
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurol. , 19 February 2021
Sec. Movement Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fneur.2021.618101
 Simon J. Schreiner1,2,3*
Simon J. Schreiner1,2,3* Lukas L. Imbach1,2
Lukas L. Imbach1,2 Philipp O. Valko1,2,3
Philipp O. Valko1,2,3 Angelina Maric1,2,3Rina Maqkaj1,2
Angelina Maric1,2,3Rina Maqkaj1,2 Esther Werth1,2,3
Esther Werth1,2,3 Christian R. Baumann1,2,3
Christian R. Baumann1,2,3 Heide Baumann-Vogel1,2
Heide Baumann-Vogel1,2Growing evidence implicates a distinct role of disturbed slow-wave sleep in neurodegenerative diseases. Reduced non-rapid eye movement (NREM) sleep slow-wave activity (SWA), a marker of slow-wave sleep intensity, has been linked with age-related cognitive impairment and Alzheimer disease pathology. However, it remains debated if SWA is associated with cognition in Parkinson disease (PD). Here, we investigated the relationship of regional SWA with cognitive performance in PD. In the present study, 140 non-demented PD patients underwent polysomnography and were administered the Montréal Cognitive Assessment (MoCA) to screen for cognitive impairment. We performed spectral analysis of frontal, central, and occipital sleep electroencephalography (EEG) derivations to measure SWA, and spectral power in other frequency bands, which we compared to cognition using linear mixed models. We found that worse MoCA performance was associated with reduced 1–4 Hz SWA in a region-dependent manner (F2, 687 =11.67, p < 0.001). This effect was driven by reduced regional SWA in the lower delta frequencies, with a strong association of worse MoCA performance with reduced 1–2 Hz SWA (F2, 687 =18.0, p < 0.001). The association of MoCA with 1–2 Hz SWA (and 1–4 Hz SWA) followed an antero-posterior gradient, with strongest, weaker, and absent associations over frontal (rho = 0.33, p < 0.001), central (rho = 0.28, p < 0.001), and occipital derivations, respectively. Our study shows that cognitive impairment in PD is associated with reduced NREM sleep SWA, predominantly in lower delta frequencies (1–2 Hz) and over frontal regions. This finding suggests a potential role of reduced frontal slow-wave sleep intensity in cognitive impairment in PD.
Parkinson disease (PD) is the second most common neurodegenerative disorder after Alzheimer disease (AD). The majority of PD patients will develop progressive cognitive impairment, likely due to ongoing neurodegeneration with Lewy body and concomitant AD pathology (1). As prevention and treatment of cognitive impairment remain major challenges, we need to identify modifiable risk factors.
Slow-wave sleep (SWS), i.e., deep non-rapid eye movement (NREM) sleep, supports cognition and could modulate neurodegeneration by influencing pathological proteins such as alpha-synuclein, tau, and beta-amyloid (2–4). Sleep induction decreases whereas sleep restriction increases pathological proteins in rodent brain tissue and interstitial fluid and human cerebrospinal fluid (2–4). These dynamics may specifically depend on SWS, as SWS disruption—but not partial sleep restriction with preserved SWS—modulates beta-amyloid levels in humans (5, 6). Slow-wave activity (SWA) quantifies sleep slow waves, the electroencephalographic hallmark of SWS. SWA likely indicates how strongly SWS could modulate neurodegeneration, as SWA correlates positively with glymphatic clearance in mice (7), and negatively with AD-related cognitive impairment and pathology in humans (8–11).
In PD, progressive deterioration of SWS has been described, with altered slow wave morphology at prodromal stages (12), reduced SWA in de novo patients (13), and reduced SWS amount in advanced stages (14). Moreover, impaired SWA has been linked with motor complications and progression (15, 16).
However, the relationship of SWA with cognition in PD remains unclear, as findings on this topic are inconsistent. A detailed sleep EEG analysis reported no association of SWA with cognitive impairment in PD patients, but instead highlighted the importance of other sleep EEG alterations over posterior derivations (17). In contrast, a recent study linked SWA, a priori selected from frontal derivations, to global cognition, but the sample included only 32 PD patients (18). Therefore, the aim of our study was to clarify the relationship of SWA with cognition in PD by analyzing SWA and its regional variation in a larger sample of non-demented PD patients. In addition, we tested the hypothesis that the relationship between cognition and SWA in PD is regional, preferentially involving frontal regions, which have been implicated with slow wave generation and altered SWA related to aging and AD pathology (9–11, 19, 20).
