 Hongying Ma1,2†
Hongying Ma1,2† Jian Qu
Jian Qu Qiang Qu
Qiang Qu- 1Department of Pharmacy, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Institute for Rational and Safe Medication Practices, Xiangya Hospital, Central South University, Changsha, China
- 3Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, China
- 4Institute of Clinical Pharmacy, Central South University, Changsha, China
- 5Department of Pharmacy, Hunan Provincial Corps Hospital of Chinese People's Armed Police Force, Changsha, China
- 6Department of Neurology, The Second Xiangya Hospital, Central South University, Changsha, China
Meige syndrome (MS) is cranial dystonia characterized by the combination of upper and lower cranial involvement and including binocular eyelid spasms (blepharospasm; BSP) and involuntary movements of the jaw muscles (oromandibular dystonia; OMD). The etiology and pathogenesis of this disorder of the extrapyramidal system are not well-understood. Neurologic and ophthalmic examinations often reveal no abnormalities, making diagnosis difficult and often resulting in misdiagnosis. A small proportion of patients have a family history of the disease, but to date no causative genes have been identified to date and no cure is available, although botulinum toxin A therapy effectively mitigates the symptoms and deep brain stimulation is gaining increasing attention as a viable alternative treatment option. Here we review the history and progress of research on MS, BSP, and OMD, as well as the etiology, pathology, diagnosis, and treatment.
Introduction
Blepharospasm (BSP), oromandibular dystonia (OMD), and Meige syndrome (MS) are different movement disorders that are different but closely related. MS is cranial dystonia characterized by the combination of upper and lower cranial involvement and includes BSP and involuntary movements of the OMD. Most researchers and clinicians would agree that MS, BSP, and OMD are not one single entity, but rather a clinical syndrome of multifactorial origin. Atypical parkinsonism patients frequently have the BSP symptom, and some patients with Huntington's disease have OMD. The causes of BSP, OMD, and MS remain elusive, but some convincing evidence suggests that these disorders are a multifactorial disease, where one or more unknown genes, as well as epigenetic and environmental factors, are combined to reach the disease threshold (1). Previously, according to etiology, dystonia has been classified as primary (dystonia is the only clinical sign and includes idiopathic or genetic disorders with no neuropathological abnormalities) or secondary [dystonia arising from neurodegeneration, acquired causes (such as lesions within the brain), or genetic conditions with a progressive course] (2). The 2013 consensus update on the phenomenology and classification of dystonia focuses on clinical characteristics and classifies dystonia as isolated (dystonia is the sole manifesting clinical feature with no other neurological or systemic signs) or combined (dystonia is combined with other neurological or systemic signs) (3). In this review, we provide a comprehensive overview of the new findings regarding the isolated/primary dystonias of BSP, OMD, and MS. in the past decades and highlight what gaps remain in our knowledge of this condition.
Relationship Between BSP, OMD, and MS
BSP is a primary adult-onset focal dystonia characterized by involuntary closure of the eyelids and spasms of the orbicularis oculi muscles (4). Over time, some patients with BSP develop OMD, which involves involuntary lower facial and masticatory movements including lip pursing, chewing, and jaw opening/clenching (5). MS is named for the French neurologist Henri Meige, who originally described a combination of BSP and involuntary movements of the lower facial and/or masticatory (jaw) muscles (6). In 1976, the British neurologist David Marsden diagnosed a case of BSP combined with oral and mandibular dystonia as Brueghel syndrome (7).
According to clinical characteristics of dystonia, dystonia is classified by age at onset, body distribution, temporal pattern, the coexistence of other movement disorders, and other neurological manifestations (3). Body distribution of dystonia comprises focal, segmental, generalized (with or without leg involvement), multifocal, and hemidystonia (3). Focal dystonia involves only one part of the body and encompasses BSP, spasmodic dysphonia, and handwriting spasm. Segmental dystonia involves two or more contiguous body regions and typical examples of segmental forms are cranial dystonia (BPS with lower facial and jaw or tongue involvement) or bibrachial dystonia (3, 8). MS is an example of the cranial subtype that manifests as a combination of BSP and OMD (9); the disorder may initially be limited to BSP or OMD only, and then later spread to other muscles (9).
Etiology classification of dystonia compromises two complementary characteristics that may be useful for classification: identifiable anatomical changes and pattern of inheritance. The term “primary” is currently used as an etiological descriptor for genetic or idiopathic cases where dystonia is isolated and there is no consistent pathologic change (3). This dual meaning does not help clarity and the use of the term primary is currently discouraged (3). Inherited dystonia forms have proven genetic origin; acquired dystonia due to a known specific cause; idiopathic dystonia has unknown causes. Therefore, MS, BSP, and OMD could be also classified into idiopathic, acquired, and inherited subtypes.
With our in-depth study of disease pathology, clinical diagnostic guidelines are constantly updated. To date, specific diagnostic guidelines of adult-onset focal dystonia have been proposed and validated for BSP and laryngeal dystonia (10, 11). Expert recommendations for diagnosing cervical, OMD, and limb dystonia were also published (12). Moreover, new clinical evaluation tools were invented such as a soft, flexible hybrid bioelectronic system that offers highly conformal, gentle lamination on the skin, while enabling wireless, quantitative detection of electrophysiological signals (13). The wearable bioelectronics outperforms the conventional manual clinical rating for BSP patients (13).
Clinical Manifestations and Epidemiology
The clinical features of MS vary widely among patients, which have different phenotypic forms, ranging from tonic spasm or prolonged closure of the eye, clonus of orbicularis oculi to complete inability to open the eyes as eyelid weakness or blepharoptosis is also very common (14). It may appear first as unilateral BSP that will later become bilateral (14). As the disease progresses, the involvement of the lower facial and masticatory muscles becomes fairly common in patients with BSP, including lip pursing, chewing, jaw thrusting, grimacing, jaw opening, and jaw closing/clenching (5). As the disease progresses, extensive multiple muscle groups show hyperactivity, with a full disease state observed after 1–4 years (15). Spasms normally last for several to tens of seconds before expanding to other areas. Dystonia can persist for several minutes while spasms become less synchronous (16–18). Clonic contraction or hyperactivity can also precede tonic contraction (16–18). Besides, some reports were showing that in elderly female patients with a history of head injury and BSP, dystonia can more easily spread to other parts of the body (5, 19, 20). The probability of spread to contiguous muscles in BSP is a very common (>50%) phenomenon. It happened highest during the initial 3–5 years of BSP onset, with nearly half of the spread occurring during the first year of illness, with rare instances recorded when the spread was delayed for a decade or more (15, 21). About 50% of patients with BSP develop dystonia in other areas, and some patients experience dry eyes and rigid pupils (22). In 155 BSP patients study by Defazio et al., previous trauma to head or face with unconsciousness, age at onset of BSP, and female sex were the predictors of the spread (23). Berman et al. analyzed 487 adult-onset isolated focal dystonia including 50% of BSP and found that the most common regions for first spread were the oromandibular region (42.2%) and neck (22.4%) for BSP (24). Increased spread risk was associated with positive family history and self-reported alcohol responsiveness (24).
Sensory tricks are common among patients with MS, which are the sensory stimuli, learned by the patients to alleviate the dystonia. Sleeping, relaxing, talking, pulling the upper eyelid, blowing cheeks, walking, exposure to cold water, yawning, or drinking beverages can all alleviate dystonia. More than half of the patients with BSP have one or more sensory tricks (14). Percham et al. reported sensory trick in 87% of BSP patients including MS patients and the majority had more than one sensory trick (15). The most common tricks were touching above the eyes, singing, humming, and talking (9). Spontaneous remissions in BSP and MS are rare and seen in <10% of the BSP and MS patients and usually tend to occur in the first 5 years of symptom onset (25, 26).
The prevalence was a range of 13–130 cases per million for BSP and 69 per million for OMD in the US (27). The prevalence of BSP accounts for 36 per million (95% CI 31–41) in Europe in 2000 (28). BSP and MS are significantly more common in females than males and the male: female ratio is 1:2 (9). The average age of MS onset is in the sixth decade and age appears to be an independent risk factor in the development of MS (9). The average age of onset for BSP is around 55 years whereas the average age of onset for OMD is only a couple of years earlier (5). Women are at more risk than men because of specific estrogen receptors influencing involuntary motor function (9, 14). In some cases, there is a family history of the disease (29–31), and MS or other forms of dystonia are present in up to 10% of first- or second-degree relatives of patients (32–35).
Pathophysiology
The pathophysiology of MS is not clear enough. A line of evidence suggests that the pathophysiology of dystonia involves the striatum, whose activity is modulated among other neurotransmitters, by the dopaminergic system (36). The most widely accepted hypothesis for the pathogenesis of MS is dopaminergic and cholinergic abnormalities (14, 36). Recent studies using neurophysiological and neuroimaging techniques have supported that environmental triggers and genetic predisposition cause plastic changes and reduced cortical inhibition (9). A voxel-based morphology analysis investigated the changes and clinical significance of brain structural abnormalities in MS patients. This study suggested an involvement of the basal ganglia and motor cortex in the pathophysiology of MS and the precuneus is involved in the development of MS (37). Positron emission tomographic scans have shown decreased blood flow to the sensorimotor area in response to lower face vibrations implies abnormal sensorimotor processing in MS patients (14).
