 Sjoukje Nutma
Sjoukje Nutma Joost le Feber
Joost le Feber Jeannette Hofmeijer
Jeannette Hofmeijer- 1Department of Neurology, Medisch Spectrum Twente, Enschede, Netherlands
- 2Clinical Neurophysiology, University of Twente, Enschede, Netherlands
- 3Department of Neurology, Rijnstate Hospital Arnhem, Arnhem, Netherlands
Postanoxic encephalopathy is the key determinant of death or disability after successful cardiopulmonary resuscitation. Animal studies have provided proof-of-principle evidence of efficacy of divergent classes of neuroprotective treatments to promote brain recovery. However, apart from targeted temperature management (TTM), neuroprotective treatments are not included in current care of patients with postanoxic encephalopathy after cardiac arrest. We aimed to review the clinical evidence of efficacy of neuroprotective strategies to improve recovery of comatose patients after cardiac arrest and to propose future directions. We performed a systematic search of the literature to identify prospective, comparative clinical trials on interventions to improve neurological outcome of comatose patients after cardiac arrest. We included 53 studies on 21 interventions. None showed unequivocal benefit. TTM at 33 or 36°C and adrenaline (epinephrine) are studied most, followed by xenon, erythropoietin, and calcium antagonists. Lack of efficacy is associated with heterogeneity of patient groups and limited specificity of outcome measures. Ongoing and future trials will benefit from systematic collection of measures of baseline encephalopathy and sufficiently powered predefined subgroup analyses. Outcome measurement should include comprehensive neuropsychological follow-up, to show treatment effects that are not detectable by gross measures of functional recovery. To enhance translation from animal models to patients, studies under experimental conditions should adhere to strict methodological and publication guidelines.
Introduction
Survival rates of out-of-hospital cardiac arrest have increased considerably over the past decades (1, 2). In contrast, neurological outcome of survivors has improved only marginally. Of patients surviving up to hospital admission, more than three quarters initially remain comatose as a result of diffuse cerebral ischaemia (3, 4). Comatose patients after circulatory arrest are treated on intensive care units to support airway, breathing, and circulation. Anoxic-ischemic encephalopathy is the key determinant of death and disability, with rates of in-hospital mortality or enduring neurologic impairment >50% (5). Targeted temperature management (TTM) at 33 or 36°C is applied as a therapeutic strategy in most hospitals to improve brain recovery, although the clinical evidence supporting efficacy is complex and mechanisms of action are incompletely understood (6–9).
Treatment strategies other than TTM that were beneficial in animal models and have been tested in clinical trials include glutamate and calcium antagonists, anti-inflammatory therapies, and anti-oxidants. None of these improved cerebral recovery or functional outcome of patients after cardiac arrest. Proposed explanations include poor extrapolation from animal models to patients, insufficient knowledge of when and where we can interfere in the complex pathophysiology of brain damage after global ischaemia, and heterogeneity of patients groups (10, 11).
In this article, we review the clinical evidence of efficacy of neuroprotective treatments in comatose patients after cardiac arrest. We discuss treatment effects, and the lack thereof, in the context of the pathophysiology of postanoxic encephalopathy, and propose future directions.
Search Strategy and Selection Criteria
For analysis of tested neuroprotective measures in comatose patients after out-of-hospital cardiac arrest (OHCA), we applied a search in the Medline and Pubmed databases until October 2019. We used several combinations of the keywords and MeSH terms. For selection of the target patient group we used “post-anoxic encephalopathy,” “hypoxic ischemic brain injury,” “coma,” “cardiopulmonary resuscitation,” and “cardiac arrest” (Figure 1). For selection of interventions we first used the general term “neuroprotective” in combination with “outcome,” later we searched more specifically on tested interventions such as “xenon,” “magnesium” etc. Review articles were used to screen reference lists. We included prospective, controlled, intervention trials with clinical neurological outcome measures. Studies that used the cerebral biomarkers NSE or S100b as a substitute for neurological outcome were also included. Technique of cardiopulmonary resuscitation was not taken into account as an in- or exclusion criterion. When a certain topic was addressed by a large number of studies, the latest meta-analysis was used, supplemented by more recent studies in that field. This applied to hypothermia and adrenaline (epinephrine). We excluded studies on in-hospital cardiac arrest, retrospective, observational, and uncontrolled studies, case studies, studies in pediatric populations, and animal studies.
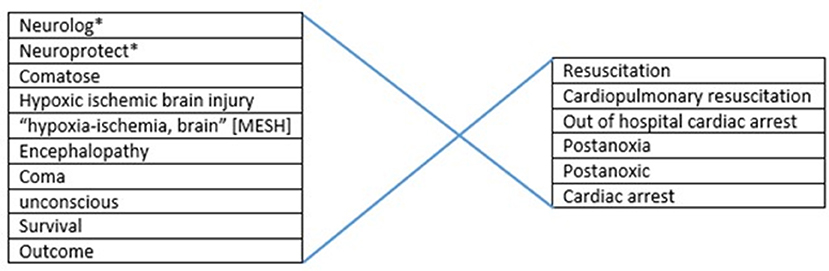
Figure 1. Overview of used search strategy.
Results
We included 53 studies on 21 different therapies. Over the past 30 years there has been a significant increase in trials on cerebral recovery after resuscitation (Figure 2). The first years were characterized by studying the effects of barbiturates, calcium antagonists, and hypothermia. Later, the focus shifted to novel therapies in this field like xenon, exenatide, and cyclosporine.