This study was conducted after ethical approval (Kantonale Ethikkommission Zürich) and aligned with the Declaration of Helsinki. We included all consecutive PD patients, who had undergone a strictly predefined diagnostic work-up within the highly specialized medicine program to evaluate eligibility for deep brain stimulation between January 2012 and July 2018. This standardized work-up included cognitive testing and polysomnography, irrespective of cognitive or sleep complaints, and results were thoroughly documented. From this documentation, we extracted demographic and clinical data, including Hoehn and Yahr stage, Unified Parkinson's Disease Rating Scale motor part (after >12 h without PD medication), motor phenotype (21), and medication at the timing of polysomnography, including the levodopa equivalent dose (LED) (22), and sleep medications, i.e., drugs that could affect sleep (antidepressants, antipsychotics, acetylcholine-esterase-inhibitors, benzodiazepines, benzodiazepine-receptor-agonists, stimulants, melatonin), which we further analyzed if taken by more than one patient.
Trained neuropsychologists or neurologists administered the Montréal Cognitive Assessment (MoCA) during medical “on” (23). For descriptive purposes, we screened patients with MoCA ≥ 26 as cognitively normal, and patients with MoCA <26 as cognitively impaired, because this MoCA cutoff has been shown to have a high diagnostic accuracy to screen for mild cognitive impairment in PD (24). Inclusion criteria were diagnosed PD (25), and available MoCA and polysomnography. Exclusion criteria were other forms of Parkinsonism, a clinical diagnosis of dementia (26), or incompatible polysomnography (e.g., no sleep).
Patients underwent a single-night video-polysomnography in the in-house sleep laboratory (Embla N7000, RemLogic v3.2). Experienced sleep experts (E. W., H. B. V., S. J. S.) manually scored sleep stages according to the AASM manual (27). The following variables were derived: total sleep time, sleep efficiency, percentages of wake after sleep onset, rapid eye movement (REM) and non-REM (NREM) sleep stages (N1, N2, N3), sleep latency (time until N2), indices (events/hour) for apnea-hypopnea, arousal, and periodic limb movements during sleep, and REM sleep behavior disorder (RBD) according to international criteria (28).
To test our hypothesis that cognitive impairment is associated with SWA, predominantly over frontal regions, we performed spectral analysis from six EEG derivations covering frontal, central, and occipital regions (F3, F4, C3, C4, O1, O2, all referenced to contralateral mastoid). Preprocessing of EEG and spectral analysis were performed using previously described routines (0.5 Hz high-pass and 40 Hz low-pass filtering, visual inspection and semi-automatic artifact removal; Fast Fourier Transformation; 5 s-epochs) (29). We documented the number of artifact-free EEG epochs from each EEG derivation, to exclude possible confounding. On artifact-free EEG, aligned with manual sleep scoring, we averaged spectral power during N2 and N3 over the entire night in the slow oscillation (0.5–1 Hz), delta (1–4 Hz), theta (4–8 Hz), alpha (8–13 Hz), and beta (13–20 Hz) frequency bands. Additional SWA bins were computed (1–2, 2–3, 3–4 Hz) because previously reported associations of AD-related cognitive impairment and pathology with SWA where particularly evident in the lower delta frequencies <2 Hz (9–11). We normalized band power to total 0.5–40 Hz power during N2 and N3, to avoid bias due to interindividual differences in EEG amplitude and capture the relative contributions of the 1-Hz segmented SWA to NREM power. For improved interpretation, we also report on absolute NREM sleep power densities.
The statistical analysis was carried out with SPSS (v23.0; IBM, Armonk, NY). We studied the relationship of cognition with regional NREM sleep EEG power within a linear mixed model framework. For each frequency band, separate linear mixed models were calculated, with NREM sleep EEG power as dependent variable, and hemisphere (left, right), region (frontal, central, occipital), MoCA performance, and the interactions of MoCA with hemisphere and region as fixed effects. A random effect was defined for patient intercept. All models were controlled for age, sex, disease duration, years of education, motor phenotype (akinetic-rigid yes/no), LED, and the sum of sleep medications. We used estimates of fixed effects for significant interactions of MoCA with region or hemisphere, to locate regional effects. All continuous variables were transformed into z scores, to allow comparisons among estimates. Skewed variables were log-transformed. We assessed normality of variables and residuals by applying the Kolmogorov-Smirnov test and plotting. To correct for multiple testing, the Bonferroni-Holm method was applied. For descriptive purposes, we also compared clinical and polysomnography data and NREM sleep EEG power densities with significant interaction effects between patients with and without cognitive impairment, using the Chi2 test for categorical data and—depending on normal distribution—the independent sample t-test or Wilcoxon rank-sum test for continuous data. Bivariate correlations were used to illustrate the strongest association of MoCA with NREM sleep EEG power. We consider corrected p-values < 0.05 as significant but report uncorrected p-values whenever informative.
Tables 1, 2 summarize clinical and polysomnography characteristics. We excluded three patients due to incompatible polysomnography (no N2 and N3) and seven derivations with poor quality on one hemisphere.
