 Mariana Y. García-Bermúdez1
Mariana Y. García-Bermúdez1 Kristine K. Freude2
Kristine K. Freude2 Zaynab A. Mouhammad1
Zaynab A. Mouhammad1 Peter van Wijngaarden3,4Keith K. Martin3,4
Peter van Wijngaarden3,4Keith K. Martin3,4 Miriam Kolko1,5*
Miriam Kolko1,5*- 1Department of Drug Design and Pharmacology, University of Copenhagen, Copenhagen, Denmark
- 2Department for Veterinary and Animal Science, University of Copenhagen, Copenhagen, Denmark
- 3Center for Eye Research Australia, Royal Victorian Eye and Ear Hospital, East Melbourne, VIC, Australia
- 4Ophthalmology, Department of Surgery, University of Melbourne, Melbourne, VIC, Australia
- 5Department of Ophthalmology, Copenhagen University Hospital, Rigshospitalet-Glostrup, Glostrup, Denmark
Glaucoma is the second leading cause of blindness worldwide, affecting ~80 million people by 2020 (1, 2). The condition is characterized by a progressive loss of retinal ganglion cells (RGCs) and their axons accompanied by visual field loss. The underlying pathophysiology of glaucoma remains elusive. Glaucoma is recognized as a multifactorial disease, and lowering intraocular pressure (IOP) is the only treatment that has been shown to slow the progression of the condition. However, a significant number of glaucoma patients continue to go blind despite intraocular pressure-lowering treatment (2). Thus, the need for alternative treatment strategies is indisputable. Accumulating evidence suggests that glial cells play a significant role in supporting RGC function and that glial dysfunction may contribute to optic nerve disease. Here, we review recent advances in understanding the role of glial cells in the pathophysiology of glaucoma. A particular focus is on the dynamic and essential interactions between glial cells and RGCs and potential therapeutic approaches to glaucoma by targeting glial cells.
Introduction
Glaucoma is a group of eye diseases that can cause vision loss and blindness. The number of people with glaucoma is increasing due to the age-related nature of the disease (3). Hence, it is estimated that more than 120 million people worldwide will suffer from glaucoma in 2040 (4). Glaucoma is characterized by progressive degeneration of retinal ganglion cells (RGCs) and is often asymptomatic until its advanced stages when vision loss is irreversible (5). Glaucoma can be classified as either primary or secondary, with secondary glaucoma attributable to known pathologies or medications. Glaucoma may be further classified as either open-angle or angle-closure according to the anatomy of the aqueous outflow pathway (2, 6). In all subtypes of glaucoma, the inner retinal degeneration, especially the gradual loss of RGCs, is a hallmark (7). RGCs are the output neurons of the retina, and their axons transfer visual information from the retina to the brain (8). RGC dysfunction and death lead to vision impairment and ultimately to blindness. To date, no approved treatments for glaucoma directly target RGCs. Instead, the only available treatments are indirectly protective for RGCs by lowering the intraocular pressure (IOP). It is, therefore, crucial to identify cellular mechanisms for the prevention of RGC degeneration, the repair of dysfunctional cells, and the promotion of axonal regeneration to limit the projected burden of vision impairment and blindness from glaucoma (9, 10). Although the most investigated risk factors for glaucoma progression include IOP, age, genetic background, thinner corneal thickness, and vascular dysregulation (11), other disease mechanisms such as oxidative stress, mitochondrial dysfunction, excitotoxicity, and immunological processes may contribute to the pathophysiology of the disease (2, 12, 13). In this context, accumulating evidence suggests that glial cells in the retina and optic nerve may play important roles in the pathogenesis of RGCs (14–16). However, despite having been studied for more than a century, there are substantial aspects of the interrelationship between glial cells and RGCs that are still to be elucidated (17, 18).
Emerging literature emphasizes the roles of glia in both the maintenance of the retina and in the pathogenesis of glaucoma (15, 19). Although Müller glia, astrocytes, oligodendrocytes, and microglia have different developmental origins, they are now known to share many functions. Although some functions are subserved simultaneously by different glia, others are performed by specific glial subtypes (14, 19–22). Similarly, attention has turned to the complex interactions between retinal glia and neurons and the centrality of these interactions to retinal homeostasis (14, 19, 23, 24). Likewise, it is evident that the glial response to injury stimuli can further perpetuate RGC damage (17, 23, 25, 26). Despite these important advances in our understanding of the interactions between glia and retinal neurons in health and in the context of glaucoma, there is still much to be learned.
Glial Cells of the Retina are Not Just Support Cells
Glial cells are named after the Greek word for glue, as it was thought that their function was simply to bind the neurons in the central nervous system (CNS) together (27). It is now understood that glial cells play a range of diverse and complex functions beyond the provision of structural support to neurons. Two basic types of glial cells are found in the human retina: macroglia and microglia. Retinal macroglia are comprised of Müller glia and astrocytes. Macroglia maintain retinal homeostasis by regulating ion exchange, glucose, and neurotransmitter transport (14). Microglia respond to retinal injury and are important in the maintenance of neuronal networks and the mediation of neuroinflammation (14, 28–30). In the optic nerve, oligodendrocytes, another type of macroglia, and astrocytes provide essential support to RGC axons as they travel to the brain (31). Accumulating evidence suggests that both types of glial cells are interacting with the retinal and optic nerves, and are important contributors to the pathophysiological processes leading to glaucomatous RGC loss (14, 15, 17, 23, 32–34).
A Partnership Between Müller Glia and Retinal Ganglion Cells
Müller glia are found throughout the retina, with processes extending from the outer limiting membrane to the inner limiting membrane and surrounding the RGCs. Their unique morphology and distribution are related to the role of Müller glia as mediators of the transport of molecules between RGCs and the vitreous humor, retinal vessels, and the subretinal space (15) (Figure 1). Müller glia have multiple functions and are symbiotically associated with RGCs.
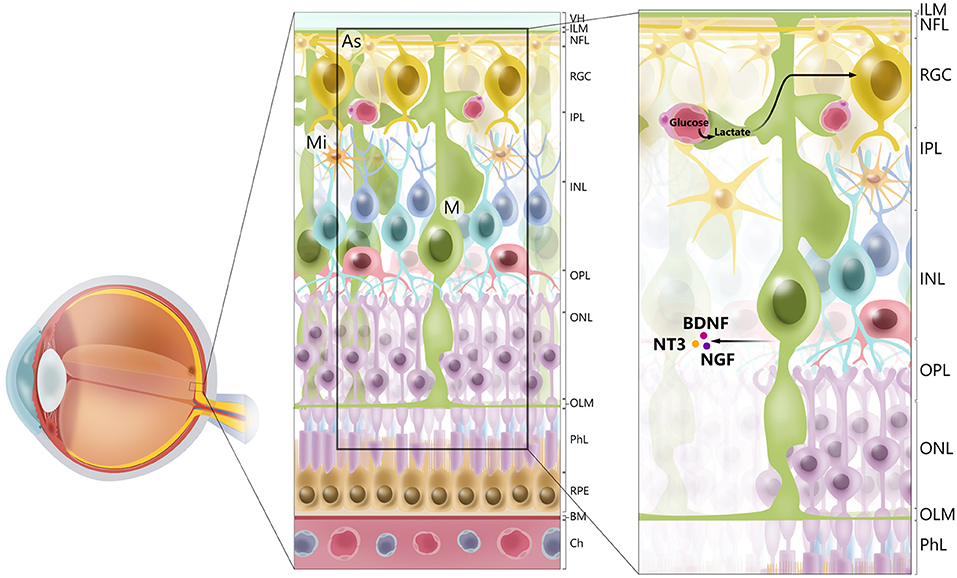
Figure 1. Cellular architecture of mammalian retina. Retina consists of 10 layers. Starting with layer furthest away from vitreous humor (VH), layers of retina are retinal pigment epithelium (RPE), photoreceptor layer (PhL), outer limiting membrane (OLM), outer nuclear layer (ONL), outer plexiform layer (OPL), inner nuclear layer (INL), inner plexiform layer (IPL), retinal ganglion cell layer (RGC), retinal nerve fiber layer (RGC axons) (NFL), and inner limiting membrane (ILM). Main types of glial cells found in mammalian retina are macroglia [Müller glia (MG) and astrocytes (As), and microglia (Mi)]. Glial cells serve to maintain retinal homeostasis. As illustrated, Müller glia traverses retina, providing structural support to neurons, mediating transport of molecules between retinal vessels and RGCs, and secreting neurotrophic factors, such as BDNF, NT3, and NGF. Furthermore, processes of Müller glia comprise outer and inner limiting membranes of retina. Müller glia, end-feet of astrocytes and vascular endothelial cells, and basal membrane constitute blood–retinal barrier. BM. basal membrane; Ch, choriocapilaris.
Glutamate Clearance to Avoid Neurotoxicity
An essential role of Müller glia is their ability to rapidly remove excess glutamate from the extracellular space by amino acid transporters (excitatory amino acid transporters), keeping it at low concentrations to avoid excitotoxicity (18, 24, 35, 36). Glutamate is converted to glutamine via the glial-specific enzyme glutamine synthetase. Glutamine subsequently serves as the precursor for glutamate in neurons. In patients with glaucoma and some animal models of the disease, an augmentation of glutamine expression in Müller glia has been shown, indicating an enhanced activation of the glutamate–glutamine cycle (37–39). In addition, it is thought that increased glutamine levels in Müller glia might be due to reduced glutamine requirement in damaged RGCs (37). In addition to removing excess glutamate from the synapse, Müller glia can also use glutamate as a metabolic substrate (16, 18, 26) (Figure 2).
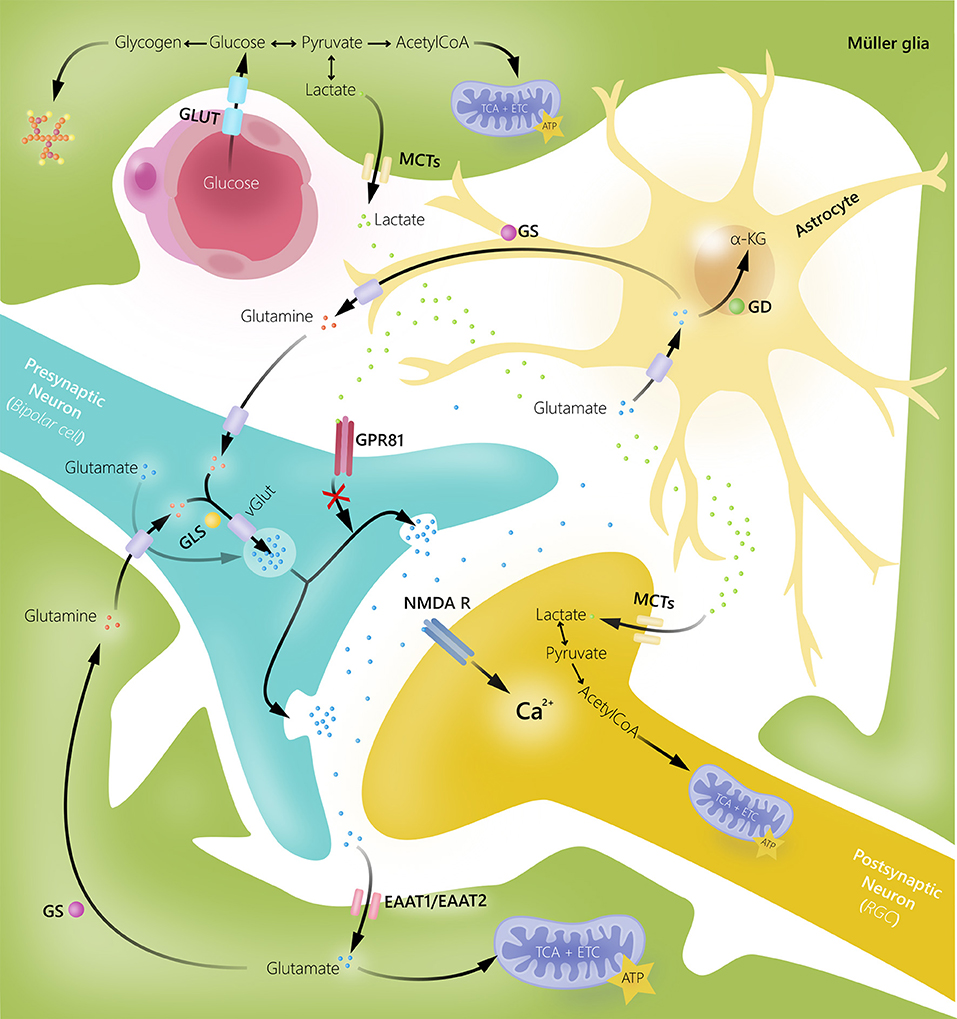
Figure 2. Glial cell and neuronal interactions in human retina. Müller glia and astrocytes take up excess extracellular glutamate to prevent glutamate-induced excitotoxic damage of retinal ganglion cells (RGC). Once glutamate is transported into glial cells, it is converted into glutamine by glutamine synthetase (GS). Glutamine can then be released by glial cells, taken up by neurons, converted into glutamate by glutaminase (GLS), and reused in synaptic neurotransmission. Glutamate can also be converted into α-ketoglutarate by glutamate dehydrogenase (GD) and used as an energy substrate. Müller glia supply bipolar cells and RGCs with energy substrates in form of lactate. Additionally, lactate released by Müller glia may function as a signaling molecule for G-protein coupled receptor 81 (GPR81) to inhibit glutamate release. EAAT, excitatory amino acid transporter; GLUT, glucose transporter; MCTs, monocarboxylate transporters; NMDAR, N-methyl-D-aspartate receptor.
