). A large cohort study called PREVAIL III compared 966 antibody-positive EVD survivors to 2,350 antibody negative close contacts with no history of EVD as a control group over a period of 5 years. EVD survivors were found to have urinary frequency, headache, fatigue, muscle pain, memory loss, and joint pain more frequently than controls. On follow up, survivors were also more likely to have an abnormal neurologic exam (47). Uveitis was also more common among survivors, and prior research has demonstrated the persistence of viral RNA in ocular fluid in these individuals, suggesting a pathologic effect of active viral replication (47, 48). Viral relapse causing meningoencephalitis was observed in a 39-year-old Scottish nurse 9 months after a full recovery from typical EVD. She was found to have multiple cranial neuropathies in addition to radiculopathy. LP was performed, with CSF RT-PCR positive for EV. Other cases have been suggestive of EVD relapse in the CNS (39). Neurologists should be aware of the range of neurologic manifestations, and remarkable capacity for the virus to relapse with neurologic symptoms.
Hendra virus (HeV) belongs to a family of viruses called Paramyxoviridae, and are negative-sense, single-stranded RNA viruses in the order Mononegavirales. Given close homology and cross-reactivity on serological tests, HeV is commonly grouped with the Nipah Virus (NiV); together, these are referred to as Henipavirus (49). Hendra virus was discovered after an equine and human outbreak in Australia that resulted in the death of 13 horses and a horse trainer in 1994 (50). The very same year, another outbreak resulted in the death of a pair of horses and their owner in Australia who was admitted to the hospital with meningitis and was found on autopsy to have neuropathological evidence of CNS viral invasion (51). Pteropid fruit bats (flying foxes) are the primary reservoir host and infected horses act as intermediate hosts after exposure to bodily fluid, such as urine, feces or saliva, from an infected bat. Veterinarians and horse trainers are at particular risk for human infection thereafter.
Studies of acute human infection have demonstrated that a wide variety of tissues may be infected by HeV, including lung, liver, blood vessel, and brain (52). Humans infected with Hendra virus tend to first develop influenza-like and respiratory symptoms, which may be followed by systemic infection, encephalitis, and meningitis (53). Neuropathological findings have confirmed that HeV is neuronotropic and can cause meningitis or acute encephalitis which, in some cases, may be relapsing. Neuropathologic specimens from patients have revealed vasculitis, vasculitis-induced thrombosis, meningeal involvement, parenchymal inflammation, necrotic plaques and neuronal invasion (53, 54). Diagnosis of the systemic condition can be accomplished with serum ELISA or RT-PCR, although no tests specific to CNS infection have been studied (55).
Outbreaks have appeared to cluster in cooler months, a finding hypothesized to be related to the breeding cycles of flying foxes, or favorable survival of the virus in cooler and drier environments (56). In 2014, the Australian government approved a plan to destroy flying fox colonies in efforts to preempt further outbreaks. However, some have argued that disrupting colonies may increase the viral shedding among bats since increased stress may facilitate shedding in bat saliva (57, 58). Risk reduction strategies include an equine vaccine that became available in 2012; post-exposure treatment with neutralizing antibodies; and ecological interventions. Proposed ecological interventions include conservation/restoration of bat feeding habitats, to reduce risk of nutritional stress (which may potentiate salivary shedding) and urban colonization, and fencing horses away from trees at night to reduce exposures (56).
Influenza are a group of RNA viruses of the Orthomyxoviridae family, which cause worldwide seasonal outbreaks of acute respiratory illness associated with significant morbidity and mortality. Seasonal cycles are occasionally dominated by pandemic strains of the virus, which cause devastating worldwide disease. There are four types of influenza viruses, A through D, which are characterized based on antigenic characteristics of viral surface proteins. Of these, types A and B primarily produce important human disease (59). The enduring infectivity of influenza comes from antigenic variation of its viral surface proteins, hemagglutinin and neuraminidase, which allow it to evade the human immune response to prior outbreaks. Influenza A is responsible for pandemic disease due to antigenic drift, in which viral surface proteins (particularly, hemagglutinin) are replaced with novel subtypes that are new to human hosts. This new genetic material often comes from waterfowl, an animal population that carry a large reservoir of influenza (60). In contrast, typical seasonal flu is a product of antigenic drift, which occurs in both influenza A and B, and refers to more subtle changes in surface proteins that come from accumulation of mutations, diminishing the antibody response elicited from prior outbreaks (60). Influenza B is carried primarily in human hosts, thus precluding major genomic changes. Though relatively rare, neurologic complications can result from both seasonal and pandemic outbreaks of influenza and compound the morbidity and mortality of the disease.
Neurological complications of influenza are widely reported in the pediatric population, but also occur in adults. Such complications can involve the CNS or PNS, and can range in severity from mild to severe, debilitating or fatal manifestations. Seizures are the most commonly reported neurologic complication, occurring in two-thirds of hospitalized pediatric patients who experience a neurologic complication. The majority of cases are febrile seizures, and in these cases the prognosis is generally good (61). Influenza-associated encephalopathy (IAE) is the second most common neurologic complication of influenza, though it is important to recognize that this term is non-specific and can present with non-focal signs such as changes in attention or arousal, as well as focal neurological findings. In most cases of IAE, there is no evidence of inflammation in the CSF and imaging studies are often unremarkable (61). RT-PCR has shown influenza virus in scattered cases, but the virus may be present in less than half of cases, suggesting a possible parainfectious mechanism of disease (62, 63). Determination of neurologic complications of influenza can thus be bolstered by demonstration of viral load in CSF, but its absence does not preclude the diagnosis.
Several IAE syndromes have been recognized and named based on specific clinical features, and in these cases, patients may be hospitalized specifically for neurologic care. Among the more specific and devastating of the IAE syndromes is acute necrotizing encephalopathy (ANE). ANE usually develops about 5 days after onset of typical viral symptoms, and is characterized by rapid deterioration in mental status, progressing to coma over the span of days. It is most strongly associated with influenza infection, although other viral infections have also been linked. Mild pleocytosis, elevated protein, and xanthochromia can be observed in the CSF (64, 65). Brain MRI characteristically shows bilateral necrosis of the thalami, although the cerebellum and brainstem can also be involved (61, 64). The prognosis is grim, and those who survive are often left with ongoing neurologic dysfunction, including motor deficits, intellectual disability, and epilepsy (64). Other important IAE syndromes reported include classical Reye's syndrome, which typically occurs in the convalescent phase of influenza and other viral illnesses, in children over 5 years of age. It is marked by rapidly progressive encephalopathy, associated with hepatic dysfunction. Affected children often develop coma and seizures. The underlying pathophysiology is thought to involve mitochondrial dysfunction (64). In some cases, the disease may be triggered by salicylate exposure. In the United States, prohibiting the use of salicylates in pediatric patients has been associated with a dramatic reduction in the incidence of Reye's syndrome (66). Imaging in Reye's syndrome is notable for diffuse cerebral edema, and laboratory tests will show evidence of liver dysfunction. CSF is characteristically non-inflammatory, thus, pleocytosis should argue against this diagnosis (61).
A spike in narcolepsy cases followed 1918 H1N1 influenza pandemic (67), and the same pattern appeared in northern Europe (68–71) and China (72) following the more recent 2009-10 H1N1 pandemic. The increase in incidence in northern Europe aligned with the pattern of Pandemrix influenza vaccine administration, which has been linked to immune-mediated narcolepsy (73, 74), although direct viral infection also has an independent effect as shown by the appearance of the syndrome after H1N1 infection in the absence of vaccine (68, 72, 75).
There are many more neurologic syndromes described following influenza infection, though the full list is beyond the scope of this article. Similar to the other presentations described, these syndromes are not singularly associated with influenza. They include acute disseminated encephalomyelitis (ADEM), transverse myelitis, acute inflammatory demyelinating polyneuropathy (AIDP) and the Miller-Fisher variant, as well as ischemic stroke. Of note, rare cases of AIDP and transverse myelitis have been associated with the influenza vaccine, though the relative risk is estimated to be much lower than the benefit of protection against active infection (61, 76).
One study of 34 children with acute necrotizing encephalopathy (including 11 influenza as prodrome) demonstrated a favorable outcome in 7 of 12 children with early steroid administration compared to 0 of 5 children without this treatment, although this was not statistically significant (77). Some case reports found benefit of steroids in pediatric influenza encephalopathy (78, 79) as well as adult influenza-associated acute necrotizing encephalitis (80). However, a larger retrospective cohort study of 692 children in Japan examined administration of corticosteroids early in the course of illness in children with influenza encephalopathy and did not show any benefit (81). The treatment of neurologic manifestations of influenza demands further research; the current evidence does not definitively support use of steroids (82).
Lassa fever is a hemorrhagic disease, caused by the single-stranded RNA Lassa virus hosted primarily by Mastomys natalensis rats and endemic to Western Africa. It initially infects immune cells in the nasopharynx, with subsequent spread to regional lymph nodes followed by multiorgan dissemination. After an incubation period of 1 to 3 weeks, its clinical course is heterogeneous; the majority of patients experience only malaise, headache, and low-grade fever, but more severe cases can include such diverse manifestations as diarrhea, hypotension, and pulmonary edema. Oozing blood from the oropharynx or, less commonly, the rectum or genitals, is seen in fewer than 20% of patients (83).
The most common neurologic complication of Lassa fever is sensorineural deafness, which has been reported in up to 25% to one-third of patients who survive the illness, although other research suggests this may be an overestimate (83, 84). Interestingly, the hearing loss usually occurs during the convalescent phase of the illness, sometimes after the patient is otherwise recovered, pointing to an immune cause of the disorder rather than a more direct viral cause (85). The hearing loss can be unilateral or bilateral and resolves spontaneously in fewer than half of cases. There appears to be no correlation between severity of illness and likelihood of hearing loss.
The less common CNS effects of Lassa fever are as heterogeneous as its systemic symptoms, including ataxia, meningitis, seizures, and coma (86, 87). The most common neuropsychiatric manifestation is depression but mania and psychosis are described (88).
Diagnosis of Lassa fever is commonly clinical in the context of appropriate signs and symptoms in an endemic region or following an appropriate exposure to a host animal. The disease can be diagnosed with high sensitivity and specificity with serum RT-PCR if logistically feasible, although the equipment required for this is expensive and requires significant operator training (89). Additionally, the high variation between lineages of the virus can make it poorly sensitive in some contexts (90).
In all cases of CNS involvement, symptomatic treatment of the disorders (i.e., antiepileptic drugs for seizures; antipsychotics for agitation in psychosis) appear to be the only indicated management. No proven treatment exists for amelioration of sensorineural deafness, and early treatment with ribavirin does not appear to protect against development of this symptom nor improve its chances of resolution.
Marburg virus forms its own genus within the family Filoviridae (91). It has a high mortality and is deemed by the U.S. Department of Homeland Security as a bioterrorism threat (92). The first recorded outbreak was in 1967 in Germany and Serbia. The most recent outbreak was in Uganda in 2017 (92). It is a hemorrhagic fever virus in the same family as Ebola virus, but it has only had three major outbreaks and a few reported cases to date (91). It is thought to be spread through mucosal transmission, breaks or abrasions of the skin, and parenterally, though the 1967 outbreak did isolate the virus in semen, and therefore, there is possibly sexual transmission (91). The virus has been isolated in blood, urine, and saliva (93).
The disease has three phases: a generalization phase, an early organ phase and a late organ or convalescence phase. The generalization phase consists of influenza-like symptoms of headache, chills, myalgias, and malaise, and the early organ phase consists of evidence of hemorrhagic disease. The late organ phase, which usually starts around day 13, is when most of the neurologic symptoms manifest, with reported convulsions, restlessness, obtundation, confusion, dementia, or coma (91). Patients in Germany were noted to have sullen, negative, and slightly aggressive behaviors (94).
Marburg virus may directly infect the CNS. Autopsies have shown glial nodule encephalitis in three of five cases in Germany (94). Neuropathology of hamster models have shown edema, enlargement of blood vessels in the brain, intracranial hemorrhage, and encephalitic lesions (91). RT-PCR can be used for diagnosis of the disease in principle, but no tests are commercially available and so, in practical terms, diagnosis is performed clinically (95).
Meningococcal meningitis, caused by the gram-negative diplococci Neisseria meningitidis, is the most common cause of bacterial meningitis in young adults, and a major cause of both endemic and epidemic bacterial meningitis in children and adults worldwide (96). In the United States and other high-income countries, serogroups B, C, and Y are prevalent, while in the sub-Saharan African region known as “the meningitis belt,” repeated pandemics of serogroup A disease have occurred every 5 to 10 years (96). Vaccines available for the prevention of meningococcal infection include quadrivalent meningococcal polysaccharide conjugate vaccines for serogroups A, C, W, and Y (ACWY vaccine) and serogroup B conjugate vaccines. The capsular polysaccharide present in serogroup B resembles a protein found in human tissue, historically making vaccine development difficult. This has resulted in many individuals receiving quadrivalent ACWY vaccination while remaining vulnerable to serogroup B, although in the past 5 years the serogroup B vaccine has been increasingly effective and recommended (97–99).
The most common clinical presentations of meningococcal disease are meningitis and meningococcemia with sepsis. The classic features of meningococcal infection include a hemorrhagic rash, altered consciousness, and meningismus, however these may frequently not be present on a patient's initial presentation (100). Patients with meningitis generally have low serum concentration of meningococci with a high concentration in the CSF, whereas patients with meningococcal septicemia have the reverse findings, with high serum concentration of meningococci and endotoxin and low concentrations in the CSF (96).