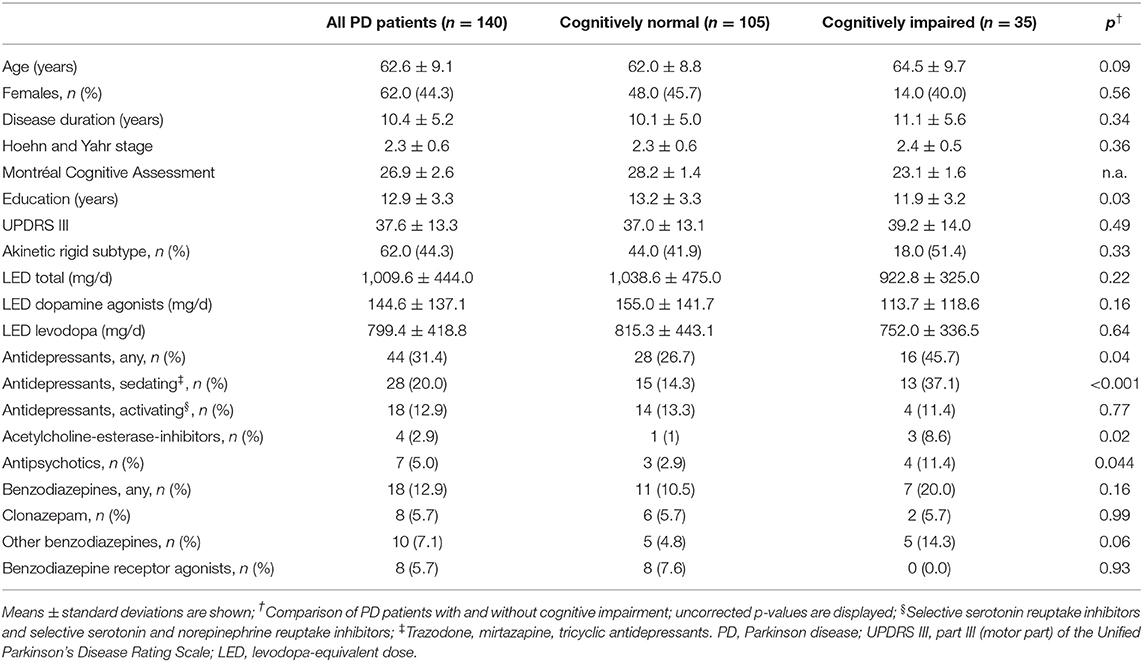
Table 1. Clinical characteristics.
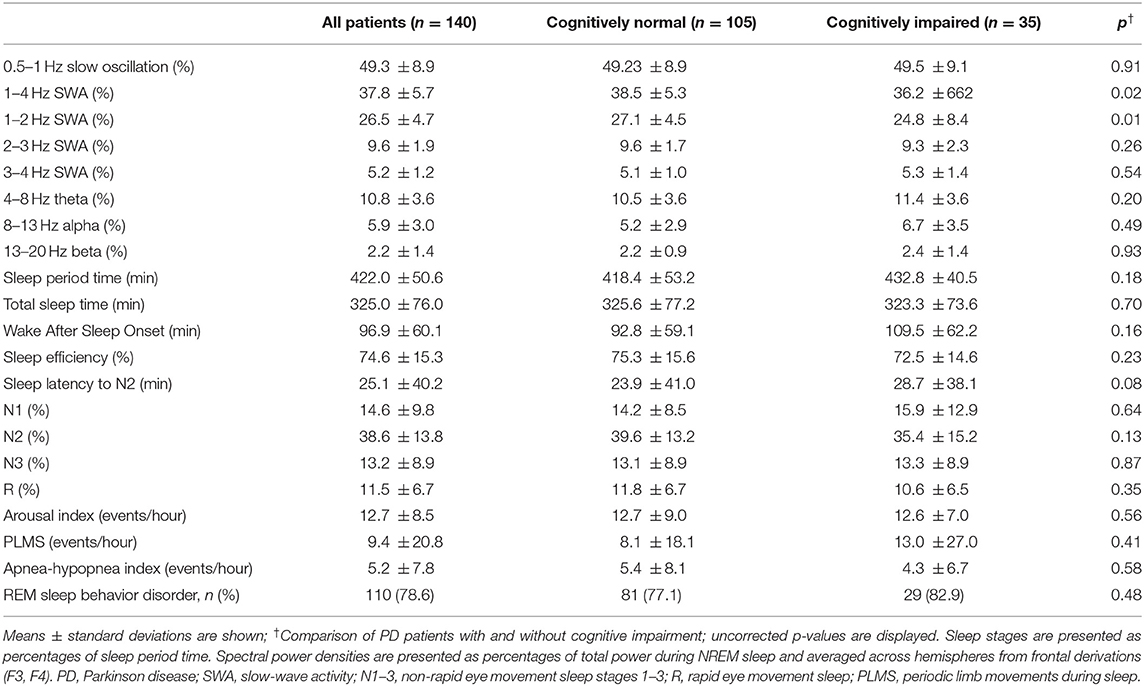
Table 2. Frontal NREM sleep EEG power and polysomnography.