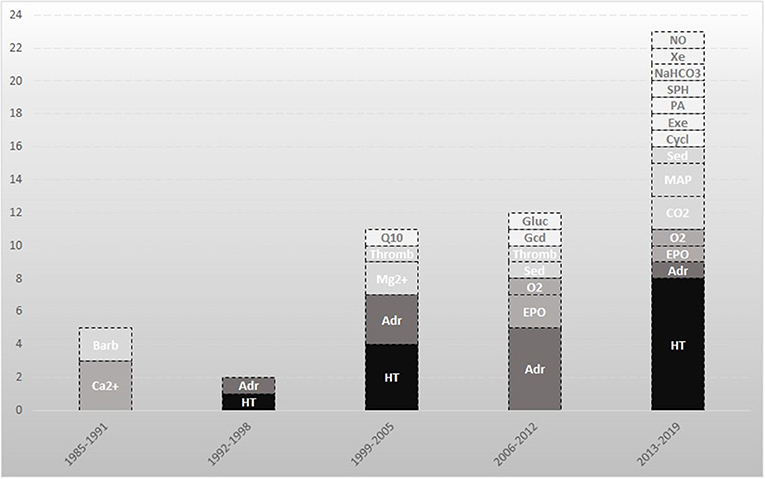
Figure 2. Neuroprotective studies directed at improvement of neurological outcome after cardiac arrest. An overview of the included studies in this review. The darker the box, the larger the amount of included studies on that topic. Barb indicates barbiturates; Ca2+, calcium antagonists; CO2, carbon dioxide; Cycl, cyclosporine; Adr, adrenaline; EPO, erythropoietin; Exe, exenatide; Gcd, glucocorticoid; Gluc, glucose; HT, hypothermia; Mg2+, magnesium; MAP, mean arterial pressure; NaHCO3, sodium bicarbonate; NO, Sodium nitrite; O2, oxygen; PA, prophylactic antibiotics; Q10, coenzyme Q10; Sed, sedation; SPH, scopolamine and penehyclidine hydrochloride; Thromb, thrombolysis; Xe, xenon.
Since we included a multitude of therapies, addressed in studies with disparate diagnostic criteria and study designs, meta-analysis was not possible. Therefore, a narrative review was chosen. We found two cohort studies on treatment of status epilepticus, but no prospective controlled studies. A randomized controlled trial is currently in progress (12). Several randomized controlled trials (RCT) on early application of extracorporeal life support in cardiac arrest are in progress [NCT03065647 (13), NCT01605409 (14), NCT02527031 (15), NCT01511666 (16), NCT03101787 (17)], no results are published yet. Also no studies could be found concerning specific nutrition after cardiac arrest. Study details and results are summarized in Supplementary Table 1.
Pathophysiological Considerations
The human brain contributes only 2% to the total body weight, yet it accounts for 20% of total body oxygen consumption and 25% of glucose utilization (18, 19). This high metabolism in combination with the lack of cerebral glucose stores makes the brain highly susceptible to blood flow interruption. Insufficient blood flow (ischaemia) and oxygen delivery (hypoxia) during cardiac arrest may lead to loss of neuronal function within seconds (20). Initially, this is reversible. Transitions from reversible to irreversible brain damage occur in minutes, hours, or days, and depend on the level of the remaining blood flow, the duration of ischaemia, and the extent of reperfusion (Figure 3) (21).
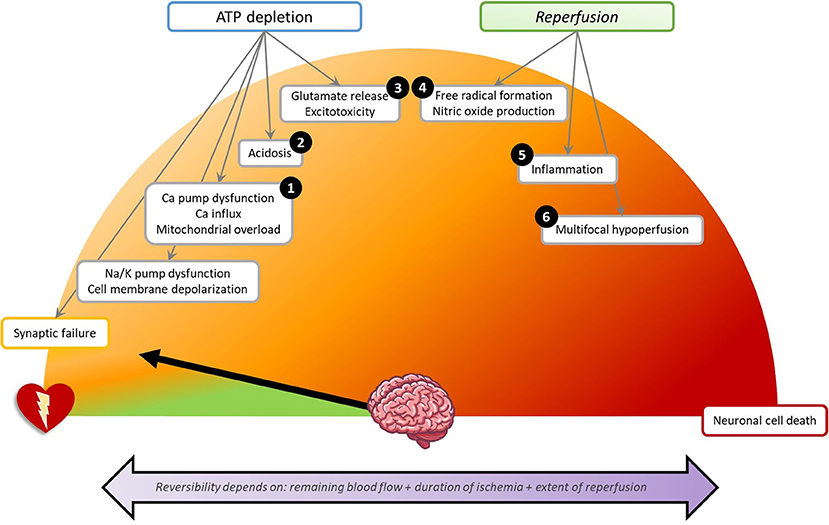
Figure 3. Schematic overview of pathophysiology of brain damage in the first 72 h after cardiac arrest. Each step can lead to direct or delayed neuronal cell death. The numbers indicate the presumed point of action of the discussed neuroprotective treatments. (1) Calcium antagonists: Nimodipine, Flunarizine, Lidoflazine. Mitigating mitochondrial damage: Cyclosporine, Coenzyme Q10. (2) Preventing acidosis: Sodium bicarbonate. (3) Glutamate antagonism: Noble gases, Exenatide, Scopolamine, and penehyclidine hydrochloride, Magnesium. (4) Antioxidants: Preventing hyperoxia, Sodium nitrite. (5) Anti-inflammation: Erythropoietin, Glucocorticoids. (6) Optimizing cerebral perfusion: Adrenaline, Mild hypocapnia, High mean arterial pressure, Thrombolysis. (1–6) Pan-inhibition: Hypothermia. Not indicated by a number: Decreasing cerebral metabolism: Barbiturates. Supportive therapies: Sedation, Glucose regulation, Prophylactic antibiotics. Na/K, sodium/potassium; Ca, calcium.
Disappearance of synaptic activity is the first effect of cerebral hypoxia or ischaemia and occurs within seconds to minutes (22). Synaptic activity indicates the process in which neurons pass chemical signals to other neurons and is also called neurotransmission. Early synaptic failure is due to impaired presynaptic neurotransmitter release (11, 23). Synaptic failure may be completely reversible. However, with lasting hypoxia or ischaemia, synaptic disturbances may become permanent, even with preserved membrane potential (24).
Depending on the remaining perfusion levels, cerebral glucose and ATP stores are depleted within minutes to hours (19). Ultimately, this results in dysfunctioning of ATP-dependent ion pumps, especially transmembrane sodium-potassium pumps. The subsequent inflow of sodium and outflow of potassium leads to loss of ion gradients across the plasma membrane, causing depolarization (i.e., loss of membrane potential) (19). This leads to inability to generate action potentials. Since the net flow of osmotically active particles from the extracellular space into the neurons (sodium, chloride) exceeds that from intracellular to extracellular (potassium), the intracellular osmolality increases. This causes inflow of water into the cells leading to cell swelling (25). Cell swelling is reversible with rapid restoration of perfusion. In the absence of reperfusion, it leads to necrotic cell death.