Neurologic sequelae of meningococcal meningitis include focal neurologic deficits, hearing loss, seizures, cognitive impairment, vision loss, and hydrocephalus (101). Focal neurologic deficits occur less commonly in meningococcal meningitis compared to pneumococcal meningitis (3% of children and 2–9% of adults in meningococcal meningitis, 3–14% of children, and 11–36% of adults in pneumococcal meningitis) (101) and are most often secondary to cerebrovascular events (102). Hearing loss is similarly less common in meningococcal meningitis than in pneumococcal meningitis, and may be transient or permanent. Studies indicate a higher rate of hearing loss secondary to meningococcal meningitis in children in low-resource countries (19–23% of children) as compared to high-resource countries (about 4%) (101). Seizures are common in patients with bacterial meningitis, most often in the acute phase of illness, although some patients may have recurrent seizures after recovery. Patients recovering from meningococcal meningitis are at risk for long-term cognitive impairment, which can present as school challenges in children (103, 104) and cognitive slowing and memory impairment in adults (105), which may not improve.
The gold standard of diagnosis for N. meningitidis meningitis is CSF gram stain and culture, although the latter can be falsely negative as soon as 1–2 h after antibiotic administration. Several RT-PCRs have been developed that show high sensitivity and specificity even after antibiotic administration (106, 107), although these are rarely used in place of culture as they do not reveal antimicrobial sensitivities.
Treatment for meningococcal infection includes early antibiotic treatment, which results in rapid clearance of meningococci from the CSF (108). Aggressive management of complications such as shock or elevated intracranial pressure may also be required. Corticosteroids have only been shown to be effective in bacterial meningitis caused by Streptococcus pneumoniae, and so their use is appropriate early in the course of illness before pathogen speciation but are not indicated once N. meningitidis has been confirmed (109). With antibiotic treatment, the mortality rate for meningococcal meningitis has decreased to 7–10% (110, 111). Mortality is generally secondary to meningococcal septic shock or cerebral edema in the case of meningococcal meningitis (96).
Middle East respiratory syndrome-related coronavirus (MERS-CoV) is an enveloped, single-stranded, positive-sense RNA virus belonging to the diverse family Coronaviridae, which also includes SARS-CoV and 2019-nCoV. MERS-CoV was first identified as a human pathogen in 2012, when it was identified in a case of fatal pneumonia and renal failure in a 60-year old man in Saudi Arabia (112). Humans acquire the virus from dromedary camels, in which the virus is highly prevalent and can cause cold-like symptoms (113–115), although MERS-CoV has also been found in bats, a common coronavirus natural reservoir (116, 117). Molecularly, MERS-CoV uses the dipeptidyl peptidase 4 cell surface receptor protein, alternatively called DPP4 or CD26, to enter and infect cells (118, 119). As of November, 2019, 2,494 cases of MERS-CoV infection have been reported in humans, including outbreaks in 27 different countries, with an estimated case-fatality rate of 34.4% (120), making MERS-CoV the deadliest of the human coronaviruses. Clinically, infection can present variably; some people remain asymptomatic while others develop multi-organ involvement, with hallmark features including acute respiratory distress syndrome, renal failure, and coagulopathy. Risk factors for a more severe clinical course, as for other coronaviruses, include older age; presence of medical comorbidities such as hypertension, renal disease, or diabetes; and initial laboratory assessment consistent with a severe inflammatory response (121, 122).
Neurologic manifestations in MERS-CoV infection have been cited in small case series. In one examination of 70 MERS-CoV positive patients, 12.9% developed headache, 25.7% confusion, and 8.6% seizures (123). One series reported three cases of depressed arousal, vomiting, or focal motor deficits, with MRI abnormalities including patchy diffusion restriction and T2 hyperintensity in the subcortical and periventricular white matter, basal ganglia, thalami, midbrain, pons, cerebellum, and even extending into the cervical spinal cord (124). CSF analysis was performed in two of the three cases, and was notable for elevated protein, but normal glucose and cell counts. MERS-CoV RT-PCR in the CSF was negative. In a second series of four cases, one developed ophthalmoplegia and ataxia, without imaging, electromyogram, nerve conduction study or CSF abnormalities, including a negative MERS-CoV RT-PCR of the CSF (125). The three remaining cases in this series made note of presumed PNS involvement, with limb weakness, paresthesias, extremity pain, and hyporeflexia. Finally, two cases have been published noting neurological complications attributed to other organ system dysfunction in MERS-CoV, one of intracranial hemorrhage secondary to coagulopathy, and the second a length-dependent axonal polyneuropathy attributed to critical illness (126). Pathophysiology of the various neurologic manifestations of MERS-CoV, however, remains speculative.
Monkeypox is a double-stranded DNA, zoonotic virus that characteristically causes a disease similar to smallpox, but with milder exanthem and lower mortality. The definitive reservoir has not been established, but it appears transmissible through monkeys as well as terrestrial rodents including rats and squirrels. Person-to-person transmission is possible but appears to occur at relatively low rates and result in a milder disease course (127, 128).
We have been able to find only two cases of neurologic complications of monkeypox in the literature. The individuals affected were both young girls who developed encephalitis, requiring intubation and mechanical ventilation. One 3-year-old girl died on day 2 of hospitalization while a 6-year-old girl survived after a 14-day course in the intensive care unit. The former patient had no CSF diagnostics performed, while the latter patient had detectable orthopoxvirus-reactive IgM in CSF (129, 130).
Of note, monkeypox appears to have a fatal course almost exclusively in infants and young children, specifically those who have not received vaccination against smallpox (129).
Nipah virus (NiV) is an enveloped, negative-sense single-stranded RNA Paramyxovirus of the genus Henipavirus, endemic to Southeast Asia and Africa (131, 132). First isolated around 1999 after a series of outbreaks in Malaysia and Singapore, it owes its name to the Sungai Nipah village in Malaysia (133). Bats of the genus Pteropus act as its primary reservoir, and NiV seropositivity has been found among Pteropus populations as widely distributed as West Africa, Madagascar, and across southeast Asia (132, 134, 135). NiV from asymptomatic bats can cause human infection either by direct exposure to bat secretions on fruit/sap or via a wide range of symptomatic intermediate host animals (pigs, horses, dogs, cats) (136).
NiV infection presents primarily with a fulminant encephalitis (55, 132, 137). After a 5–21 day incubation period, a patient typically presents with high fever, headache and meningismus (132, 138). Although rare during the initial Malaysian outbreak, an accompanying viral pneumonia has been noted in more than half of cases during recent outbreaks in Bangladesh and India (139). After neurologic symptom onset, mental status may decline rapidly, often progressing in just 24–48 h to severe encephalopathy and coma; convulsive seizures are common, and mortality is high (132). Outbreak case fatality rates have varied widely between 40 and 90%, notably with lower rates with initial Malaysian outbreaks and higher rates with recent outbreaks in Bangladesh and India (138, 140, 141). Infection is typically monophasic, but reports describe cases of relapsing fulminant encephalitis years after the initial infection in some patients, even those whose initial symptoms were mild (142). CSF chemistry profile generally resembles other non-hemorrhagic viral meningitides. MRI shows diffuse and patchy, often punctate, subcortical and deep white matter lesions, less often with cortical and brainstem involvement, typically with lesions less than 1 cm in diameter (143). Pathology demonstrates a necrotizing small-vessel-predominant vasculitis present both diffusely in the CNS as well as in the heart, lung, kidney, and most other organs sampled (144). Diagnosis can be performed with RT-PCR or ELISA; while the gold standard is detection of neutralizing antibodies against NiV in serum, the virus is classified as a level 4 biosafety agent and so this is not generally viable in a clinical setting (55, 145). Treatment is largely supportive, as antiviral trials to date have shown equivocal or no therapeutic effects (146–148).
The first recognized outbreak is thought to have occurred on Malaysian pig farms, where asymptomatic bats in tree branches hanging above farms transmitted NiV in their saliva and other secretions to pigs below leading to rampant pig-pig and pig-to-human transmission via respiratory and other secretions (149). More than 300 human cases and 105 patient deaths occurred (150). With the exception of one nurse with no direct pig contact demonstrating asymptomatic NiV seropositivity and MRI brain findings typical for infected persons (151, 152), evidence of human-to-human transmission during initial outbreaks was extremely limited.
Since this, outbreaks have been recognized in Bangladesh and India nearly annually since 2001 from a related and potentially more dangerous strain of NiV, with additional outbreaks in the Philippines stemming from NiV-infected horses (132, 149, 153, 154). Human case clustering in these recent NiV outbreaks supports a concerning new feature of person-to-person transmission (132, 155–157). In some recent outbreaks, such as the 2018 Kerala, India outbreak during which 16 of 18 confirmed cases died, the overwhelming majority of cases and fatalities were due to human-to-human transmission after one or a few index animal-human transmissions (155, 158, 159). These developments raise concern for an increased risk of large-scale and lethal pandemic spread, prompting designation of NiV by the WHO to a high-risk “priority disease” (160, 161).
No NiV-protective vaccines are currently commercially available, although host animal and human Nipah virus vaccines are currently in development. Recent small-scale preclinical studies with these vaccines have reported effective immunity after VZV-platform vaccination of non-human primates (131, 162, 163). Ribavirin treatment was associated with a significantly lowered risk of mortality in one cohort study of 140 individuals in Malaysia (146), although a smaller study in Kerala showed no difference (164). Until and potentially even after any future success in widespread vaccination, the Nipah Virus will likely continue to threaten human populations for the foreseeable future given the stable, geographically widespread and genetically diverse range of animal (bat) reservoirs, which themselves serve important ecological roles, as well as the wide array of susceptible intermediate hosts including pigs, horses, dogs, and cats (132, 140).
In December 2019, a series of viral pneumonia cases of unknown cause appeared in Wuhan, Wubei, China, and deep sequencing analysis from lower respiratory tract samples indicated that the infection was due to 2019 novel coronavirus (2019-nCoV) (165). The 2019-nCoV (COVID-19) is a betacoronavirus in the same subgenus as the severe acute respiratory syndrome (SARS) virus (see below), and therefore, also referred to as SARS-CoV-2. The structure of COVID-19's receptor-binding gene region is similar to that of the SARS coronavirus, and COVID-19 uses the same angiotensin-converting enzyme 2 (ACE2) receptor for cell entry (166).
It is too early to tell if there is increased prevalence of neurological complications as a result of COVID-19 when compared to other respiratory viruses. Among a subset of patients with COVID-19 in three hospitals in Wuhan, China: 36% of patients had neurologic manifestations with CNS manifestations in 25%, PNS manifestations in 9%, and skeletal muscle injury in 11% (167). Multiple retrospective case series in hospitalized patients report CNS manifestations that include dizziness, headache, and encephalopathy (168). Additionally, smell and taste disturbances are characteristic of COVID-19, leading some to speculate CNS entry via the olfactory mucosa as a possible route of central nervous system entry (167, 169). Given the severity of pulmonary disease seen in some patients with COVID-19, there is also a theory as to whether the brainstem could be affected through infection of the nucleus of the solitary fascicle, which modulates respiratory effort in response to atmospheric carbon dioxide and oxygen concentration, although there has been no concrete evidence of this to date (170).
Some case series and case reports have described peripheral nerve and muscle complications of COVID-19 that can be seen in other viral infections. GBS was reported in both Wuhan (171) and Italy, without identification of COVID-19 in the CSF, and of note, not all patients had elevated cells in the CSF (172). A case of acute myelitis in COVID-19 infection in Wuhan is reported, but no CSF profile or spine imaging are included (173). A case of COVID-19-associated myopathy caused by type I interferonopathy has been reported (174). Rhabdomyolysis has also been reported as an atypical presentation of COVID-19 (175): 20% of patients with severe respiratory disease and 5% with non-severe respiratory illness had evidence of skeletal muscle injury in a retrospective cohort of COVID-19 patients (167).
Diagnosis of systemic COVID-19 is most commonly performed with RT-PCR on nasopharyngeal swabs, serology tests, and antibody-based rapid tests (176, 177). Some case reports and case series have reported detection of SARS-CoV-2 via RT-PCR in CSF of COVID-19-positive individuals with neurologic symptoms (178–180), although this finding has also been absent in many individuals who have severe COVID-19 infection with neurological symptoms (181–183). Furthermore, a reanalysis using the Xpert®–Xpress SARS-CoV-2 test of stored CSF samples performed in three individuals who initially had virus detected in CSF by RT-PCR showed no subsequent evidence of virus (184). It thus remains to be seen what role measurement of virus in CSF plays in clinical management of individuals with COVID-19.
We anticipate studies focused on the impact of SARS-CoV-2 infection on neurological function and the long-term neurological sequelae will continue to be published and our understanding will deepen over the coming years. At this point, it is obvious that extrapulmonary manifestation of SARS-CoV-2 includes neurological symptoms and disease, and will be crucial for clinicians to recognize and treat.
Yersinia pestis is a zoonotic, gram-negative, non-motile organism transmitted by the tropical rat flea (Xenopsylla cheopis). The plague disease it causes has three clinical subtypes: bubonic, septicemic, and pneumonic. The most common bubonic form is characterized by sudden onset of malaise, dizziness, high fever, and lymphadenitis. This can progress to septicemic plague, which includes a heterogeneous phenotype of hypotension plus other systemic manifestations; approximately 80–90% of septicemic plague cases occur with preceding buboes (enlarged lymph nodes) (185). The most commonly reported neurological complication of plague is meningitis, which appears to arise in the context of delayed treatment of primary bubonic or septicemic plague.