Cognitive impairment as indicated by worse MoCA performance was associated with reduced regional 1–4 Hz SWA (MoCA*region: F2, 687 = 11.67, p-corrected < 0.001). This effect was driven by a reduction in the lower delta frequencies, as indicated by a strong association of worse MoCA performance with reduced regional 1–2 Hz SWA (MoCA*region interaction: F2, 687 =18.0, p-corrected < 0.001) but not with SWA in higher delta frequencies (Table 3). Further, worse MoCA performance was weakly associated with increased regional alpha power. An association of worse MoCA performance with increased regional theta power did not survive correction for multiple testing (Table 3). Main effects of MoCA were not significant (only a trend was observed for 1–2 Hz SWA: F2, 687 = 3.7, p-uncorrected = 0.05), indicating that cognition was associated with NREM sleep EEG power in a region-dependent manner.
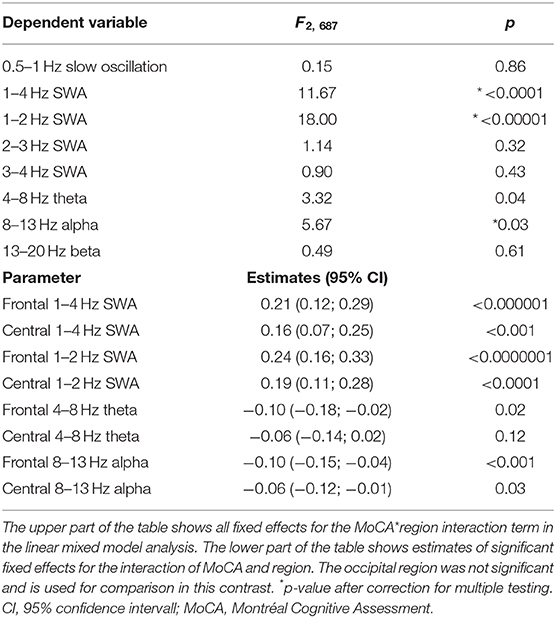
Table 3. Effects of cognitive performance (MoCA) on regional NREM sleep EEG power.
The association of MoCA performance with SWA followed an antero-posterior gradient, with strongest, weaker, and absent associations over frontal, central, and occipital derivations, respectively (Table 3). Figure 1A provides a binary visual representation of this relationship across patients with and without cognitive impairment. Figure 1B displays a continuous visual representation of the strongest effect, i.e., the association of worse MoCA performance with reduced frontal 1–2 Hz SWA (frontal: rho = 0.33, p < 0.001; central: rho = 0.28, p < 0.001). Similar but weaker findings were obtained when absolute instead of relative NREM sleep power was used (results not shown).
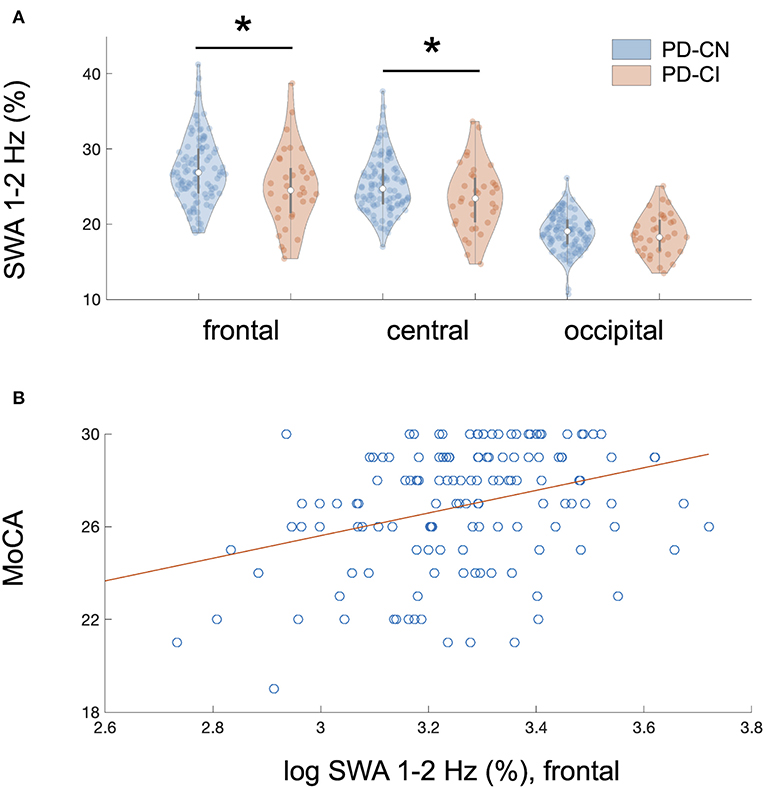
Figure 1. Regional association of reduced NREM sleep SWA with cognitive impairment in PD patients. (A) Cognitive impairment showed a regional and frequency-specific association with reduced NREM sleep SWA, predominantly in the lower delta frequencies (1–2 Hz), in 140 non-demented patients with Parkinson disease (PD); linear mixed model analysis: MoCA*region interaction, F2, 687 = 18.0, p < 0.00001 after correction for multiple testing and controlling for relevant covariates. The association of MoCA performance with SWA followed an antero-posterior gradient with strongest, weaker, and absent associations over frontal, central, and occipital derivations, respectively. Plot A represents a binary visual representation of regional 1–2 Hz SWA across cognitively impaired (n = 35, labeled as PD-CI, red color) and cognitively normal PD patients (n = 105, labeled as PD-CN, blue color). (B) Cognitive impairment and regional NREM sleep 1–2 Hz SWA showed the strongest association over frontal EEG derivations. This relationship is illustrated for averaged bifrontal 1–2 Hz SWA in all 140 PD patients; bivariate correlation: rho = 0.33, p < 0.0001. The Montréal Cognitive Assessment (MoCA) was used to screen for cognitive impairment. Slow-wave activity (SWA) was normalized to total spectral power during NREM sleep (0.5–40 Hz) and log-transformed, yet image A displays raw data.*significant after controlling for multiple testing.