Deoxyglucose metabolism in the striatum and thalamus was shown to be increased in MS by positron emission tomography, which was proposed to be related to hyperactivity in the striatum and hypothalamus (38, 39). MS is thought to develop as a result of damage to the brain base, causing an imbalance in dopamine receptor sensitivity (40). MS was also found to be associated with decreased inhibition in the cerebral cortex caused by environmental factors and genetic susceptibility (9, 14, 40). Animal studies have demonstrated that abnormal interactions among trigeminal blink circuits, basal ganglia, and the cerebellum contribute to the disease (41). Although there is no definitive evidence for the existence of MS susceptibility genes, some studies indicate that it is a low penetrance autosomal dominant disorder (33, 42–45).
Silent functional magnetic resonance imaging (MRI) has shown decreased activation of the primary motor cortex (Brodmann Area 4) and premotor cortex (Brodmann Area 6) in the mouth representing areas in MS patients having isolated BSP (14). It might be caused by abnormal control of cranial nerve nuclei in the brain stem by basal ganglia. Resting-state functional magnetic resonance imaging (MRI) has shown altered functional connectivity at rest in widespread brain regions including basal ganglia, cerebellar, primary/secondary sensorimotor, and visual areas in BSP and MS patients. This may reflect a predisposition for defective movement inhibition and sensorimotor integration (46). Transcranial magnetic stimulation electromyographic (EMG) responses study showed that MS had shorter SPs than BSP alone. The shortened SP in facial muscles reflects hypoexcitability of cortical inhibitory neurons in MS (47). The analysis of the semiautomatic rhythmic movements required for chewing and swallowing could reveal the action-related dystonic features. MS patients had an excess duration of muscle activity, frequent cocontraction, loss of rhythmicity during chewing, and abnormalities in the chewing to swallowing transition phase. These abnormalities, similar in type to those encountered in other forms of focal dystonia, maybe the expression of abnormal motor control of basal ganglia over mastication-related movement pattern generators of the brainstem (48).
The roles of the basal ganglia and thalamus are essential for coordination of eyelid movements, and the blink reflex, as well as the cerebello-thalamo-cortical pathways and cortico-striato-pallido-thalamo-cortical pathways, as demonstrated on functional MRI. Blink reflex and masseter inhibitory reflex have also been studied in BSP and MS patients (47, 49–53). Reflexive blinking in BPS is associated with increased activation in the caudate nucleus and sensorimotor cortices and the association between decreasing neural response during reflexive blinking in the cerebellum and disease duration suggests a loss of inhibition within the sensorimotor corticobasal ganglia network (51). Studies found that BSP is associated with a lesion of a complex neural network-cortex-thalamus-globus pallidus-cortex-and does not correspond to a single, unique lesion. This network is connected with ascending and descending sensory-motor pathways and motor nuclei (54). The blink reflex consists of an early, pontine R1-component and a late, medullary R2-component (55). Studies found that the latency of the early (R1) and late (R2) components of the blink reflex and the corneal reflex was normal in MS patients. However, the amplitude and the duration of the R1 and R2 and the duration of the corneal reflex were increased (56, 57). This has been demonstrated in BSP by coupling a train of electric shocks to the supraorbital nerve during the R2 (the second and major response) of the blink reflex (58). This causes an increase in the R2 amplitude that is enhanced in BSP patients relative to control subjects (4, 59). The studies about the blink reflex circuit in MS and BSP supported that the loss of inhibition within the blink reflex circuit contributed to the pathophysiology of these diseases.
The pathogenesis and the pathophysiology of OMD are not well-known (60). The study compared the “movement-related cortical potentials” (MRCPs) between 6 OMD patients and 8 normal subjects and found that MRCP amplitudes over central and parietal areas for mouth opening and lateral movements were significantly reduced compared to normal subjects, which implies that impaired cortical preparatory process for jaw movements exist in OMD (61). Although scientists attempted to elucidate the pathophysiology of OMD using several neuroimaging techniques, its etiology remains unclear (61, 62). Further research is needed to explore the pathophysiology of OMD to develop better treatments.
Genetics
To date there have been no specific pathogenic genes identified for MS, BSP, and OMD; however, given the close relationship between MS, BSP, OMD, and dystonia, dystonia susceptibility genes may play an important role in disease development.
To summarize the mutations related to MS, BSP, and OMD, we searched the PubMed database using the following search strategies: (Meige syndrome*[text word] OR blepharospasm [text word] OR oromandibular dystonia [text word] OR “Meige syndrome” [MESH] OR “blepharospasm” [MESH] OR “oromandibular dystonia” [MESH]) AND (mutation* [text word] OR polymorphism*[text word] OR variant*[text word]). Candidate genes that have been previously linked to MS, BSP, and OMD are shown in Table 1 (42, 45, 63, 64, 68, 86).
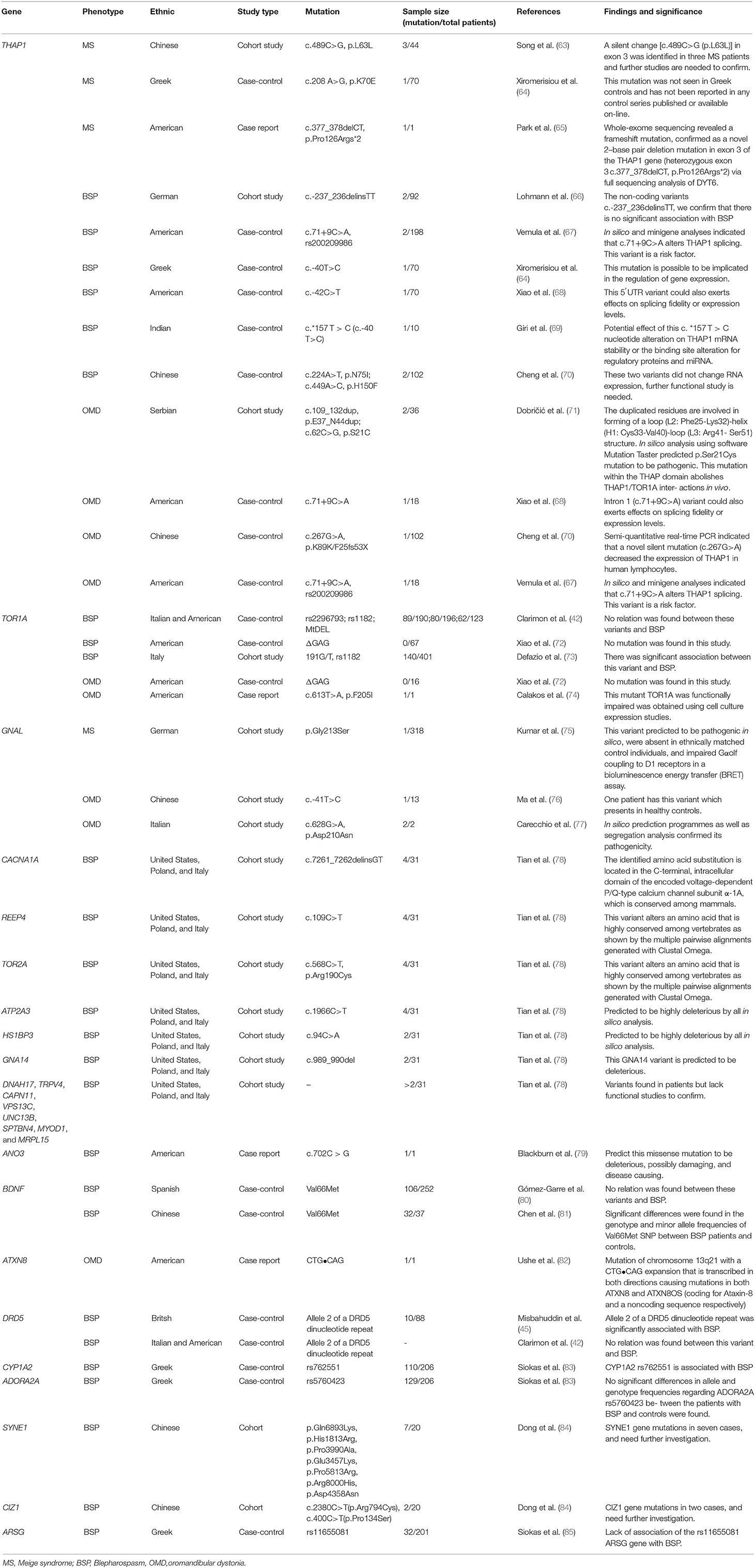
Table 1. The susceptibility genes of MS, BSP, OMD.
Mutations in torsion dystonia (DYT)1 (TOR1A encoding torsion (Tor)A, Online Mendelian Inheritance in Man [OMIM] ID: 605204) and DYT6 (THAP1 encoding THAP domain-containing 1, OMIM ID: 609520) often affect the craniocervical muscle and may contribute to the pathophysiology of MS (87–89). Evidence from gene expression studies in rodents and brain functional imaging suggests that DYT1 dystonia is a disorder of neural network development. At the cellular level, TorA mediates the interaction between the nuclear envelope and cytoskeleton, and mutations in TorA can indirectly prevent transcription factor entry into or exit from the nucleus (50, 90). TOR1A was shown to be critical for synapse formation and hence, for organizing connectivity in the spinal sensorimotor circuit (91); TOR1A mutations are observed in cases of early-onset torsion dystonia (92) and have been linked to late-onset focal, segmental, and multifocal dystonia including BSP, OMD, and MS (42, 72, 73). TOR1A mutations were investigated in BSP and OMD patients in four publications (42, 72–74). ΔGAG of TOR1A mutation was not identified in 67 BSP and 16 OMD patients, which implies that this mutation may not be associated with BSP and OMD (72). A case report found c.613T>A (p.F205I) mutation in OMD and this mutation produces frequent inclusions when expressed in cultured cells, a phenotype shared by the Δ E mutant TOR1A, but not wildtype protein (68). A case-control study found a significant association between rs1182 of TOR1A and BSP (62). While another study suggested that rs2296793, rs1182, and MtDEL of TOR1A were not associated with BSP (42). Up to now, limited evidence showed the role of TOR1A mutation in MS, BSP, and OMD.