Dysfunctioning of ATP-dependent calcium channels causes influx of calcium into the intracellular space, which activates pathways leading up to apoptosis (19). High intracellular calcium activates the mitochondrial permeability transition pore. This protein in the inner membrane of mitochondria only opens under pathological conditions and releases cytochrome C, an activator of various cascades leading to apoptosis (26, 27) Very high calcium leads to direct destruction of mitochondria (27). In addition, calcium mediates release of glutamate, resulting in overexcitation of the NMDA receptor (“excitotoxicity”), leading to neuronal damage and cell death (28, 29).
Restoration of perfusion may cause additional (secondary) brain damage. First, free radical or reactive oxygen species may cause cellular lipid and protein degradation. Second, reperfusion is associated with inflammatory responses and microvascular damage (19). Third, reperfusion is often unevenly distributed due to cerebral vasospasm, increased blood viscosity, and platelet aggregation. This causes focal or multifocal hypoperfusion, which is called “no-reflow phenomenon” (30).
The timescales of the various pathophysiological processes define the therapeutic windows of opportunity for neuroprotective treatments interacting with these processes. The first minutes to hours after diffuse cerebral ischaemia by cardiac arrest are characterized by massive cortical synaptic failure. This is reflected by suppressed EEG patterns. With timely reperfusion this is, in principle, reversible. However, synaptic failure lasting over 6–24 h is associated with transitions toward permanent brain damage (31, 32). With deep or persistent hypoperfusion, cell swelling occurs, which is reversible with rapid restoration of perfusion, but may lead to cell death within minutes (33). Pathways leading up to programmed cell death (apoptosis) are activated on timescales up to 72 h after cardiac arrest (34). This suggests that time windows of opportunity for interventions to prevent the transition from reversible to irreversible brain damage lie between hours and days, with largest effects in the first hours after cardiac arrest (20). Interventions to improve brain network and functional recovery by enhancement of connectivity may be effective on longer timescales (35).
Measuring Neurological Outcome
Clinical Outcome Scales
We included studies that used functional recovery according to the Cerebral Perfomance Category (CPC) (36) or the extended version of the Glasgow Outcome Scale (GOS-E) (37) as measures of outcome. CPC is a five-point scale ranging from brain death (CPC 5) to full recovery or mild disability (CPC 1). CPC 1–2 is mostly considered as good and CPC 3–5 as poor neurological outcome. The GOS-E uses 8 different levels of disability in which a score of 1 equals death and 8 good recovery. A score of 5–8 is considered good neurological outcome (Figure 4). Both outcome scales are criticized for limited discrimination (38, 39) and GOS-E is only validated in patients with traumatic (and not in anoxic) brain injury (40, 41).
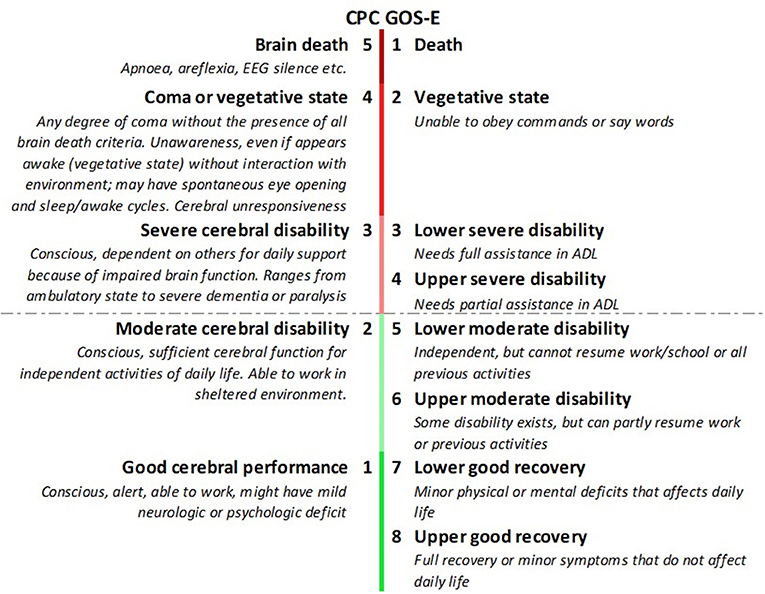
Figure 4. Cerebral Performance Category (CPC) compared to extended Glasgow Outcome Scale (GOS-E). CPC 1-2 and GOS-E 5-8 are considered good neurological outcome.
Biochemical Biomarkers
We also included studies that used blood levels of neuron specific enolase (NSE) or S-100b as measures of outcome. NSE and S-100b are proteins that are released in the blood and cerebrospinal fluid with damage of neurons and glial cells, respectively (42). Higher NSE blood values at day 1 and 3 are associated with poorer outcome after out-of-hospital cardiac arrest (43, 44). Reported cut-off values for reliable prediction of poor outcome range from 20 to 65 μg/L (45). Higher S-100b blood values are associated with poorer neurological outcome, but the predictive values for individual patients are limited (46). S-100b is also present in muscle and adipose tissue and can therefore be increased by a thoracic trauma due to cardiopulmonary resuscitation (47). Because of the extra-cerebral sources, heterogeneous measurement techniques, and divergent proposed cut-off threshold for prediction, the use of these biomarkers for prediction of outcome after cardiac arrest is controversial (45–47).
Neuroprotective Treatments of Comatose Patients After Cardiac Arrest
Pan-Inhibition
Hypothermia
The working mechanism of induced hypothermia is presumed to be pan-inhibition, by reducing ATP depletion (7, 48) and anoxic depolarization (8, 49). Also glutamate antagonism (50, 51), anti-inflammatory effects (52, 53), reduction of free radical production (9) and anti-apoptotic effects (54, 55) have been described. In 2002, in two relatively small RCT's (n = 77 and n = 273), TTM at 32–34°C was associated with a higher probability of a good outcome of comatose patients after cardiac arrest (respectively, 21/43 vs. 9/34 and 75/136 vs. 54/137) (56, 57). However, control groups contained relatively many patients with hyperthermia which is associated with secondary damage after an anoxic insult (58). This led to the believe that the beneficial factor was not the induced hypothermia, but the prevention of fever. The Targeted Temperature Management trial (TTM) compared controlled normothermia at 36°C with hypothermia at 33°C for 28 h and showed no significant differences in mortality (50% in 33°C group vs. 48% in 36 °C) and no differences in neurological outcome measured by CPC (CPC 1–2 46.5 vs. 47.8%) (6).