The pathogenesis of plague meningitis is presumed due to hematogenous dissemination from buboes, and in light of this some studies have proposed that axillary buboes predispose to meningitis given easier access to lymphatic tracts leading to intracranial compartments (186). The case series published to date have not found a statistically significant effect for this. Development of septicemic plague has also been cited as a risk factor for meningitis, but similarly no statistically significant findings have been published.
The first documented case of CNS involvement of plague was in 1940, when a 3-year-old with no known immunosuppression developed headache, cough, and fever over 6 days, subsequently developing signs of meningismus and with microscopic evaluation of CSF consistent with Y. pestis (187). Subsequent reports have also included a significant number of pediatric patients. One case series detailed a 7-year-old girl who presented with malaise and buboes, and subsequently developed encephalopathy and meningismus followed by death; as well as a 47 year-old woman who presented with malaise and fever, then progressed to develop a waxing-waning delirium and Kernig's sign in the setting of fever (188). A retrospective analysis examined a cohort of 105 patients from 1970 to 1979 in whom 6 patients (6%) developed neurologic complications; 5 of those 6 patients were children aged 10 to 15 years old, while the remaining patient was a 35 year-old woman (189). A case series of 27 plague patients in Arizona documented 3 individuals (11%) with meningitis, all between 2 and 8 years of age (190).
In most cases studied, clinical signs and symptoms of meningitis appeared after onset of systemic symptoms, with a delay ranging from a few days to two or more weeks. Diagnosis is generally with Y. pestis culture from bubo aspirate, sputum, or blood, depending on the type of plague with which an individual presents, although rapid antigen-based tests are also available (185). Since the majority of case reports examining plague meningitis are from the 1970s or earlier, imaging features of neurologic disease are not well characterized.
Rift Valley Fever Virus (RVFV) is a negative-sense, single-stranded RNA virus that belongs to the genus Phlebovirus (191). RVFV is primarily transmitted to livestock through arthropods, primarily the Aedes aegypti mosquito. While humans can be infected by mosquito bite, the primary route of infection is due to direct or indirect contact with infected animal blood and products, possibly through aerosolization (192).
Rift Valley Fever (RVF) was first identified in East Africa in 1931 with nine recognized outbreaks between 1999 and 2010 (193) and a spreading geographic distribution that extends from Madagascar to Saudi Arabia. RVFV was identified as a potential biologic weapon by the USA prior to termination of a biologic weapons program in 1969 (194). There have been no cases of human-to-human transmission (195).
In humans, most cases of RVF are mild and can cause a non-specific flu-like illness with fever, muscle pain, joint pain and headache. However, severe cases include ocular, meningoencephalitis or hemorrhagic fever forms. Neurologic manifestations are identified in up to 17% of patients in some outbreaks, which can manifest with encephalopathy, ataxia, vertigo, seizures, and coma (196). Retinitis can occur in one or both eyes with the affected eye showing macular edema and exudates. Some cases demonstrate vasculitis, retinal hemorrhage or infarcts (192). Meningoencephalitis has a delayed onset of presentation (2–60 days) with fever, headache, cranial nerve deficits, seizures, hemiparesis, visual hallucinations, choreiform movements, and locked-in syndrome all reported (192, 197). Histopathology of brain lesions shows focal necrosis with infiltration of lymphocytes and macrophages with perivascular cuffing (192). A case report described MRI brain findings of multiple bilateral asymmetrical cortical hyperintense areas as well as CSF characterized by elevated protein and lymphocytic pleiocytosis (198). RVFV has not been isolated in human CSF, but has been found in animal studies (199).
Diagnosis for RFV is through RT-PCR of viral RNA or ELISA for IgM antibodies against RVF virus (200). Treatment for RVF is mainly supportive. Ribavirin was tried during outbreaks. Newer antivirals are being tested on animal models. A live attenuated vaccine exists primarily for veterinary use. A human inactivated vaccine was trialed for high risk exposures, but is not widely approved for commercial use (191).
Severe acute respiratory distress syndrome (SARS) is a viral respiratory illness caused by a type of coronavirus called SARS-associated coronavirus (SARS-CoV) (201). Coronaviruses are enveloped non-segmented positive-sense RNA viruses belonging to the family Coronaviridae (165). Infections are usually mild but two betacoronaviruses, SARS-CoV and MERS-CoV (see above) are associated with higher mortality rates, with SARS-CoV, estimated by the WHO, to have had a global case fatality of around 9.6% (202). SARS was the first major novel infectious disease to affect countries internationally in the twenty-first century, and is reported to originate from southern China in 2002 (203).
There has been concern that SARS may translocate and infect the CNS, with reports of isolation of the virus in the brain (204) and specifically in neurons from autopsies of eight confirmed SARS patients (205). Isolation of the virus through RT-PCR of the CSF was performed in a case of a woman with SARS who experienced convulsions (203). An additional case of RT-PCR detection occurred when a woman with SARS presented with status epilepticus; of note, in this case, the CSF showed no white cells and normal protein, i.e., a non-inflammatory picture (206). SARS has also been isolated in the CSF of a child with ADEM. There are reports of increased rates of depression, anxiety, and posttraumatic symptoms after SARS infection (207).
In murine models, SARS has been correlated to a possible chronic demyelinating condition that resembles multiple sclerosis (208). Murine models have also shown SARS enters the brain primarily through the olfactory bulb. The infection results in transneuronal spread to the brain (209).
The most sensitive modality for SARS diagnosis is ELISA for IgM against the virus, although this can remain negative in infected patients for weeks into illness (210). RT-PCR has been tested for patients earlier in the course of disease, although sensitivity on many of these tests may be as low as 50% (211, 212).
Similar to COVID-19, there are neuromuscular complications in patients with SARS, including skeletal muscle injury and rhabdomyolysis, although these manifestations are theorized to be related to a cytokine storm rather than direct infection by the virus (213).
Smallpox is caused by the variola virus, which is a double-stranded DNA virus in the Orthopoxvirus genus. Although it was eradicated in 1980, there is concern that smallpox could reemerge as a bioterrorism agent. Human to human transmission predominantly occurs through airborne droplets, but can also occur through infected vesicles, scabs, and fomites. It is highly contagious. Transmission in a household can be up to 60% (214). Diagnosis is with serum RT-PCR (215).
Smallpox can incubate for 1–2 weeks and patients are most infectious within 10 days of developing lesions (216). The most typical manifestation of smallpox, termed Variola major, starts with a prodrome of fever, malaise, headache, and delirium before development of skin lesions. Lesions occur after 24 h, initially on mucous membranes (enanthem) followed by a cutaneous rash (exanthem) starting in the face and spreading to the extremities that becomes vesicular then pustular. Mortality is estimated at 30–50% (217). Variola minor, cause by a similar strain of Variola virus, leads to milder disease with estimated 1% mortality (217, 218).
Encephalopathy is common in clinical presentations of smallpox (216). Acute perivenular demyelination was found in patients who have died of smallpox (217, 219). Encephalitis can develop around 6–10 days after presentation and occur in ~1 in 500 patients with variola major and ~1 in every 2,000 patients with variola minor (219, 220). Patients can also report severe headaches, hallucinations and delirium. Smallpox has also been associated with ocular complications, most often binocular, which can lead to blindness (217). CSF initially shows a lymphocytic or neutrophilic pleocytosis, which becomes a lymphocytic pleocytosis with moderately elevated protein and normal glucose (219).
Post-vaccination encephalitis is a rare but severe complication (221). In patients less than 2 years of age, encephalitis incidence is estimated at 0 to 103 in 100,000 person-years and occurs 6–10 days after infection. In patients over 2 years old, the incidence is estimated at 2 to 1,219 per 100,000 person-years and occurs 11–15 days after infection (217). Mortality from post-vaccine encephalitis can be as high as 25% (222). Other neurologic complications associated with the vaccine include headache, seizures, cranial nerve palsy, GBS, hemiplegia, and coma (222). Pathology shows hyperemia, lymphocytic infiltration of the meninges, and degeneration of the basal ganglia (217).
The Francisella genus is a group of gram negative bacilli, of which F. tularensis is a highly virulent subtype responsible for the majority of the human disease known as tularemia. It is a fastidiously growing bacterium, which was first isolated after an outbreak of a rodent-borne illness that occurred in 1911 in Tulare County, California following the San Francisco Earthquake. During the outbreak, the organism was grown from cultures obtained from infected ground squirrels. F. tularensis is found almost exclusively in North America; disease outside North America occurs in the Northern Hemisphere, and is attributed to the holarctica subspecies (223). Though human disease is rare, F. tularensis is recognized to be among the most infectious of pathogens, with a dose of only 10 organisms capable of causing infection with a high mortality rate when untreated. Given its potential lethality and ease for dissemination through airborne delivery, the Centers for Disease Control and Prevention (CDC) has recognized F. tularensis as a potential bioweapon (224).
Transmission largely occurs through two possible disease cycles: terrestrial or aquatic. Terrestrial cycles rely on rabbits and hares as hosts, with arthropods such as ticks and flies as vectors. Aquatic life cycles rely on mammals, such as beavers and muskrats, who shed the live organism into bodies of water, where it can infect exposed humans (223). The disease can also be directly transmitted from close contact with host mammals, and multiple mechanisms have been recognized: the processing and consuming of contaminated meat, hamster bites, and lawn care (running over infectious material from an animal, or the animal itself with a lawn mower or weed-whacker can disseminate the organism through aerosolization) are a few reported scenarios (223). In Martha's Vineyard, Massachusetts, an outbreak of tularemia was observed during the summer of 2000, prompting case-control studies of prior cases that showed lawn mowing and brush cutting as risk factors (225). Importantly, healthcare and laboratory workers who process infected specimens are also at risk for disease (223).
Tularemia infection can vary in presentation based on host factors, bacterial virulence, and how the disease was transmitted. The incubation period can vary considerably, though typically lasts 3 to 6 days. Clinical manifestations are most simply categorized as ulceroglandular or typhoidal, though pneumonic presentations are increasingly common and can occur with both types. Ulceroglandular disease is the more common form, composing about 75% of cases, and has several overlapping subtypes. Classically, it is characterized by a skin lesion associated with regional, tender lymphadenopathy. History often reveals recent contact with possible host mammals or arthropod vectors through insect bites. The skin lesion is thought to represent the site of inoculation, and can appear as an eschar with surrounding erythema (226). Exposure to the eye and pharynx can cause localized manifestations with conjunctivitis or pharyngitis, which may be accompanied by lymphadenopathy. Typhoidal tularemia is less common and is characterized by high fever with hepatosplenomegaly. There is usually an absence of skin or lymph node involvement, although often there is associated gastrointestinal and pulmonary disease (223). Diagnosis is through antibody titers in serum, with a 4-fold increase between acute and convalescent values representing a positive result. Specific titer cutoffs in the acute phase of illness can also be used to diagnose the disease even without matching serum samples from convalescence (227).
Given the overall rarity of tularemia infection, its neurologic complications are largely the subject of case reports. Both classic and Miller Fisher variants of GBS are described (228, 229), though some cases are disputed to represent a manifestation of infectious polyneuritis, critical illness polyneuropathy, or myopathy (230). Meningitis is more commonly reported. It is rarely complicated by cerebral abscesses (231). A 61-year-old patient with gait difficulties and ataxia on examination was found to have tularemia and was diagnosed with cerebellitis. His neurologic difficulties completely improved with treatment of infection (232). Ventriculoperitoneal shunt (VPS) infection due to tularemia has been identified in a child with medical history of myelomeningocele requiring VPS who later disclosed that, 2 weeks prior to becoming ill, he had helped his friends skin a rabbit (233).
All clinicians must maintain a high index of clinical suspicion for tularemia, as serologic testing depends on the identification of antibodies against the organism, which are typically not detectable until at least 2 weeks after the onset of illness. Gram stain and culture rarely detect the organism (234).
Yellow fever virus (YFV) is a positive-sense, single-stranded RNA virus that belongs in the Flavivirus genus. It is predominantly transmitted by mosquitoes and is endemic to tropical regions of Sub-Saharan Africa and South America. While an effective live-attenuated vaccine has been developed that protects against YFV, there continue to be occasional outbreaks predominantly in Africa, South America, and Asia where immunization levels are low (235).
YFV incubates for 3–6 days after transmission and can lead to a mild non-specific febrile illness. However, a portion of patients (~14%) can develop severe disease leading to hemorrhagic fever and multi-organ failure (236). Diagnosis can be performed with either RT-PCR or ELISA of serum, depending on when during the time course of infection the testing takes place (237, 238). Neurologic manifestations are rare, but early studies described delirium, seizures, and coma. Elevated intracranial pressure can also occur with CSF demonstrating elevated protein and absence of inflammatory cells. The brain tissue does not show severe inflammatory changes, but can show petechiae, perivascular hemorrhages, and edema (239). As a result, the CNS changes were thought to be due to cerebral edema or metabolic factors rather than direct inflammation (236, 239). Neuroinvasion leading to encephalitis appears to be exceedingly rare with only case reports of optic neuritis, cranial nerve palsy, and paralysis (236). However, YFV has been detected in human CSF (240). A later case series examining severe yellow fever cases admitted to the ICU found that seizures occurred in 24% of patients without evidence of significant cerebral edema on CT imaging (241). There is a paucity of literature describing neurologic manifestations of yellow fever, especially given an effective vaccine.
Severe neurologic manifestations have been associated with the yellow fever vaccine. While rare, neurotropic disease associated with the vaccine 17D is estimated at 0.8 per 100,000 people vaccinated, especially in individuals >60 years old (242). Neurotropic disease manifestations range from headache and encephalopathy to encephalitis, demyelinating disease, and GBS (243).