Glial Cell Line-Derived Neurotrophic Factor in Glutamate Homeostasis
Another crucial Müller glia feature is the ability to release neurotrophic factors. In this context, studies have shown that ischemia-induced glial cell activation results in the release of glial cell line-derived neurotrophic factor (GDNF), which increases glutamate uptake, thereby potentially facilitating neuroprotection by reducing glutamate-induced excitotoxicity (37, 40). The potential neuroprotective role of GDNF has been supported in a rat ocular hypertension model, where an intravitreal injection of GDNF-containing microspheres was shown to increase RGC survival while reducing glia cell activation (41). The neuroprotective effect of GDNF has furthermore been associated with reduced activation of the L-glutamate receptor, N-methyl-D-aspartate receptor (NMDAR) (42), by receptor desensitization and downregulation in both neocortical neurons and astrocytes through activation of mitogen-activated protein kinase (MAPK) (43, 44). However, there are conflicting results regarding the effect of GNDF on glutamate homeostasis, with one study suggesting that GDNF pre-treatment can increase neuronal cell death via upregulation of glutamate transporters with a consequent increased excitotoxic concentration of glutamate (45).
N-Methyl-D-Aspartate Receptor Activation Is Crucial in Retinal Homeostasis
In general, safeguards against glutamate excitotoxicity have been proposed to be important treatment targets to prevent retinal neurodegeneration. In particular, NMDAR activation has been extensively studied and found to be essential for retinal homeostasis but, at the same time, to cause neurodegeneration when overactivated (46). To activate NMDARs, D-serine, or glycine along with glutamate are required. D-serine is a physiological coagonist of the NMDA subtype of glutamate receptor (47). The enzyme serine racemase has been shown to catalyze the conversion of L-serine to D-serine in rats and mouse Müller glia and in cortical astrocytes (46, 48). Furthermore, D-serine has been shown to play an important regulatory role in NMDAR response to light-evoked activity in retinal neurons (49). D-serine and serine racemase are mainly localized in Müller glia and retinal astrocytes (48), and in this regard, glia dysregulation of D-serine metabolism has been associated with retinal neurodegeneration, including glaucoma (47, 50).
Glycolysis Is the Energetic Support of Müller Glia
Bioenergetic support is central to retinal homeostasis, as the retina is one of the most metabolically active tissues in the body (51). Although retinal neurons are highly dependent on mitochondrial phosphorylation to produce adenosine triphosphate (ATP), aerobic glycolysis has been shown to be the major provider of ATP in Müller glia (52). It is still unclear why glucose is not completely oxidized under aerobic glycolysis conditions in Müller glia. Some studies have suggested that the absence of the aspartate glutamate carrier (AGC) in Müller glia may explain the predominance of glycolysis in these cells (53, 54). Thus, AGC is a major component of the malate-aspartate shuttle (MAS) that translocates electrons produced during glycolysis to mitochondria to oxidize glucose (54). Despite some studies claiming that oxidative phosphorylation is less likely in Müller glia, we and others have observed that Müller glia switch primarily to mitochondrial respiration under glucose-deprived conditions (26, 55–58), whereas in the presence of sufficient intracellular glucose levels, Müller glia mainly rely on a combination of aerobic and anaerobic glycolysis (59, 60).
Lactate Can Act as a Primary Energy Source
The predominant glycolysis during normal conditions results in the aerobic conversion of glucose into lactate, which is thought to be shuttled to the RGCs. The shuttling of lactate from glia to retinal neurons was originally hypothesized by Pellerin et al. (61). Their hypothesis predicted that uptake of glutamate would trigger glycolytic production of lactate, which in turn would be released and taken up by the surrounding neurons to fuel oxidative metabolism (62). It is clear that the lactate shuttle alone cannot explain the complex partnership between Müller glia and RGCs. Although lactate is highly produced in Müller glia, the overall role of lactate is likely to be greater in the retina compared with other tissues. Hence, the content of lactate is much higher in the retina compared with other tissues (16), and studies have reported a preference for lactate as a primary energy source in both Müller glia and RGCs compared with glucose (58, 63, 64).
It is clear that lactate is at the crossroads between glycolytic and oxidative energy metabolism and that more studies are necessary to understand further the roles of lactate in retinal homeostasis and pathology (65).
Mitochondrial Dysfunction Is Associated With Glaucoma
Nevertheless, lactate metabolism is tightly associated with mitochondrial function, and disturbances in such have been identified as important to numerous retinal and optic nerve diseases, including glaucoma (52, 66, 67). Thus, age-related impairments of mitochondrial function may exacerbate these diseases (67–70). Mitochondrial genetic variations have been associated with primary open-angle glaucoma (POAG) in large genetic studies (71). In particular, genes involved in mitochondrial lipid and carbohydrate metabolism pathways have been implicated in the pathogenesis of POAG and normal-tension glaucoma (72). In line with these findings, metabolomic studies have also identified dysfunctional carbohydrate metabolism in POAG (73). In parallel, Müller glia seems to be vulnerable to the effects of metabolic stress and mitochondrial dysfunction (52, 54, 59, 74, 75). Because many of the essential functions of Müller glia are energy-dependent, mitochondrial dysfunction leaves these cells vulnerable during glucose restriction (52, 59).
Retinal Diseases Cause Müller Gliosis and Nitric Oxide Production
As Müller glia span the entire depth of the neural retina, they are vulnerable to most forms of retinal injury. Accordingly, Müller gliosis, characterized by increased expression of the glial fibrillary acidic protein and activation of extracellular signal-regulated kinases, occurs in a wide range of retinal diseases (23, 76–78). Immediately after injury, Müller gliosis may be neuroprotective due to the production and release of antioxidants and trophic factors, including expression of ciliary body-derived neurotrophic factor (CNTF) (79). In contrast, later-stage gliosis has been associated with cell death and the establishment of a glial scar that inhibits neuronal regeneration (15, 80, 81). A particular pathological event during gliosis is the accumulation of nitric oxide (NO). Thus, NO has been shown to cause intracellular damage by inhibiting mitochondrial function, lowering ATP, and via direct damage to DNA. Furthermore, in a rat experimental glaucoma model, NO synthase (NOS) levels were found to be increased at the optic nerve head in response to IOP increase, resulting in increased NO levels (82). Despite the substantial literature on the neurotoxic effects of NO, NO also plays an important role in regulating retinal vascular tone to match neuronal activity (83). Thus, although NO contributes to neurovascular coupling and retinal homeostasis, it may become injurious in excess and in the context of retinal injury.
Overall, there is a growing body of evidence indicating that Müller glia is essential for RGC survival and that Müller glial dysfunction and stress are important factors in the pathogenesis of glaucoma (26, 80).
Astrocytes and Their Role in Retinal Ganglion Cell Homeostasis and Glaucoma
Retinal astrocytes, also called astroglia, link neurons to blood vessels and are located almost exclusively in the retinal nerve fiber layer (84). They have been found to provide structural and physiological support to optic nerve head axons (85) and modulate remodeling of the extracellular matrix in response to IOP elevation (86, 87). During retinal injury or in response to elevated IOP, astrocytes are activated, followed by morphological changes, such as cell body hypertrophy and loss of thick processes (17, 87). In addition, astrocyte processes have been shown to lose their parallel orientation and distribution once axons are lost (88).
Astrocytes Have Several and Different Functions in the Retina
Retinal astrocytes supply bioenergetic substrates to RGCs via the glutamate/glutamine cycle and via the transport of lactate and pyruvate (88, 89). It is thought that astrocytes account for more than 70% of the mitochondria in the optic nerve head (90, 91). In this context, astrocytes have been shown to engulf and degrade dysfunctional axonal mitochondria, a process known as transmitophagy (92, 93). Moreover, astrocytes have been shown to remove ions and recycle neurotransmitters from the extracellular space (28). During various pathological events, including elevated IOP or simply during aging, astrocytes undergo gliosis, a process of neurochemical and morphological remodeling (90, 94). Astroglial activation in glaucoma has been shown to increase the expression of many factors, including endothelin-1 (ET-1), tumor necrosis factor-α (TNF-α), oxidative stress molecules, and trophic factors, e.g., CNTF, with varied neuroprotective and harmful properties (17, 25, 32, 95, 96).
Activated Astrocytes Play a Role in Glaucoma
In the course of progressive glaucoma, reactive gliosis and inflammation may potentially promote regeneration and remodeling in the optic nerve (90, 94). Within this era, IOP-induced mechanical stress has been shown to upregulate epidermal growth factor receptor after activation of astrocytes and finally leading to a neurodegenerative response with the upregulation of TNF-α, matrix metalloproteinases (MMPs), and endothelin and nitric oxide synthase-2 (NOS-2) (97–100). Upregulated expression of the phagocytosis-related gene Mac-2 (101) and ET-1 have also been described in experimental glaucoma as well as in the plasma and aqueous humor of glaucoma patients, indicating an association between these molecules and phagocytic degeneration of myelin in the optic nerve head transition zone in glaucoma patients (14, 92).
Astrocytes Are Involved in Retinal Homeostasis
Overall, there is considerable evidence that astrocytes are essential for the maintenance of retinal homeostasis by clearing debris from RGC axons, supporting RGCs with energy substrates, and finally by removing excess neurotransmitters from the synaptic cleft (89, 95, 102). In contrast, activated astrocytes may have detrimental properties such as secretion of neurotoxic molecules and induction of inflammatory responses (14, 17, 95, 103, 104). Despite the lower abundance of astrocytes compared with Müller glia in the retina, the two macroglia subtypes have multiple overlapping functions but also separate properties that, in most cases, remain unknown. Future studies are highly needed to investigate the individual and the similar functions of both astrocytes and Müller glia. Moreover, their potential partnership is also important to understand. Current knowledge of such a partnership is discussed later.
Oligodendrocyte and Their Essential Support of Retinal Ganglion Cell Axons
Oligodendrocytes myelinate axons in the CNS (31), significantly reducing the energy requirements for the propagation of action potential and further protecting against various cytotoxic and excitotoxic factors (105, 106). A single oligodendrocyte can produce numerous myelin segments on multiple axons (107). Myelinating oligodendrocytes express neurotransmitter receptors, ion channels, transporters, and gap junctions (108). RGC axons remain unmyelinated until they reach the retrolaminar portion of the optic nerve. At this point, the axons are ensheathed by and supported by oligodendrocytes (31).
Oligodendrocytes Support Metabolic Transport
Oligodendrocytes are great examples of metabolic coupling between glia, which furnishes essential metabolic substrates to RGC axons (108, 109). Thus, oligodendrocytes have been shown to shuttle lactate to axons, thereby promoting axonal function and survival (107, 110). Both lactate and pyruvate are transported via monocarboxylate transporters (MCTs). MCT-1 transports lactate out of the oligodendrocyte membrane, whereas MCT-2 transporters are located on the RGC-axons and transport lactate into the RGC-axon (111). Astrocytes are also involved in lactate transport, as glucose is taken up in astrocyte processes from the blood vessels and metabolized to lactate/pyruvate, which is then transported to the oligodendroctyes via gap junctions and subsequently to the axons (107).
Poor Myelination Is Involved in Neurodegeneration
An experimental link to the potential importance of impaired bioenergetic supply to axons in glaucoma has been provided. Thus, Rindholm et al. performed a study on mutant and transgenic mice with deficient proteolipid protein, a principal component of myelin, and showed that these mice had axonal degeneration, whereas the action potential propagation remained intact. Rindholm et al. further investigated the impact of myelin basic protein deficiency and reported that these mice lacked both compacted myelin and action potential propagation in the absence of axonal degeneration (112–114). Overall, these studies indicated that oligodendrocytes provide axons with support for survival and action potential propagation and that a dysbalance in oligodendrocyte myelination can be detrimental for RGC axon function and survival (112).
Mitochondrial Dysfunction Is Associated With Glaucoma
Mitochondrial dysfunction has also been shown to play crucial roles in the homeostasis of oligodendrocytes. Thus, mitochondria are essential for the development of myelin sheaths and the development of carbon skeletons and lipid metabolism (115). Oxygen starvation and glucose deprivation have furthermore been shown to inhibit myelin development, and added together, multiple stressors have been found to have a detrimental impact on oligodendrocytes and thus associated with retinal neurodegeneration, such as seen in glaucoma (116).