Several trials focussed on timing of inducing hypothermia and alternative cooling techniques. A 2018 RCT on alternative cooling techniques (59) showed no differences between the different techniques, but did find a significantly better survival in the intervention compared to historical controls receiving normothermia. Information on incidence of hyperthermia in this group, was lacking. A Cochrane review and later published trials comparing pre-hospital and in-hospital start of cooling also showed no certain benefit of a pre-hospital start for survival or neurological outcome (60–63), and even showed higher rates of pulmonary edema when rapid infusion of cold saline fluids were used for induction of cooling (63). A study on longer duration of hypothermia at 33°C also showed no differences in outcome (64), but did report on impaired thrombin without an increase of bleeding complications (65). The ongoing TTM2 trial compares target temperature of 33°C and standard care avoiding fever [NCT02908308 (66)]. If beneficial at all, optimal timing and duration of targeted temperature management remain to be elucidated.
The association of decreased mortality when using neuromuscular blockade during hypothermia (67–69), could not be confirmed by two small RCT's comparing hypothermia treatment with and without neuromuscular blockade (70, 71).
Decreasing Cerebral Metabolism
Barbiturates
After several feasibility trials using barbiturates in comatose patients after cardiac arrest (72), these substances were tested in efficacy trials on neuroprotection after cardiac arrest in the 1980s. The presumed working mechanism involves global depression of cerebral metabolism (73), depression of release of ROS, and inhibition of lipid degradation (74, 75). A randomized trial tested a single thiopental dose in comatose patients after cardiac arrest and found no significant effects on survival (77% in intervention group vs. 80% in standard-therapy group) or in good cerebral recovery (20 vs. 15%) (76). A smaller study with a control group of matched historical cases also found no differences in survival, despite a non-significant higher mortality in the first 6 h in the thiopental group in patients with ischemic heart disease. However, after these 6 h, good neurological recovery was observed significantly more frequent in the thiopental group (61 vs. 37%, p < 0.03) (77).
Glutamate Antagonism
(Noble) Gases
Various in vitro and animal studies showed beneficial effects of noble gases on hypoxic-ischemic brain damage (78, 79). This resulted in feasibility and safety studies on xenon, helium, and hydrogen in patients with postanoxic encephalopathy after cardiac arrest (80–82). The presumed mechanism of action is competitive inhibition at the glycine co-activation site of the NMDA receptor, thereby preventing toxic overexcitation. A single-blinded, randomized study in 110 patients studied the effect of inhaled xenon on white matter damage assessed by diffusion tensor magnetic resonance imaging (MRI). Fractional anisotropy was higher in the intervention than in the control group, suggesting less damage of white matter tracts. However, functional recovery as expressed by CPC and mRS scores at 6 months showed no differences between the groups (83). An RCT on the effects of inhalation of hydrogen on neurological outcome is in progress [UMIN000019820 (84)].
Exenatide
The glucagon-like peptide-1 (GLP-1) exenatide is used for treatment of type 2 diabetes mellitus. It showed neuroprotective and anti-inflammatory capacities in several in vivo and in vitro studies (85). Exenatide is a mediator of glutamate release and can prevent toxic over-excitation by inhibiting glutamate release (86, 87). Exenatide given in the first 6 h after return of spontaneous circulation (ROSC) had no statistically significant effect on NSE levels or clinical outcome in an RCT with 118 patients (88).
Scopolamine and Penehyclidine Hydrochloride
Another possible therapy targeting the NMDA-receptor is penehyclidine hydrochloride (PHC). In a study with 80 patients randomized to either scopolamine or PHC, PHC was associated with lower intracranial pressures, higher cerebral perfusion pressures and lower NSE. However, clinical outcome measures and a control group without experimental treatment were lacking, hampering interpretation of the data (89).
Magnesium
In vitro and in vivo studies showed beneficial effects of magnesium on neuronal and neurological recovery after hypoxic-ischemic damage, due to reduction in glutamate response and calcium entry blocking capacities. Two randomized placebo controlled studies focussed on the effects of magnesium sulfate, both in a pre-hospital setting. The first (n = 105), used magnesium in patients with refractory ventricular fibrillation and showed no differences in gaining ROSC or in neurological outcome of the three surviving patients (90). The second (n = 300), studied effects of diazepam with or without magnesium. Neurological outcome expressed as awakening and having comprehensible speech was not significantly different between both groups (91). Other studies on magnesium in cardiac arrest did not address neurological recovery or were focussed on in-hospital cardiac arrest.
Calcium Antagonists
Nimodipine
Of all the calcium antagonists nimodipine is studied most in comatose patients with postanoxic encephalopathy. After a safety study of nimodipine in 22 OHCA patients in 1987 (92), a randomized double-blind study in 155 patients administered nimodipine or placebo in a prehospital setting. No effects were demonstrated on long term survival or probability of good neurological outcome (93). Another randomized study in 51 patients showed significantly higher cerebral blood flow in the nimodipine group, without differences in intracranial pressure measured on several time points, compared to placebo. Neurological outcome did not differ between the groups (94). A smaller randomized trial measured the intracranial pressure continuously and found lower mean pressures in the nimodipine group. Neurological outcome nor survival was taken into account (95).
Lidoflazine
The Brain Resuscitation Clinical Trial II Study Group included 520 comatose patients after cardiac arrest and randomized them to lidoflazine or placebo. At 6 months there was no difference in mortality (82% in lidoflazine group vs. 83% in placebo group), or in proportion of patients with good outcome (15 vs. 13%) (96).
Preventing Acidosis
Sodium Bicarbonate
Sodium bicarbonate (NaHCO3) has been used to reverse acidosis and treat hyperkalaemia in cardiopulmonary resuscitation. Both favorable and unfavorable effects of administering NaHCO3 during CPR have been reported. Unfavorable effects include paradoxical respiratory acidosis due to increased carbon dioxide tension (97). Observational studies showed a possible role for NaHCO3 in prolonged cardiopulmonary resuscitation (CPR), to compensate the consequent severe acidosis that is associated with an impaired responsiveness to catecholamines (98). A large randomized study on prehospital NaHCO3 administration (n = 792) found no differences in survival (13.8% vs. 13.9%), but confirmed a possible beneficial effect in prolonged CPR with a trend toward increased survival (32.8% in the intervention group vs. 15.4%, p = 0.07) (99). The latest RCT in 50 patients focussed only on prolonged cardiac arrest (>10 min) and treated with NaHCO3 or placebo when there was evidence of severe metabolic acidosis. Despite a significant difference in pH (6.99 in intervention group vs. 6.9 in placebo group) there were no differences in survival and neurological outcome (100).