THAP1 is a transcription factor expressed in the central nervous system; mutations in this protein lead to aberrant eukaryotic initiation factor 2α (eIF2α) signaling, mitochondrial dysfunction, defects in neuronal projection and axon guidance, and long-term synaptic depression, which are common features of various forms of primary dystonia (93, 94). THAP1 regulates cell proliferation and the G1/S checkpoint by modulating retinoblastoma/E2F target genes (95, 96), and THAP1 overexpression and knockdown in endothelial cells inhibit cell proliferation (95). Some mutations commonly found in BSP, OMD, and MS are shown in Table 1 (63, 64, 66, 68, 70). Now THAP1 was the most studied gene for the diseases and was the most likely susceptibility gene of these diseases. A silent change [c.489C>G (p.L63L)] in exon 3 was identified in three MS patients and further studies are needed to confirm its effects on gene function (53). A case-control study found the c.208 A>G (p.K70E) mutation in one of 70 MS patients, and this mutation was not seen in Greek controls and has not been reported in any control series published or available on-line (52). Whole-exome sequencing revealed a frameshift mutation in one MS patient, confirmed as a novel 2–base pair deletion mutation in exon 3 of the THAP1 gene (heterozygous exon 3 c.377_378delCT, p.Pro126Args*2) via full sequencing analysis of DYT6 (65). In silico and minigene analyses indicated that c.71+9C>A alters THAP1 splicing and this variant is a risk factor (67). c.-40T>C was identified in two BSP patients in two publications, the potential effect of this nucleotide alteration on THAP1 mRNA stability or the binding site alteration for regulatory proteins and miRNA (64, 69). c.-42C>T of THAP1 was found in one BSP patient, which is a 5′UTR variant which could also exert effects on splicing fidelity or expression levels (68). p.N75I and p.H150F mutations were identified in two BSP patients and these two variants did not change RNA expression, the further functional study is needed (70). Moreover, a series of case-control studies in OMD patients also identified some mutations, although the frequencies are rare. The mutation c.109_132dup(p.E37_N44dup) duplicated residues that involved in the forming of a loop (L2: Phe25-Lys32)-helix (H1: Cys33-Val40)-loop (L3: Arg41- Ser51) structure (71). Semi-quantitative real-time PCR indicated that a novel silent mutation (c.267G>A) decreased the expression of THAP1 in human lymphocytes (70). In silico and minigene analyses indicated that c.71+9C>A alters THAP1 splicing or expression levels in two publications (67, 68). Based on the current study, we conclude that this gene mutation is rare in patients, and more large-scale case-control studies are needed to search for the mutation and the related functional studies of the mutation.
GNAL encodes guanine nucleotide-binding protein G(olf) subunit α, which mediates odorant signaling in the olfactory epithelium; it is located on chromosome 18p centromeric to the DYT7 locus of focal dystonia (97, 98). GNAL mutations have been linked to abnormalities in dopamine type 1 and/or adenosine A2A receptor transmission that is thought to result in dystonia (98, 99), and have been detected in patients with OMD and MS (76, 77, 100). Kumar et al. carried out a cohort study and found p.Gly213Ser mutation in one MS patient. This variant was predicted to be pathogenic in silico, was absent in ethnically matched control individuals, and impaired Gαolf coupling to D1 receptors in a bioluminescence energy transfer (BRET) assay (75). One patient has c.-41T>C mutation which also presents in healthy controls (76). In silico prediction programmers as well as segregation analysis confirmed the pathogenicity of p.Asp210Asn mutation in two OMD patients (77).
Brain-derived neurotrophic factor (BDNF) is a member of the nerve growth factor family (101) that is necessary for the survival of striatal neurons and for synaptic plasticity in the adult brain (101). The BDNF Val66Met mutation is associated with an increased risk of BSP development and may protect against BSP (81), although other studies have found no association between the BDNF Val66Met variant and BSP (80, 102).
ANO3 encodes anoctamin-3, which belongs to the anoctamin family of Ca2+-activated chloride channels and is highly expressed in the striatum, hippocampus, and cortex. ANO3 modulates neuronal excitability (79, 103), and c.702C > G mutation has been observed in one BSP patient (79). Functional predicting showed this missense mutation be deleterious, possibly damaging, and disease-causing (79). A recent case-control study showed the rs11655081 of ARSG was not associated with BSP (104). SYNE1 gene mutations in seven BSP, CIZ1 gene mutations in two BSP patients were identified and the role of these mutations in the etiology of BSP needs further investigation (84, 85).
A case reported a mutation in ATXN8 in a BSP patient (82). This mutation of chromosome 13q21 with a CTG•CAG expansion that is transcribed in both directions causing mutations in both ATXN8 and ATXN8OS (coding for Ataxin-8 and a noncoding sequence, respectively) (82). Whole-exome sequencing of 31 subjects with BSP from 21 independent pedigrees identified mutations in several genes including CACNA1A encoding calcium voltage-gated channel subunit α1 A (NM_001127222.1: c.7261_7262delinsGT, p.Pro2421Val), REEP4 encoding receptor accessory protein 4 (NM_025232.3: c.109C>T, p.Arg37Trp), TOR2A (NM_130459.3: c.568C>T, p.Arg190Cys), and ATP2A3 encoding ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 3 (NM_005173.3: c.1966C>T, p.Arg656Cys). Deleterious mutations in HS1BP3 encoding hematopoietic cell-specific Lyn substrate 1-binding protein 3 (NM_022460.3: c.94C>A, p.Gly32Cys) and GNA14 encoding G protein subunit α14 (NM_004297.3: c.989_990del, p.Thr330ArgfsTer67) were present in a father and son with segmental CCD that initially presented as BSP. Additionally, deleterious variants of genes encoding dynein axonemal heavy chain (DNAH17); transient receptor potential cation channel subfamily V member 4 (TRPV4); calpain 11 (CAPN11); vacuolar protein sorting 13 homolog C (VPS13C); unc-13 homolog B (UNC13B); spectrin β, non-erythrocytic 4 (SPTBN4); myogenic differentiation 1 (MYOD1); and mitochondrial ribosomal protein L15 (MRPL15) have been found in two or more unrelated BSP patients (78). Despite the identification of many gene mutations in BSP, OMD, and MS patients, the mutation rates of these genes are low and it is a lack of functional experiments of mutant genes. More large-scale whole-exome sequencing studies are needed to search for the mutation and the related functional studies of the pathogenic mutations also need to confirm their roles in these diseases.
Clinical Management
There is no curative treatment for MS, BSP, or OMD. Current treatments include botulinum neurotoxin (BoNT) therapy, oral medication, and surgical intervention.
BoNT Therapy
The most common drug treatment for MS, BSP, and OMD is the injection of BoNT. BoNT-A was first used for the treatment of idiopathic BSP and is currently the preferred therapeutic approach owing to its high efficacy and few side effects (105). BoNT-A has been approved by the U.S. Food and Drug Administration for the treatment of BSP and CD while BoNT-B has been approved for CD (106). The first-time BoNT injections should use minimum effective starting doses, which helps to prevent side effects such as excessive focal muscle weakness (107). The starting doses for each toxin were the same (1.25–2.5 U per site), but the maximum doses used within MS, BSP, and OMD are variable. In common practice, minimal side effects are seen when starting at 5 U per site (107). Some large safety studies have shown efficacy and no significant long-term side effects. Recent guidelines summarized the levels of evidence for BSP and OMD (106). OnaBoNT/A and incoBoNT/A were approved by the U.S. Federal Drug Administration (FDA) for BSP treatment. OnaBoNT/A and aboBoNT/A were approved by FDA for OMD treatment (107). Recent long-term safety and efficacy in daily clinical practice study found that the treatment of BSP and MS with onaBoNT/A and aboBoNT/A is safe and effective, also over a long observation period of up to 29 years (27).
The most common side effect is excessive focal muscle weakness, including ptosis and lagophthalmos in BSP patients while eye dryness and diplopia are less common adverse events (107). The most common adverse events in treating OMD are chewing weakness, dysphagia, dysarthria (tongue injections), and dry mouth (diffusion into salivary glands), while generalized weakness, allergic reactions, and flu-like symptoms are rare side effects (107). The main point for the excessive use of BoNT injection is therapeutic resistance that occurs due to antibody production after recurrent and long-term use (14).