We did not observe any associations of MoCA with spectral power in relation to hemisphere (no significant MoCA*hemisphere interaction), suggesting that the relationship of MoCA with NREM sleep EEG did not depend on hemispheric differences. Thus, averaged regional NREM sleep power densities across hemispheres were used in further analyses. We observed an additional significant fixed effect of sleep medications, which related to reduced 1–4 Hz SWA (F1, 132 = 9.8, p-corrected = 0.02) and 1–2 Hz SWA (F1, 132 = 8.5, p-corrected = 0.03).
Out of the 140 patients, 35 (25%) screened positive and 105 (75%) negative for cognitive impairment. Conventional polysomnography characteristics did not differ between patients with and without cognitive impairment. Compared to cognitively normal patients, patients with cognitive impairment more frequently took sedating antidepressants, acetylcholine-esterase-inhibitors, antipsychotics, and (at trend-level significance) other benzodiazepines, and showed fewer education years (see Table 1).
In 140 non-demented PD patients, we investigated whether cognitive impairment is associated with SWA, i.e., objectively measured SWS intensity. We found that worse MoCA performance was associated with reduced SWA, particularly in the lower delta frequencies (1–2 Hz) and over frontal EEG derivations. This finding adds evidence that reduced SWA is associated with cognitive impairment in PD. Moreover, the association of SWA with cognition was regional and frequency-specific, which resembles previously reported associations of frontal SWA—especially low-frequency SWA—with cognitive impairment and pathology in a context of early AD (8–11, 20). Thus, reduced SWA, predominantly over frontal derivations, may represent an overlapping marker of early cognitive impairment in neurodegenerative diseases such as PD and AD. However, it remains unclear whether neurodegeneration impacts SWS, or vice versa, or both.
In PD, shared etiologies could cause sleep and cognitive impairments, as both show overlapping patho-anatomical correlates, with alpha-synuclein and tau accumulation in brain regions regulating cognition and sleep, including brainstem, hypothalamic and limbic areas (30). Moreover, several candidate mechanisms underlying cognitive impairment in PD overlap with those linking disturbed sleep with neurodegeneration, including glial alterations, oxidative stress, or pathological protein accumulation (1, 31, 32).
Although not a primary outcome of this study, we did not observe differences in polysomnographic parameters other than reduced SWA when comparing cognitively impaired with cognitively normal patients. In contrast, previous studies linked cognitive impairment in PD with various sleep abnormalities, including reduced sleep efficiency, sleep apnea, RBD, REM sleep slowing, or reduced spindles (17, 33–38). However, these studies are not directly comparable due to differences in methods and patient characteristics. For example, some previous studies used actigraphy, assessed cognitive subdomains (33, 34), or included patients with higher degrees of sleep and cognitive impairments (36, 37). Although our study is cross-sectional, our finding on reduced NREM SWA resonates with longitudinal studies linking cognitive decline in PD with other NREM sleep abnormalities such as reduced spindles or SWS amount (17, 38, 39). Importantly, compared to SWS amount, SWA may be more sensitive to neurodegenerative cognitive impairment, as previously discussed (8).