In humans, optic nerve oligodendrocytes from glaucoma patients have been found to have smaller mitochondria compared with age-matched controls (116), supporting findings in the DBA/2J mouse model of glaucoma in which bioenergetic impairment was associated with axonal degeneration (114). Overall, there is some evidence for the involvement of oligodendrocytes in the pathogenesis of axonal dysfunction and loss. However, future studies are needed to investigate further how these glial cells predispose to axonal metabolic compromise and loss in glaucoma (117).
Microglia and Their Impact on Retinal Ganglion Cell Survival and Function
Microglia are innate immune cells of the CNS. Activation of microglia may be triggered by multiple events (20), such as ATP release from nerve terminals, activated immune cells or damaged cells (118), neurotransmitter accumulation (119), the release of growth factors or cytokines (120), and changes in ion homeostasis (121). Activation of microglia is highly regulated. Hence, beneficial activation of microglia leads to the secretion of anti-inflammatory cytokines, such as interleukin (IL)-10 vs interleukin-10 (IL-10) that inhibits the production of pro-inflammatory cytokines by microglia (122–124). In contrast, marked activation of microglia in the setting of major insults results in the release of pro-inflammatory cytokines and cytotoxic agents, such as TNF-α, IL-1β, IL-6, inducible NOS, and NO, which in turn kill potential pathogens (125, 126) (Table 1).
Table 1. Effector molecules of activated microglia.
Activated microglia migrate toward sites of injury due to the expression of β-integrin CD11α (126) in a process mediated by the transcription factor nuclear factor-kappa-light-chain-enhancer of activated B cells (2). In addition, microglia support myelination, oligodendrogenesis, and neurogenesis, as well as stimulate synaptic formation and maturation (144).
Activated Microglia and Glaucoma
Substantial evidence has shown microglial involvement in several eye diseases, including glaucoma (34, 145–148). In glaucoma, activated microglia have been described in clusters around blood vessels in the injured optic nerve and choriocapillaris, indicating an innate immune activation (149, 150). Dual roles for activated microglia have been proposed in glaucoma. On the one hand, activated microglia can phagocytose degenerated or dying RGCs, thereby maintaining a retinal environment free of toxic molecules (33), as well as the ability to secrete neurotrophic factors, such as brain-derived neurotrophic factor (BDNF) and CNTF (151), which provide neuroprotection and possibly promote neuroregeneration (95). On the other hand, chronic microglial activation leads to the release of neurotoxic and pro-inflammatory molecules, as mentioned earlier (28). In rodent glaucoma models, where mice were exposed to either hypoxic damage or ocular hypertension, a release of TNF-α and IL-1β from activated microglia accompanied by apoptosis of RGCs was found, supporting the involvement of microglia in glaucomatous neurodegeneration (152, 153). In addition, in an experimental rat model of glaucoma, microglial activation was shown to increase inducible NOS expression, NO production, and RGC injury (154). Activated microglia have furthermore been shown to release reactive oxygen species and prostaglandin E2, which predispose to RGC apoptosis (155). In glaucoma, microglial cell activation at the optic nerve head has been shown to be associated with altered cellular morphology, protein expression, and antigen-presentation (149, 156). Moreover, damage-associated molecular patterns (DAMPs), released by RGCs or by astroglia in the optic nerve head, which can trigger an inflammatory response, have been shown in response to elevated IOP (32). Finally, microglial activation has been correlated with axonal degeneration in an experimental glaucoma model (33).
Microglia, Friend, or Foe?
As with macroglia, significant research has suggested that microglia can be considered either friend and foe depending on the degree of activation and context (21, 77, 143, 157, 158). One mechanism by which microglial activation is controlled is via a series of cell surface receptors (33). Thus, microglia are usually activated only when necessary to minimize safety damage to neighboring cells (159). Consequently, damage to RGCs may occur as observed in glaucoma when microglial homeostasis is disrupted. The expression of several inhibitory receptors decreases with age. Examples of such receptors are CX3CR1 and CD200. Ligand binding to CX3CR1 is crucial for the elimination of damaged or dying cells, whereas ligand binding to CD200 receptors leads to modulation of activated microglia during chronic as well as acute inflammation (138, 159). Other receptors vary with sex and age, such as the purinergic receptors P2 that bind to ATP to mediate intercellular communication (159). In a mice model of glaucoma, it has been demonstrated that a deficient activation of CX3CR1 enhances microglia activation and leads to neurotoxic loss of RGCs (34).
Emerging evidence exists on the impact of autophagy in microglia activation (160). In this context, autophagy modulation is thought to regulate microglia phagocytosis and inflammatory response (160, 161). Autophagy can either be considered as pro-inflammatory or anti-inflammatory depending on the acute or chronic stage of the injury (161). Potentially, the balance between autophagy and microglia can be regulated by pharmacological inhibition of, e.g., 3-methyladenine. Finally, studies indicate that some humans are genetically impaired of basal autophagy, which can impact retinal homeostasis and potentially promote retinal neurodegeneration, hereunder glaucoma (162–164). Future studies are needed to investigate further the partnership between autophagy and microglia as well as to elucidate further the interaction between microglia as well as other glia subtypes and RGCs.
Retinal Glial Interactions
Emerging evidence has identified the importance of cross talk between retinal glial cells in health and disease (14, 165–167) (Figure 3). In general, glia interactions attempt to maintain retinal homeostasis and regulate each other's activity. However, glia interactions can also create imbalance and thus contribute to retinal neurodegeneration.
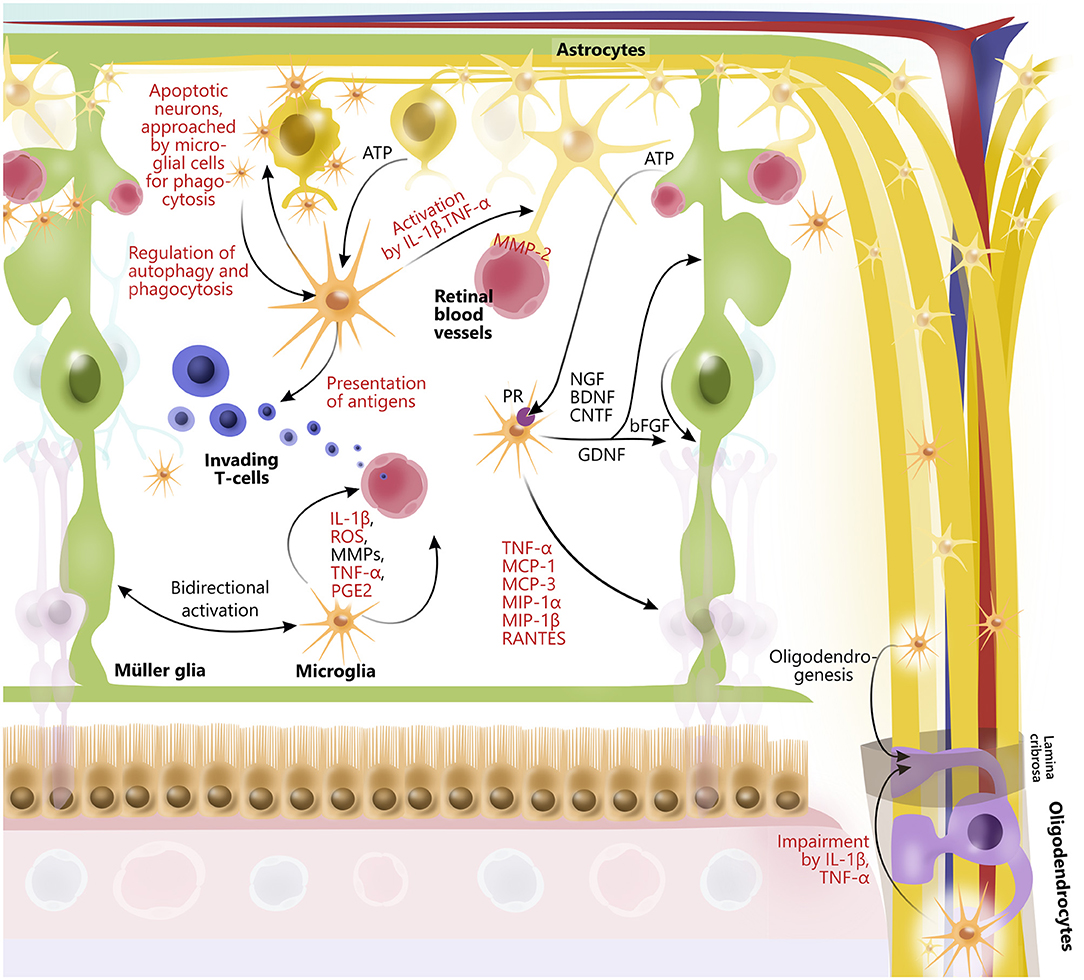
Figure 3. Glial interactions in retina and optic nerve. In retina, astrocytes, Müller glia, oligodendrocytes, and microglia widely interact to maintain retinal homeostasis by release of trophic factors and cytokines, ATP-exchange, phagocytosis of neuronal debris, antigen presentation, and by promoting activity of each other. Furthermore, retinal glia cells are interactively involved in maintaining retinal vessels as well as blood–retinal barrier, for example, through interactions between microglia and astrocytes. Some glial interactions also impair function of other glial cells. In particular, microglia via release of cytokines (IL-1β and TNF-alpha) affect function of oligodendrocytes, which myelinate axons of retinal ganglion cells in optic nerve. However, microglia can also contribute to oligodendrogenesis. Although glial interactions attempt to maintain retinal homeostasis, they can also promote retinal neurodegeneration. Prodegenerative factors released from glial cells to interact with one another are highlighted in red.
An example of a glia interaction is the modulation of T cell response due to microglia antigen presentation (165), which in turn regulate the inflammatory cytokine levels, including TNF-α and IL-1β, followed by astrocyte activation (28, 168). Furthermore, microglial migration and immune cell recruitment have been correlated with Müller glial activation (14), indicating tight coordination of retinal immune responses (169). In line with this, activated microglia secrete TNF-α, CCL2 (MCP-1), MCP-3, MIP-1α, MIP-1β, and CCL5 (RANTES), which mediate activation and recruitment of additional microglia, amplifying the inflammatory response (84, 165, 170). In addition, microglia-derived IL-1β has been shown to upregulate the expression of Ccl2, Cxcl1, and Cxcl10 in Müller glia, which has been associated with retinal neurodegeneration (171). Another example of microglia and Müller glia interaction is the secretion of microglia-derived nerve growth factor (NGF), BDNF, and CNTF from microglia that in themselves protect RGCs (172) but also modulate the production of basic fibroblast growth factor and GDNF in Müller glia, conferring neuroprotection (167, 173). Both astrocytes, microglia, oligodendrocytes, and neurons secrete MMPs (174). Under normal conditions, astrocytes and microglia express MMP-2 (gelatinase A) in the foot processes near blood vessels (175). Upon astrocyte and microglial activation, MMP-2 is, however, increased, thereby causing increased permeability of the blood–retinal barrier (176), angiogenesis, and glial scar formation (177). Activated microglia also express MMP-3 (stromelysin-1), which in turn activates proMMP-9 (178). MMP-9 has been found to be elevated when there is an increase in the blood–retinal barrier permeability. In line with this, an MMP-9 increase has been shown in diabetic rat retinas when glucose levels rise (176). MMP-9 has also been implicated in myelin basic protein degradation, and it has therefore been suggested that MMP-9 is associated with demyelination and axonal injury (178–180). Finally, both the effects of MMP-9 and MMP-3 have been shown to be enhanced by TNF-α and IL-1, further indicating complex interactions between molecules secreted by different retinal glia (181–183).
Glia–glia interactions have also been shown between oligodendrocytes and microglia as well as astrocytes. In this context, studies have shown that microglial activation can cause astrogliosis and apoptosis of oligodendrocytes via TNF-α and IL-1β, causing an increase in MMP production in astrocytes and microglia as well as a possible induction of oxidative stress, inflammation, and glutamate toxicity (183–185).
Although there is currently relatively sparse literature on glia–glia interactions, it can be expected that this interaction will prove to be very relevant for both the understanding of retinal homeostasis and for neurodegenerative diseases such as glaucoma (186).
Therapeutic Approaches
Targeting Retinal and Optic Nerve Glial Cells to Treat Glaucoma
Glial cells in the retina and the optic nerve play an important role in supporting RGCs and their axons, and because glial cell dysfunction has been implicated in glaucoma, it is conceivable that future treatments for glaucoma may target glial cells.