Oral Medications
While BoNT is first-line therapy, clinicians should consider using oral medications as primary therapy when relative contraindications exist, although there have been few multi-center, placebo-controlled, double-blind studies evaluating their clinical utility in MS, BSP, and OMD. Evidence-based reviews have been published (14, 40, 63), but none of the available agents has been tested in rigorously controlled clinical trials. Oral medications that have been used to treat MS, BSP, and OMD include the anticholinergics (e.g., trihexyphenidyl and benztropine), benzodiazepines (e.g., clonazepam, diazepam, and lorazepam), GABA receptor agonist (e.g., baclofen), dopamine precursor such as levodopa, dopamine receptor agonist (e.g., bromocriptine, and tiapride), vesicular monoamine transporter 2 inhibitor (e.g., tetrabenazine), anticonvulsant such as levetiracetam. Eszopiclone and nitrazepam could alleviate the BSP via reacting at those specific subunits (omega-1 and omega-2) of the GABA receptor complex (108). Some case reports found that zolpidem is effective in such patients as it is highly specific for a GABA omega-1 receptor (109, 110). However, some studies found limited efficacy of oral medications such as zolpidem, levetiracetam, and valproate (111, 112). A randomized controlled trial concluded that Levetiracetam does not appear to be efficacious in patients with OMD or cranial dystonia (111). A single-center, double-blind cross-sectional study revealed that valproate had low efficacy in the treatment of MS (112). Zolpidem and Levetiracetam were slightly effective in patients with MS (109, 113, 114). Moreover, the magnitude of improvement typically obtained with commonly used drugs is often modest, such as the anticholinergics (e.g., trihexyphenidyl and benztropine), benzodiazepines (e.g., clonazepam and lorazepam), baclofen, and tetrabenazine (5). Compare to BoNT, oral medication efficacy is at best modest and does not show the same level. Oral medication therapies are further limited by systemic side effects which are not usually seen with BoNT therapy (107). For oral medication in OMD, the recent system review pointed out that no definitive conclusions can be drawn about the type of patients that may benefit, nor about the preferred type or mode of the appliance (115).
Surgical Treatment
Surgical treatment is an option for patients who are unresponsive to the conventional drugs used to treat MS, BSP, and OMD. Partial resection of the periorbital muscle resulted in long-term improvement; however, this is not the favored therapeutic strategy owing to postoperative complications such as inflammation, aesthetic issues, hematoma, and exposure keratitis among others and the efficacy of BoNT (116–118). Deep brain stimulation (DBS) has gained increasing attention in recent years as a treatment option for intractable dystonia including MS and DYT1 dystonia (18, 119–122). Some studies have demonstrated that DBS is ineffective in a subset of patients with craniocervical segmental dystonia (123–126), but it was found to improve symptoms of MS (104, 127–129). DBS of the globus pallidus internal and subthalamic nucleus was effective in patients with medically refractory MS, including those exhibiting severe preoperative symptoms (130). A recent meta-analysis showed that DBS of the globus pallidus internal and subthalamic nucleus may be an effective therapy for even refractory MS. Higher preoperative scores probably indicate larger improvement and stimulation targets or other clinical factors do not constitute the outcome predictive factors (131). Reported cases of MS treated with DBS are summarized in Table 2; however, given their limited number, more in-depth studies are needed to validate the clinical utility of this method. BoNT is still the main treatment option for BSP. When it fails, there are not many options. Given the experience of BSP improving with DBS of the globus pallidus internal and subthalamic nucleus in patients with MS, DBS surgery can be an acceptably effective therapy for patients with isolated BSP. The risk of the procedure should be weighed cautiously against the potential benefit.
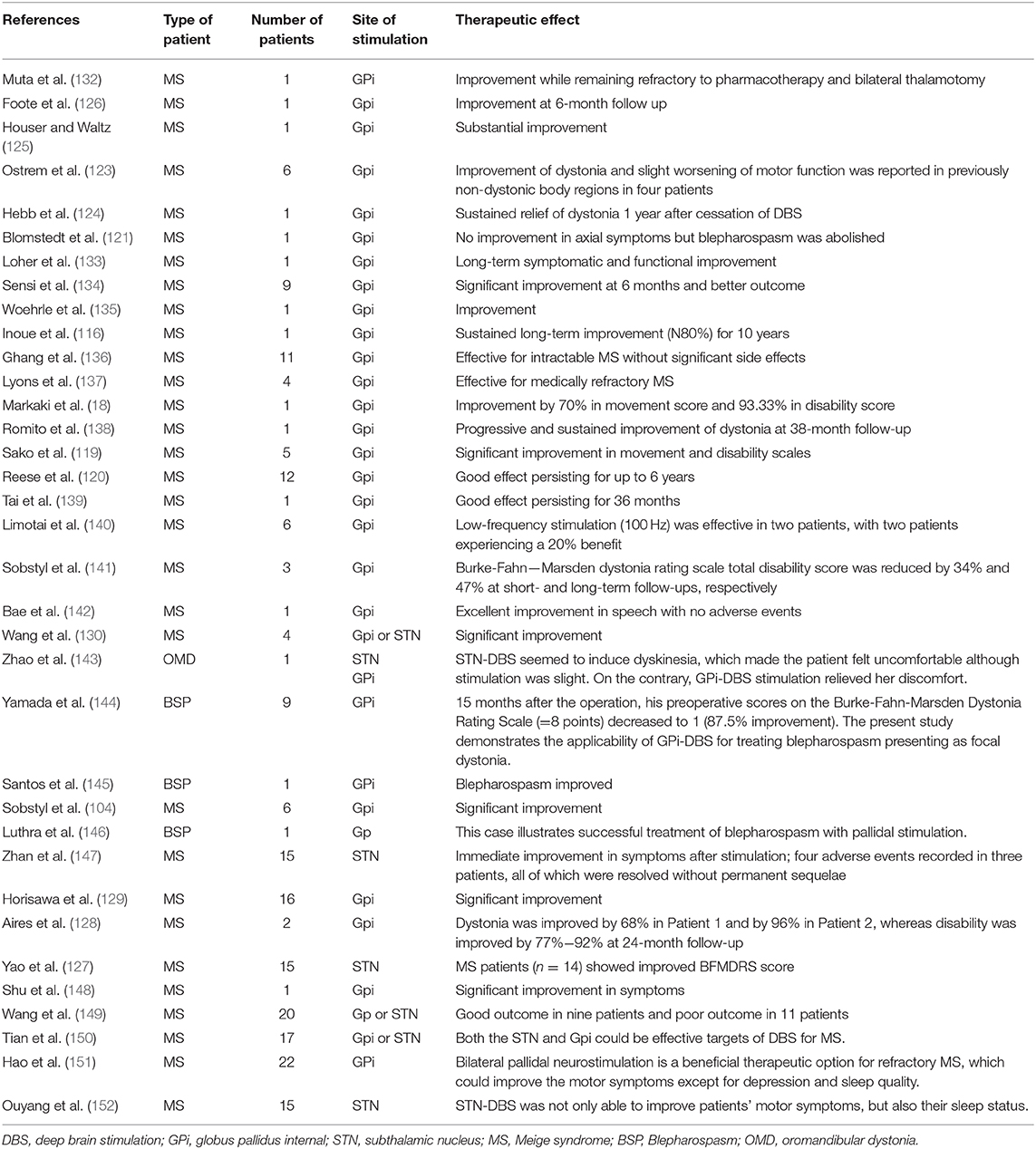
Table 2. Case series of MS, BSP, and OMD patients undergoing DBS.
Conclusions
MS is complex dystonia that includes BSP and OMD. Due to its low incidence, possible genetic heterogeneity, and late age of onset, it is difficult to obtain complete case data in families and, consequently, to identify genetic markers and susceptibility genes. Indeed, no causative genes have been confirmed to date. The viable treatment option that is currently available for MS is repeated injections of BoNT. DBS is an option for patients who are unresponsive to conventional drugs. The application of high-throughput, genome-wide analytical approaches is expected to reveal novel disease markers and potential therapeutic targets, thereby providing a basis for the development of more effective drugs that can bring clinical relief of symptoms and improve the quality of life of BSP, OMD, and MS patients.
Author Contributions
JQ and QQ: conceptualization. HM, YS, and LY: collection and analysis of bibliography. JQ, QQ, HM, YS, and LY: writing original draft. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 82073944 and 81603208), Hunan Provincial Natural Science Foundation of China (2018JJ3718), and WUJIEPING Medical Foundation (LCYX-Q001, 320.6750.19090-11).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Defazio G, Hallett M, Jinnah HA, Conte A, Berardelli A. Blepharospasm 40 years later. Mov Disord. (2017) 32:498–509. doi: 10.1002/mds.26934
3. Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. (2013) 28:863–73. doi: 10.1002/mds.25475
4. Hallett M, Evinger C, Jankovic J, Stacy M, Workshop BI. Update on blepharospasm: report from the BEBRF international workshop. Neurology. (2008) 71:1275–82. doi: 10.1212/01.wnl.0000327601.46315.85
5. LeDoux MS. Meige syndrome: what's in a name? Parkinsonism Relat Disord. (2009) 15:483–9. doi: 10.1016/j.parkreldis.2009.04.006
6. H M. Les convulsions de la face. Ue forme clinique de convulsion faciale bilaterale et mediane. Rev Neurol. (1910) 21:437–43.