Antioxidants
Preventing Hyperoxia
Animal studies demonstrated that hyperoxia in the first 24 h after cardiac arrest is associated with poor neurological outcome (101). A prospective observational study on patients after cardiac arrest showed an independent association between early hyperoxia and poor neurological outcome (102). Four studies addressed the feasibility of lowering oxygen levels in a pre-hospital setting (103–106). Three of the four studies showed feasibility. The biggest risk was inadvertent hypoxaemia (103, 105). The groups were small and often neurological outcome measures were not taken into account, so conclusions on efficacy cannot be drawn. Another study on early reduction of oxygen levels post cardiac arrest is in progress (NCT03138005) (107). A larger study by the COMACARE study group compared systemic arterial normoxia (PaO2 10–15 kPa) to moderate hyperoxia (PaO2 20–25 kPa) and found no significant differences in the primary endpoint NSE serum concentration at 48 h after cardiac arrest or in neurological outcome measured by CPC (108).
Sodium Nitrite
During hypoxia nitrite is converted to nitric oxide via several pathways (109). This free radical has shown to be neuroprotective by reducing production of reactive oxygen species in animal studies (110, 111). A pilot study intravenously administered sodium nitrite to 120 patients during resuscitation from OHCA. Due to insufficient serum levels, the dose of 25 mg was halfway adjusted to 60 mg. Despite adequate serum levels, there were no differences in rate to ROSC, survival or neurological outcome at discharge compared to matched controls. There were no differences in systolic blood pressure or use of noradrenaline (norepinephrine) between both groups (112).
Anti-inflammation
Erythropoietin
Promising pre-clinical studies suggested neuroprotective effects of erythropoietin (EPO) by inhibition of neuronal apoptosis (113) and anti-inflammatory qualities (114). This gave rise to three clinical studies. A prospective study with case matched controls (n = 58) showed no significant difference in survival on day 28. One case of arterial vascular thrombosis was observed as adverse event in the EPO-group (115). The second study (n = 72) also used case matched controls and found a significantly higher rate of survival up to hospital admission and 24h survival in the intervention group (92 vs. 65% and 83 vs. 52%), but no difference in CPC at hospital discharge (116). The third and largest study was an RCT in 476 patients and found no significant difference in CPC-scores at any time point. Serious adverse events consisting of thrombotic complications, were more frequent in the EPO-treated group (117).
Glucocorticoids
Since cardiac arrest is associated with an impaired cortisol release from the adrenal cortex, two clinical trials focused on the effects of glucocorticoid suppletion on outcome. A pilot study compared hydrocortisone with placebo during resuscitation, resulting in a large increase in attaining ROSC in the hydrocortisone group (61 vs. 39%, p = 0.038), but a comparable median CPC of 4 at discharge in four surviving patients of each group (118). Other studies on glucocorticoid administration focussed on in-hospital cardiac arrests (119).
Mitigating Mitochondrial Damage
Cyclosporine
This immunosuppressant substance used in the treatment of for example Crohn's disease got new attention when in vitro and in vivo studies showed promising effects on preventing mitochondrial permeability (120). In an RCT in 794 patients, cyclosporine was administered during resuscitation of non-shockable out-of-hospital cardiac arrest. Survival, neurological outcome, and the primary endpoint of multi-organ failure were essentially the same in the two groups (121).
Coenzyme Q10
(CoQ10) is an electron transporter between mitochondrial complexes. Dysfunction compromises mitochondrial function. Administration of CoQ10 has shown neuroprotective effects in for example Parkinson's disease (122). One observational study showed an association between low CoQ10 levels and increased mortality after cardiac arrest (123). An RCT in 49 patients compared CoQ10 suppletion with placebo within 6 h after cardiac arrest during 5 days, survival at 3 months was significantly increased in the CoQ10 group (17 of 25 vs. 7 of 24 patients). Persistent vegetative state was more frequent in the CoQ10 group (7 vs. 3 patients) (124).
Optimizing Cerebral Perfusion
Adrenaline
Adrenaline has been an established medicine in advanced life support protocols for many years (125). By stimulating the α-adrenergic receptors it causes vasoconstriction and thereby a higher coronary blood flow and a bigger chance of ROSC after cardiac arrest (126). A positive effect on neurological outcome was never found, which has been attributed to platelet activation mediated by adrenaline induced thrombosis, with impairment of cerebral blood flow (127). A large randomized trial (n = 8,014) compared adrenaline to placebo and found significantly higher survival rates at 30 days, but worse neurological functioning in the surviving intervention group (128). A recent meta-analysis concluded that a standard dose of adrenaline compared to pooled treatments (defined as placebo, no drugs, high dose of adrenaline or adrenaline + vasopressin) improves survival to hospital and increases the chances of a good neurological outcome. However, when standard dosed adrenaline was compared with just placebo or no drugs, no significant differences in neurological outcome were found (129). It can be concluded that optimal dosing and effects on neurological outcome are still unclear.
Carbon Dioxide Levels
Mild hypercapnia should compensate the compromised cerebral blood flow after cardiac arrest by augmenting cerebral perfusion due to vasodilation (130). However, higher carbon dioxide levels carry the risk of increased intracranial pressure and of pulmonary vasoconstriction. On the other hand, hypocapnia is associated with worse neurological outcomes (131). Probably, preventing hypocapnia and inducing mild hypercapnia is beneficial. A feasibility trial in 83 patients compared normocapnia (PaCO2 35–45 mmHg) with mild hypercapnia (PaCO2 50–55 mmHg) and found no differences in GOS. The increase in NSE was significantly lower in the hypercapnia group 24, 48, and 72 h (132). A large RCT comparing these same PaCO2 levels (TAME) is in progress (133). Another RCT (n = 123) compared low-normal PaCO2 (33–35 mmHg) with high-normal PaCO2 (43–45 mmHg), maintained this for 36 h. There were no significant differences in NSE at 48 h or in neurological outcome. In the high-normal PaCO2 group there was one case of unexplained cerebral oedema on CT scanning. Two patients had severe ARDS (108).