Zika Virus (ZIKV) is a positive-sense RNA flavivirus first isolated and identified in Uganda in 1947 (244). ZIKV is widely distributed in warm climates with separate African and Asian lineages, though few cases in humans were reported before the spread of the Asian lineage occurred in Yap in 2007, French Polynesia in 2013, and the Americas in 2015 (245). The ZIKV virus is primarily transmitted by Aedes mosquitoes (246), although it can also be spread by sexual contact (247), blood transfusion, or maternal-fetal transmission (246). Symptomatic ZIKV infection occurs in 20–50% of infected patients and usually consists of a non-specific rash, low-grade fever, arthralgia, myalgia, and conjunctivitis (248). Diagnosis is generally with RT-PCR followed by ELISA for IgM in patients with equivocal results or those with negative results and sufficient clinical suspicion (249, 250). ZIKV gained significant attention during the American pandemic from 2015 to 2017 due to rapid spread of the American subclade of the Asian lineage throughout a susceptible population and the recognition of severe neurological complications.
Neurologic symptoms of ZIKV infection in adults can be divided into peripheral and central manifestations. Peripherally, ZIKV infection is associated with GBS, which was estimated to affect 2–3 patients per 10,000 ZIKV infections (251). Symptoms of GBS began 5–10 days after ZIKV illness. Acute inflammatory demyelinating polyneuropathy, acute motor sensory axonal neuropathy, Bickerstaff's encephalitis, and the Miller-Fisher syndrome subtypes of GBS have all been reported after infection (252). CNS ZIKV disease is less common compared to ZIKV-associated GBS and may manifest as an encephalomyelitis (253) or vasculitis leading to strokes.
Fetuses of women infected with ZIKV during pregnancy are at risk of congenital Zika syndrome, and ZIKV-associated microcephaly. Four to seven percentage of infections will result in fetal loss. Twenty to thirty percentage of the fetuses of women infected with ZIKV will develop infection (254). Infected fetuses are at risk for congenital ZIKV syndrome, which consists of a disruption in cortical development and subsequent collapse of the skull, resulting in microcephaly. Affected infants may also have subcortical calcifications, chorio-retinal atrophy, pigmented mottling of the retina, arthrogryposis, and hearing loss (255).
Diseases with the potential for epidemic and pandemic spread have a variety of neurological manifestations, ranging from rare presentations involving the CNS to fulminant manifestations that define the disease outbreak. While some diseases represent regional epidemics, several reflect pandemic diseases. The role of the neurologist and neurologically-focused clinician is critical for early identification of outbreaks and characterization of the range of features of systemic disease-causing pathogens.
Each disease listed includes CNS involvement although in some cases the situation is atypical (e.g., plague) or the data reflect few cases in vulnerable groups (e.g., cholera). Six of the diseases (Chikungunya, COVID-19, F. tularensis, influenza, MERS, and Zika virus) additionally include a recognized element of PNS symptom burden. The most common CNS manifestation is a non-specific encephalopathy, although a number of diseases cause more syndromic neurologic features that are pivotal in the disease diagnosis such as in the fulminant encephalitis of Nipah virus, the ophthalmoplegia and ataxia of MERS-CoV-2, and the ICH of Crimean-Congo hemorrhagic fever or Marburg virus disease. The most common PNS manifestation of the diseases on this list is post-infectious AIDP.
As climate change continues to disrupt host and vector habitats, diseases with regional epidemic potential have risk of reaching new populations and locations. One example of this is Crimean-Congo hemorrhagic fever but others are also recognized (26, 27). It is thus crucial that healthcare providers globally are able to recognize the clinical manifestations of these diseases so that appropriate treatment can be initiated promptly at an individual level while pathogen spread is attenuated at a population level.
The range of evidence for the pathogens is variable and tends to focus on case reports and case series. While two diseases (monkeypox, tularemia) are studied in only a handful of individual case reports, all other pathogens include at least moderately sized case series and often large, population-scale reviews indicating a significant burden of neurologic disease.
The mechanism of neurological involvement is variable among the diseases. Twelve of the associated pathogens have potential evidence of direct neurotropism as shown either through identification in CSF during active infection or neuropathologic findings (V. cholerae, ebolavirus, influenza virus, Hendra virus, Lassa virus, N. meningitidis, Nipah virus, Y. pestis, SARS, F. tularensis, yellow fever virus, Zika virus). Four pathogens have possible neurotropism as shown either through presence in the brains of animal models but not humans (Chikungunya virus, Crimean-Congo hemorrhagic fever virus, Rift Valley fever virus) or by presence of antibodies but not the pathogen itself in CSF of affected individuals (Marburg virus). In four pathogens, the question of direct neurotropism remains unclear (monkeypox, MERS, smallpox, SARS-CoV-2). Many of the diseases show evidence of parainfectious neurologic syndromes in addition to any direct neurotropism they may have (Chikungunya, influenza, Lassa fever, COVID-19, smallpox, tularemia, Zika).
Of the 20 diseases on the list, seven (Chikungunya, COVID-19, influenza, MERS, Neisseria meningitis, plague, tularemia, and Zika virus), have spread to all or nearly all WHO world regions, often despite having well-identified origins in particular locations. Vaccines are presently available for only eight of the diseases (cholera, COVID-19, ebola virus disease, influenza, Neisseria meningitis, plague, smallpox, yellow fever), while two others have a vaccine available for animal hosts (Hendra virus) or under development for humans (rift valley fever virus) (Table 1). Of the remaining 10 diseases, none have curative treatment and many have potentially fatal outcomes, highlighting the importance of control of disease spread following recognition.
Our work has several limitations. We provide a scoping review but not a systematic review. Our summary is limited by the review being performed of English language articles with searches performed through PubMed and related medical search engines online. We did not include historical literature, for example on plague or smallpox, that may be earlier than our search was able to extend. By the nature of this topic, the data reviewed came from a variety of regions with differing availability of neurological expertise, testing techniques, disease surveillance, and access to healthcare which led to a lack of standardization of some results. Although we attempted to be exhaustive in reporting the range of infections, due to the broad scope of our task and high number of manuscripts available on some pathogens, we highlight the most critical information for neurologists in the field.
Future efforts at prevention of the spread of these pathogens—and the epidemics and pandemics that they are capable of causing—should span investments in science, education, clinical care, and public health. Directions for the medical community could include increased funding for vaccine research as well as strengthening medical facilities with the ability to address and evaluate disease surges where and when they arise. Pragmatic efforts include public education on the best methods to prevent the spread of disease including face masks, hand hygiene, quarantine, and proper handling of biospecimens, burials, and autopsies. Effective and unified population-level messaging could decrease morbidity and mortality of the individual patient and prevent further spread while reducing the burden on healthcare systems (256–261).
While the diseases on the WHO list have the potential to cause widespread harm, many of them fortunately have distinct clinical and diagnostic features that make it possible to identify outbreaks when they are suspected, and early interventions by providers can significantly impact the trajectory of a pathogen's spread. This review has outlined the neurological features of all of the currently listed diseases with epidemic and pandemic potential by the WHO with the goal of assisting clinicians who see neurological conditions with the prompt identification of diseases. This list may evolve and expand over time as new pathogens are discovered, pathogenicity changes, or hosts and conditions for spread continue to change. One lesson that is crucial to glean from the high morbidity and mortality of the COVID-19 pandemic is that the interconnectedness of the modern world makes the threat of any of these 20 diseases real and imminent, and the medical community must be vigilant in their surveillance to ensure they do not spread.
FM was additionally involved with project supervision. All authors contributed to the manuscript conception, drafting, and editing for critical content.
The authors thank the Bev Mahfuz fund, supporting neurology resident research in the Massachusetts General Brigham Neurology Residency Program, for funding the open access publication charges of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. World Health Organization. (2020). Available online at: https://www.who.int/emergencies/diseases/en/
2. Deeba IM, Hasan MM, Al Mosabbir A, Siam MHB, Islam MS, Raheem E, et al. Manifestations of Atypical Symptoms of Chikungunya during the Dhaka Outbreak (2017) in Bangladesh. Am J Trop Med Hyg. (2019) 100:1545–8. doi: 10.4269/ajtmh.19-0122
3. Gutierrez-Saravia E, Gutierrez CE. Chikungunya virus in the Caribbean: a threat for all of the Americas. J Pediatric Infect Dis Soc. (2015) 4:1–3. doi: 10.1093/jpids/piv002
4. Pinheiro TJ, Guimarães LF, Silva MTT, Soares CN. Neurological manifestations of Chikungunya and Zika infections. Arq Neuropsiquiatr. (2016) 74:937–43. doi: 10.1590/0004-282X20160138
5. Mehta R, Gerardin P, de Brito CAA, Soares CN, Ferreira MLB, Solomon T. The neurological complications of chikungunya virus: a systematic review. Rev Med Virol. (2018) 28:e1978. doi: 10.1002/rmv.1978
6. Chandak NH, Kashyap RS, Kabra D, Karandikar P, Saha SS, Morey SH, et al. Neurological complications of Chikungunya virus infection. Neurol India. (2009) 57:177–80. doi: 10.4103/0028-3886.51289
7. Crosby L, Perreau C, Madeux B, Cossic J, Armand C, Herrmann-Storke C, et al. Severe manifestations of chikungunya virus in critically ill patients during the 2013–2014 Caribbean outbreak. Int J Infect Dis. (2016) 48:78–80. doi: 10.1016/j.ijid.2016.05.010
8. Lemant J, Boisson V, Winer A, Thibault L, André H, Tixier F, et al. Serious acute chikungunya virus infection requiring intensive care during the Reunion Island outbreak in 2005–2006. Crit Care Med. (2008) 36:2536–41. doi: 10.1097/CCM.0b013e318183f2d2
9. Panning M, Grywna K, Van Esbroeck M, Emmerich P, Drosten C. Chikungunya fever in travelers returning to Europe from the Indian Ocean region, 2006. Emerg Infect Dis. (2008) 14:416. doi: 10.3201/eid1403.070906
10. Edwards T, Signor CC, Williams C, Larcher C, Espinel M, Theaker J, et al. Analytical and clinical performance of a Chikungunya qRT-PCR for Central and South America. Diagn Microbiol Infect Dis. (2017) 89:35–9. doi: 10.1016/j.diagmicrobio.2017.06.001
11. Arpino C, Curatolo P, Rezza G. Chikungunya and the nervous system: what we do and do not know. Rev Med Virol. (2009) 19:121–9. doi: 10.1002/rmv.606
12. World Health Organization. Cholera vaccines: WHO position paper. Wkly Epidemiol Rec. (2010) 85:117–28.
13. Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. (2011) 477:462–5. doi: 10.1038/nature10392
14. Morris JG Jr, Black RE. Cholera and other vibrioses in the United States. N Engl J Med. (1985) 312:343–50. doi: 10.1056/NEJM198502073120604
15. Naidu LS, Bakerman PR, Saubolle MA, Lewis K. Vibrio cholerae non-0:1 meningitis in an infant. Pediatr Infect Dis J. (1993) 12:879–81. doi: 10.1097/00006454-199310000-00016
16. Bose A, Philip JK, Jesudason M. Neonatal septicemia caused by Vibrio cholerae O:139. Pediatr Infect Dis J. (2000) 19:166. doi: 10.1097/00006454-200002000-00017
17. Kerketta JA, Paul AC, Kirubakaran VBC, Jesudason MV, Moses PD. Non-01 Vibrio cholerae septicemia and meningitis in a neonate. Indian J Pediatr. (2002) 69:909–10. doi: 10.1007/BF02723720
18. Hao Y, Wang Y, Bi Z, Sun B, Jin Y, Bai Y, et al. A case of non-O1/non-O139 Vibrio cholerae septicemia and meningitis in a neonate. Int J Infect Dis. (2015) 35:117–9. doi: 10.1016/j.ijid.2015.05.004
19. Liou CW, Lui CC, Cheng MH. A case of intracerebral abscess caused by non-O1 Vibrio cholerae. Eur J Clin Microbiol Infect Dis. (2001) 20:678–80. doi: 10.1007/s100960100569
20. Dick MH, Guillerm M, Moussy F, Chaignat C-L. Review of two decades of cholera diagnostics–how far have we really come? PLoS Negl Trop Dis. (2012) 6:e1845. doi: 10.1371/journal.pntd.0001845
21. Rubin LG, Altman J, Epple LK, Yolken RH. Vibrio cholerae meningitis in a neonate. Journal of Pediatrics. (1981) 98:940–2.
22. Naidu LS, Bakerman PR, Saubolle MA, Lewis K. Vibrio cholerae non-0: 1 meningitis in an infant. Pediatr Infect Dis J. (1993) 12:879–80.