We are aware of two previous studies that compared SWA to cognition in PD, with inconsistent findings. While a detailed sleep EEG analysis in 68 PD patients reported no association of cognitive impairment or decline with SWA (NREM delta power) (17), a recent study linked frontal SWA to global cognition in 32 PD patients (18). Compared to these studies, our analysis includes a larger sample (n = 140) of non-demented PD patients and 1-Hz- segmentation of regional SWA. Thus, our data adds evidence that SWA is linked to cognition in PD. In addition, our findings suggest that the relationship of cognition and SWA in PD is regional and frequency-specific. The pattern of specific involvement of frontal regions and predominant alteration of lower delta frequencies is consistent with a physiological antero-posterior gradient of SWA <2 Hz (40), local slow wave generation (19) and altered SWA related to aging and AD pathology (9–11, 20). Thus, a possible explanation for the discrepant findings on SWA and cognition are the distinct cognitive phenotypes and related trajectories in PD (41, 42). Reduced frontal SWA could be a possible marker of frontal-executive (18) rather than posterior cognitive dysfunction and incipient dementia in PD (17).
Our finding should be interpreted with caution given several limitations. We only cross-sectionally screened for cognitive impairment, without assessing cognitive self-reports, subdomains or longitudinal trajectories. On the other hand, the fact that simple cognitive screening revealed our finding might indicate that the link between SWS and cognition in PD is rather strong. Lack of a habituation polysomnography introduces a “first night effect” as possible bias. The retrospective study design is a limitation, but all procedures and documentations followed strictly predefined routines as part of a diagnostic work-up prior to deep brain stimulation. Thus, patient selection may limit generalization. On the other hand, our sample is representative for many non-demented PD patients with moderate disease severity, who could benefit from interventions to prevent cognitive decline. We were unable to systematically consider all factors that may influence cognition and sleep, such as depression, nocturia, or nocturnal “off” symptoms. Patients continued medication during polysomnography, and cognitively impaired patients more frequently took several medications with possible effects on sleep and cognition. Although this may simply reflect pharmacological management in PD patients with more impaired cognition and sleep, we also observed that these medications were additionally associated with less SWA. Importantly, though, accounting for these pharmacological effects did not attenuate the association of MoCA performance with SWA. Future studies may investigate pharmacological influences on SWS and cognition in more depth.
Taken together, the present study suggests that cognitive impairment in PD patients is associated with reduced SWA, particularly in the lower delta frequencies (1–2 Hz) and in frontal regions. Our study adds evidence that SWS could play a beneficial role in PD.
Future research should relate SWS characteristics to longitudinal trajectories, cognitive sub-domains in larger samples, and neuroimaging or fluid biomarkers, and explore the potential of SWS as a biomarker and therapeutic target in PD and related disorders.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The study was reviewed and approved by the local ethics committee (Kantonale Ethikkommission Zürich, Switzerland). The patients provided written informed consent for further use of health-related data.
SS: conceptualization, data acquisition, data analysis, data visualization, writing of the draft, and funding acquisition. LI and PV: data analysis, review and editing of the draft. AM: conceptualization, review and editing of the draft. RM: data acquisition, review and editing of the draft. EW: data analysis, resources, review and editing of the draft. CB: conceptualization, review and editing of the draft, funding acquisition, supervision, and resources. HB-V: conceptualization, data acquisition, review and editing of the draft, and supervision. All authors contributed to the article and approved the submitted version.
The Swiss Neurological Society supported this work (stipend to SS). The Swiss National Science Foundation supported this work (Grant No. 188790 to CB). Parkinson Schweiz supported this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the Swiss Neurological Society, the Swiss National Science Foundation, and Parkinson Schweiz for their support.