7. Marsden CD. Blepharospasm-oromandibular dystonia syndrome (Brueghel's syndrome). A variant of adult-onset torsion dystonia? J Neurol Neurosurg Psychiatry. (1976) 39:1204–9. doi: 10.1136/jnnp.39.12.1204
8. Balint B, Mencacci NE, Valente EM, Pisani A, Rothwell J, Jankovic J, et al. Dystonia. Nat Rev Dis Primers. (2018) 4:25. doi: 10.1038/s41572-018-0023-6
9. Pandey S, Sharma S. Meige's syndrome: history, epidemiology, clinical features, pathogenesis and treatment. J Neurol Sci. (2017) 372:162–70. doi: 10.1016/j.jns.2016.11.053
10. Defazio G, Hallett M, Jinnah HA, Berardelli A. Development and validation of a clinical guideline for diagnosing blepharospasm. Neurology. (2013) 81:236–40. doi: 10.1212/WNL.0b013e31829bfdf6
11. Ludlow CL, Adler CH, Berke GS, Bielamowicz SA, Blitzer A, Bressman SB, et al. Research priorities in spasmodic dysphonia. Otolaryngol Head Neck Surg. (2008) 139:495–505. doi: 10.1016/j.otohns.2008.05.624
12. Defazio G, Albanese A, Pellicciari R, Scaglione CL, Esposito M, Morgante F, et al. Expert recommendations for diagnosing cervical, oromandibular, and limb dystonia. Neurol Sci. (2019) 40:89–95. doi: 10.1007/s10072-018-3586-9
13. Mahmood M, Kwon S, Berkmen GK, Kim YS, Scorr L, Jinnah HA, et al. Soft nanomembrane sensors and flexible hybrid bioelectronics for wireless quantification of blepharospasm. IEEE Trans Biomed Eng. (2020) 67:3094–100. doi: 10.1109/TBME.2020.2975773
15. Peckham EL, Lopez G, Shamim EA, Richardson SP, Sanku S, Malkani R, et al. Clinical features of patients with blepharospasm: a report of 240 patients. Eur J Neurol. (2011) 18:382–6. doi: 10.1111/j.1468-1331.2010.03161.x
16. Defazio G, Abbruzzese G, Aniello MS, Bloise M, Crisci C, Eleopra R, et al. Environmental risk factors and clinical phenotype in familial and sporadic primary blepharospasm. Neurology. (2011) 77:631–7. doi: 10.1212/WNL.0b013e3182299e13
17. Miao J, Liu R, Li J, Du Y, Zhang W, Li Z. Meige's syndrome and hemichorea associated with hyperthyroidism. J Neurol Sci. (2010) 288:175–7. doi: 10.1016/j.jns.2009.10.018
18. Markaki E, Kefalopoulou Z, Georgiopoulos M, Paschali A, Constantoyannis C. Meige's syndrome: a cranial dystonia treated with bilateral pallidal deep brain stimulation. Clin Neurol Neurosurg. (2010) 112:344–6. doi: 10.1016/j.clineuro.2009.12.005
19. Kranz G, Shamim EA, Lin PT, Kranz GS, Voller B, Hallett M. Blepharospasm and the modulation of cortical excitability in primary and secondary motor areas. Neurology. (2009) 73:2031–6. doi: 10.1212/WNL.0b013e3181c5b42d
20. Kenney C, Jankovic J. Botulinum toxin in the treatment of blepharospasm and hemifacial spasm. J Neural Trans. (2008) 115:585–91. doi: 10.1007/s00702-007-0768-7
21. Svetel M, Pekmezovic T, Tomic A, Kresojevic N, Kostic VS. The spread of primary late-onset focal dystonia in a long-term follow up study. Clin Neurol Neurosurg. (2015) 132:41–3. doi: 10.1016/j.clineuro.2015.02.015
23. Defazio G, Berardelli A, Abbruzzese G, Coviello V, Carella F, De Berardinis MT, et al. Risk factors for spread of primary adult onset blepharospasm: a multicentre investigation of the Italian movement disorders study group. J Neurol Neurosurg Psychiatry. (1999) 67:613–9. doi: 10.1136/jnnp.67.5.613
24. Berman BD, Groth CL, Sillau SH, Pirio Richardson S, Norris SA, Junker J, et al. Risk of spread in adult-onset isolated focal dystonia: a prospective international cohort study. J Neurol Neurosurg Psychiatry. (2020) 91:314–20. doi: 10.1136/jnnp-2019-321794
25. Castelbuono A, Miller NR. Spontaneous remission in patients with essential blepharospasm and Meige syndrome. Am J Ophthalmol. (1998) 126:432–5. doi: 10.1016/S0002-9394(98)00099-3
26. Levey AS, Eckardt KU, Dorman NM, Christiansen SL, Hoorn EJ, Ingelfinger JR, et al. Nomenclature for kidney function and disease: report of a kidney disease: improving global outcomes (KDIGO) consensus conference. Kidney Int. (2020) 97:1117–29. doi: 10.1016/j.kint.2020.02.010
27. Jochim A, Meindl T, Huber C, Mantel T, Zwirner S, Castrop F, et al. Treatment of blepharospasm and Meige's syndrome with abo- and onabotulinumtoxinA: long-term safety and efficacy in daily clinical practice. J Neurol. (2020) 267:267–75. doi: 10.1007/s00415-019-09581-w
28. Epidemiological Study of Dystonia in Europe Collaborative G. A prevalence study of primary dystonia in eight European countries. J Neurol. (2000) 247:787–92. doi: 10.1007/s004150070094
29. Tolosa ES. Clinical features of Meige's disease (idiopathic orofacial dystonia): a report of 17 cases. Arch Neurol. (1981) 38:147–51. doi: 10.1001/archneur.1981.00510030041005
30. Defazio G, Berardelli A, Hallett M. Do primary adult-onset focal dystonias share aetiological factors? Brain. (2007) 130 (Pt 5):1183–93. doi: 10.1093/brain/awl355
31. Grandas F, Elston J, Quinn N, Marsden CD. Blepharospasm: a review of 264 patients. J Neurosurg Psychiatry. (1988) 51:767–72. doi: 10.1136/jnnp.51.6.767
32. Diaz Neira W, Lousa M, Millan JM. New magnetic resonance findings in the Meige syndrome. Neurologia. (1993) 8:127–8.
33. Defazio G, Livrea P, Guanti G, Lepore V, Ferrari E. Genetic contribution to idiopathic adult-onset blepharospasm and cranial-cervical dystonia. Eur Neurol. (1993) 33:345–50. doi: 10.1159/000116969
34. Morrison PJ, Patterson VH. Cranial dystonia (Meige syndrome) in postencephalitic parkinsonism. Mov Disord. (1992) 7:90–1. doi: 10.1002/mds.870070121
35. Jimenez-Jimenez FJ, Molina-Arjona JA, Roldan-Montaud A, Agulla A, Santos J, Fernandez-Ballesteros A. Blepharospasm associated with neurocysticercosis. Acta Neurol. (1992) 14:56–9.
36. Ribot B, Aupy J, Vidailhet M, Mazere J, Pisani A, Bezard E, et al. Dystonia and dopamine: from phenomenology to pathophysiology. Prog Neurobiol. (2019) 182:101678. doi: 10.1016/j.pneurobio.2019.101678
37. Liu J, Li L, Chen L, Liu R, Jiang Y, Fang J, et al. Grey matter changes in Meige syndrome: a voxel-based morphology analysis. Sci Rep. (2020) 10:14533. doi: 10.1038/s41598-020-71479-9
38. Nutt JG, Hammerstad JP. Blepharospasm and oromandibular dystonia (Meige's syndrome) in sisters. Ann Neurol. (1981) 9:189–91. doi: 10.1002/ana.410090214
39. Casey DE. Pharmacology of blepharospasm-oromandibular dystonia syndrome. Neurology. (1980) 30 (7 Pt 1):690–5. doi: 10.1212/WNL.30.7.690
40. Hallett M. Blepharospasm: recent advances. Neurology. (2002) 59:1306–12. doi: 10.1212/01.WNL.0000027361.73814.0E
41. Evinger C. Animal models for investigating benign essential blepharospasm. Curr Neuropharmacol. (2013) 11:53–8. doi: 10.2174/157015913804999441
42. Clarimon J, Brancati F, Peckham E, Valente EM, Dallapiccola B, Abruzzese G, et al. Assessing the role of DRD5 and DYT1 in two different case-control series with primary blepharospasm. Mov Disord. (2007) 22:162–6. doi: 10.1002/mds.21182
43. Defazio G, Brancati F, Valente EM, Caputo V, Pizzuti A, Martino D, et al. Familial blepharospasm is inherited as an autosomal dominant trait and relates to a novel unassigned gene. Mov Disord. (2003) 18:207–12. doi: 10.1002/mds.10314
44. Maniak S, Sieberer M, Hagenah J, Klein C, Vieregge P. Focal and segmental primary dystonia in north-western Germany–a clinico-genetic study. Acta Neurol Scand. (2003) 107:228–32. doi: 10.1034/j.1600-0404.2003.01362.x
45. Misbahuddin A, Placzek MR, Chaudhuri KR, Wood NW, Bhatia KP, Warner TT. A polymorphism in the dopamine receptor DRD5 is associated with blepharospasm. Neurology. (2002) 58:124–6. doi: 10.1212/WNL.58.1.124