Targeting Müller Glia and Astrocytes
Modulation of Glutamate via N-Methyl-D-Aspartate Receptors
RGC damage leads to elevated glutamate levels, causing NMDARs to be overstimulated with a consequent increase in Ca2+ influx and excitotoxicity (187). Impaired clearance of glutamate by Müller glia is involved in the pathogenesis of both glaucoma and diabetic retinopathy (37, 188, 189). Amidation and oxidation are the two routes of glutamate disposal. In a cultured rat retinal Müller cell line, treatment with hydrocortisone was shown to increase the amidation of glutamate to glutamine, whereas the addition of branched-chain keto acids was found to enhance oxidation of glutamate, suggesting that intracellular levels of glutamate play a role in the removal of extracellular glutamate (35). In a rodent model of ocular hypertension, the use of memantine nanoparticles, a non-competitive NMDAR antagonist, conferred RGCs neuroprotection (190). Unfortunately, oral memantine did not show any significant prevention of glaucoma progression when tested in a clinical trial (191). Another modulator of glutamate-induced toxicity, brimonidine, has been proposed to slow the rate of glaucoma progression in glaucoma patients in an additive manner other than its IOP-lowering effect (192). In a rodent model, brimonidine was shown to modulate glutamate uptake by glial cells after induction of ocular hypertension, suggesting neuroprotection through modulation of macroglia (193).
Neurotrophin Administration
Neurotrophins are important in the development, differentiation, and survival of RGCs. Many of these neurotrophins are produced by glial cells during normal conditions. In this context, BDNF has been shown to protect RGCs in mice with ocular hypertension (194). Furthermore, it has been shown that GDNF combined with BDNF convey synergic protective effects (195). Finally, glial cells are important producers of neurotrophins, including NGF, BDNF, CNTF, neurotrophin-3, and neurotrophin-4/5, all having potential as neuroprotective properties (196). Among the mentioned neurotrophins, more have already been tested in preclinical settings. An example is mature BDNF, which has been targeted to secretory vesicles within RGCs by adeno-associated virus gene therapy, increasing BDNF production and long-term BDNF receptor expression in a mouse model of optic nerve damage and in a rat model of chronic IOP, which provides neuroprotection against RGCs (197). In addition, increased release of CNTF by Müller glia has been shown to provide endogenous neuroprotection of RGCs after both ischemias and in response to the induced ocular hypertension (194). Recently, an encapsulated cell technology has allowed a controlled, continuous, and prolonged administration of CNTF in animal models that provide photoreceptor protection (198). In general, the administration of exogenous neurotrophins or the augmentation of endogenous production has been shown to have a protective effect on RGCs in several experimental models, highlighting this as a potential therapeutic strategy for glaucoma (199). The efficacy of such treatments may, however, decrease over time as treatment with chronic neurotrophin administration can lead to downregulation of the relevant receptors (197).
Targeting Astrocytes and Astrocyte Activation
Astrocyte activation may be an important factor in the pathogenesis of glaucoma. In this context, inhibition of astrocyte activation has been shown to increase neuronal survival in experimental glaucoma models via modulation of a tyrosine kinase inhibitor of epidermal growth factor receptor (200) or blocking of endothelin-1 (201). Similarly, the neuroprotective effects of calcium channel blockers and endothelin blockers in humans with glaucoma are thought to act via this mechanism (202).
In addition to the previously mentioned involvement of TNF-α in microglia activation, TNF-α has also been shown to mediate both astrocyte, Müller glia, and oligodendrocyte activation followed by RGC death (203–205). In a rodent glaucoma model, this detrimental effect of TNF-α was demonstrated after intravitreal TNF-α injections and reversed by an antibody neutralizing TNF-α activity or by deleting the genes encoding TNF-α or its receptor, TNF-R2 (204). Future studies are needed to define further the potential role of TNF-α inhibitors as a treatment target for neuroprotection (206).
Nutrition may affect retinal homeostasis and, in particular, mitochondrial function. Hence, a ketogenic diet was shown to increase mitochondrial respiration, hereunder mitochondrial respiration in astrocytes (207, 208). In addition, the ketogenic diet has been reported to restore monocarboxylate transporters to boost the antioxidant response followed by preservation of RGC function and structure without affecting glycogen stores (117). Finally, a low-carbohydrate diet has been shown to reduce the risk of POAG in a US cohort (209). In summary, nutrition and ketogenic diets may increase the resistance toward glaucomatous neurodegeneration. Future studies are, however, necessary to further investigate such potential.
Targeting Oligodendrocytes
Inhibition of Inflammatory Mediators
Oligodendrocyte degeneration has been correlated to TNF-α release followed by increased IOP and optic nerve damage. In this context, suppression of TNF-α, with an anti-TNF-α blocking antibody or the deletion of the gene encoding TNF-α, was shown to elicit neuroprotective effects in the optic nerve and RGCs in a mouse model of glaucoma (204).
Inhibiting Lingo-1
LINGO-1 is a leucine-rich repeat, expressed on oligodendrocytes and neurons. It negatively regulates differentiation and myelination, neuronal survival, and axonal regeneration. In glaucoma models and human CNS diseases, LINGO-1 expression has been found to be upregulated (210). LINGO-1 has furthermore been shown to have negative regulatory functions in axonal regeneration, neuronal survival, and oligodendrocyte differentiation as well as myelination. In addition, LINGO-1 was found to be increased in models of spinal cord injury and glaucoma (211). The LINGO-1 monoclonal antibody, BIIB033, has shown promise as a neuroprotective and neuroregenerative strategy in clinical studies, but continued evaluations are needed to confirm this promising effect in glaucoma patients (211).
Targeting Microglia
Inhibition of Microglial Activation
Microglia have increasingly been identified as targets in glaucoma neuroprotection and neuroregeneration (22, 28, 212, 213). The blockade of the microglial adenosine A2A receptor has been shown to protect RGCs from elevated IOP in murine glaucoma models by controlling the microglial activation and inhibiting reactive oxygen species (214). Minocycline has also been shown to inhibit microglial activation and upregulate pro-survival genes in experimental glaucoma (215, 216). Although minocycline has proven oral safety and has been found to cross the blood–brain barrier, long-term randomized control trials with the necessary high doses of minocycline are needed (217).
Modulation of Microglial Activation
In addition to the harmful effects of microglia, the activation of these cells can also benefit the healthy eye, where such activation, e.g., acute inflammation, has been shown to protect against neuronal damage (22). Microglia activation is regulated by several inhibitory pathways, such as the ligand fractalkine (FXN or Cx3cl1) in neurons and the receptors Cx3cr1 and CD200R in microglia (218). The loss of Cx3xl1 signaling has been shown to exacerbate dysfunctional axon transport in RGCs, increased CCR2+ macrophages infiltration (219), and upregulation of NOS-2 in myeloid cells from DBA/2J mice (219).
CD200 is expressed in the vascular endothelium of the retina, photoreceptors, and RGCs (34) and interacts with CD200R to modulate microglial activation (220). Breakdown of CD200-CD200R is involved in RGC loss in experimental glaucoma (221). Thus, in a rat model with optic nerve crush, the agonist of CD200R known as CD200Fc was shown to increase CD200R expression and inhibit CD200 expression, thereby assisting in the neuroprotection of RGCs (222, 223). Overall, microglial activation may be beneficial for RGC function and survival, and numerous studies emphasize the dual roles of microglia activation. However, more studies are needed to further understand such potential beneficial functions of microglia in glaucoma.
Inhibition of Inflammatory Mediators
In advanced glaucoma, inflammatory mediators, including TNF-α, are increased. Inhibition or genetic deletion of TNF-α reduces the activation of microglia (32, 224), and the blockade of TNF-α signaling has been shown to protect RGCs in an experimental glaucoma mouse model (204, 225).
The Fas ligand (FasL), a member of the TNF protein family, links microglia activation and the induction of apoptosis of RGCs through the Fas receptor. In the eye, FasL can be expressed as the membrane-bound pro-apoptotic and pro-inflammatory protein (mFasL) or as the soluble, non-apoptotic, and non-inflammatory form sFasL (226). Previous studies have shown that FasL is constitutively expressed in ocular tissue, where the ligand helps maintain the immune-privileged state of the eye and helps prevent neoangiogenesis. However, FasL has also been shown to play an important role in retinal neurotoxicity. In this context, FasL has been shown to accelerate RGC death in an experimental glaucoma model (227). In line with this, the peptide inhibitor of the Fas receptor, ONL1204, has been found to halt mFasL activation by inhibition of microglial activation, inflammation, and apoptosis of RGCs in a mice model (228). In contrast to the neurotoxic effects of FasL, FasL administration has been shown to protect RGCs from cell death (227). Overall, the contradictive roles of FasL and mFasL require more studies to investigate the potential roles of FasL/mFasL modulation in future neuroprotective treatments.
Reducing Oxidative Stress
Orally administered docosahexaenoic acid and intravitreal injection of polysialic acid has been shown to reduce microglial activation by decreasing oxidative stress and inflammation in rodent models of glaucoma and experimental glaucoma models (146). Moreover, the natural resin mixture, propolis, produced by honeybees, has been shown to reduce neuroinflammatory responses and reduce oxidative stress in microglia cell cultures by inhibiting nuclear factor-κB when cultures were exposed to hypoxia (229).
Macrophage Pro-Inflammatory Cytokines
Glial cell activation triggers macrophage infiltration and the release of pro-inflammatory cytokines that further activate glial cells. Such pro-inflammatory cytokines have been found to be upregulated in both the blood and in the aqueous humor of patients with POAG (133, 230). Accordingly, treatments targeting macrophage-derived pro-inflammatory cytokines, such as IL-1β, may be used in the future treatment of glaucoma (133, 194).
Conclusion
Glial cells play complex and multifactorial roles in glaucoma. Advances in our understanding of the nature and regulation of these various roles in health and disease have enabled the identification of novel therapeutic strategies to protect RGCs in glaucoma. Therapies targeting glial cells or those emulating the protective effects of these cells on RGCs will likely go hand-in-hand with conventional therapies to lower IOP and with emerging approaches that aim to augment neuronal bioenergetic resilience and promote axonal repair.
Author Contributions
MG-B has search the literature and written the first draft. MK has been main responsible for the process. ZM has designed the figures. KM, PW, and KF have all been participating in critical commenting and proof reading. All authors have contributed to the writing process.
Funding
The study was supported by the Velux Foundation, Denmark; Bagenkop-Nielsens Eye-Foundation, Denmark and the Hørslev Foundation, Denmark.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Victorian Government for the Operational Infrastructure Support to the Center for Eye Research Australia and the Innovation Foundation Denmark BrainStem.
References
1. Rieck J. The pathogenesis of glaucoma in the interplay with the immune system. Invest Ophthalmol Vis Sci. (2013) 54:2393–409. doi: 10.1167/iovs.12-9781
2. Tsai T, Reinehr S, Maliha AM, Joachim SC. Immune mediated degeneration and possible protection in glaucoma. Front Neurosci. (2019) 13:931. doi: 10.3389/fnins.2019.00931
3. Kolko M, Horwitz A, Thygesen J, Jeppesen J, Torp-Pedersen C. The prevalence and incidence of glaucoma in Denmark in a fifteen year period: a nationwide study. PLoS ONE. (2015) 10:e0132048. doi: 10.1371/journal.pone.0132048
4. Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. (2014) 121:2081–90. doi: 10.1016/j.ophtha.2014.05.013
5. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. (2014) 311:1901–11. doi: 10.1001/jama.2014.3192
6. Ohnell H, Bengtsson B, Heijl A. Making a correct diagnosis of glaucoma: data from the EMGT. J Glaucoma. (2019) 28:859–64. doi: 10.1097/IJG.0000000000001342
7. Tribble JR, Cross SD, Samsel PA, Sengpiel F, Morgan JE. A novel system for the classification of diseased retinal ganglion cells. Vis Neurosci. (2014) 31:373–80. doi: 10.1017/S0952523814000248
8. Tian N. Developmental mechanisms that regulate retinal ganglion cell dendritic morphology. Dev Neurobiol. (2011) 71:1297–309. doi: 10.1002/dneu.20900
9. Wareham LK, Risner ML, Calkins DJ. Protect, repair, and regenerate: towards restoring vision in glaucoma. Curr Ophthalmol Rep. (2020) 8:301–10. doi: 10.1007/s40135-020-00259-5
10. Peters D, Bengtsson B, Heijl A. Lifetime risk of blindness in open-angle glaucoma. Am J Ophthalmol. (2013) 156:724–30. doi: 10.1016/j.ajo.2013.05.027
11. Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. (2012) 31:152–81. doi: 10.1016/j.preteyeres.2011.11.002
12. Aires ID, Ambrosio AF, Santiago AR. Modeling human glaucoma: lessons from the in vitro models. Ophthalmic Res. (2017) 57:77–86. doi: 10.1159/000448480
13. Kolko M. Mitochondria and the eye diseases - editorial. Mitochondrion. (2017) 36:1–3. doi: 10.1016/j.mito.2017.08.010
14. Chong RS, Martin KR. Glial cell interactions and glaucoma. Curr Opin Ophthalmol. (2015) 26:73–7. doi: 10.1097/ICU.0000000000000125
15. Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. (2006) 25:397–424. doi: 10.1016/j.preteyeres.2006.05.003
16. Vohra R, Kolko M. Neuroprotection of the inner retina: Muller cells and lactate. Neural Regen Res. (2018) 13:1741–2. doi: 10.4103/1673-5374.238612
17. Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC. Glia-neuron interactions in the mammalian retina. Prog Retin Eye Res. (2016) 51:1–40. doi: 10.1016/j.preteyeres.2015.06.003
18. Skytt DM, Toft-Kehler AK, Braendstrup CT, Cejvanovic S, Gurubaran IS, Bergersen LH. Glia-neuron interactions in the retina can be studied in cocultures of muller cells and retinal ganglion cells. Biomed Res Int. (2016) 2016:1087647. doi: 10.1155/2016/1087647
19. Bringmann A, Wiedemann P. Muller glial cells in retinal disease. Ophthalmologica. (2012) 227:1–19. doi: 10.1159/000328979
20. Bosco A, Steele MR, Vetter ML. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol. (2011) 519:599–620. doi: 10.1002/cne.22516
21. Ghosh A, Streit WJ, Minghetti L, Basu A. Microglia in development and disease. Clin Dev Immunol. (2013) 2013:736459. doi: 10.1155/2013/736459
22. Adornetto A, Russo R, Parisi V. Neuroinflammation as a target for glaucoma therapy. Neural Regen Res. (2019) 14:391–4. doi: 10.4103/1673-5374.245465
23. de Hoz R, Rojas B, Ramirez AI, Salazar JJ, Gallego BI, Trivino A, et al. Retinal macroglial responses in health and disease. Biomed Res Int. (2016) 2016:2954721. doi: 10.1155/2016/2954721
24. Kawasaki A, Otori Y, Barnstable CJ. Muller cell protection of rat retinal ganglion cells from glutamate and nitric oxide neurotoxicity. Invest Ophthalmol Vis Sci. (2000) 41:3444–50.