Mean Arterial Pressure
Many observational studies showed an association between a higher mean arterial pressure (MAP) and an increase in survival and improvement in neurological outcome (134). On the other hand vasoactive medication is associated with increased mortality (135). The first prospective trial on this topic dates from 2018 and compared low-normal (65–75 mmHg) to high normal (80–100 mmHg) MAP maintained for 36 h after cardiac arrest in 120 patients. There were no significant differences in the primary outcome measure of NSE at 48 h, nor in neurological outcome (136). A more recent study randomized 112 post-cardiac arrest patients to a protocol focussed on haemodynamic optimization (MAP 85–100 mmHg and SvO2 65–75%) or a MAP of 65 mmHg. Their primary outcome measure, cerebral damage according to DW-MRI, showed no differences between the two groups. Neurological outcome at discharge and after 6 months was the same in both groups (137).
Thrombolysis
To target microthrombi involved in the no-reflow phenomenon, fibrinolytic therapy has been studied to ameliorate cerebral damage. Studies in humans were often ambivalent on the point of action of thrombolysis, applying thrombolysis mainly for resolving pulmonary embolism or coronary thrombosis, and not primarily to improve cerebral blood flow. After some feasibility studies without data on neurological outcome (138, 139), a long term follow-up study in a small population suggested beneficial effects of thrombolytic therapy on neurological outcome (140). The first randomized controlled trial studied the effects of thrombolysis in patients with pulseless electrical activity (PEA). Of the 233 included patients only one survived (141), so no conclusions on effects on neurological outcome can be drawn. A larger trial enrolled 1,050 patients with a witnessed arrest of presumed cardiac origin. At 30 days there was no differences in survival (77 in thrombolysis group, 89 in placebo group) or neurological outcome. Intracranial hemorrhage occurred more often in the intervention group (14 vs. 2 patients) (142). Guidelines on cardiac arrest treatment now state that thrombolysis should only be considered in case of suspected pulmonary embolism (4).
Supportive Therapies
Sedation
Several studies addressed sedation techniques targeting rapid awakening after discontinuation of sedation in comatose patients after cardiac arrest. Two prospective studies took neurological outcome into account as a secondary endpoint. The first randomized study (n = 59) compared propofol/remifentanil (PR) with midazolam/fentanyl (MF) and concluded that the time to extubation was significantly shorter in the PR group. There was no difference in neurological outcome (143). A later cohort study compared two sedation regimens used in different time blocks (2008–2013 vs. 2014–2016) and also found a smaller delay in awakening in the PR group, with no significant changes in good neurological outcome (144). No studies compared sedation with no sedation in this population.
Glucose Regulation
Large fluctuations in blood glucose levels and hyperglycaemia are associated with poor neurological outcome and death in comatose patients after cardiac arrest (145). In accordance with studies in critically ill patients in general, maintenance of normoglycemia is advised in patients after cardiac arrest. In an RCT in 90 patients two different glucose regulation regimes where compared, with no differences in mortality between strict (blood glucose of 4–6 mmol/L) vs. moderate (blood glucose of 6–8 mmol/L) glucose control (146).
Prophylactic Antibiotics
A pilot trial compared prophylactic vs. clinically-driven treatment with antibiotics after cardiac arrest. The main hypothesis was that prevention of early onset pneumonia should decrease the severity of the systemic inflammatory response after resuscitation. There was no significant difference in survival or neurological outcome (147). The results of another trial on antibiotherapy to prevent infectious complications after cardiac arrest are still pending [NCT02186951 (148)].
Discussion
None of the neuroprotective treatments that effectively reduced brain damage after global cerebral ischemia in animal models improved outcome of patients with postanoxic encephalopathy after cardiac arrest in clinical trials, unequivocally. This includes TTM at 33 or 36°C (6). Although, compelling evidence shows that hyperthermia is associated with poor neurological outcome (58, 149, 150), the evidence of efficacy of lowering brain temperature to 32–34°C is complex.
An important limitation of previous and ongoing trials on neuroprotective treatments after cardiac arrest is the lack of subgroup analyses according to measures of encephalopathy severity. It is unlikely that the divergent pathophysiological scenarios ranging from reversible synaptic failure to severe cell swelling and inflammation all warrant the same neuroprotective strategy. International guidelines on treatment of comatose patients after cardiac arrest recognize that “whether certain subpopulations may benefit from lower or higher temperatures remains unclear” (4). To fill that knowledge gap, previous and ongoing clinical trials, such as TTM2 [NCT02908308 (66)] and TAME [NCT03114033 (133)], include predefined subgroup analyses according to widely-accepted factors, such as reflow times, causes of arrest, and initial cardiac arrest rhythm. Although relevant, these are mostly indirect indicators of encephalopathy severity.
A recent analysis of 1,319 comatose patients after cardiac arrest demonstrated divergent effects of TTM at 33°C in mild vs. severe encephalopathy both with and without cardiopulmonary failure (151). This is supported by experimental studies in animal models, showing interaction between cooling and severity of encephalopathy (152, 153). Over the past decade, a multitude of studies on outcome prediction of comatose patients after cardiac arrest have identified reliable and easily retrievable direct measures of encephalopathy severity, such as EEG (32, 154), imaging (155) and biochemical measures (44). Systematic collection of such measures at baseline, with sufficiently powered predefined subgroup analyses, provides an opportunity to identify treatment effects in relatively homogeneous subgroups of patients with postanoxic encephalopathy.
Another factor hampering detection of treatment effects after cardiac arrest is the choice of outcome measures. Traditionally, for pragmatic reasons, 5 or 6 point scales of functional recovery are used, such as the CPC scale or the GOS. These measure gross neurological recovery, but cannot detect small differences in cognitive or behavioral functioning. Several studies used NSE (89, 104, 108, 132, 156), like intracranial pressure (89, 94, 95), near-infrared spectroscopy (108), cerebral blood flow (94), and MRI (83, 89, 137), as a surrogate outcome measures. However, it is largely unclear how these correlate with neurological outcome. Instead of using global outcome scales or indirect parameters of cerebral damage, detailed neuropsychological testing at 6 or 12 months after cardiac arrest holds potential to detect small, but meaningful, cognitive effects of new therapies under study.