23. Ismail E, Shafik M, Al-Mutairi G. A case of non-O: 1 Vibrio cholerae septicemia with meningitis, cerebral abscess and unilateral hydrocephalus in a preterm baby. Eur J Clin Microbiol Infect Dis. (2001) 20:598. doi: 10.1007/s100960100553
24. Shayan S, Bokaean M, Shahrivar MR, Chinikar S. Crimean-Congo hemorrhagic fever. Lab Med. (2015) 46:180–9. doi: 10.1309/LMN1P2FRZ7BKZSCO
25. Bente DA, Forrester NL, Watts DM, McAuley AJ, Whitehouse CA, Bray M. Crimean-Congo hemorrhagic fever: history, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antiviral Res. (2013) 100:159–89. doi: 10.1016/j.antiviral.2013.07.006
26. Estrada-Pena A, Ayllon N, de la Fuente J. Impact of climate trends on tick-borne pathogen transmission. Front Physiol. (2012) 3:64. doi: 10.3389/fphys.2012.00064
27. Hawman DW, Feldmann H. Recent advances in understanding Crimean-Congo hemorrhagic fever virus. F1000Res. (2018) 7:F1000 Faculty Rev-1715. doi: 10.12688/f1000research.16189.1
28. Yilmaz GR, Buzgan T, Irmak H, Safran A, Uzun R, Cevik MA, et al. The epidemiology of Crimean-Congo hemorrhagic fever in Turkey, 2002-2007. Int J Infect Dis. (2009) 13:380–6. doi: 10.1016/j.ijid.2008.07.021
29. Swanepoel R, Shepherd AJ, Leman PA, Shepherd SP, Miller GB. A common-source outbreak of Crimean-Congo haemorrhagic fever on a dairy farm. S Afr Med J. (1985) 68:635–7.
30. Alavi-Naini R, Moghtaderi A, Metanat M. An unusual intracerebral hemorrhage. Can J Infect Dis Med Microbiol. (2004) 15:175–7. doi: 10.1155/2004/769260
31. Kleib AS, Salihy SM, Ghaber SM, Sidiel BW, Sidiya KC, Bettar ES. Crimean-Congo hemorrhagic fever with acute subdural Hematoma, Mauritania, 2012. Emerg Infect Dis. (2016) 22:1305–6. doi: 10.3201/eid2207.151782
32. Ozsoy S, Gokmen A, Ozdemir M, Akduman B, Korkusuz I, Javan GT. Medical examiners and Crimean-Congo hemorrhagic fever contamination risk. J For Leg Med. (2015) 36:32–6. doi: 10.1016/j.jflm.2015.08.010
33. Conger NG, Paolino KM, Osborn EC, Rusnak JM, Gunther S, Pool J, et al. Health care response to CCHF in US soldier and nosocomial transmission to health care providers, Germany, 2009. Emerg Infect Dis. (2015) 21:23–31. doi: 10.3201/eid2101.141413
34. Oztoprak B, Oztoprak I, Engin A. Is the brain spared in Crimean-Congo haemorrhagic fever? An MR-SWI study to reveal CNS involvement. Eur Radiol. (2018) 28:3893–901. doi: 10.1007/s00330-018-5310-9
35. Spengler JR, Kelly Keating M, McElroy AK, Zivcec M, Coleman-McCray JD, Harmon JR, et al. Crimean-Congo hemorrhagic fever in humanized mice reveals glial cells as primary targets of neurological infection. J Infect Dis. (2017) 216:1386–97. doi: 10.1093/infdis/jix215
36. Duh D, Saksida A, Petrovec M, Dedushaj I, Avšič-Županc T. Novel one-step real-time RT-PCR assay for rapid and specific diagnosis of Crimean-Congo hemorrhagic fever encountered in the Balkans. J Virol Methods. (2006) 133:175–9. doi: 10.1016/j.jviromet.2005.11.006
37. Atkinson B, Chamberlain J, Logue CH, Cook N, Bruce C, Dowall SD, et al. Development of a real-time RT-PCR assay for the detection of Crimean-Congo hemorrhagic fever virus. VectorBorne Zoonot Dis. (2012) 12:786–93. doi: 10.1089/vbz.2011.0770
38. Franz DR, Jahrling PB, McClain DJ, Hoover DL, Byrne WR, Pavlin JA, et al. Clinical recognition and management of patients exposed to biological warfare agents. Clin Lab Med. (2001) 21:435–73. doi: 10.1016/S0272-2712(18)30018-0
39. Billioux BJ. Neurological complications and sequelae of Ebola virus disease. Curr Infect Dis Rep. (2017) 19:19. doi: 10.1007/s11908-017-0573-x
40. Baseler L, Chertow DS, Johnson KM, Feldmann H, Morens DM. The pathogenesis of Ebola virus disease. Annu Rev Pathol. (2017) 12:387–418. doi: 10.1146/annurev-pathol-052016-100506
41. Holmes EC, Dudas G, Rambaut A, Andersen KG. The evolution of Ebola virus: insights from the 2013-2016 epidemic. Nature. (2016) 538:193–200. doi: 10.1038/nature19790
42. World Health Organization (2020). Available online at: https://www.who.int/news-room/fact-sheets/detail/ebola-virus-disease
43. Billioux BJ, Smith B, Nath A. Neurological complications of Ebola virus infection. Neurotherapeutics. (2016) 13:461–70. doi: 10.1007/s13311-016-0457-z
44. de Greslan T, Billhot M, Rousseau C, Mac Nab C, Karkowski L, Cournac JM, et al. Ebola virus-related encephalitis. Clin Infect Dis. (2016) 63:1076–8. doi: 10.1093/cid/ciw469
45. Sagui E, Janvier F, Baize S, Foissaud V, Koulibaly F, Savini H, et al. Severe Ebola virus infection with encephalopathy: evidence for direct virus involvement. Clin Infect Dis. (2015) 61:1627–8. doi: 10.1093/cid/civ606
46. Clark DV, Kibuuka H, Millard M, Wakabi S, Lukwago L, Taylor A, et al. Long-term sequelae after Ebola virus disease in Bundibugyo, Uganda: a retrospective cohort study. Lancet Infect Dis. (2015) 15:905–12. doi: 10.1016/S1473-3099(15)70152-0
47. Prevail III Study Group, Sneller MC, Reilly C, Badio M, Bishop RJ, Eghrari AO, et al. A longitudinal study of Ebola sequelae in Liberia. N Engl J Med. (2019) 380:924–34. doi: 10.1056/NEJMoa1805435
48. Varkey JB, Shantha JG, Crozier I, Kraft CS, Lyon GM, Mehta AK, et al. Persistence of Ebola virus in ocular fluid during convalescence. N Engl J Med. (2015) 372:2423–7. doi: 10.1056/NEJMoa1500306
49. Maes P, Amarasinghe GK, Ayllón MA, Basler CF, Bavari S, Blasdell KR, et al. Taxonomy of the order Mononegavirales: second update 2018. Arch Virol. (2019) 164:1233–44. doi: 10.1007/s00705-018-04126-4
50. Halpin K, Young PL, Field H, Mackenzie J. Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. J Gen Virol. (2000) 81:1927–32. doi: 10.1099/0022-1317-81-8-1927
51. Mahalingam S, Herrero LJ, Playford EG, Spann K, Herring B, Rolph MS, et al. Hendra virus: an emerging paramyxovirus in Australia. Lancet Infect Dis. (2012) 12:799–807. doi: 10.1016/S1473-3099(12)70158-5
52. Mire CE, Satterfield BA, Geisbert TW. Pathogenesis of Hendra virus in humans. Hum Emerg Re-Emerg Infect. (2015) 2:207–26. doi: 10.1002/9781118644843.ch11
53. Dawes BE, Freiberg AN. Henipavirus infection of the central nervous system. Pathog Dis. (2019) 77:ftz023. doi: 10.1093/femspd/ftz023
54. Wong KT, Robertson T, Ong BB, Chong JW, Yaiw KC, Wang LF, et al. Human Hendra virus infection causes acute and relapsing encephalitis. Neuropathol Appl Neurobiol. (2009) 35:296–305. doi: 10.1111/j.1365-2990.2008.00991.x
55. Ong KC, Wong KT. Henipavirus encephalitis: recent developments and advances. Brain Pathol. (2015) 25:605–13. doi: 10.1111/bpa.12278
56. Plowright RK, Eby P, Hudson PJ, Smith IL, Westcott D, Bryden WL, et al. Ecological dynamics of emerging bat virus spillover. Proc Biol Sci. (2015) 282:20142124. doi: 10.1098/rspb.2014.2124
57. Choisy M, Rohani P. Harvesting can increase severity of wildlife disease epidemics. Proc R Soc B Biol Sci. (2006) 273:2025–34. doi: 10.1098/rspb.2006.3554
58. Kessler MK, Becker DJ, Peel AJ, Justice NV, Lunn T, Crowley DE, et al. Changing resource landscapes and spillover of henipaviruses. Ann N Y Acad Sci. (2018) 1429:78. doi: 10.1111/nyas.13910
59. World Health Organization (2020). Available online at: http://www.euro.who.int/en/health-topics/communicable-diseases/influenza/data-and-statistics/virology-of-human-influenza
60. Treanor J. Influenza vaccine–outmaneuvering antigenic shift and drift. N Engl J Med. (2004) 350:218–20. doi: 10.1056/NEJMp038238
61. Ekstrand JJ. Neurologic complications of influenza. Semin Pediatr Neurol. (2012) 19:96–100. doi: 10.1016/j.spen.2012.02.004
62. Fujimoto S, Kobayashi M, Uemura O, Iwasa M, Ando T, Katoh T, et al. PCR on cerebrospinal fluid to show influenza-associated acute encephalopathy or encephalitis. Lancet. (1998) 352:873–5. doi: 10.1016/S0140-6736(98)12449-2
63. Ito Y, Ichiyama T, Kimura H, Shibata M, Ishiwada N, Kuroki H, et al. Detection of influenza virus RNA by reverse transcription-PCR and proinflammatory cytokines in influenza-virus-associated encephalopathy. J Med Virol. (1999) 58:420–5. doi: 10.1002/(SICI)1096-9071(199908)58:4<420::AID-JMV16>3.0.CO;2-T
64. Mizuguchi M, Yamanouchi H, Ichiyama T, Shiomi M. Acute encephalopathy associated with influenza and other viral infections. Acta Neurol Scand Suppl. (2007) 186:45–56. doi: 10.1111/j.1600-0404.2007.00809.x
65. Tsai JP, Baker AJ. Influenza-associated neurological complications. Neurocrit Care. (2013) 18:118–30. doi: 10.1007/s12028-012-9796-8
66. Pugliese A, Beltramo T, Torre D. Reye's and Reye's-like syndromes. Cell Biochem Funct. (2008) 26:741–6. doi: 10.1002/cbf.1465
68. Dauvilliers Y, Montplaisir J, Cochen V, Desautels A, Einen M, Lin L, et al. Post-H1N1 Narcolepsy-Cataplexy. Oxford: Oxford University Press (2010).
69. Sweden M. Occurrence of Narcolepsy With Cataplexy Among Children and Adolescents in Relation to the H1N1 Pandemic and Pandemrix Vaccinations—Results of a Case Inventory Study by the MPA in Sweden During 2009-2010. Uppsala: Lakemedelsverket (2011).
70. Miller E, Andrews N, Stellitano L, Stowe J, Winstone AM, Shneerson J, et al. Risk of narcolepsy in children and young people receiving AS03 adjuvanted pandemic A/H1N1 2009 influenza vaccine: retrospective analysis. Bmj. (2013) 346:f794. doi: 10.1136/bmj.f794
71. Oberle D, Drechsel-Bäuerle U, Schmidtmann I, Mayer G, Keller-Stanislawski B. Incidence of narcolepsy in Germany. Sleep. (2015) 38:1619–28. doi: 10.5665/sleep.5060
72. Han F, Lin L, Warby SC, Faraco J, Li J, Dong SX, et al. Narcolepsy onset is seasonal and increased following the 2009 H1N1 pandemic in China. Ann Neurol. (2011) 70:410–7. doi: 10.1002/ana.22587
73. Verstraeten T, Cohet C, Dos Santos G, Ferreira GL, Bollaerts K, Bauchau V, et al. Pandemrix™ and narcolepsy: a critical appraisal of the observational studies. Hum Vaccines Immunother. (2016) 12:187–93. doi: 10.1080/21645515.2015.1068486
74. Sarkanen T, Alakuijala A, Julkunen I, Partinen M. Narcolepsy associated with Pandemrix vaccine. Curr Neurol Neurosci Rep. (2018) 18:43. doi: 10.1007/s11910-018-0851-5
75. Partinen M, Kornum BR, Plazzi G, Jennum P, Julkunen I, Vaarala O. Narcolepsy as an autoimmune disease: the role of H1N1 infection and vaccination. Lancet Neurol. (2014) 13:600–13. doi: 10.1016/S1474-4422(14)70075-4
76. Lehmann HC, Hartung HP, Kieseier BC, Hughes RA. Guillain-Barre syndrome after exposure to influenza virus. Lancet Infect Dis. (2010) 10:643–51. doi: 10.1016/S1473-3099(10)70140-7
77. Okumura A, Mizuguchi M, Kidokoro H, Tanaka M, Abe S, Hosoya M, et al. Outcome of acute necrotizing encephalopathy in relation to treatment with corticosteroids and gammaglobulin. Brain Dev. (2009) 31:221–7. doi: 10.1016/j.braindev.2008.03.005
78. Ohtsuki N, Kimura S, Nezu A, Aihara Y. Effects of mild hypothermia and steroid pulse combination therapy on acute encephalopathy associated with influenza virus infection: report of two cases. Brain Dev. (2000) 32:318. doi: 10.1155/2020/6616805
79. Yoganathan S, Sudhakar SV, James EJ, Thomas MM. Acute necrotising encephalopathy in a child with H1N1 influenza infection: a clinicoradiological diagnosis and follow-up. Bmj Case Rep. (2016) 2016:bcr2015213429. doi: 10.1136/bcr-2015-213429
80. Alsolami A, Shiley K. Successful Treatment of Influenza-Associated Acute Necrotizing Encephalitis in an Adult Using High-Dose Oseltamivir and Methylprednisolone: Case Report and Literature Review. New York, NY: Open Forum Infectious Diseases; Oxford University Press US (2017). p. ofx145.