46. Jochim A, Li Y, Gora-Stahlberg G, Mantel T, Berndt M, Castrop F, et al. Altered functional connectivity in blepharospasm/orofacial dystonia. Brain Behav. (2018) 8:e00894. doi: 10.1002/brb3.894
47. Curra A, Romaniello A, Berardelli A, Cruccu G, Manfredi M. Shortened cortical silent period in facial muscles of patients with cranial dystonia. Neurology. (2000) 54:130–5. doi: 10.1212/WNL.54.1.130
48. Mascia MM, Valls-Sole J, Marti MJ, Sanz S. Chewing pattern in patients with Meige's syndrome. Mov Disord. (2005) 20:26–33. doi: 10.1002/mds.20272
49. Martino D, Defazio G, Alessio G, Abbruzzese G, Girlanda P, Tinazzi M, et al. Relationship between eye symptoms and blepharospasm: a multicenter case-control study. Mov Disord. (2005) 20:1564–70. doi: 10.1002/mds.20635
50. Carbon M, Eidelberg D. Abnormal structure-function relationships in hereditary dystonia. Neuroscience. (2009) 164:220–9. doi: 10.1016/j.neuroscience.2008.12.041
51. Nguyen P, Kelly D, Glickman A, Argaw S, Shelton E, Peterson DA, et al. Abnormal neural responses during reflexive blinking in blepharospasm: an event-related functional MRI study. Mov Disord. (2020) 35:1173–80. doi: 10.1002/mds.28042
52. Benbir G, Kiziltan ME. Blink reflex studies in postparalytic facial syndrome and blepharospasm: trigeminal and extratrigeminal somatosensory stimulation. J Clin Neurophysiol. (2014) 31:535–40. doi: 10.1097/WNP.0000000000000095
53. Ferrazzano G, Conte A, Fabbrini G, Bologna M, Macerollo A, Defazio G, et al. Botulinum toxin and blink rate in patients with blepharospasm and increased blinking. J Neurol Neurosurg Psychiatry. (2015) 86:336–40. doi: 10.1136/jnnp-2014-307821
54. Girard B, Davoudi O, Tatry M, Tassart M. Secondary blepharospasm, analysis and pathophysiology of blepharospasm. J Fr Ophtalmol. (2020) 44:e1–12. doi: 10.1016/j.jfo.2020.11.001
55. Ellrich J, Treede RD. Characterization of blink reflex interneurons by activation of diffuse noxious inhibitory controls in man. Brain Res. (1998) 803:161–8. doi: 10.1016/S0006-8993(98)00646-5
56. Berardelli A, Rothwell JC, Day BL, Marsden CD. Pathophysiology of blepharospasm and oromandibular dystonia. Brain. (1985) 108 (Pt 3):593–608. doi: 10.1093/brain/108.3.593
57. Schwingenschuh P, Katschnig P, Edwards MJ, Teo JT, Korlipara LV, Rothwell JC, et al. The blink reflex recovery cycle differs between essential and presumed psychogenic blepharospasm. Neurology. (2011) 76:610–4. doi: 10.1212/WNL.0b013e31820c3074
58. Quartarone A, Sant'Angelo A, Battaglia F, Bagnato S, Rizzo V, Morgante F, et al. Enhanced long-term potentiation-like plasticity of the trigeminal blink reflex circuit in blepharospasm. J Neurosci. (2006) 26:716–21. doi: 10.1523/JNEUROSCI.3948-05.2006
59. Valls-Sole J, Defazio G. Blepharospasm: update on epidemiology, clinical aspects, and pathophysiology. Front Neurol. (2016) 7:45. doi: 10.3389/fneur.2016.00045
60. Raoofi S, Khorshidi H, Najafi M. Etiology, diagnosis and management of oromandibular dystonia: an update for stomatologists. J Dent. (2017) 18:73–81.
61. Yoshida K, Kaji R, Kohara N, Murase N, Ikeda A, Shibasaki H, et al. Movement-related cortical potentials before jaw excursions in oromandibular dystonia. Mov Disord. (2003) 18:94–100. doi: 10.1002/mds.10296
62. Yoshida K. Development and validation of a disease-specific oromandibular dystonia rating scale (OMDRS). Front Neurol. (2020) 11:583177. doi: 10.3389/fneur.2020.583177
63. Song W, Chen Y, Huang R, Chen K, Pan P, Yang Y, et al. Novel THAP1 gene mutations in patients with primary dystonia from southwest China. J Neurol Sci. (2011) 309:63–7. doi: 10.1016/j.jns.2011.07.023
64. Xiromerisiou G, Dardiotis E, Tsironi EE, Hadjigeorgiou G, Ralli S, Kara E, et al. THAP1 mutations in a Greek primary blepharospasm series. Parkinsonism Relat Disord. (2013) 19:404–5. doi: 10.1016/j.parkreldis.2012.08.015
65. Park JE, Vanegas-Arroyave N, Hallett M, Lungu C. A woman with a novel mutation of THAP1 with a prominent response to deep brain stimulation of the globus pallidus internus. JAMA Neurol. (2015) 72:1369. doi: 10.1001/jamaneurol.2015.1954
66. Lohmann K, Uflacker N, Erogullari A, Lohnau T, Winkler S, Dendorfer A, et al. Identification and functional analysis of novel THAP1 mutations. Eur J Hum Genet. (2012) 20:171–5. doi: 10.1038/ejhg.2011.159
67. Vemula SR, Xiao J, Zhao Y, Bastian RW, Perlmutter JS, Racette BA, et al. A rare sequence variant in intron 1 of THAP1 is associated with primary dystonia. Mol Genet Genomic Med. (2014) 2:261–72. doi: 10.1002/mgg3.67
68. Xiao J, Zhao Y, Bastian RW, Perlmutter JS, Racette BA, Tabbal SD, et al. Novel THAP1 sequence variants in primary dystonia. Neurology. (2010) 74:229–38. doi: 10.1212/WNL.0b013e3181ca00ca
69. Giri S, Naiya T, Equbal Z, Sankhla CS, Das SK, Ray K, et al. Genetic screening of THAP1 in primary dystonia patients of India. Neurosci Lett. (2017) 637:31–7. doi: 10.1016/j.neulet.2016.11.060
70. Cheng FB, Ozelius LJ, Wan XH, Feng JC, Ma LY, Yang YM, et al. THAP1/DYT6 sequence variants in non-DYT1 early-onset primary dystonia in China and their effects on RNA expression. J Neurol. (2012) 259:342–7. doi: 10.1007/s00415-011-6196-5
71. Dobricic VS, Kresojevic ND, Svetel MV, Jankovic MZ, Petrovic IN, Tomic AD, et al. Mutation screening of the DYT6/THAP1 gene in Serbian patients with primary dystonia. J Neurol. (2013) 260:1037–42. doi: 10.1007/s00415-012-6753-6
72. Xiao J, Bastian RW, Perlmutter JS, Racette BA, Tabbal SD, Karimi M, et al. High-throughput mutational analysis of TOR1A in primary dystonia. BMC Med Genet. (2009) 10:24. doi: 10.1186/1471-2350-10-24
73. Defazio G, Matarin M, Peckham EL, Martino D, Valente EM, Singleton A, et al. The TOR1A polymorphism rs1182 and the risk of spread in primary blepharospasm. Mov Disord. (2009) 24:613–6. doi: 10.1002/mds.22471
74. Calakos N, Patel VD, Gottron M, Wang G, Tran-Viet KN, Brewington D, et al. Functional evidence implicating a novel TOR1A mutation in idiopathic, late-onset focal dystonia. J Med Genet. (2010) 47:646–50. doi: 10.1136/jmg.2009.072082
75. Kumar KR, Lohmann K, Masuho I, Miyamoto R, Ferbert A, Lohnau T, et al. Mutations in GNAL: a novel cause of craniocervical dystonia. JAMA Neurol. (2014) 71:490–4. doi: 10.1001/jamaneurol.2013.4677
76. Ma LY, Wang L, Yang YM, Wan XH. Mutations in GNAL gene in 214 cases with isolated dystonia. Parkinsonism Relat Disord. (2015) 21:1367–8. doi: 10.1016/j.parkreldis.2015.08.026
77. Carecchio M, Panteghini C, Reale C, Barzaghi C, Monti V, Romito L, et al. Novel GNAL mutation with intra-familial clinical heterogeneity: expanding the phenotype. Parkinsonism Relat Disord. (2016) 23:66–71. doi: 10.1016/j.parkreldis.2015.12.012
78. Tian J, Vemula SR, Xiao J, Valente EM, Defazio G, Petrucci S, et al. Whole-exome sequencing for variant discovery in blepharospasm. Mol Genet Genomic Med. (2018) 6:601–26. doi: 10.1002/mgg3.411
79. Blackburn PR, Zimmermann MT, Gass JM, Harris KG, Cousin MA, Boczek NJ, et al. A novel ANO3 variant identified in a 53-year-old woman presenting with hyperkinetic dysarthria, blepharospasm, hyperkinesias, and complex motor tics. BMC Med Genet. (2016) 17:93. doi: 10.1186/s12881-016-0354-7
80. Gomez-Garre P, Huertas-Fernandez I, Caceres-Redondo MT, Alonso-Canovas A, Bernal-Bernal I, Blanco-Ollero A, et al. BDNF Val66Met polymorphism in primary adult-onset dystonia: a case-control study and meta-analysis. Mov Disord. (2014) 29:1083–6. doi: 10.1002/mds.25938