25. Guttenplan KA, Stafford BK, El-Danaf RN, Adler DI, Munch AE, Weigel MK, et al. Neurotoxic reactive astrocytes drive neuronal death after retinal injury. Cell Rep. (2020) 31:107776. doi: 10.1016/j.celrep.2020.107776
26. Toft-Kehler AK, Skytt DM, Kolko M. A perspective on the muller cell-neuron metabolic partnership in the inner retina. Mol Neurobiol. (2018) 55:5353–61. doi: 10.1007/s12035-017-0760-7
27. Gonzalez-Perez O, Lopez-Virgen V, Quinones-Hinojosa A. Astrocytes: everything but the glue. Neuroimmunol Neuroinflamm. (2015) 2:115–7. doi: 10.4103/2347-8659.153979
28. Wang JW, Chen SD, Zhang XL, Jonas JB. Retinal microglia in glaucoma. J Glaucoma. (2016) 25:459–65. doi: 10.1097/IJG.0000000000000200
29. Kolb H, Fernandez E, Nelson R (editors). Webvision: The Organization of the Retina and Visual System. Salt Lake City, UT: University of Utah Health Sciences Center (1995).
30. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
31. Simons M, Nave KA. Oligodendrocytes: myelination and axonal support. Cold Spring Harb Perspect Biol. (2015) 8:a020479. doi: 10.1101/cshperspect.a020479
32. Wei X, Cho KS, Thee EF, Jager MJ, Chen DF. Neuroinflammation and microglia in glaucoma: time for a paradigm shift. J Neurosci Res. (2019) 97:70–6. doi: 10.1002/jnr.24256
33. Silverman SM, Wong WT. Microglia in the retina: roles in development, maturity, and disease. Annu Rev Vis Sci. (2018) 4:45–77. doi: 10.1146/annurev-vision-091517-034425
34. Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, et al. The role of microglia in retinal neurodegeneration: Alzheimer's disease, parkinson, and glaucoma. Front Aging Neurosci. (2017) 9:214. doi: 10.3389/fnagi.2017.00214
35. Ola MS, Hosoya K, LaNoue KF. Regulation of glutamate metabolism by hydrocortisone and branched chain keto acids in cultured rat retinal Muller cells (TR-MUL). Neurochem Int. (2011) 59:656–63. doi: 10.1016/j.neuint.2011.06.010
36. Reichenbach A, Bringmann A. New functions of Muller cells. Glia. (2013) 61:651–78. doi: 10.1002/glia.22477
37. Carter-Dawson L, Shen F, Harwerth RS, Smith EL 3rd, Crawford ML, Chuang A. Glutamine immunoreactivity in Muller cells of monkey eyes with experimental glaucoma. Exp Eye Res. (1998) 66:537–45. doi: 10.1006/exer.1997.0447
38. Shen F, Chen B, Danias J, Lee KC, Lee H, Su Y, et al. Glutamate-induced glutamine synthetase expression in retinal Muller cells after short-term ocular hypertension in the rat. Invest Ophthalmol Vis Sci. (2004) 45:3107–12. doi: 10.1167/iovs.03-0948
39. Dkhissi O, Chanut E, Wasowicz M, Savoldelli M, Nguyen-Legros J, Minvielle F, et al. Retinal TUNEL-positive cells and high glutamate levels in vitreous humor of mutant quail with a glaucoma-like disorder. Invest Ophthalmol Vis Sci. (1999) 40:990–5.
40. Vohra R, Tsai JC, Kolko M. The role of inflammation in the pathogenesis of glaucoma. Surv Ophthalmol. (2013) 58:311–20. doi: 10.1016/j.survophthal.2012.08.010
41. Jiang C, Moore MJ, Zhang X, Klassen H, Langer R, Young M. Intravitreal injections of GDNF-loaded biodegradable microspheres are neuroprotective in a rat model of glaucoma. Mol Vis. (2007) 13:1783–92.
42. Bonde C, Kristensen BW, Blaabjerg M, Johansen TE, Zimmer J, Meyer M. GDNF and neublastin protect against NMDA-induced excitotoxicity in hippocampal slice cultures. Neuroreport. (2000) 11:4069–73. doi: 10.1097/00001756-200012180-00032
43. Gegelashvili G, Dehnes Y, Danbolt NC, Schousboe A. The high-affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signalling mechanisms. Neurochem Int. (2000) 37:163–70. doi: 10.1016/S0197-0186(00)00019-X
44. Nicole O, Ali C, Docagne F, Plawinski L, MacKenzie ET, Vivien D, et al. Neuroprotection mediated by glial cell line-derived neurotrophic factor: involvement of a reduction of NMDA-induced calcium influx by the mitogen-activated protein kinase pathway. J Neurosci. (2001) 21:3024–33. doi: 10.1523/JNEUROSCI.21-09-03024.2001
45. Bonde C, Sarup A, Schousboe A, Gegelashvili G, Noraberg J, Zimmer J. GDNF pre-treatment aggravates neuronal cell loss in oxygen-glucose deprived hippocampal slice cultures: a possible effect of glutamate transporter up-regulation. Neurochem Int. (2003) 43:381–8. doi: 10.1016/S0197-0186(03)00025-1
46. Wolosker H. The neurobiology of d-Serine signaling. Adv Pharmacol. (2018) 82:325–48. doi: 10.1016/bs.apha.2017.08.010
47. Zhang X, Zhang R, Zhou X, Wu J. Decreased d-Serine levels prevent retinal ganglion cell apoptosis in a glaucomatous animal model. Invest Ophthalmol Vis Sci. (2018) 59:5045–52. doi: 10.1167/iovs.18-24691
48. Dun Y, Duplantier J, Roon P, Martin PM, Ganapathy V, Smith SB. Serine racemase expression and D-serine content are developmentally regulated in neuronal ganglion cells of the retina. J Neurochem. (2008) 104:970–8. doi: 10.1111/j.1471-4159.2007.05015.x
49. Stevens ER, Esguerra M, Kim PM, Newman EA, Snyder SH, Zahs KR, et al. D-serine and serine racemase are present in the vertebrate retina and contribute to the physiological activation of NMDA receptors. Proc Natl Acad Sci USA. (2003) 100:6789–94. doi: 10.1073/pnas.1237052100
50. Wolosker H, Balu DT. D-Serine as the gatekeeper of NMDA receptor activity: implications for the pharmacologic management of anxiety disorders. Transl Psychiatry. (2020) 10:184. doi: 10.1038/s41398-020-00870-x
51. Casson RJ, Chidlow G, Crowston JG, Williams PA, Wood JPM. Retinal energy metabolism in health and glaucoma. Prog Retin Eye Res. (2020) 100881. doi: 10.1016/j.preteyeres.2020.100881. [Epub ahead of print].
52. Winkler BS, Arnold MJ, Brassell MA, Puro DG. Energy metabolism in human retinal Muller cells. Invest Ophthalmol Vis Sci. (2000) 41:3183–90.