81. Hatachi T, Michihata N, Takeuchi M, Matsui H, Fushimi K, Yasunaga H. Early steroid pulse therapy among children with influenza virus-associated encephalopathy. J Intens Care. (2020) 8:1–8. doi: 10.1186/s40560-020-00479-8
82. Meijer WJ, Linn FH, Wensing AM, Leavis HL, van Riel D, GeurtsvanKessel CH, et al. Acute influenza virus-associated encephalitis and encephalopathy in adults: a challenging diagnosis. JMM Case Rep. (2016) 3:e005076. doi: 10.1099/jmmcr.0.005076
83. Khan SH, Goba A, Chu M, Roth C, Healing T, Marx A, et al. New opportunities for field research on the pathogenesis and treatment of Lassa fever. Antiviral Res. (2008) 78:103–15. doi: 10.1016/j.antiviral.2007.11.003
84. Okokhere PO, Ibekwe TS, Akpede GO. Sensorineural hearing loss in Lassa fever: two case reports. J Med Case Rep. (2009) 3:36. doi: 10.1186/1752-1947-3-36
86. Macher AM, Wolfe MS. Historical Lassa fever reports and 30-year clinical update. Emerg Infect Dis. (2006) 12:835–7. doi: 10.3201/eid1205.050052
87. Okokhere PO, Bankole IA, Akpede GO. Central nervous system manifestations of lassa fever in Nigeria and the effect on mortality. J Neurol Sci. (2013) 333:e604. doi: 10.1016/j.jns.2013.07.2107
88. Solbrig MV. Lassa virus and central nervous system diseases. In: The Arenaviridae. (1993). p. 325–30.
89. Ölschläger S, Lelke M, Emmerich P, Panning M, Drosten C, Hass M, et al. Improved detection of Lassa virus by reverse transcription-PCR targeting the 5′ region of S RNA. J Clin Microbiol. (2010) 48:2009–13. doi: 10.1128/JCM.02351-09
90. Wiley MR, Fakoli L, Letizia AG, Welch SR, Ladner JT, Prieto K, et al. Lassa virus circulating in Liberia: a retrospective genomic characterisation. Lancet Infect Dis. (2019) 19:1371–8. doi: 10.1016/S1473-3099(19)30486-4
91. Mehedi M, Groseth A, Feldmann H, Ebihara H. Clinical aspects of Marburg hemorrhagic fever. Future Virol. (2011) 6:1091–106. doi: 10.2217/fvl.11.79
92. Rubin R. HHS Funds development of vaccine against marburg virus. JAMA. (2019) 321:1557. doi: 10.1001/jama.2019.4035
93. Centers for Disease Control Prevention. Outbreak of Marburg virus hemorrhagic fever–Angola, October 1, 2004-March 29, 2005. MMWR Morb Mortal Wkly Rep. (2005) 54:308–9.
94. Shifflett K, Marzi A. Marburg virus pathogenesis - differences and similarities in humans and animal models. Virol J. (2019) 16:165. doi: 10.1186/s12985-019-1272-z
95. Wang Y, Zhang X, Wei H. Laboratory detection and diagnosis of filoviruses. Virol Sin. (2011) 26:73. doi: 10.1007/s12250-011-3186-9
96. Stephens DS, Greenwood B, Brandtzaeg P. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet. (2007) 369:2196–210. doi: 10.1016/S0140-6736(07)61016-2
97. Serruto D, Bottomley MJ, Ram S, Giuliani MM, Rappuoli R. The new multicomponent vaccine against meningococcal serogroup B, 4CMenB: immunological, functional and structural characterization of the antigens. Vaccine. (2012) 30:B87–97. doi: 10.1016/j.vaccine.2012.01.033
98. Read RC, Baxter D, Chadwick DR, Faust SN, Finn A, Gordon SB, et al. Effect of a quadrivalent meningococcal ACWY glycoconjugate or a serogroup B meningococcal vaccine on meningococcal carriage: an observer-blind, phase 3 randomised clinical trial. Lancet. (2014) 384:2123–31. doi: 10.1016/S0140-6736(14)60842-4
99. Folaranmi T, Rubin L, Martin SW, Patel M, MacNeil JR. Use of serogroup B meningococcal vaccines in persons aged≥ 10 years at increased risk for serogroup B meningococcal disease: recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb Mortal Wkly Rep. (2015) 64:608. doi: 10.15585/mmwr.mm6441a3
100. Thompson MJ, Ninis N, Perera R, Mayon-White R, Phillips C, Bailey L, et al. Clinical recognition of meningococcal disease in children and adolescents. Lancet. (2006) 367:397–403. doi: 10.1016/S0140-6736(06)67932-4
101. Lucas MJ, Brouwer MC, van de Beek D. Neurological sequelae of bacterial meningitis. J Infect. (2016) 73:18–27. doi: 10.1016/j.jinf.2016.04.009
102. Schut ES, Lucas MJ, Brouwer MC, Vergouwen MD, van der Ende A, van de Beek D. Cerebral infarction in adults with bacterial meningitis Neurocrit Care. (2012) 16:421–7. doi: 10.1007/s12028-011-9634-4
103. Koomen I, Grobbee DE, Jennekens-Schinkel A, Roord JJ, van Furth AM. Parental perception of educational, behavioural and general health problems in school-age survivors of bacterial meningitis. Acta Paediatr. (2003) 92:177–85. doi: 10.1111/j.1651-2227.2003.tb00523.x
104. Roed C, Omland LH, Skinhoj P, Rothman KJ, Sorensen HT, Obel N. Educational achievement and economic self-sufficiency in adults after childhood bacterial meningitis. JAMA. (2013) 309:1714–21. doi: 10.1001/jama.2013.3792
105. Hoogman M, van de Beek D, Weisfelt M, de Gans J, Schmand B. Cognitive outcome in adults after bacterial meningitis. J Neurol Neurosurg Psychiatry. (2007) 78:1092–6. doi: 10.1136/jnnp.2006.110023
106. Bryant PA, Li HY, Zaia A, Griffith J, Hogg G, Curtis N, et al. Prospective study of a real-time PCR that is highly sensitive, specific, and clinically useful for diagnosis of meningococcal disease in children. J Clin Microbiol. (2004) 42:2919–25. doi: 10.1128/JCM.42.7.2919-2925.2004
107. Wu HM, Cordeiro SM, Harcourt BH, Maria da Gloria SC, Azevedo J, Oliveira TQ, et al. Accuracy of real-time PCR, Gram stain and culture for Streptococcus pneumoniae, Neisseria meningitidis and Haemophilus influenzae meningitis diagnosis. BMC Infect Dis. (2013) 13:26. doi: 10.1186/1471-2334-13-26
108. Kanegaye JT, Soliemanzadeh P, Bradley JS. Lumbar puncture in pediatric bacterial meningitis: defining the time interval for recovery of cerebrospinal fluid pathogens after parenteral antibiotic pretreatment. Pediatrics. (2001) 108:1169–74.
109. Brouwer MC, McIntyre P, de Gans J, Prasad K, van de Beek D. Corticosteroids for acute bacterial meningitis. Cochrane Database Syst Rev. (2015) CD004405. doi: 10.1002/14651858.CD004405.pub3
110. Goldacre MJ, Roberts SE, Yeates D. Case fatality rates for meningococcal disease in an English population, 1963-98: database study. BMJ. (2003) 327:596–7. doi: 10.1136/bmj.327.7415.596
111. Heckenberg SG, de Gans J, Brouwer MC, Weisfelt M, Piet JR, Spanjaard L, et al. Clinical features, outcome, and meningococcal genotype in 258 adults with meningococcal meningitis: a prospective cohort study. Medicine. (2008) 87:185–92. doi: 10.1097/MD.0b013e318180a6b4
112. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. (2012) 367:1814–20. doi: 10.1056/NEJMoa1211721
113. Perera RA, Wang P, Gomaa MR, El-Shesheny R, Kandeil A, Bagato O, et al. Seroepidemiology for MERS coronavirus using microneutralisation and pseudoparticle virus neutralisation assays reveal a high prevalence of antibody in dromedary camels in Egypt. Euro Surveill. (2013) 18: 20574. doi: 10.2807/1560-7917.ES2013.18.36.20574
114. Reusken CB, Haagmans BL, Muller MA, Gutierrez C, Godeke GJ, Meyer B, et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect Dis. (2013) 13:859–66. doi: 10.1016/S1473-3099(13)70164-6
115. Azhar EI, El-Kafrawy SA, Farraj SA, Hassan AM, Al-Saeed MS, Hashem AM, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med. (2014) 370:2499–505. doi: 10.1056/NEJMoa1401505
116. Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, Epstein JH, et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg Infect Dis. (2013) 19:1819. doi: 10.3201/eid1911.131172
117. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. (2019) 17:181–92. doi: 10.1038/s41579-018-0118-9
118. Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. (2013) 495:251–4. doi: 10.1038/nature12005
119. Lu L, Zhong W, Bian Z, Li Z, Zhang K, Liang B, et al. (2020). A comparison of mortality-related risk factors of COVID-19, SARS, and MERS: a systematic review and meta-analysis. J Infect. (2020) 81:e18–25. doi: 10.1016/j.jinf.2020.07.002
120. World Health Organization (2020). Available online at: https://www.who.int/emergencies/mers-cov/en/
121. Matsuyama R, Nishiura H, Kutsuna S, Hayakawa K, Ohmagari N. Clinical determinants of the severity of Middle East respiratory syndrome (MERS): a systematic review and meta-analysis. BMC Public Health. (2016) 16:1203. doi: 10.1186/s12889-016-3881-4
122. Park JE, Jung S, Kim A, Park JE. MERS transmission and risk factors: a systematic review. BMC Public Health. (2018) 18:574. doi: 10.1186/s12889-018-5484-8
123. Saad M, Omrani AS, Baig K, Bahloul A, Elzein F, Matin MA, et al. Clinical aspects and outcomes of 70 patients with Middle East respiratory syndrome coronavirus infection: a single-center experience in Saudi Arabia. Int J Infect Dis. (2014) 29:301–6. doi: 10.1016/j.ijid.2014.09.003
124. Arabi YM, Harthi A, Hussein J, Bouchama A, Johani S, Hajeer AH, et al. Severe neurologic syndrome associated with Middle East respiratory syndrome corona virus (MERS-CoV). Infection. (2015) 43:495–501. doi: 10.1007/s15010-015-0720-y
125. Kim JE, Heo JH, Kim HO, Song SH, Park SS, Park TH, et al. Neurological Complications during Treatment of Middle East Respiratory Syndrome. J Clin Neurol. (2017) 13:227–33. doi: 10.3988/jcn.2017.13.3.227
126. Algahtani H, Subahi A, Shirah B. Neurological complications of middle East Respiratory Syndrome coronavirus: a report of two cases and review of the literature. Case Rep Neurol Med. (2016) 2016:3502683. doi: 10.1155/2016/3502683
127. Fine PE, Jezek Z, Grab B, Dixon H. The transmission potential of monkeypox virus in human populations. Int J Epidemiol. (1988) 17:643–50.
128. Heymann DL, Szczeniowski M, Esteves K. Re-emergence of monkeypox in Africa: a review of the past six years. Br Med Bull. (1998) 54:693–702.
129. Jezek Z, Szczeniowski M, Paluku KM, Mutombo M. Human monkeypox: clinical features of 282 patients. J Infect Dis. (1987) 156:293–8.
130. Sejvar JJ, Chowdary Y, Schomogyi M, Stevens J, Patel J, Karem K, et al. Human monkeypox infection: a family cluster in the midwestern United States. J Infect Dis. (2004) 190:1833–40. doi: 10.1086/425039
131. Broder CC, Weir DL, Reid PA. Hendra virus and Nipah virus animal vaccines. Vaccine. (2016) 34:3525–34. doi: 10.1016/j.vaccine.2016.03.075
132. Singh RK, Dhama K, Chakraborty S, Tiwari R, Natesan S, Khandia R, et al. Nipah virus: epidemiology, pathology, immunobiology and advances in diagnosis, vaccine designing and control strategies - a comprehensive review. Vet Q. (2019) 39:26–55. doi: 10.1080/01652176.2019.1580827
133. Centers for Disease Control and Prevention. Update: outbreak of Nipah virus–Malaysia and Singapore, 1999. MMWR Morb Mortal Wkly Rep. (1999) 48:335.
134. Iehle C, Razafitrimo G, Razainirina J, Andriaholinirina N, Goodman SM, Faure C, et al. Henipavirus and Tioman virus antibodies in pteropodid bats, Madagascar. Emerg Infect Dis. (2007) 13:159–61. doi: 10.3201/eid1301.060791
135. Sun B, Jia L, Liang B, Chen Q, Liu D. Phylogeography, transmission, and viral proteins of nipah virus. Virol Sin. (2018) 33:385–93. doi: 10.1007/s12250-018-0050-1
136. Mackenzie JS, Chua KB, Daniels PW, Eaton BT, Field HE, Hall RA, et al. Emerging viral diseases of Southeast Asia and the Western Pacific. Emerg Infect Dis. (2001) 7(3 Suppl.):497–504. doi: 10.3201/eid0707.017703
137. Chua KB, Goh KJ, Wong KT, Kamarulzaman A, Tan PS, Ksiazek TG, et al. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet. (1999) 354:1257–9.