81. Chen Y, Song W, Yang J, Chen K, Huang R, Zhao B, et al. Association of the Val66Met polymorphism of the BDNF gene with primary cranial-cervical dystonia patients from South-west China. Parkinsonism Relat Disord. (2013) 19:1043–5. doi: 10.1016/j.parkreldis.2013.06.004
82. Ushe M, Perlmutter JS. Oromandibular and lingual dystonia associated with spinocerebellar ataxia type 8. Mov Disord. (2012) 27:1741–2. doi: 10.1002/mds.25295
83. Siokas V, Kardaras D, Aloizou AM, Liampas I, Papageorgiou E, Drakoulis N, et al. CYP1A2 rs762551 and ADORA2A rs5760423 polymorphisms in patients with blepharospasm. J Mol Neurosci. (2020) 70:1370–5. doi: 10.1007/s12031-020-01553-4
84. Dong H, Luo Y, Fan S, Yin B, Weng C, Peng B. Screening gene mutations in Chinese patients with benign essential blepharospasm. Front Neurol. (2019) 10:1387. doi: 10.3389/fneur.2019.01387
85. Siokas V, Kardaras D, Aloizou AM, Asproudis I, Boboridis KG, Papageorgiou E, et al. Lack of association of the rs11655081 ARSG gene with blepharospasm. J Mol Neurosci. (2019) 67:472–6. doi: 10.1007/s12031-018-1255-3
86. Xiao J, Zhao Y, Bastian RW, Perlmutter JS, Racette BA, Tabbal SD, et al. The c.-237_236GA>TT THAP1 sequence variant does not increase risk for primary dystonia. Mov Disord. (2011) 26:549–52. doi: 10.1002/mds.23551
87. Bressman SB, Sabatti C, Raymond D, de Leon D, Klein C, Kramer PL, et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology. (2000) 54:1746–52. doi: 10.1212/WNL.54.9.1746
88. Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saunders-Pullman R. Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol. (2009) 8:441–6. doi: 10.1016/S1474-4422(09)70081-X
89. Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet. (2007) 80:393–406. doi: 10.1086/512129
90. Xiao J, Gong S, Zhao Y, LeDoux MS. Developmental expression of rat torsinA transcript and protein. Brain Res Dev Brain Res. (2004) 152:47–60. doi: 10.1016/j.devbrainres.2004.05.012
91. Monje MHG, Sanchez-Ferro A. Tor1a gene in GABApre interneurons: the new player in the “impaired inhibition” game of dystonia? Mov Disord. (2018) 33:1408. doi: 10.1002/mds.114
92. Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. (1997) 17:40–8. doi: 10.1038/ng0997-40
93. Vander Heyden AB, Naismith TV, Snapp EL, Hodzic D, Hanson PI. LULL1 retargets TorsinA to the nuclear envelope revealing an activity that is impaired by the DYT1 dystonia mutation. Mol Biol Cell. (2009) 20:2661–72. doi: 10.1091/mbc.e09-01-0094
94. Zakirova Z, Fanutza T, Bonet J, Readhead B, Zhang W, Yi Z, et al. Mutations in THAP1/DYT6 reveal that diverse dystonia genes disrupt similar neuronal pathways and functions. PLoS Genet. (2018) 14:e1007169. doi: 10.1371/journal.pgen.1007169
95. Cayrol C, Lacroix C, Mathe C, Ecochard V, Ceribelli M, Loreau E, et al. The THAP-zinc finger protein THAP1 regulates endothelial cell proliferation through modulation of pRB/E2F cell-cycle target genes. Blood. (2007) 109:584–94. doi: 10.1182/blood-2006-03-012013
96. Ledoux MS, Dauer WT, Warner TT. Emerging common molecular pathways for primary dystonia. Mov Disord. (2013) 28:968–81. doi: 10.1002/mds.25547
97. Leube B, Rudnicki D, Ratzlaff T, Kessler KR, Benecke R, Auburger G. Idiopathic torsion dystonia: assignment of a gene to chromosome 18p in a German family with adult onset, autosomal dominant inheritance and purely focal distribution. Hum Mol Genet. (1996) 5:1673–7. doi: 10.1093/hmg/5.10.1673
98. Jones DT, Reed RR. Golf: an olfactory neuron specific-G protein involved in odorant signal transduction. Science. (1989) 244:790–5. doi: 10.1126/science.2499043
99. Fuchs T, Saunders-Pullman R, Masuho I, Luciano MS, Raymond D, Factor S, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet. (2013) 45:88–92. doi: 10.1038/ng.2496
100. Saunders-Pullman R, Fuchs T, San Luciano M, Raymond D, Brashear A, Ortega R, et al. Heterogeneity in primary dystonia: lessons from THAP1, GNAL, and TOR1A in Amish-Mennonites. Mov Disord. (2014) 29:812–8. doi: 10.1002/mds.25818
101. Kleim JA, Chan S, Pringle E, Schallert K, Procaccio V, Jimenez R, et al. BDNF val66met polymorphism is associated with modified experience-dependent plasticity in human motor cortex. Nat Neurosci. (2006) 9:735–7. doi: 10.1038/nn1699
102. Martino D, Muglia M, Abbruzzese G, Berardelli A, Girlanda P, Liguori M, et al. Brain-derived neurotrophic factor and risk for primary adult-onset cranial-cervical dystonia. Eur J Neurol. (2009) 16:949–52. doi: 10.1111/j.1468-1331.2009.02633.x
103. Charlesworth G, Plagnol V, Holmstrom KM, Bras J, Sheerin UM, Preza E, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet. (2012) 91:1041–50. doi: 10.1016/j.ajhg.2012.10.024
104. Sobstyl M, Brzuszkiewicz-Kuzmicka G, Zaczynski A, Pasterski T, Aleksandrowicz M, Zabek M. Long-term clinical outcome of bilateral pallidal stimulation for intractable craniocervical dystonia (Meige syndrome). Report of 6 patients. J Neurol Sci. (2017) 383:153–7. doi: 10.1016/j.jns.2017.10.017
105. Fante RG, Frueh BR. Differential section of the seventh nerve as a tertiary procedure for the treatment of benign essential blepharospasm. Ophthalmic Plast Reconstr Surg. (2001) 17:276–80. doi: 10.1097/00002341-200107000-00007
106. Simpson DM, Hallett M, Ashman EJ, Comella CL, Green MW, Gronseth GS, et al. Practice guideline update summary: botulinum neurotoxin for the treatment of blepharospasm, cervical dystonia, adult spasticity, and headache: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. (2016) 86:1818–26. doi: 10.1212/WNL.0000000000002560
107. Hassell TJW, Charles D. Treatment of blepharospasm and oromandibular dystonia with botulinum toxins. Toxins. (2020) 12:269. doi: 10.3390/toxins12040269
108. Zhao S, Zhang Y, Xu L, Wei L, Chen W. A case report of psychoactive drugs aggravating and alleviating Meige syndrome. Shanghai Arch Psychiatry. (2016) 28:222–6. doi: 10.11919/j.issn.1002-0829.216038
109. An JY, Kim JS, Kim YI, Lee KS. Successful treatment of the Meige syndrome with oral zolpidem monotherapy. Mov Disord. (2008) 23:1619–21. doi: 10.1002/mds.22179
110. Miyazaki Y, Sako W, Asanuma K, Izumi Y, Miki T, Kaji R. Efficacy of zolpidem for dystonia: a study among different subtypes. Front Neurol. (2012) 3:58. doi: 10.3389/fneur.2012.00058
111. Park JE, Srivanitchapoom P, Maurer CW, Mathew P, Sackett J, Paine R, et al. Lack of efficacy of levetiracetam in oromandibular and cranial dystonia. Acta Neurol Scand. (2017) 136:103–8. doi: 10.1111/ane.12701
112. Snoek JW, van Weerden TW, Teelken AW, van den Burg W, Lakke JP. Meige syndrome: double-blind crossover study of sodium valproate. J Neurol Neurosurg Psychiatry. (1987) 50:1522–5. doi: 10.1136/jnnp.50.11.1522
113. Zesiewicz TA, Louis ED, Sullivan KL, Menkin M, Dunne PB, Hauser RA. Substantial improvement in a Meige's syndrome patient with levetiracetam treatment. Mov Disord. (2004) 19:1518–21. doi: 10.1002/mds.20233
114. Yardimci N, Karatas M, Kilinc M, Benli S. Levetiracetam in Meige's syndrome. Acta Neurol Scand. (2006) 114:63–6. doi: 10.1111/j.1600-0404.2006.00624.x
115. De Meyer M, Vereecke L, Bottenberg P, Jacquet W, Sims AB, Santens P. Oral appliances in the treatment of oromandibular dystonia: a systematic review. Acta Neurol Belg. (2020) 120:831–6. doi: 10.1007/s13760-020-01404-4
116. Inoue N, Nagahiro S, Kaji R, Goto S. Long-term suppression of Meige syndrome after pallidal stimulation: a 10-year follow-up study. Mov Disord. (2010) 25:1756–8. doi: 10.1002/mds.23166