1. Aarsland D, Creese B, Politis M, Chaudhuri KR, Ffytche DH, Weintraub D, et al. Cognitive decline in Parkinson disease. Nat Rev Neurol. (2017) 13:217–31. doi: 10.1038/nrneurol.2017.27
2. Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. (2009) 326:1005–7. doi: 10.1126/science.1180962
3. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. (2013) 342:373–7. doi: 10.1126/science.1241224
4. Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. (2019) 363:eaav2546. doi: 10.1126/science.aav2546
5. Ju Y-ES, Ooms SJ, Sutphen C, Macauley SL, Zangrilli MA, Jerome G, et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain. (2017) 140:2104–11. doi: 10.1093/brain/awx148
6. Olsson M, Arlig J, Hedner J, Blennow K, Zetterberg H. Sleep deprivation and cerebrospinal fluid biomarkers for Alzheimer's disease. Sleep. (2018) 41. doi: 10.1093/sleep/zsy025
7. Hablitz LM, Vinitsky HS, Sun Q, Stæger FF, Sigurdsson B, Mortensen KN, et al. Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci Adv. (2019) 5:eaav5447. doi: 10.1126/sciadv.aav5447
8. Westerberg CE, Mander BA, Florczak SM, Weintraub S, Mesulam MM, Zee PC, et al. Concurrent impairments in sleep and memory in amnestic mild cognitive impairment. J Int Neuropsychol Soc. (2012) 18:490–500. doi: 10.1017/S135561771200001X
9. Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, et al. beta-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. (2015) 18:1051–7. doi: 10.1038/nn.4035
10. Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, et al. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer's disease. Sci Transl Med. (2019) 11:eaau6550. doi: 10.1126/scitranslmed.aau6550
11. Winer JR, Mander BA, Kumar S, Reed M, Baker SL, Jagust WJ, et al. Sleep Disturbance Forecasts β-Amyloid Accumulation across Subsequent Years. Curr Biol. (2020) 30:4291–8.e3. doi: 10.1016/j.cub.2020.08.017
12. Sunwoo J-S, Cha KS, Byun J-I, Jun J-S, Kim T-J, Shin J-W, et al. NREM sleep EEG oscillations in idiopathic REM sleep behavior disorder: a study of sleep spindles and slow oscillations. Sleep. (2020) zsaa160. doi: 10.1093/sleep/zsaa160
13. Brunner H, Wetter TC, Hogl B, Yassouridis A, Trenkwalder C, Friess E. Microstructure of the non-rapid eye movement sleep electroencephalogram in patients with newly diagnosed Parkinson's disease: effects of dopaminergic treatment. Mov Disord. (2002) 17:928–33. doi: 10.1002/mds.10242
14. Diederich NJ, Vaillant M, Mancuso G, Lyen P, Tiete J. Progressive sleep 'destructuring' in Parkinson's disease. A polysomnographic study in 46 patients. Sleep Med. (2005) 6:313–8. doi: 10.1016/j.sleep.2005.03.011
15. Amato N, Manconi M, Moller JC, Sarasso S, Stanzione P, Staedler C, et al. Levodopa-induced dyskinesia in Parkinson disease: sleep matters. Ann Neurol. (2018) 84:905–17. doi: 10.1002/ana.25360
16. Schreiner SJ, Imbach LL, Werth E, Poryazova R, Baumann-Vogel H, Valko PO, et al. Slow-wave sleep and motor progression in Parkinson disease. Ann Neurol. (2019) 85:765–70. doi: 10.1002/ana.25459
17. Latreille V, Carrier J, Gaudet-Fex B, Rodrigues-Brazete J, Panisset M, Chouinard S, et al. Electroencephalographic prodromal markers of dementia across conscious states in Parkinson's disease. Brain. (2016) 139 (Pt 4):1189–99. doi: 10.1093/brain/aww018
18. Wood KH, Memon AA, Memon RA, Joop A, Pilkington J, Catiul C, et al. Slow wave sleep and EEG delta spectral power are associated with cognitive function in Parkinson's disease. J Parkinsons Dis. (2020). doi: 10.3233/JPD-202215
19. Murphy M, Riedner BA, Huber R, Massimini M, Ferrarelli F, Tononi G. Source modeling sleep slow waves. Proc Natl Acad Sci USA. (2009) 106:1608–13. doi: 10.1073/pnas.0807933106
20. Varga AW, Wohlleber ME, Gimenez S, Romero S, Alonso JF, Ducca EL, et al. Reduced slow-wave sleep is associated with high cerebrospinal fluid Abeta42 levels in cognitively normal elderly. Sleep. (2016) 39:2041–8. doi: 10.5665/sleep.6240
21. Baumann CR, Held U, Valko PO, Wienecke M, Waldvogel D. Body side and predominant motor features at the onset of Parkinson's disease are linked to motor and nonmotor progression. Mov Disord. (2014) 29:207–13. doi: 10.1002/mds.25650
22. Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Mov Disord. (2010) 25:2649–53. doi: 10.1002/mds.23429
23. Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, et al. The montreal cognitive assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. (2005) 53:695–9. doi: 10.1111/j.1532-5415.2005.53221.x
24. Dalrymple-Alford JC, MacAskill MR, Nakas CT, Livingston L, Graham C, Crucian GP, et al. The MoCA: well-suited screen for cognitive impairment in Parkinson disease. Neurology. (2010) 75:1717–25. doi: 10.1212/WNL.0b013e3181fc29c9
25. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. (1992) 55:181–4. doi: 10.1136/jnnp.55.3.181
26. Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord. (2007) 22:1689–707. doi: 10.1002/mds.21507
27. Berry RB, Brooks R, Gamaldo CE, Harding SM, Marcus C, Vaughn BV. The AASM Scoring Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications, Version 2.2. Darien, Illinois: American Academy of Sleep Medicine (2015).