138. Hossain MJ, Gurley ES, Montgomery JM, Bell M, Carroll DS, Hsu VP, et al. Clinical presentation of nipah virus infection in Bangladesh. Clin Infect Dis. (2008) 46:977–84. doi: 10.1086/529147
139. Ang BSP, Lim TCC, Wang L. Nipah virus infection. J Clin Microbiol. (2018) 56. doi: 10.1128/JCM.01875-17
140. Goh KJ, Tan CT, Chew NK, Tan PS, Kamarulzaman A, Sarji SA, et al. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N Engl J Med. (2000) 342:1229–35. doi: 10.1056/NEJM200004273421701
141. Arunkumar G, Chandni R, Mourya D, Singh S, Sadanandan R, Sudan P. Nipah investigators people and health study group. Outbreak investigation of Nipah virus disease in Kerala, India. J Infect Dis. (2018). p. 1867–78.
142. Tan CT, Goh KJ, Wong KT, Sarji SA, Chua KB, Chew NK, et al. Relapsed and late-onset Nipah encephalitis. Ann Neurol. (2002) 51:703–8. doi: 10.1002/ana.10212
143. Lim CC, Sitoh YY, Hui F, Lee KE, Ang BS, Lim E, et al. Nipah viral encephalitis or Japanese encephalitis? MR findings in a new zoonotic disease. AJNR Am J Neuroradiol. (2000) 21:455–61.
144. Wong KT, Shieh WJ, Kumar S, Norain K, Abdullah W, Guarner J, et al. Nipah virus infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am J Pathol. (2002) 161:2153–67. doi: 10.1016/S0002-9440(10)64493-8
145. Daniels P, Ksiazek T, Eaton BT. Laboratory diagnosis of Nipahand Hendra virus infections. Microbes Infect. (2001) 3:289–95. doi: 10.1016/S1286-4579(01)01382-X
146. Chong HT, Kamarulzaman A, Tan CT, Goh KJ, Thayaparan T, Kunjapan SR, et al. Treatment of acute Nipah encephalitis with ribavirin. Ann Neurol. (2001) 49:810–3. doi: 10.1002/ana.1062
147. Pallister J, Middleton D, Crameri G, Yamada M, Klein R, Hancock TJ, et al. Chloroquine administration does not prevent Nipah virus infection and disease in ferrets. J Virol. (2009) 83:11979–82. doi: 10.1128/JVI.01847-09
148. Freiberg AN, Worthy MN, Lee B, Holbrook MR. Combined chloroquine and ribavirin treatment does not prevent death in a hamster model of Nipah and Hendra virus infection. J Gen Virol. (2010) 91:765–72. doi: 10.1099/vir.0.017269-0
149. Soman Pillai V, Krishna G, Valiya Veettil M. Nipah virus: past outbreaks and future containment. Viruses. (2020) 12:465. doi: 10.3390/v12040465
150. Aljofan M. Hendra and Nipah infection: emerging paramyxoviruses. Virus Res. (2013) 177:119–26. doi: 10.1016/j.virusres.2013.08.002
151. Tan K, Sarji SA, Tan C-T, Abdullah B, Chong H, Thayaparan T, et al. Patients with asymptomatic Nipah virus infection may have abnormal cerebral MR imaging. Neurol J Southeast Asia. (2000) 5:69–73.
152. Tan CT, Tan KS. Nosocomial transmissibility of Nipah virus. J Infect Dis. (2001) 184:1367. doi: 10.1086/323996
153. Luby SP, Hossain MJ, Gurley ES, Ahmed BN, Banu S, Khan SU, et al. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001-2007. Emerg Infect Dis. (2009) 15:1229–35. doi: 10.3201/eid1508.081237
154. Luby SP. The pandemic potential of Nipah virus. Antiviral Res. (2013) 100:38–43. doi: 10.1016/j.antiviral.2013.07.011
155. Gurley ES, Montgomery JM, Hossain MJ, Bell M, Azad AK, Islam MR, et al. Person-to-person transmission of Nipah virus in a Bangladeshi community. Emerg Infect Dis. (2007) 13:1031–7. doi: 10.3201/eid1307.061128
156. Sazzad HM, Hossain MJ, Gurley ES, Ameen KM, Parveen S, Islam MS, et al. Nipah virus infection outbreak with nosocomial and corpse-to-human transmission, Bangladesh. Emerg Infect Dis. (2013) 19:210–7. doi: 10.3201/eid1902.120971
157. Ching PK, de los Reyes VC, Sucaldito MN, Tayag E, Columna-Vingno AB, Malbas FF Jr, et al. Outbreak of henipavirus infection, Philippines, 2014. Emerg Infect Dis. (2015) 21:328–31. doi: 10.3201/eid2102.141433
158. Thomas B, Chandran P, Lilabi MP, George B, Sivakumar CP, Jayadev VK, et al. Nipah virus infection in Kozhikode, Kerala, South India, in 2018: epidemiology of an outbreak of an emerging disease. Indian J Community Med. (2019) 44:383–7. doi: 10.4103/ijcm.IJCM_198_19
159. Wilson A, Warrier A, Rathish B. Contact tracing: a lesson from the Nipah virus in the time of COVID-19. Trop Doct. (2020) 50:174–5. doi: 10.1177/0049475520928217
160. Román RG, Wang L-F, Lee B, Halpin K, de Wit E, Broder CC, et al. Nipah@ 20: lessons learned from another virus with pandemic potential. mSphere. (2020) 5:e00602-20. doi: 10.1128/mSphere.00602-20
161. World Health Organization. Prioritizing Diseases for Research and Development in Emergency Contexts. Geneva: World Health Organization (2020).
162. Prescott J, DeBuysscher BL, Feldmann F, Gardner DJ, Haddock E, Martellaro C, et al. Single-dose live-attenuated vesicular stomatitis virus-based vaccine protects African green monkeys from Nipah virus disease. Vaccine. (2015) 33:2823–9. doi: 10.1016/j.vaccine.2015.03.089
163. Bird BH, Mazet JAK. Detection of emerging zoonotic pathogens: an integrated one health approach. Annu Rev Anim Biosci. (2018) 6:121–39. doi: 10.1146/annurev-animal-030117-014628
164. Chandni R, Renjith T, Fazal A, Yoosef N, Ashhar C, Thulaseedharan N, et al. Clinical manifestations of nipah virus–infected patients who presented to the emergency department during an outbreak in Kerala State in India, May 2018. Clin Infect Dis. (2020) 71:152–7. doi: 10.1093/cid/ciz789
165. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
166. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
167. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. (2020) 77:683–90. doi: 10.1001/jamaneurol.2020.1127
168. Collantes MEV, Espiritu AI, Sy MCC, Anlacan VMM, Jamora RDG. Neurological manifestations in COVID-19 infection: a systematic review and meta-analysis. Can J Neurol Sci. (2020) 48:66–76. doi: 10.1017/cjn.2020.146
169. Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. (2020) 24:1–8. doi: 10.1101/2020.06.04.135012
170. Conde G, Pájaro LDQ, Marzola IDQ, Villegas YR, Salazar LRM. Neurotropism of SARS-CoV 2: mechanisms and manifestations. J Neurol Sci. (2020) 412:116824. doi: 10.1016/j.jns.2020.116824
171. Zhao H, Shen D, Zhou H, Liu J, Chen S. Guillain-Barre syndrome associated with SARS-CoV-2 infection: causality or coincidence? Lancet Neurol. (2020) 19:383–4. doi: 10.1016/S1474-4422(20)30109-5
172. Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, et al. Guillain-Barre Syndrome Associated with SARS-CoV-2. N Engl J Med. (2020) 382:2574–6. doi: 10.1056/NEJMc2009191
173. Zhao K, Huang J, Dai D, Feng Y, Liu L, Nie S. Acute myelitis after SARS-CoV-2 infection: a case report. MedRxiv. (2020) 5:1–3. doi: 10.1101/2020.03.16.20035105
174. Manzano GS, Woods JK, Amato AA. Covid-19–associated myopathy caused by type I interferonopathy. N Engl J Med. (2020) 383:2389–90. doi: 10.1056/NEJMc2031085
175. Suwanwongse K, Shabarek N. Rhabdomyolysis as a presentation of 2019 novel coronavirus disease. Cureus. (2020) 12:e10123. doi: 10.7759/cureus.7561
176. Fang FC, Naccache SN, Greninger AL. The laboratory diagnosis of COVID-19–frequently-asked questions. Clin Infect Dis. (2020) 71:2996–3001. doi: 10.1093/cid/ciaa742
177. Patel A, Jernigan DB. Initial public health response and interim clinical guidance for the 2019 novel coronavirus outbreak—United States, December 31, 2019–February 4, 2020. Morb Mort Wkly Rep. (2020) 69:140. doi: 10.15585/mmwr.mm6908e1
178. Kremer S, Lersy F, de Sèze J, Ferré J-C, Maamar A, Carsin-Nicol B, et al. Brain MRI findings in severe COVID-19: a retrospective observational study. Radiology. (2020) 297:E242–51. doi: 10.1148/radiol.2020202222
179. Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis. (2020) 94:55–8. doi: 10.1016/j.ijid.2020.03.062
180. Virhammar J, Kumlien E, Fällmar D, Frithiof R, Jackmann S, Sköld MK, et al. Acute necrotizing encephalopathy with SARS-CoV-2 RNA confirmed in cerebrospinal fluid. Neurology. (2020) 95:445–9. doi: 10.1212/WNL.0000000000010250
181. Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. (2020) 382:2268–70. doi: 10.1056/NEJMc2008597
182. Miller EH, Namale VS, Kim C, Dugue R, Waldrop G, Ciryam P, et al. Cerebrospinal Analysis in Patients With COVID-19. Oxford: Open forum infectious diseases; Oxford University Press US (2020). p. ofaa501.
183. Placantonakis DG, Aguero-Rosenfeld M, Flaifel A, Colavito J, Inglima K, Zagzag D, et al. SARS-CoV-2 is not detected in the cerebrospinal fluid of encephalopathic COVID-19 patients. Front Neurol. (2020) 11:587384. doi: 10.3389/fneur.2020.587384
184. Edén A, Kanberg N, Gostner J, Fuchs D, Hagberg L, Andersson L-M, et al. CSF biomarkers in patients with COVID-19 and neurological symptoms: a case series. Neurology. (2020) 96:e294–300. doi: 10.1212/WNL.0000000000010977
185. Prentice MB, Rahalison L. Plague. Lancet. (2007) 369:1196–207. doi: 10.1016/S0140-6736(07)60566-2
186. Butler T. Yersinia infections: centennial of the discovery of the plague bacillus. Clin Infect Dis. (1994) 19:655–61; quiz 62–3.
187. Burton E, Hennessey RSF. An unusual case of plague with meningitis. East Afr Med J. (1940) 17:554–7.
189. Becker TM, Poland JD, Quan TJ, White ME, Mann JM, Barnes AM. Plague meningitis–a retrospective analysis of cases reported in the United States, 1970-1979. West J Med. (1987) 147:554–7.
190. Crook LD, Tempest B. Plague: a clinical review of 27 cases. Arch Intern Med. (1992) 152:1253–6.
192. Ikegami T, Makino S. The pathogenesis of Rift Valley fever. Viruses. (2011) 3:493–519. doi: 10.3390/v3050493
193. Dar O, McIntyre S, Hogarth S, Heymann D. Rift Valley fever and a new paradigm of research and development for zoonotic disease control. Emerg Infect Dis. (2013) 19:189–93. doi: 10.3201/eid1902.120941
194. Treble A. Chemical and Biological Weapons: Possession and Programs Past and Present. Monterey, CA: Center for Nonproliferation Studies (2002).
195. Jouan A, Coulibaly I, Adam F, Philippe B, Riou O, Leguenno B, et al. Analytical study of a Rift Valley fever epidemic. Res Virol. (1989) 140:175–86.
196. Madani TA, Al-Mazrou YY, Al-Jeffri MH, Mishkhas AA, Al-Rabeah AM, Turkistani AM, et al. Rift Valley fever epidemic in Saudi Arabia: epidemiological, clinical, and laboratory characteristics. Clin Infect Dis. (2003) 37:1084–92. doi: 10.1086/378747
197. Al-Hazmi M, Ayoola EA, Abdurahman M, Banzal S, Ashraf J, El-Bushra A, et al. Epidemic Rift Valley fever in Saudi Arabia: a clinical study of severe illness in humans. Clin Infect Dis. (2003) 36:245–52. doi: 10.1086/345671
198. Alrajhi AA, Al-Semari A, Al-Watban J. Rift Valley fever encephalitis. Emerg Infect Dis. (2004) 10:554–5. doi: 10.3201/eid1003.020817
199. Rippy MK, Topper MJ, Mebus CA, Morrill JC. Rift Valley fever virus-induced encephalomyelitis and hepatitis in calves. Vet Pathol. (1992) 29:495–502.