Lack of extrapolation from animal models to patients has been discussed extensively. In addition to obvious disparities between animal models and patients (157), reasons include methodological flaws of animal studies, like the lack of sample size calculations, lack of randomization, and unblinded outcome assessments (10, 158). This, in combination with a presumed large publication bias, leads to an overstatement of efficacy of at least 30% (158). To improve meaningful extrapolation from animal models to patients, experimental animal studies should adhere to methodological quality guidelines, and journals are encouraged to use strict publication criteria (159, 160)
This is a narrative review. We included all prospective, controlled, intervention trials, without a systematic analysis of the quality of the included studies and resulting evidence according to the PRISMA guidelines. A multitude of factors hampers interpretation of data. In general, populations were small and heterogeneous, without sufficient details on in-hospital treatment. Therefore, our appreciation of the evidence, and the lack thereof, is qualitative. However, previous meta-analyses of effects of hypothermia (150), adrenaline (129), or erythropoietin (161) led to essentially the same conclusions.
Conclusion
Promising results from animal studies on neuroprotective treatments in postanoxic encephalopathy could not be extrapolated to patients after cardiac arrest. This lack of extrapolation is related to overestimation of pre-clinical evidence, and critical disparities between animal models and patients. Almost all previous studies focussed on neuronal inhibition, but brain stimulation possibly holds a larger potential to improve brain recovery after cardiac arrest. Future clinical trials should be conducted with sufficiently large, well-described populations. Outcome measurement should include comprehensive neuropsychological follow-up, to show treatment effects that are not detectable by gross measures of functional recovery.
Author Contributions
SN: conceptualization, methodology, investigation, writing—original draft, writing—review & editing, and visualization. JF: writing—review & editing. JH: conceptualization, writing—review & editing, visualization, and supervision. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by funding of ZonMW (Grant number 95105001). Ph.D., candidate, Sjoukje Nutma, was funded by this grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.614698/full#supplementary-material
References
1. Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Böttiger BW, et al. Post–cardiac arrest syndrome. Circulation. (2008) 118:2452–83. doi: 10.1161/CIRCULATIONAHA.108.190652
2. Geri G, Mongardon N, Daviaud F, Empana JP, Dumas F, Cariou A. Neurological consequences of cardiac arrest: where do we stand? Ann Fr Anesth Reanim. (2014) 33:98–101. doi: 10.1016/j.annfar.2013.11.003
3. Deakin CD, Fothergill R, Moore F, Watson L, Whitbread M. Level of consciousness on admission to a heart attack centre is a predictor of survival from out-of-hospital cardiac arrest. Resuscitation. (2014) 85:905–9. doi: 10.1016/J.RESUSCITATION.2014.02.020
4. Soar J, Nolan JP, Böttiger BW, Perkins GD, Lott C, Carli P, et al. European resuscitation council guidelines for resuscitation 2015: section 3. Adult advanced life support. Resuscitation. (2015) 95:100–47. doi: 10.1016/j.resuscitation.2015.07.016
5. Laver S, Farrow C, Turner D, Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med. (2004) 30:2126–8. doi: 10.1007/s00134-004-2425-z
6. Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, et al. Targeted temperature management at 33°C vs. 36°C after cardiac arrest. N Engl J Med. (2013) 369:2197–206. doi: 10.1056/NEJMoa1310519
7. Sutton LN, Clark BJ, Norwood CR, Woodford EJ, Welsh FA. Global cerebral ischemia in piglets under conditions of mild and deep hypothermia. Stroke. (1991) 22:1567–73. doi: 10.1161/01.STR.22.12.1567
8. Nakashima K, Todd MM. Effects of hypothermia, pentobarbital, and isoflurane on postdepolarization amino acid release during complete global cerebral ischemia. Anesthesiology. (1996) 85:161–8. doi: 10.1097/00000542-199607000-00022
9. Maier CM, Sun GH, Cheng D, Yenari MA, Chan PH, Steinberg GK. Effects of mild hypothermia on superoxide anion production, superoxide dismutase expression, and activity following transient focal cerebral ischemia. Neurobiol Dis. (2002) 11:28–42. doi: 10.1006/nbdi.2002.0513
10. van der Worp HB, Macleod MR. Preclinical studies of human disease: time to take methodological quality seriously. J Mol Cell Cardiol. (2011) 51:449–50. doi: 10.1016/j.yjmcc.2011.04.008
11. Hofmeijer J, van Putten MJAM. Ischemic cerebral damage: an appraisal of synaptic failure. Stroke. (2012) 43:607–15. doi: 10.1161/STROKEAHA.111.632943
12. Ruijter BJ, van Putten MJ, Horn J, Blans MJ, Beishuizen A, van Rootselaar A-F, et al. Treatment of electroencephalographic status epilepticus after cardiopulmonary resuscitation (TELSTAR): study protocol for a randomized controlled trial. Trials. (2014) 15:433. doi: 10.1186/1745-6215-15-433
13. Robert W Neumar, MD P. ECPR for Refractory Out-Of-Hospital Cardiac Arrest-Full Text View - ClinicalTrials.gov. (2020). Available online at: https://clinicaltrials.gov/ct2/show/NCT03065647 (accessed November 8, 2019).
14. NIH. Emergency Cardiopulmonary Bypass for Cardiac Arrest-Full Text View-ClinicalTrials.gov. (2020). Available online at: https://clinicaltrials.gov/ct2/show/NCT01605409 (accessed November 8, 2019).
15. NIH. A Comparative Study Between a Pre-hospital and an In-hospital Circulatory Support Strategy (ECMO) in Refractory Cardiac Arrest (APACAR2)-Full Text View-ClinicalTrials.gov. (2020). Available online at: https://clinicaltrials.gov/ct2/show/NCT02527031 (accessed November 8, 2019).
16. NIH. Hyperinvasive Approach in Cardiac Arrest-Full Text View-ClinicalTrials.gov. (2020). Available online at: https://clinicaltrials.gov/ct2/show/NCT01511666 (accessed November 8, 2019).