28. Medicine AAoS. International Classification of Sleep Disorders: Darien, IL: Medicine AAoS (2014).
29. Baumann-Vogel H, Imbach LL, Surucu O, Stieglitz L, Waldvogel D, Baumann CR, et al. The impact of subthalamic deep brain stimulation on sleep-wake behavior: a prospective electrophysiological study in 50 Parkinson patients. Sleep. (2017) 40. doi: 10.1093/sleep/zsx033
30. Kalaitzakis ME, Gentleman SM, Pearce RK. Disturbed sleep in Parkinson's disease: anatomical and pathological correlates. Neuropathol Appl Neurobiol. (2013) 39:644–53. doi: 10.1111/nan.12024
31. Cedernaes J, Osorio RS, Varga AW, Kam K, Schiöth HB, Benedict C. Candidate mechanisms underlying the association between sleep-wake disruptions and Alzheimer's disease. Sleep Med Rev. (2017) 31:102–11. doi: 10.1016/j.smrv.2016.02.002
32. Bohnen NI, Hu MTM. Sleep disturbance as potential risk and progression factor for Parkinson's disease. J Parkinsons Dis. (2019) 9:603–14. doi: 10.3233/JPD-191627
33. Stavitsky K, Neargarder S, Bogdanova Y, McNamara P, Cronin-Golomb A. The impact of sleep quality on cognitive functioning in Parkinson's disease. J Int Neuropsychol Soc. (2012) 18:108–17. doi: 10.1017/S1355617711001482
34. Gunn DG, Naismith SL, Terpening Z, Lewis SJ. The relationships between poor sleep efficiency and mild cognitive impairment in Parkinson disease. J Geriatr Psychiatry Neurol. (2014) 27:77–84. doi: 10.1177/0891988713509135
35. Chahine LM, Xie SX, Simuni T, Tran B, Postuma R, Amara A, et al. Longitudinal changes in cognition in early Parkinson's disease patients with REM sleep behavior disorder. Parkinsonism Relat Disord. (2016) 27:102–6. doi: 10.1016/j.parkreldis.2016.03.006
36. Mery VP, Gros P, Lafontaine AL, Robinson A, Benedetti A, Kimoff RJ, et al. Reduced cognitive function in patients with Parkinson disease and obstructive sleep apnea. Neurology. (2017) 88:1120–8. doi: 10.1212/WNL.0000000000003738
37. Sobreira EST, Sobreira-Neto MA, Pena-Pereira MA, Chagas MHN, Fernandes RMF, Eckeli AL, et al. Global cognitive performance is associated with sleep efficiency measured by polysomnography in patients with Parkinson's disease. Psychiatry Clin Neurosci. (2019) 73:248–53. doi: 10.1111/pcn.12819
38. Latreille V, Carrier J, Lafortune M, Postuma RB, Bertrand JA, Panisset M, et al. Sleep spindles in Parkinson's disease may predict the development of dementia. Neurobiol Aging. (2015) 36:1083–90. doi: 10.1016/j.neurobiolaging.2014.09.009
39. Bugalho P, Ladeira F, Barbosa R, Marto JP, Borbinha C, da Conceição L, et al. Polysomnographic predictors of sleep, motor and cognitive dysfunction progression in Parkinson's disease: a longitudinal study. Sleep Med. (2020). doi: 10.1016/j.sleep.2020.06.020
40. Werth E, Achermann P, Borbély AA. Brain topography of the human sleep EEG: antero-posterior shifts of spectral power. Neuroreport. (1996) 8:123–7. doi: 10.1097/00001756-199612200-00025
41. Kehagia AA, Barker RA, Robbins TW. Cognitive impairment in Parkinson's disease: the dual syndrome hypothesis. Neurodegener Dis. (2013) 11:79–92. doi: 10.1159/000341998
Keywords: Parkinson disease, sleep, slow-wave sleep, cognition, cognitive impairment, polysomnography, EEG, spectral analysis
Citation: Schreiner SJ, Imbach LL, Valko PO, Maric A, Maqkaj R, Werth E, Baumann CR and Baumann-Vogel H (2021) Reduced Regional NREM Sleep Slow-Wave Activity Is Associated With Cognitive Impairment in Parkinson Disease. Front. Neurol. 12:618101. doi: 10.3389/fneur.2021.618101
Received: 16 October 2020; Accepted: 25 January 2021;
Published: 19 February 2021.
Edited by:
Salvatore Galati, Neurocenter of Southern Switzerland, SwitzerlandReviewed by:
Veronique Latreille, McGill University, CanadaCopyright © 2021 Schreiner, Imbach, Valko, Maric, Maqkaj, Werth, Baumann and Baumann-Vogel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon J. Schreiner, c2ltb24uc2NocmVpbmVyQHVzei5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.