200. Nguku P, Sharif S, Omar A, Nzioka C, Muthoka P, Njau J, et al. Rift Valley fever outbreak-Kenya, November 2006-January 2007. Morb Mortal Wkly Rep. (2007) 56:73–6. Available online at: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm5604a3.htm
201. Centers for Disease Control and Prevention. SARS | Home | Severe Acute Respiratory Syndrome | SARS-CoV Disease. (2020). Available online at: https://www.cdc.gov/sars/index.html (accessed April 15, 2020)
202. World Health Organization. Summary of Probable SARS Cases With Onset of Illness From 1 November 2002 to 31 July 2003. (2003). Available online at: https://www.who.int/publications/m/item/summary-of-probable-sars-cases-with-onset-of-illness-from-1-november-2002-to-31-july-2003
203. Lam WK, Zhong NS, Tan WC. Overview on SARS in Asia and the world. Respirology. (2003) 8(Suppl.):S2–5. doi: 10.1046/j.1440-1843.2003.00516.x
204. Li YC, Bai WZ, Hashikawa T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol. (2020) 92:552–5. doi: 10.1002/jmv.25728
205. Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. (2005) 202:415–24. doi: 10.1084/jem.20050828
206. Hung EC, Chim SS, Chan PK, Tong YK, Ng EK, Chiu RW, et al. Detection of SARS coronavirus RNA in the cerebrospinal fluid of a patient with severe acute respiratory syndrome. Clin Chem. (2003) 49:2108–9. doi: 10.1373/clinchem.2003.025437
207. Lee AM, Wong JG, McAlonan GM, Cheung V, Cheung C, Sham PC, et al. Stress and psychological distress among SARS survivors 1 year after the outbreak. Can J. Psychiatry. (2007) 52:233–40. doi: 10.1177/070674370705200405
208. Yeh EA, Collins A, Cohen ME, Duffner PK, Faden H. Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics. (2004) 113: e73–6. doi: 10.1542/peds.113.1.e73
209. Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol. (2008) 82:7264–75. doi: 10.1128/JVI.00737-08
210. Chen X, Zhou B, Li M, Liang X, Wang H, Yang G, et al. Serology of severe acute respiratory syndrome: implications for surveillance and outcome. J Infect Dis. (2004) 189:1158–63. doi: 10.1086/380397
211. Ng EK, Hui DS, Chan KA, Hung EC, Chiu RW, Lee N, et al. Quantitative analysis and prognostic implication of SARS coronavirus RNA in the plasma and serum of patients with severe acute respiratory syndrome. Clin Chem. (2003) 49:1976–80. doi: 10.1373/clinchem.2003.024125
212. Poon L, Guan Y, Nicholls J, Yuen K, Peiris J. The aetiology, origins, and diagnosis of severe acute respiratory syndrome. Lancet Infect Dis. (2004) 4:663–71. doi: 10.1016/S1473-3099(04)01172-7
213. Chen LL, Hsu CW, Tian YC, Fang JT. Rhabdomyolysis associated with acute renal failure in patients with severe acute respiratory syndrome. Int J Clin Pract. (2005) 59:1162–6. doi: 10.1111/j.1368-5031.2005.00540.x
214. Ralston MSA, Murray MBP, Vela-Duarte D, Orjuela KD, Pastula DM. Neuroterrorism Preparedness for the Neurohospitalist. Neurohospitalist. (2019) 9:151–9. doi: 10.1177/1941874418806668
215. Olson VA, Laue T, Laker MT, Babkin IV, Drosten C, Shchelkunov SN, et al. Real-time PCR system for detection of orthopoxviruses and simultaneous identification of smallpox virus. J Clin Microbiol. (2004) 42:1940–6. doi: 10.1128/JCM.42.5.1940-1946.2004
216. Breman JG, Henderson DA. Diagnosis and management of smallpox. N Engl J Med. (2002) 346:1300–8. doi: 10.1056/NEJMra020025
217. Cleri DJ, Villota FJ, Porwancher RB. Smallpox, bioterrorism, and the neurologist. Arch Neurol. (2003) 60:489–94. doi: 10.1001/archneur.60.4.489
218. Shchelkunov SN, Totmenin AV, Loparev VN, Safronov PF, Gutorov VV, Chizhikov VE, et al. Alastrim smallpox variola minor virus genome DNA sequences. Virology. (2000) 266:361–86. doi: 10.1006/viro.1999.0086
219. Marsden JP. Acute perivascular myelinoclasis “acute disseminated encephalomyelitis”: a report of two further cases associated with exanthemata. Lancet. (1934) 224:871–2.
220. Han MH, Zunt JR. Bioterrorism and the nervous system. Curr Neurol Neurosci Rep. (2003) 3:476–82. doi: 10.1007/s11910-003-0050-9
221. Bartlett J, Borio L, Radonovich L, Mair JS, O'Toole T, Mair M, et al. Smallpox vaccination in 2003: key information for clinicians. Clin Infect Dis. (2003) 36:883–902. doi: 10.1086/374792
222. Lane JM, Ruben FL, Neff JM, Millar JD. Complications of smallpox vaccination, 1968: results of ten statewide surveys. J Infect Dis. (1970) 122:303–9.
223. Nigrovic LE, Wingerter SL. Tularemia. Infect Dis Clin North Am. (2008) 22:489–504, ix. doi: 10.1016/j.idc.2008.03.004
224. Centers for Disease Control and Prevention. (2020). Available online at: https://emergency.cdc.gov/agent/tularemia/facts.asp
225. Feldman KA, Enscore RE, Lathrop SL, Matyas BT, McGuill M, Schriefer ME, et al. An outbreak of primary pneumonic tularemia on Martha's Vineyard. N Engl J Med. (2001) 345:1601–6. doi: 10.1056/NEJMoa011374
226. Evans ME, Gregory DW, Schaffner W, McGee ZA. Tularemia: a 30-year experience with 88 cases. Medicine. (1985) 64:251–69.
227. Petersen JM, Schriefer ME. Francisella. Manual of Clinical Microbiology. 11 ed. Washington, DC: American Society of Microbiology (2015). p. 851–62.
228. Mushinski JF, Taniguchi RM, Stiefel JW. Guillain-Barre syndrome associated with ulceroglandular tularemia. Neurology. (1964) 14:877–9.
229. Blech B, Christiansen M, Asbury K, Orenstein R, Ross M, Grill M. Polyneuritis cranialis after acute tularemia infection: a case study. Muscle Nerve. (2020) 61:E1–2. doi: 10.1002/mus.26725
230. Zhou C, Yang Y, Zhang H. Guillain-Barre syndrome and ulceroglandular tularemia. Infection. (2013) 41:1199. doi: 10.1007/s15010-013-0515-y
231. Gangat N. Cerebral abscesses complicating tularemia meningitis. Scand J Infect Dis. (2007) 39:258–61. doi: 10.1080/00365540600823243
232. LeDoux MS. Tularemia presenting with ataxia. Clin Infect Dis. (2000) 30:211–2. doi: 10.1086/313621
233. Pittman T, Williams D, Friedman AD. A shunt infection caused by Francisella tularensis. Pediatr Neurosurg. (1996) 24:50–1.
234. Hepburn MJ, Simpson AJ. Tularemia: current diagnosis and treatment options. Expert Rev Anti Infect Ther. (2008) 6:231–40. doi: 10.1586/14787210.6.2.231
235. Garske T, Van Kerkhove MD, Yactayo S, Ronveaux O, Lewis RF, Staples JE, et al. Yellow Fever in Africa: estimating the burden of disease and impact of mass vaccination from outbreak and serological data. PLoS Med. (2014) 11:e1001638. doi: 10.1371/journal.pmed.1001638
236. Monath TP, Barrett AD. Pathogenesis and pathophysiology of yellow fever. Adv Virus Res. (2003) 60:343–95. doi: 10.1016/S0065-3527(03)60009-6
237. Monath TP. Yellow fever: an update. Lancet Infect Dis. (2001) 1:11–20. doi: 10.1016/S1473-3099(01)00016-0
238. Domingo C, Charrel RN, Schmidt-Chanasit J, Zeller H, Reusken C. Yellow fever in the diagnostics laboratory. Emerg Microbes Infect. (2018) 7:1–15. doi: 10.1038/s41426-018-0128-8
239. Stevenson LD. Pathologic changes in the nervous system in yellow fever. Arch Pathol. (1939) 27:249–66.
240. Marinho PES, Alvarenga PPM, Crispim APC, Candiani TMS, Alvarenga AM, Bechler IM, et al. Wild-type yellow fever virus RNA in cerebrospinal fluid of child. Emerg Infect Dis. (2019) 25:1567–70. doi: 10.3201/eid2508.181479
241. Ho YL, Joelsons D, Leite GFC, Malbouisson LMS, Song ATW, Perondi B, et al. Severe yellow fever in Brazil: clinical characteristics and management. J Travel Med. (2019) 26:taz040. doi: 10.1093/jtm/taz040
242. Monath TP, Vasconcelos PF. Yellow fever. J Clin Virol. (2015) 64:160–73. doi: 10.1016/j.jcv.2014.08.030
243. McMahon AW, Eidex RB, Marfin AA, Russell M, Sejvar JJ, Markoff L, et al. Neurologic disease associated with 17D-204 yellow fever vaccination: a report of 15 cases. Vaccine. (2007) 25:1727–34. doi: 10.1016/j.vaccine.2006.11.027
244. Dick GW, Kitchen SF, Haddow AJ. Zika virus. I Isolations and serological specificity. Trans R Soc Trop Med Hyg. (1952) 46:509–20.
245. Hayes EB. Zika virus outside Africa. Emerg Infect Dis. (2009) 15:1347–50. doi: 10.3201/eid1509.090442
246. Gutierrez-Bugallo G, Piedra LA, Rodriguez M, Bisset JA, Lourenco-de-Oliveira R, Weaver SC, et al. Vector-borne transmission and evolution of Zika virus. Nat Ecol Evol. (2019) 3:561–9. doi: 10.1038/s41559-019-0836-z
247. Russell K, Hills SL, Oster AM, Porse CC, Danyluk G, Cone M, et al. Male-to-female sexual transmission of Zika virus—United States, January–April (2016) 2016. Clin Infect Dis. (2017) 64:211–3. doi: 10.1093/cid/ciw692
248. Musso D, Gubler DJ. Zika virus. Clin Microbiol Rev. (2016) 29:487–524. doi: 10.1128/CMR.00072-15
249. Calvet GA, dos Santos FB, Sequeira PC. Zika virus infection: epidemiology, clinical manifestations and diagnosis. Curr Opin Infect Dis. (2016) 29:459–66. doi: 10.1097/QCO.0000000000000301
250. Landry ML, St. George K. Laboratory diagnosis of Zika virus infection Arch Pathol Lab Med. (2017) 141:60–7. doi: 10.5858/arpa.2016-0406-SA
251. Mier-y-Teran-Romero L, Delorey MJ, Sejvar JJ, Johansson MA. Guillain–Barré syndrome risk among individuals infected with Zika virus: a multi-country assessment. BMC Med. (2018) 16:67. doi: 10.1186/s12916-018-1052-4
252. Munoz LS, Parra B, Pardo CA, Neuroviruses Emerging in the Americas S. Neurological implications of zika virus infection in adults. J Infect Dis. (2017) 216(Suppl_10):S897–905. doi: 10.1093/infdis/jix511
253. da Silva IRF, Frontera JA, de Filippis AMB, do Nascimento OJM. Neurologic complications associated with the Zika virus in Brazilian adults. JAMA Neurol. (2017) 74:1190–8. doi: 10.1001/jamaneurol.2017.1703
254. Musso D, Ko AI, Baud D. Zika virus infection - After the pandemic. N Engl J Med. (2019) 381:1444–57. doi: 10.1056/NEJMra1808246
255. Moore CA, Staples JE, Dobyns WB, Pessoa A, Ventura CV, Fonseca EB, et al. Characterizing the pattern of anomalies in congenital zika syndrome for pediatric clinicians. JAMA Pediatr. (2017) 171:288–95. doi: 10.1001/jamapediatrics.2016.3982
256. Alpuche-Aranda C, Lazcano-Ponce E. Public health literacy in the face of the COVID-19 pandemic emergency. Salud Publ Mexico. (2020) 62:331–40. doi: 10.21149/11408
257. Cheng VC, Wong S-C, Chuang VW, So SY, Chen JH, Sridhar S, et al. The role of community-wide wearing of face mask for control of coronavirus disease 2019 (COVID-19) epidemic due to SARS-CoV-2. J Infect. (2020) 81:107–114. doi: 10.1016/j.jinf.2020.04.024
258. Lyu W, Wehby GL. Community use of face masks and COVID-19: evidence from a natural experiment of state mandates in the US: study examines impact on COVID-19 growth rates associated with state government mandates requiring face mask use in public. Health Affairs. (2020) 39:1419–25. doi: 10.1377/hlthaff.2020.00818
259. Pradhan D, Biswasroy P, Ghosh G, Rath G. A review of current interventions for COVID-19 prevention. Arch Med Res. (2020) 51:363–74. doi: 10.1016/j.arcmed.2020.04.020
260. Song P, Karako T. COVID-19: real-time dissemination of scientific information to fight a public health emergency of international concern. Biosci Trends. (2020) 14:1–2. doi: 10.5582/bst.2020.01056
Keywords: epidemic, pandemic, neuroviral epidemics, clinical neurology, tropical medicine
Citation: McEntire CRS, Song K-W, McInnis RP, Rhee JY, Young M, Williams E, Wibecan LL, Nolan N, Nagy AM, Gluckstein J, Mukerji SS and Mateen FJ (2021) Neurologic Manifestations of the World Health Organization's List of Pandemic and Epidemic Diseases. Front. Neurol. 12:634827. doi: 10.3389/fneur.2021.634827
Received: 29 November 2020; Accepted: 25 January 2021;
Published: 22 February 2021.
Edited by:
Peter R. Williamson, National Institutes of Health (NIH), United StatesReviewed by:
Sophie Cho, National Institutes of Health Clinical Center (NIH), United StatesCopyright © 2021 McEntire, Song, McInnis, Rhee, Young, Williams, Wibecan, Nolan, Nagy, Gluckstein, Mukerji and Mateen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farrah J. Mateen, Zm1hdGVlbkBtZ2guaGFydmFyZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.