53. Amoedo ND, Punzi G, Obre E, Lacombe D, De Grassi A, Pierri CL, et al. AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim Biophys Acta. (2016) 1863:2394–412. doi: 10.1016/j.bbamcr.2016.04.011
54. Ola MS, LaNoue KF. Molecular basis for increased lactate formation in the Muller glial cells of retina. Brain Res Bull. (2019) 144:158–63. doi: 10.1016/j.brainresbull.2018.11.023
55. Winkler BS, Starnes CA, Sauer MW, Firouzgan Z, Chen SC. Cultured retinal neuronal cells and Muller cells both show net production of lactate. Neurochem Int. (2004) 45:311–20. doi: 10.1016/j.neuint.2003.08.017
56. Winkler BS, Sauer MW, Starnes CA. Modulation of the Pasteur effect in retinal cells: implications for understanding compensatory metabolic mechanisms. Exp Eye Res. (2003) 76:715–23. doi: 10.1016/S0014-4835(03)00052-6
57. Vohra R, Aldana BI, Waagepetersen H, Bergersen LH, Kolko M. Dual properties of lactate in muller cells: the effect of GPR81 activation. Invest Ophthalmol Vis Sci. (2019) 60:999–1008. doi: 10.1167/iovs.18-25458
58. Vohra R, Aldana BI, Skytt DM, Freude K, Waagepetersen H, Bergersen LH, et al. Essential roles of lactate in Muller cell survival and function. Mol Neurobiol. (2018) 55:9108–21. doi: 10.1007/s12035-018-1056-2
59. Toft-Kehler AK, Skytt DM, Svare A, Lefevere E, Van Hove I, Moons L, et al. Mitochondrial function in Muller cells - does it matter? Mitochondrion. (2017) 36:43–51. doi: 10.1016/j.mito.2017.02.002
60. Hurley JB, Lindsay KJ, Du J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J Neurosci Res. (2015) 93:1079–92. doi: 10.1002/jnr.23583
61. Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, et al. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci. (1998) 20:291–9. doi: 10.1159/000017324
62. Magistretti PJ, Pellerin L. Metabolic coupling during activation. a cellular view. Adv Exp Med Biol. (1997) 413:161–6. doi: 10.1007/978-1-4899-0056-2_18
63. Vohra R, Aldana BI, Bulli G, Skytt DM, Waagepetersen H, Bergersen LH, et al. Lactate-mediated protection of retinal ganglion cells. J Mol Biol. (2019) 431:1878–88. doi: 10.1016/j.jmb.2019.03.005
64. Vohra R, Kolko M. Lactate: more than merely a metabolic waste product in the inner retina. Mol Neurobiol. (2020) 57:2021–37. doi: 10.1007/s12035-019-01863-8
65. Glancy B, Kane DA, Kavazis AN, Goodwin ML, Willis WT, Gladden LB. Mitochondrial lactate metabolism: history and implications for exercise and disease. J Physiol. (2020) 599:863–88. doi: 10.1113/JP278930
66. Chrysostomou V, Rezania F, Trounce IA, Crowston JG. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol. (2013) 13:12–5. doi: 10.1016/j.coph.2012.09.008
67. Ito YA, Di Polo A. Mitochondrial dynamics, transport, and quality control: a bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion. (2017) 36:186–92. doi: 10.1016/j.mito.2017.08.014
68. Lefevere E, Toft-Kehler AK, Vohra R, Kolko M, Moons L, Van Hove I. Mitochondrial dysfunction underlying outer retinal diseases. Mitochondrion. (2017) 36:66–76. doi: 10.1016/j.mito.2017.03.006
69. Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. J Alzheimers Dis. (2010) 20(Suppl. 2):S369–79. doi: 10.3233/JAD-2010-100543
70. Osborne NN. Mitochondria: their role in ganglion cell death and survival in primary open angle glaucoma. Exp Eye Res. (2010) 90:750–7. doi: 10.1016/j.exer.2010.03.008
71. Van Bergen NJ, Crowston JG, Craig JE, Burdon KP, Kearns LS, Sharma S, et al. Measurement of systemic mitochondrial function in advanced primary open-angle glaucoma and leber hereditary optic neuropathy. PLoS ONE. (2015) 10:e0140919. doi: 10.1371/journal.pone.0140919
72. Khawaja AP, Cooke Bailey JN, Kang JH, Allingham RR, Hauser MA, Brilliant M, et al. Assessing the association of mitochondrial genetic variation with primary open-angle glaucoma using gene-set analyses. Invest Ophthalmol Vis Sci. (2016) 57:5046–52. doi: 10.1167/iovs.16-20017
73. Leruez S, Marill A, Bresson T, de Saint Martin G, Buisset A, Muller J, et al. A metabolomics profiling of glaucoma points to mitochondrial dysfunction, senescence, and polyamines deficiency. Invest Ophthalmol Vis Sci. (2018) 59:4355–61. doi: 10.1167/iovs.18-24938
74. Toft-Kehler AK, Gurubaran IS, Desler C, Rasmussen LJ, Skytt DM, Kolko M. Oxidative stress-induced dysfunction of muller cells during starvation. Invest Ophthalmol Vis Sci. (2016) 57:2721–8. doi: 10.1167/iovs.16-19275
75. Vohra R, Gurubaran IS, Henriksen U, Bergersen LH, Rasmussen LJ, Desler C. Disturbed mitochondrial function restricts glutamate uptake in the human Muller glia cell line, MIO-M1. Mitochondrion. (2017) 36:52–9. doi: 10.1016/j.mito.2017.02.003
76. Baumann B, Sterling J, Song Y, Song D, Fruttiger M, Gillies M, et al. Conditional Muller cell ablation leads to retinal iron accumulation. Invest Ophthalmol Vis Sci. (2017) 58:4223–34. doi: 10.1167/iovs.17-21743
77. Johnson EC, Morrison JC. Friend or foe? Resolving the impact of glial responses in glaucoma. J Glaucoma. (2009) 18:341–53. doi: 10.1097/IJG.0b013e31818c6ef6
78. Gallego BI, Salazar JJ, de Hoz R, Rojas B, Ramirez AI, Salinas-Navarro M, et al. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J Neuroinflammation. (2012) 9:92. doi: 10.1186/1742-2094-9-92
79. Dahlmann-Noor AH, Vijay S, Limb GA, Khaw PT. Strategies for optic nerve rescue and regeneration in glaucoma and other optic neuropathies. Drug Discov Today. (2010) 15:287–99. doi: 10.1016/j.drudis.2010.02.007
80. Shinozaki Y, Koizumi S. [Pathogenic roles of retinal glia in glaucoma]. Nihon Yakurigaku Zasshi. (2020) 155:87–92. doi: 10.1254/fpj.19120
81. Nomura-Komoike K, Saitoh F, Fujieda H. Phosphatidylserine recognition and Rac1 activation are required for Muller glia proliferation, gliosis and phagocytosis after retinal injury. Sci Rep. (2020) 10:1488. doi: 10.1038/s41598-020-58424-6
82. Morgan J, Caprioli J, Koseki Y. Nitric oxide mediates excitotoxic and anoxic damage in rat retinal ganglion cells cocultured with astroglia. Arch Ophthalmol. (1999) 117:1524–9. doi: 10.1001/archopht.117.11.1524
83. Jacoby J, Nath A, Jessen ZF, Schwartz GW. A self-regulating gap junction network of amacrine cells controls nitric oxide release in the retina. Neuron. (2018) 100:1149–62e5. doi: 10.1016/j.neuron.2018.09.047
84. Rashid K, Akhtar-Schaefer I, Langmann T. Microglia in retinal degeneration. Front Immunol. (2019) 10:1975. doi: 10.3389/fimmu.2019.01975
85. Tehrani S, Davis L, Cepurna WO, Delf RK, Lozano DC, Choe TE, et al. Optic nerve head astrocytes display axon-dependent and -independent reactivity in response to acutely elevated intraocular pressure. Invest Ophthalmol Vis Sci. (2019) 60:312–21. doi: 10.1167/iovs.18-25447
86. Jones EV, Bouvier DS. Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neural Plast. (2014) 2014:321209. doi: 10.1155/2014/321209
87. Tehrani S, Davis L, Cepurna WO, Choe TE, Lozano DC, Monfared A, et al. Astrocyte structural and molecular response to elevated intraocular pressure occurs rapidly and precedes axonal tubulin rearrangement within the optic nerve head in a rat model. PLoS ONE. (2016) 11:e0167364. doi: 10.1371/journal.pone.0167364
88. Cooper ML, Collyer JW, Calkins DJ. Astrocyte remodeling without gliosis precedes optic nerve Axonopathy. Acta Neuropathol Commun. (2018) 6:38. doi: 10.1186/s40478-018-0542-0
89. Nehlig A, Coles JA. Cellular pathways of energy metabolism in the brain: is glucose used by neurons or astrocytes? Glia. (2007) 55:1238–50. doi: 10.1002/glia.20376
90. Cooper ML, Crish SD, Inman DM, Horner PJ, Calkins DJ. Early astrocyte redistribution in the optic nerve precedes axonopathy in the DBA/2J mouse model of glaucoma. Exp Eye Res. (2016) 150:22–33. doi: 10.1016/j.exer.2015.11.016
91. Perge JA, Koch K, Miller R, Sterling P, Balasubramanian V. How the optic nerve allocates space, energy capacity, and information. J Neurosci. (2009) 29:7917–28. doi: 10.1523/JNEUROSCI.5200-08.2009
92. Boya P, Esteban-Martinez L, Serrano-Puebla A, Gomez-Sintes R, Villarejo-Zori B. Autophagy in the eye: Development, degeneration, and aging. Prog Retin Eye Res. (2016) 55:206–45. doi: 10.1016/j.preteyeres.2016.08.001
93. Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci USA. (2014) 111:9633–8. doi: 10.1073/pnas.1404651111
94. Calkins DJ, Pekny M, Cooper ML, Benowitz L, Lasker IIoA, Glaucomatous Neurodegeneration P. The challenge of regenerative therapies for the optic nerve in glaucoma. Exp Eye Res. (2017) 157:28–33. doi: 10.1016/j.exer.2017.01.007
95. Alqawlaq S, Flanagan JG, Sivak JM. All roads lead to glaucoma: Induced retinal injury cascades contribute to a common neurodegenerative outcome. Exp Eye Res. (2019) 183:88–97. doi: 10.1016/j.exer.2018.11.005
96. Prasanna G, Krishnamoorthy R, Yorio T. Endothelin, astrocytes and glaucoma. Exp Eye Res. (2011) 93:170–7. doi: 10.1016/j.exer.2010.09.006
97. Huang TY, Chu HC, Lin YL, Lin CK, Hsieh TY, Chang WK, et al. Minocycline attenuates experimental colitis in mice by blocking expression of inducible nitric oxide synthase and matrix metalloproteinases. Toxicol Appl Pharmacol. (2009) 237:69–82. doi: 10.1016/j.taap.2009.02.026
98. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
99. Shareef S, Sawada A, Neufeld AH. Isoforms of nitric oxide synthase in the optic nerves of rat eyes with chronic moderately elevated intraocular pressure. Invest Ophthalmol Vis Sci. (1999) 40:2884–91.
100. Lin FL, Cheng YW, Yu M, Ho JD, Kuo YC, Chiou GCY, et al. The fungus-derived retinoprotectant theissenolactone C improves glaucoma-like injury mediated by MMP-9 inhibition. Phytomedicine. (2019) 56:207–14. doi: 10.1016/j.phymed.2018.11.002
101. Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. (2012) 35:153–79. doi: 10.1146/annurev.neuro.051508.135728
102. Jha MK, Morrison BM. Glia-neuron energy metabolism in health and diseases: new insights into the role of nervous system metabolic transporters. Exp Neurol. (2018) 309:23–31. doi: 10.1016/j.expneurol.2018.07.009
103. Wang R, Seifert P, Jakobs TC. Astrocytes in the optic nerve head of glaucomatous mice display a characteristic reactive phenotype. Invest Ophthalmol Vis Sci. (2017) 58:924–32. doi: 10.1167/iovs.16-20571
104. Davis CH, Marsh-Armstrong N. Discovery and implications of transcellular mitophagy. Autophagy. (2014) 10:2383–4. doi: 10.4161/15548627.2014.981920
105. Butt AM, Pugh M, Hubbard P, James G. Functions of optic nerve glia: axoglial signalling in physiology and pathology. Eye. (2004) 18:1110–21. doi: 10.1038/sj.eye.6701595
106. Gao L, Macklin W, Gerson J, Miller RH. Intrinsic and extrinsic inhibition of oligodendrocyte development by rat retina. Dev Biol. (2006) 290:277–86. doi: 10.1016/j.ydbio.2005.11.007
107. Philips T, Rothstein JD. Oligodendroglia: metabolic supporters of neurons. J Clin Invest. (2017) 127:3271–80. doi: 10.1172/JCI90610
108. Butt AM, Papanikolaou M, Rivera A. Physiology of oligodendroglia. Adv Exp Med Biol. (2019) 1175:117–28. doi: 10.1007/978-981-13-9913-8_5
109. Chamberlain KA, Sheng ZH. Mechanisms for the maintenance and regulation of axonal energy supply. J Neurosci Res. (2019) 97:897–913. doi: 10.1002/jnr.24411
110. Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J Neurosci. (2011) 31:538–48. doi: 10.1523/JNEUROSCI.3516-10.2011
111. Gerhart DZ, Leino RL, Drewes LR. Distribution of monocarboxylate transporters MCT1 and MCT2 in rat retina. Neuroscience. (1999) 92:367–75. doi: 10.1016/S0306-4522(98)00699-X
112. Takeda K, Dezawa M, Kitada M. The expression of PLP/DM-20 mRNA is restricted to the oligodendrocyte-lineage cells in the adult rat spinal cord. Histochem Cell Biol. (2016) 145:147–61. doi: 10.1007/s00418-015-1384-5
113. Loers G, Aboul-Enein F, Bartsch U, Lassmann H, Schachner M. Comparison of myelin, axon, lipid, and immunopathology in the central nervous system of differentially myelin-compromised mutant mice: a morphological and biochemical study. Mol Cell Neurosci. (2004) 27:175–89. doi: 10.1016/j.mcn.2004.06.006
114. Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. (1998) 280:1610–3. doi: 10.1126/science.280.5369.1610
115. Rinholm JE, Vervaeke K, Tadross MR, Tkachuk AN, Kopek BG, Brown TA, et al. Movement and structure of mitochondria in oligodendrocytes and their myelin sheaths. Glia. (2016) 64:810–25. doi: 10.1002/glia.22965
116. Rosko L, Smith VN, Yamazaki R, Huang JK. Oligodendrocyte bioenergetics in health and disease. Neuroscientist. (2019) 25:334–43. doi: 10.1177/1073858418793077
117. Harun-Or-Rashid M, Pappenhagen N, Palmer PG, Smith MA, Gevorgyan V, Wilson GN. Structural and functional rescue of chronic metabolically stressed optic nerves through respiration. J Neurosci. (2018) 38:5122–39. doi: 10.1523/JNEUROSCI.3652-17.2018
118. Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, et al. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem. (2000) 75:965–72. doi: 10.1046/j.1471-4159.2000.0750965.x
119. Domercq M, Vazquez-Villoldo N, Matute C. Neurotransmitter signaling in the pathophysiology of microglia. Front Cell Neurosci. (2013) 7:49. doi: 10.3389/fncel.2013.00049
120. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. (2014) 32:367–402. doi: 10.1146/annurev-immunol-032713-120240
121. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. (2007) 10:1387–94. doi: 10.1038/nn1997
122. Lively S, Schlichter LC. Microglia responses to pro-inflammatory stimuli (LPS, IFNgamma+TNFalpha) and reprogramming by resolving cytokines (IL-4, IL-10). Front Cell Neurosci. (2018) 12:215. doi: 10.3389/fncel.2018.00215
123. Yi S, Jiang X, Tang X, Li Y, Xiao C, Zhang J, et al. IL-4 and IL-10 promotes phagocytic activity of microglia by up-regulation of TREM2. Cytotechnology. (2020) 72:589–602. doi: 10.1007/s10616-020-00409-4