117. Coscarelli JM. Essential blepharospasm. Semin Ophthalmol. (2010) 25:104–8. doi: 10.3109/08820538.2010.488564
118. Leon-Sarmiento FE, Gutierrez C, Bayona-Prieto J. Functional neurology of blepharospasm. Medicina. (2008) 68:318–24.
119. Sako W, Morigaki R, Mizobuchi Y, Tsuzuki T, Ima H, Ushio Y, et al. Bilateral pallidal deep brain stimulation in primary Meige syndrome. Parkinsonism Relat Disord. (2011) 17:123–5. doi: 10.1016/j.parkreldis.2010.11.013
120. Reese R, Gruber D, Schoenecker T, Bazner H, Blahak C, Capelle HH, et al. Long-term clinical outcome in meige syndrome treated with internal pallidum deep brain stimulation. Mov Disord. (2011) 26:691–8. doi: 10.1002/mds.23549
121. Blomstedt P, Tisch S, Hariz MI. Pallidal deep brain stimulation in the treatment of Meige syndrome. Acta Neurol Scand. (2008) 118:198–202. doi: 10.1111/j.1600-0404.2008.00999.x
122. Moro E, LeReun C, Krauss JK, Albanese A, Lin JP, Walleser Autiero S, et al. Efficacy of pallidal stimulation in isolated dystonia: a systematic review and meta-analysis. Eur J Neurol. (2017) 24:552–60. doi: 10.1111/ene.13255
123. Ostrem JL, Marks WJ Jr, Volz MM, Heath SL, Starr PA. Pallidal deep brain stimulation in patients with cranial-cervical dystonia (Meige syndrome). Mov Disord. (2007) 22:1885–91. doi: 10.1002/mds.21580
124. Hebb MO, Chiasson P, Lang AE, Brownstone RM, Mendez I. Sustained relief of dystonia following cessation of deep brain stimulation. Mov Disord. (2007) 22:1958–62. doi: 10.1002/mds.21616
125. Houser M, Waltz T. Meige syndrome and pallidal deep brain stimulation. Mov Disord. (2005) 20:1203–5. doi: 10.1002/mds.20522
126. Foote KD, Sanchez JC, Okun MS. Staged deep brain stimulation for refractory craniofacial dystonia with blepharospasm: case report and physiology. Neurosurgery. (2005) 56:E415. doi: 10.1227/01.NEU.0000147978.67424.42
127. Yao C, Horn A, Li N, Lu Y, Fu Z, Wang N, et al. Post-operative electrode location and clinical efficacy of subthalamic nucleus deep brain stimulation in Meige syndrome. Parkinsonism Relat Disord. (2019) 58:40–5. doi: 10.1016/j.parkreldis.2018.05.014
128. Aires A, Gomes T, Linhares P, Cunha F, Rosas MJ, Vaz R. The impact of deep brain stimulation on health related quality of life and disease-specific disability in Meige Syndrome (MS). Clin Neurol Neurosurg. (2018) 171:53–7. doi: 10.1016/j.clineuro.2018.05.012
129. Horisawa S, Ochiai T, Goto S, Nakajima T, Takeda N, Kawamata T, et al. Long-term outcome of pallidal stimulation for Meige syndrome. J Neurosurg. (2018) 130:84–9. doi: 10.3171/2017.7.JNS17323
130. Wang X, Zhang C, Wang Y, Liu C, Zhao B, Zhang JG, et al. Deep brain stimulation for craniocervical dystonia (Meige Syndrome): a report of four patients and a literature-based analysis of its treatment effects. Neuromodulation. (2016) 19:818–23. doi: 10.1111/ner.12345
131. Wang X, Zhang Z, Mao Z, Yu X. Deep brain stimulation for Meige syndrome: a meta-analysis with individual patient data. J Neurol. (2019) 266:2646–56. doi: 10.1007/s00415-019-09462-2
132. Muta D, Goto S, Nishikawa S, Hamasaki T, Ushio Y, Inoue N, et al. Bilateral pallidal stimulation for idiopathic segmental axial dystonia advanced from Meige syndrome refractory to bilateral thalamotomy. Mov Disord. (2001) 16:774–7. doi: 10.1002/mds.1122
133. Loher TJ, Capelle HH, Kaelin-Lang A, Weber S, Weigel R, Burgunder JM, et al. Deep brain stimulation for dystonia: outcome at long-term follow-up. J Neurol. (2008) 255:881–4. doi: 10.1007/s00415-008-0798-6
134. Sensi M, Cavallo MA, Quatrale R, Sarubbo S, Biguzzi S, Lettieri C, et al. Pallidal stimulation for segmental dystonia: long term follow up of 11 consecutive patients. Mov Disord. (2009) 24:1829–35. doi: 10.1002/mds.22686
135. Woehrle JC, Blahak C, Kekelia K, Capelle HH, Baezner H, Grips E, et al. Chronic deep brain stimulation for segmental dystonia. Stereotact Funct Neurosurg. (2009) 87:379–84. doi: 10.1159/000249819
136. Ghang JY, Lee MK, Jun SM, Ghang CG. Outcome of pallidal deep brain stimulation in meige syndrome. J Korean Neurosurg Soc. (2010) 48:134–8. doi: 10.3340/jkns.2010.48.2.134
137. Lyons MK, Birch BD, Hillman RA, Boucher OK, Evidente VG. Long-term follow-up of deep brain stimulation for Meige syndrome. Neurosurg Focus. (2010) 29:E5. doi: 10.3171/2010.4.FOCUS1067
138. Romito LM, Elia AE, Franzini A, Bugiani O, Albanese A. Low-voltage bilateral pallidal stimulation for severe meige syndrome in a patient with primary segmental dystonia: case report. Neurosurgery. (2010) 67 (3 Suppl. Operative):onsE308. doi: 10.1227/01.NEU.0000381768.04640.46
139. Tai CH, Wu RM, Liu HM, Tsai CW, Tseng SH. Meige syndrome relieved by bilateral pallidal stimulation with cycling mode: case report. Neurosurgery. (2011) 69:E1333–7. doi: 10.1227/NEU.0b013e31822a9ad2
140. Limotai N, Go C, Oyama G, Hwynn N, Zesiewicz T, Foote K, et al. Mixed results for GPi-DBS in the treatment of cranio-facial and cranio-cervical dystonia symptoms. J Neurol. (2011) 258:2069–74. doi: 10.1007/s00415-011-6075-0
141. Sobstyl M, Zabek M, Mossakowski Z, Zaczynski A. Pallidal deep brain stimulation in the treatment of Meige syndrome. Neurol Neurochir Pol. (2014) 48:196–9. doi: 10.1016/j.pjnns.2014.05.008
142. Bae DW, Son BC, Kim JS. Globus pallidus interna deep brain stimulation in a patient with medically intractable meige syndrome. J Mov Disord. (2014) 7:92–4. doi: 10.14802/jmd.14013
143. Zhao XM, Zhang JG, Meng FG. Internal pallidum and subthalamic nucleus deep brain stimulation for oromandibular dystonia. Chin Med J. (2016) 129:1619–20. doi: 10.4103/0366-6999.184475
144. Yamada K, Shinojima N, Hamasaki T, Kuratsu J. Pallidal stimulation for medically intractable blepharospasm. BMJ Case Rep. (2016) 2016:bcr2015214241. doi: 10.1136/bcr-2015-214241
145. Santos AF, Veiga A, Augusto L, Vaz R, Rosas MJ, Volkmann J. Successful treatment of blepharospasm by pallidal neurostimulation. Mov Disord Clin Pract. (2016) 3:409–11. doi: 10.1002/mdc3.12297
146. Luthra NS, Mitchell KT, Volz MM, Tamir I, Starr PA, Ostrem JL. Intractable blepharospasm treated with bilateral pallidal deep brain stimulation. Tremor Other Hyperkinet Mov. (2017) 7:472. doi: 10.5334/tohm.361
147. Zhan S, Sun F, Pan Y, Liu W, Huang P, Cao C, et al. Bilateral deep brain stimulation of the subthalamic nucleus in primary Meige syndrome. J Neurosurg. (2018) 128:897–902. doi: 10.3171/2016.12.JNS16383
148. Shu W, Li Y, Li J, Zhang Y. Interleaving programming in pallidal deep brain stimulation improves outcomes in a patient with Meige syndrome. Br J Neurosurg. (2018) 32:661–2. doi: 10.1080/02688697.2018.1504883
149. Wang X, Mao Z, Cui Z, Xu X, Pan L, Liang S, et al. Predictive factors for long-term clinical outcomes of deep brain stimulation in the treatment of primary Meige syndrome. J Neurosurg. (2019) 132:1367–75. doi: 10.3171/2019.1.JNS182555
150. Tian H, Xiong NX, Xiong N, Liu XM, Rao J, Xiang W, et al. Similar long-term clinical outcomes of deep brain stimulation with different electrode targets for primary Meige syndrome: one institution's experience of 17 cases. Neuromodulation. (2020) 24:300–6. doi: 10.1111/ner.13304
151. Hao Q, Wang D, OuYang J, Ding H, Wu G, Liu Z, et al. Pallidal deep brain stimulation in primary Meige syndrome: clinical outcomes and psychiatric features. J Neurol Neurosurg Psychiatry. (2020) 91:1343–8. doi: 10.1136/jnnp-2020-323701
Keywords: Meige syndrome, blepharospasm, oromandibular dystonia, pathogenic gene, variants
Citation: Ma H, Qu J, Ye L, Shu Y and Qu Q (2021) Blepharospasm, Oromandibular Dystonia, and Meige Syndrome: Clinical and Genetic Update. Front. Neurol. 12:630221. doi: 10.3389/fneur.2021.630221
Received: 24 November 2020; Accepted: 08 March 2021;
Published: 29 March 2021.
Edited by:
Sanjay Pandey, University of Delhi, IndiaReviewed by:
Brian D. Berman, Virginia Commonwealth University, United StatesPrachaya Srivanitchapoom, Mahidol University, Thailand
Copyright © 2021 Ma, Qu, Ye, Shu and Qu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Qu, cXVxaWFuZ0Bjc3UuZWR1LmNu
†These authors have contributed equally to this work