124. Shemer A, Scheyltjens I, Frumer GR, Kim JS, Grozovski J, Ayanaw S, et al. Interleukin-10 prevents pathological microglia hyperactivation following peripheral endotoxin challenge. Immunity. (2020) 53:1033–49e7. doi: 10.1016/j.immuni.2020.09.018
125. Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Curr Med Chem. (2007) 14:1189–97. doi: 10.2174/092986707780597961
126. Hou L, Bao X, Zang C, Yang H, Sun F, Che Y, et al. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. (2018) 14:600–8. doi: 10.1016/j.redox.2017.11.010
127. Presta M, Urbinati C, Dell'era P, Lauro GM, Sogos V, Balaci L, et al. Expression of basic fibroblast growth factor and its receptors in human fetal microglia cells. Int J Dev Neurosci. (1995) 13:29–39. doi: 10.1016/0736-5748(94)00065-B
128. Shimojo M, Nakajima K, Takei N, Hamanoue M, Kohsaka S. Production of basic fibroblast growth factor in cultured rat brain microglia. Neurosci Lett. (1991) 123:229–31. doi: 10.1016/0304-3940(91)90937-O
129. Walker DG, Kim SU, McGeer PL. Complement and cytokine gene expression in cultured microglial derived from postmortem human brains. J Neurosci Res. (1995) 40:478–93. doi: 10.1002/jnr.490400407
130. Braunger BM, Leimbeck SV, Schlecht A, Volz C, Jagle H, Tamm ER. Deletion of ocular transforming growth factor beta signaling mimics essential characteristics of diabetic retinopathy. Am J Pathol. (2015) 185:1749–68. doi: 10.1016/j.ajpath.2015.02.007
131. Baptista FI, Aveleira CA, Castilho AF, Ambrosio AF. Elevated glucose and interleukin-1beta differentially affect retinal microglial cell proliferation. Mediators Inflamm. (2017) 2017:4316316. doi: 10.1155/2017/4316316
132. Hetier E, Ayala J, Bousseau A, Prochiantz A. Modulation of interleukin-1 and tumor necrosis factor expression by beta-adrenergic agonists in mouse ameboid microglial cells. Exp Brain Res. (1991) 86:407–13. doi: 10.1007/BF00228965
133. Wooff Y, Man SM, Aggio-Bruce R, Natoli R, Fernando N. IL-1 family members mediate cell death, inflammation and angiogenesis in retinal degenerative diseases. Front Immunol. (2019) 10:1618. doi: 10.3389/fimmu.2019.01618
134. Frei K, Malipiero UV, Leist TP, Zinkernagel RM, Schwab ME, Fontana A. On the cellular source and function of interleukin 6 produced in the central nervous system in viral diseases. Eur J Immunol. (1989) 19:689–94. doi: 10.1002/eji.1830190418
135. Gottschall PE, Yu X, Bing B. Increased production of gelatinase B (matrix metalloproteinase-9) and interleukin-6 by activated rat microglia in culture. J Neurosci Res. (1995) 42:335–42. doi: 10.1002/jnr.490420307
136. Sheng WS, Hu S, Kravitz FH, Peterson PK, Chao CC. Tumor necrosis factor alpha upregulates human microglial cell production of interleukin-10 in vitro. Clin Diagn Lab Immunol. (1995) 2:604–8. doi: 10.1128/CDLI.2.5.604-608.1995
137. Lee YS, Amadi-Obi A, Yu CR, Egwuagu CE. Retinal cells suppress intraocular inflammation (uveitis) through production of interleukin-27 and interleukin-10. Immunology. (2011) 132:492–502. doi: 10.1111/j.1365-2567.2010.03379.x
138. Chao CC, Hu S, Peterson PK. Modulation of human microglial cell superoxide production by cytokines. J Leukoc Biol. (1995) 58:65–70. doi: 10.1002/jlb.58.1.65
139. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer's disease. J Cell Biol. (2018) 217:459–72. doi: 10.1083/jcb.201709069
140. Colton C, Wilt S, Gilbert D, Chernyshev O, Snell J, Dubois-Dalcq M. Species differences in the generation of reactive oxygen species by microglia. Mol Chem Neuropathol. (1996) 28:15–20. doi: 10.1007/BF02815200
141. Vielma AH, Retamal MA, Schmachtenberg O. Nitric oxide signaling in the retina: what have we learned in two decades? Brain Res. (2012) 1430:112–25. doi: 10.1016/j.brainres.2011.10.045
142. Quintas C, Pinho D, Pereira C, Saraiva L, Goncalves J, Queiroz G. Microglia P2Y(6) receptors mediate nitric oxide release and astrocyte apoptosis. J Neuroinflammation. (2014) 11:141. doi: 10.1186/s12974-014-0141-3
143. Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol. (1998) 54:99–125. doi: 10.1016/S0301-0082(97)00052-X
144. Lenz KM, Nelson LH. Microglia and beyond: innate immune cells as regulators of brain development and behavioral function. Front Immunol. (2018) 9:698. doi: 10.3389/fimmu.2018.00698
145. Madeira MH, Boia R, Santos PF, Ambrosio AF, Santiago AR. Contribution of microglia-mediated neuroinflammation to retinal degenerative diseases. Mediators Inflamm. (2015) 2015:673090. doi: 10.1155/2015/673090
146. Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: just bystander or target for therapy? Prog Retin Eye Res. (2015) 45:30–57. doi: 10.1016/j.preteyeres.2014.11.004
147. Zeng HY, Green WR, Tso MO. Microglial activation in human diabetic retinopathy. Arch Ophthalmol. (2008) 126:227–32. doi: 10.1001/archophthalmol.2007.65
148. Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. (2006) 147(Suppl. 1):S232–40. doi: 10.1038/sj.bjp.0706400
149. Yuan L, Neufeld AH. Activated microglia in the human glaucomatous optic nerve head. J Neurosci Res. (2001) 64:523–32. doi: 10.1002/jnr.1104
150. van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L, et al. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflammation. (2012) 9:156. doi: 10.1186/1742-2094-9-156
151. Quigley HA, McKinnon SJ, Zack DJ, Pease ME, Kerrigan-Baumrind LA, Kerrigan DF, et al. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. (2000) 41:3460–6.
152. Sivakumar V, Foulds WS, Luu CD, Ling EA, Kaur C. Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. J Pathol. (2011) 224:245–60. doi: 10.1002/path.2858
153. Jassim AH, Inman DM. Evidence of hypoxic glial cells in a model of ocular hypertension. Invest Ophthalmol Vis Sci. (2019) 60:1–15. doi: 10.1167/iovs.18-24977
154. Hvozda Arana AG, Lasagni Vitar RM, Reides CG, Lerner SF, Ferreira SM. Glaucoma causes redox imbalance in the primary visual cortex by modulating NADPH oxidase-4, iNOS, and Nrf2 pathway in a rat experimental model. Exp Eye Res. (2020) 200:108225. doi: 10.1016/j.exer.2020.108225
155. Gao F, Ji M, Wu JH, Wang ZF. [Roles of retinal Muller cells in health and glaucoma]. Sheng Li Xue Bao. (2013) 65:654–63.
156. Tezel G, Yang X, Luo C, Peng Y, Sun SL, Sun D. Mechanisms of immune system activation in glaucoma: oxidative stress-stimulated antigen presentation by the retina and optic nerve head glia. Invest Ophthalmol Vis Sci. (2007) 48:705–14. doi: 10.1167/iovs.06-0810
157. Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. (2006) 20:714–6. doi: 10.1096/fj.05-4882fje
158. Banati RB, Graeber MB. Surveillance, intervention and cytotoxicity: is there a protective role of microglia? Dev Neurosci. (1994) 16:114–27. doi: 10.1159/000112098
159. Hu X, Liou AK, Leak RK, Xu M, An C, Suenaga J, et al. Neurobiology of microglial action in CNS injuries: receptor-mediated signaling mechanisms and functional roles. Prog Neurobiol. (2014) 119-120:60–84. doi: 10.1016/j.pneurobio.2014.06.002
160. Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci. (2017) 18:598. doi: 10.3390/ijms18030598
161. Su P, Zhang J, Wang D, Zhao F, Cao Z, Aschner M, et al. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience. (2016) 319:155–67. doi: 10.1016/j.neuroscience.2016.01.035
162. Rodriguez-Muela N, Germain F, Marino G, Fitze PS, Boya P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. (2012) 19:162–9. doi: 10.1038/cdd.2011.88
163. Russo R, Varano GP, Adornetto A, Nazio F, Tettamanti G, Girardello R, et al. Rapamycin and fasting sustain autophagy response activated by ischemia/reperfusion injury and promote retinal ganglion cell survival. Cell Death Dis. (2018) 9:981. doi: 10.1038/s41419-018-1044-5
164. Adornetto A, Parisi V, Morrone LA, Corasaniti MT, Bagetta G, Tonin P, et al. The role of autophagy in glaucomatous optic neuropathy. Front Cell Dev Biol. (2020) 8:121. doi: 10.3389/fcell.2020.00121
165. Li L, Eter N, Heiduschka P. The microglia in healthy and diseased retina. Exp Eye Res. (2015) 136:116–30. doi: 10.1016/j.exer.2015.04.020
166. Clemente D, Ortega MC, Melero-Jerez C, de Castro F. The effect of glia-glia interactions on oligodendrocyte precursor cell biology during development and in demyelinating diseases. Front Cell Neurosci. (2013) 7:268. doi: 10.3389/fncel.2013.00268
167. Harada T, Harada C, Kohsaka S, Wada E, Yoshida K, Ohno S, et al. Microglia-Muller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J Neurosci. (2002) 22:9228–36. doi: 10.1523/JNEUROSCI.22-21-09228.2002
168. Posfai B, Cserep C, Orsolits B, Denes A. New insights into microglia-neuron interactions: a neuron's perspective. Neuroscience. (2019) 405:103–17. doi: 10.1016/j.neuroscience.2018.04.046
169. Wang M, Ma W, Zhao L, Fariss RN, Wong WT. Adaptive Muller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J Neuroinflammation. (2011) 8:173. doi: 10.1186/1742-2094-8-173
170. Zeng HY, Zhu XA, Zhang C, Yang LP, Wu LM, Tso MO. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in rd mice. Invest Ophthalmol Vis Sci. (2005) 46:2992–9. doi: 10.1167/iovs.05-0118
171. Natoli R, Fernando N, Madigan M, Chu-Tan JA, Valter K, Provis J, et al. Microglia-derived IL-1beta promotes chemokine expression by Muller cells and RPE in focal retinal degeneration. Mol Neurodegener. (2017) 12:31. doi: 10.1186/s13024-017-0175-y
172. Kirsch M, Lee MY, Meyer V, Wiese A, Hofmann HD. Evidence for multiple, local functions of ciliary neurotrophic factor (CNTF) in retinal development: expression of CNTF and its receptors and in vitro effects on target cells. J Neurochem. (1997) 68:979–90. doi: 10.1046/j.1471-4159.1997.68030979.x
173. Zack DJ. Neurotrophic rescue of photoreceptors: are Muller cells the mediators of survival? Neuron. (2000) 26:285–6. doi: 10.1016/S0896-6273(00)81160-5
174. Gottschall PE, Deb S. Regulation of matrix metalloproteinase expressions in astrocytes, microglia and neurons. Neuroimmunomodulation. (1996) 3:69–75. doi: 10.1159/000097229
175. Lorenzl S, Albers DS, Narr S, Chirichigno J, Beal MF. Expression of MMP-2, MMP-9, and MMP-1 and their endogenous counterregulators TIMP-1 and TIMP-2 in postmortem brain tissue of Parkinson's disease. Exp Neurol. (2002) 178:13–20. doi: 10.1006/exnr.2002.8019
176. Giebel SJ, Menicucci G, McGuire PG, Das A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest. (2005) 85:597–607. doi: 10.1038/labinvest.3700251
177. Hsu JY, McKeon R, Goussev S, Werb Z, Lee JU, Trivedi A, et al. Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. J Neurosci. (2006) 26:9841–50. doi: 10.1523/JNEUROSCI.1993-06.2006
178. Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, Grossetete M, et al. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. (2001) 893:104–12. doi: 10.1016/S0006-8993(00)03294-7
179. Li Y, Zhou GM. MMP-9 inhibition facilitates amacrine cell loss after ouabain-induced retinal damage. Exp Eye Res. (2015) 135:174–81. doi: 10.1016/j.exer.2015.03.003
180. Manabe S, Gu Z, Lipton SA. Activation of matrix metalloproteinase-9 via neuronal nitric oxide synthase contributes to NMDA-induced retinal ganglion cell death. Invest Ophthalmol Vis Sci. (2005) 46:4747–53. doi: 10.1167/iovs.05-0128
181. De Groef L, Van Hove I, Dekeyster E, Stalmans I, Moons L. MMPs in the trabecular meshwork: promising targets for future glaucoma therapies? Invest Ophthalmol Vis Sci. (2013) 54:7756–63. doi: 10.1167/iovs.13-13088
182. Vernazza S, Tirendi S, Bassi AM, Traverso CE, Sacca SC. Neuroinflammation in primary open-angle glaucoma. J Clin Med. (2020) 9:3172. doi: 10.3390/jcm9103172
183. Jalal FY, Yang Y, Thompson J, Lopez AC, Rosenberg GA. Myelin loss associated with neuroinflammation in hypertensive rats. Stroke. (2012) 43:1115–22. doi: 10.1161/STROKEAHA.111.643080
184. Deng Y, Lu J, Sivakumar V, Ling EA, Kaur C. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathol. (2008) 18:387–400. doi: 10.1111/j.1750-3639.2008.00138.x
185. Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. (2017) 157:247–72. doi: 10.1016/j.pneurobio.2016.01.005
186. Wang M, Wong WT. Microglia-Muller cell interactions in the retina. Adv Exp Med Biol. (2014) 801:333–8. doi: 10.1007/978-1-4614-3209-8_42
187. Kolko M. Present and new treatment strategies in the management of glaucoma. Open Ophthalmol J. (2015) 9:89–100. doi: 10.2174/1874364101509010089
188. Carter-Dawson L, Shen FF, Harwerth RS, Crawford ML, Smith EL III, Whitetree A. Glutathione content is altered in Muller cells of monkey eyes with experimental glaucoma. Neurosci Lett. (2004) 364:7–10. doi: 10.1016/j.neulet.2004.03.082
189. Coughlin BA, Feenstra DJ, Mohr S. Muller cells and diabetic retinopathy. Vision Res. (2017) 139:93–100. doi: 10.1016/j.visres.2017.03.013
190. Sanchez-Lopez E, Egea MA, Davis BM, Guo L, Espina M, Silva AM, et al. Memantine-Loaded PEGylated biodegradable nanoparticles for the treatment of glaucoma. Small. (2018) 14. doi: 10.1002/smll.201701808
191. Weinreb RN, Liebmann JM, Cioffi GA, Goldberg I, Brandt JD, Johnson CA, et al. Oral memantine for the treatment of glaucoma: design and results of 2 randomized, placebo-controlled, phase 3 studies. Ophthalmology. (2018) 125:1874–85. doi: 10.1016/j.ophtha.2018.06.017
192. Cvenkel B, Kolko M. Current medical therapy and future trends in the management of glaucoma treatment. J Ophthalmol. (2020) 2020:6138132. doi: 10.1155/2020/6138132
193. Jung KI, Kim JH, Park CK. alpha2-Adrenergic modulation of the glutamate receptor and transporter function in a chronic ocular hypertension model. Eur J Pharmacol. (2015) 765:274–83. doi: 10.1016/j.ejphar.2015.08.035
194. Chen SD, Wang L, Zhang XL. Neuroprotection in glaucoma: present and future. Chin Med J. (2013) 126:1567–77.
195. Koeberle PD, Ball AK. Neurturin enhances the survival of axotomized retinal ganglion cells in vivo: combined effects with glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor. Neuroscience. (2002) 110:555–67. doi: 10.1016/S0306-4522(01)00557-7
196. Claes M, De Groef L, Moons L. Target-derived neurotrophic factor deprivation puts retinal ganglion cells on death row: cold hard evidence and caveats. Int J Mol Sci. (2019) 20:4314. doi: 10.3390/ijms20174314
197. Osborne A, Khatib TZ, Songra L, Barber AC, Hall K, Kong GYX, et al. Neuroprotection of retinal ganglion cells by a novel gene therapy construct that achieves sustained enhancement of brain-derived neurotrophic factor/tropomyosin-related kinase receptor-B signaling. Cell Death Dis. (2018) 9:1007. doi: 10.1038/s41419-018-1041-8
198. Tao W. Application of encapsulated cell technology for retinal degenerative diseases. Expert Opin Biol Ther. (2006) 6:717–26. doi: 10.1517/14712598.6.7.717
199. Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. (2003) 4:299–309. doi: 10.1038/nrn1078
200. Liu B, Neufeld AH. Activation of epidermal growth factor receptor causes astrocytes to form cribriform structures. Glia. (2004) 46:153–68. doi: 10.1002/glia.10358
201. Gugleta K. [Significance of Endothelin-1 in Glaucoma - a Short Overview]. Klin Monbl Augenheilkd. (2018) 235:140–5. doi: 10.1055/s-0043-124084
202. Mozaffarieh M, Flammer J. Is there more to glaucoma treatment than lowering IOP? Surv Ophthalmol. (2007) 52(Suppl. 2):S174–9. doi: 10.1016/j.survophthal.2007.08.013
203. Dvoriantchikova G, Ivanov D. Tumor necrosis factor-alpha mediates activation of NF-kappaB and JNK signaling cascades in retinal ganglion cells and astrocytes in opposite ways. Eur J Neurosci. (2014) 40:3171–8. doi: 10.1111/ejn.12710
204. Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, et al. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. (2006) 26:12633–41. doi: 10.1523/JNEUROSCI.2801-06.2006
205. Hu X, Xu MX, Zhou H, Cheng S, Li F, Miao Y, et al. Tumor necrosis factor-alpha aggravates gliosis and inflammation of activated retinal Muller cells. Biochem Biophys Res Commun. (2020) 531:383–9. doi: 10.1016/j.bbrc.2020.07.102
206. Tezel G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog Brain Res. (2008) 173:409–21. doi: 10.1016/S0079-6123(08)01128-X
207. Hertz L, Chen Y, Waagepetersen HS. Effects of ketone bodies in Alzheimer's disease in relation to neural hypometabolism, beta-amyloid toxicity, and astrocyte function. J Neurochem. (2015) 134:7–20. doi: 10.1111/jnc.13107
208. Hohnholt MC, Blumrich EM, Waagepetersen HS, Dringen R. The antidiabetic drug metformin decreases mitochondrial respiration and tricarboxylic acid cycle activity in cultured primary rat astrocytes. J Neurosci Res. (2017) 95:2307–20. doi: 10.1002/jnr.24050
209. Hanyuda A, Rosner BA, Wiggs JL, Willett WC, Tsubota K, Pasquale LR, et al. Low-carbohydrate-diet scores and the risk of primary open-angle glaucoma: data from three US cohorts. Eye (Lond). (2020) 34:1465–75. doi: 10.1038/s41433-020-0820-5
210. Mi S, Miller RH, Tang W, Lee X, Hu B, Wu W, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol. (2009) 65:304–15. doi: 10.1002/ana.21581
211. Mi S, Pepinsky RB, Cadavid D. Blocking LINGO-1 as a therapy to promote CNS repair: from concept to the clinic. CNS Drugs. (2013) 27:493–503. doi: 10.1007/s40263-013-0068-8
212. Williams PA, Marsh-Armstrong N, Howell GR, Lasker IIoA, Glaucomatous Neurodegeneration P. Neuroinflammation in glaucoma: A new opportunity. Exp Eye Res. (2017) 157:20–7. doi: 10.1016/j.exer.2017.02.014
213. Baudouin C, Kolko M, Melik-Parsadaniantz S, Messmer EM. Inflammation in Glaucoma: from the back to the front of the eye, and beyond. Prog Retin Eye Res. (2020) 100916. doi: 10.1016/j.preteyeres.2020.100916. [Epub ahead of print].
214. Aires ID, Boia R, Rodrigues-Neves AC, Madeira MH, Marques C, Ambrosio AF, et al. Blockade of microglial adenosine A2A receptor suppresses elevated pressure-induced inflammation, oxidative stress, and cell death in retinal cells. Glia. (2019) 67:896–914. doi: 10.1002/glia.23579
215. Levkovitch-Verbin H, Waserzoog Y, Vander S, Makarovsky D, Piven I. Minocycline upregulates pro-survival genes and downregulates pro-apoptotic genes in experimental glaucoma. Graefes Arch Clin Exp Ophthalmol. (2014) 252:761–72. doi: 10.1007/s00417-014-2588-4
216. Bell K, Und Hohenstein-Blaul NVT, Teister J, Grus F. Modulation of the immune system for the treatment of glaucoma. Curr Neuropharmacol. (2018) 16:942–58. doi: 10.2174/1570159X15666170720094529
217. Kernt M, Neubauer AS, Eibl KH, Wolf A, Ulbig MW, Kampik A, et al. Minocycline is cytoprotective in human trabecular meshwork cells and optic nerve head astrocytes by increasing expression of XIAP, survivin, and Bcl-2. Clin Ophthalmol. (2010) 4:591–604. doi: 10.2147/OPTH.S11216
218. Zeng HL, Shi JM. The role of microglia in the progression of glaucomatous neurodegeneration- a review. Int J Ophthalmol. (2018) 11:143–9. doi: 10.18240/ijo.2018.01.22
219. Breen KT, Anderson SR, Steele MR, Calkins DJ, Bosco A, Vetter ML. Loss of fractalkine signaling exacerbates axon transport dysfunction in a chronic model of glaucoma. Front Neurosci. (2016) 10:526. doi: 10.3389/fnins.2016.00526
220. Neumann H. Control of glial immune function by neurons. Glia. (2001) 36:191–9. doi: 10.1002/glia.1108
221. Taylor S, Calder CJ, Albon J, Erichsen JT, Boulton ME, Morgan JE. Involvement of the CD200 receptor complex in microglia activation in experimental glaucoma. Exp Eye Res. (2011) 92:338–43. doi: 10.1016/j.exer.2011.01.012
222. Huang R, Lan Q, Chen L, Zhong H, Cui L, Jiang L, et al. CD200Fc attenuates retinal glial responses and RGCs Apoptosis after optic nerve crush by modulating CD200/CD200R1 interaction. J Mol Neurosci. (2018) 64:200–10. doi: 10.1007/s12031-017-1020-z
223. Copland DA, Calder CJ, Raveney BJ, Nicholson LB, Phillips J, Cherwinski H, et al. Monoclonal antibody-mediated CD200 receptor signaling suppresses macrophage activation and tissue damage in experimental autoimmune uveoretinitis. Am J Pathol. (2007) 171:580–8. doi: 10.2353/ajpath.2007.070272
224. Mirshahi A, Hoehn R, Lorenz K, Kramann C, Baatz H. Anti-tumor necrosis factor alpha for retinal diseases: current knowledge and future concepts. J Ophthalmic Vis Res. (2012) 7:39–44.
225. Bras JP, Bravo J, Freitas J, Barbosa MA, Santos SG, Summavielle T, et al. TNF-alpha-induced microglia activation requires miR-342: impact on NF-kB signaling and neurotoxicity. Cell Death Dis. (2020) 11:415. doi: 10.1038/s41419-020-2626-6
226. Krishnan A, Fei F, Jones A, Busto P, Marshak-Rothstein A, Ksander BR, et al. Overexpression of soluble fas ligand following adeno-associated virus gene therapy prevents retinal ganglion cell death in chronic and acute murine models of glaucoma. J Immunol. (2016) 197:4626–38. doi: 10.4049/jimmunol.1601488
227. Gregory MS, Hackett CG, Abernathy EF, Lee KS, Saff RR, Hohlbaum AM, et al. Opposing roles for membrane bound and soluble Fas ligand in glaucoma-associated retinal ganglion cell death. PLoS ONE. (2011) 6:e17659. doi: 10.1371/journal.pone.0017659
228. Krishnan A, Kocab AJ, Zacks DN, Marshak-Rothstein A, Gregory-Ksander M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J Neuroinflammation. (2019) 16:184. doi: 10.1186/s12974-019-1576-3
229. Wu Z, Zhu A, Takayama F, Okada R, Liu Y, Harada Y, et al. Brazilian green propolis suppresses the hypoxia-induced neuroinflammatory responses by inhibiting NF-kappaB activation in microglia. Oxid Med Cell Longev. (2013) 2013:906726. doi: 10.1155/2013/906726
230. Markiewicz L, Pytel D, Mucha B, Szymanek K, Szaflik J, Szaflik JP, et al. Altered expression levels of MMP1, MMP9, MMP12, TIMP1, and IL-1beta as a risk factor for the elevated IOP and optic nerve head damage in the primary open-angle glaucoma patients. Biomed Res Int. (2015) 2015:812503. doi: 10.1155/2015/812503
Keywords: Glaucoma, glia, retinal ganglion cells, Müller glial cells, microglia, astrocytes, oligodendrocytes, retinal glia interactions
Citation: García-Bermúdez MY, Freude KK, Mouhammad ZA, van Wijngaarden P, Martin KK and Kolko M (2021) Glial Cells in Glaucoma: Friends, Foes, and Potential Therapeutic Targets. Front. Neurol. 12:624983. doi: 10.3389/fneur.2021.624983
Received: 02 November 2020; Accepted: 26 January 2021;
Published: 16 March 2021.
Edited by:
Gabor Petzold, Helmholtz Association of German Research Centers (HZ), GermanyReviewed by:
Lies De Groef, KU Leuven, BelgiumEd Levine, Vanderbilt University Medical Center, United States
Copyright © 2021 García-Bermúdez, Freude, Mouhammad, van Wijngaarden, Martin and Kolko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam Kolko, bWlyaWFta0BzdW5kLmt1LmRr