17. Bol ME, Suverein MM, Lorusso R, Delnoij TSR, Brandon Bravo Bruinsma GJ, Otterspoor L, et al. Early initiation of extracorporeal life support in refractory out-of-hospital cardiac arrest: design and rationale of the INCEPTION trial. Am Heart J. (2019) 210:58–68. doi: 10.1016/j.ahj.2018.12.008
18. Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. (2012) 75:762–77. doi: 10.1016/j.neuron.2012.08.019
19. Busl KM, Greer DM. Hypoxic-ischemic brain injury: pathophysiology, neuropathology and mechanisms. NeuroRehabilitation. (2010) 26:5–13. doi: 10.3233/NRE-2010-0531
20. Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci. (2009) 10:861–72. doi: 10.1038/nrn2735
21. Buunk G, van der Hoeven JG, Meinders AE. Cerebral blood flow after cardiac arrest. Neth J Med. (2000) 57:106–12. doi: 10.1016/s0300-2977(00)00059-0
22. Sun M-K, Xu H, Alkon DL. Pharmacological protection of synaptic function, spatial learning, and memory from transient hypoxia in rats. J Pharmacol Exp Ther. (2002) 300:408–16. doi: 10.1124/jpet.300.2.408
23. Lipton P, Whittingham TS. Reduced ATP concentration as a basis for synaptic transmission failure during hypoxia in the in vitro guinea-pig hippocampus. J Physiol. (1982) 325:51–65. doi: 10.1113/jphysiol.1982.sp014135
24. Bolay H, Gürsoy-özdemir Y, Sara Y, Onur R, Can A, Dalkara T. Persistent defect in transmitter release and synapsin phosphorylation in cerebral cortex after transient moderate Ischemic injury. Stroke. (2002) 33:1369–75. doi: 10.1161/01.STR.0000013708.54623.DE
25. Dijkstra K, Hofmeijer J, van Gils SA, van Putten MJAM. A biophysical model for cytotoxic cell swelling. J Neurosci. (2016) 36:11881–90. doi: 10.1523/JNEUROSCI.1934-16.2016
26. Ayoub IM, Radhakrishnan J, Gazmuri RJ. Targeting mitochondria for resuscitation from cardiac arrest. Crit Care Med. (2008) 36:S440–6. doi: 10.1097/ccm.0b013e31818a89f4
27. Thornton C, Hagberg H. Role of mitochondria in apoptotic and necroptotic cell death in the developing brain. Clin Chim Acta. (2015) 451:35–8. doi: 10.1016/j.cca.2015.01.026
28. Lai M-C, Yang S-N. Perinatal hypoxic-ischemic encephalopathy. J Biomed Biotechnol. (2011) 2011:609813. doi: 10.1155/2011/609813
29. Mayor D, Tymianski M. Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology. (2018) 134:178–88. doi: 10.1016/j.neuropharm.2017.11.050
30. Ames A, Wright RL, Kowada M, Thurston JM, Majno G. Cerebral ischemia. II. The no-reflow phenomenon. Am J Pathol. (1968) 52:437–53.
31. le Feber J, Erkamp N, van Putten MJAM, Hofmeijer J. Loss and recovery of functional connectivity in cultured cortical networks exposed to hypoxia. J Neurophysiol. (2017) 118:394–403. doi: 10.1152/jn.00098.2017
32. Ruijter BJ, Tjepkema-Cloostermans MC, Tromp SC, van den Bergh WM, Foudraine NA, Kornips FHM, et al. Early electroencephalography for outcome prediction of postanoxic coma: A prospective cohort study. Ann Neurol. (2019) 86:203–14. doi: 10.1002/ana.25518
33. Amir G, Ramamoorthy C, Riemer RK, Reddy VM, Hanley FL. Neonatal brain protection and deep hypothermic circulatory arrest: pathophysiology of ischemic neuronal injury and protective strategies. Ann Thorac Surg. (2005) 80:1955–64. doi: 10.1016/j.athoracsur.2004.12.040
34. Ditsworth D, Priestley MA, Loepke AW, Ramamoorthy C, McCann J, Staple L, et al. Apoptotic neuronal death following deep hypothermic circulatory arrest in piglets. Anesthesiology. (2003) 98:1119–27. doi: 10.1097/00000542-200305000-00014
35. O'Brien AT, Bertolucci F, Torrealba-Acosta G, Huerta R, Fregni F, Thibaut A. Non-invasive brain stimulation for fine motor improvement after stroke: a meta-analysis. Eur J Neurol. (2018) 25:1017–26. doi: 10.1111/ene.13643
36. Jastremski M, Sutton-Tyrrell K, Vaagenes P, Abramson N, Heiselman D, Safar P. Glucocorticoid treatment does not improve neurological recovery following cardiac arrest. Brain resuscitation clinical trial I study group. JAMA. (1989) 262:3427–30.
37. Jennett B, Snoek J, Bond MR, Brooks N. Disability after severe head injury: observations on the use of the glasgow outcome scale. J Neurol Neurosurg Psychiatry. (1981) 44:285–93. doi: 10.1136/jnnp.44.4.285
38. Ajam K, Gold LS, Beck SS, Damon S, Phelps R, Rea TD. Reliability of the cerebral performance category to classify neurological status among survivors of ventricular fibrillation arrest: a cohort study. Scand J Trauma Resusc Emerg Med. (2011) 19:38. doi: 10.1186/1757-7241-19-38
39. Grossestreuer AV, Abella BS, Sheak KR, Cinousis MJ, Perman SM, Leary M, et al. Inter-rater reliability of post-arrest cerebral performance category (CPC) scores. Resuscitation. (2016) 109:21–4. doi: 10.1016/j.resuscitation.2016.09.006
40. Moulaert VRMP, Verbunt JA, van Heugten CM, Wade DT. Cognitive impairments in survivors of out-of-hospital cardiac arrest: a systematic review. Resuscitation. (2009) 80:297–305. doi: 10.1016/j.resuscitation.2008.10.034
41. Oberholzer M, Müri RM. Neurorehabilitation of traumatic brain injury (TBI): a clinical review. Med Sci. (2019) 7:47. doi: 10.3390/medsci7030047
42. Haque A, Polcyn R, Matzelle D, Banik NL, Johnson RH. New insights into the role of neuron-specific enolase in neuro-inflammation, neurodegeneration, and neuroprotection. (2018) 8:33. doi: 10.3390/brainsci8020033
43. Zandbergen EGJ, Hijdra A, Koelman JHTM, Hart AAM, Vos PE, Verbeek MM, et al. Prediction of poor outcome within the first 3 days of postanoxic coma. Neurology. (2006) 66:62–8. doi: 10.1212/01.wnl.0000191308.22233.88
44. Stammet P, Collignon O, Hassager C, Wise MP, Hovdenes J, Åneman A, et al. Neuron-specific enolase as a predictor of death or poor neurological outcome after out-of-hospital cardiac arrest and targeted temperature management at 33 C and 36 C. J Am Coll Cardiol. (2015) 65:2104–14. doi: 10.1016/j.jacc.2015.03.538
45. Wijdicks EFM Hijdra A Young GB Bassetti CL Wiebe S Quality Standards Subcommittee of the American academy of neurology. Practice parameter: prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): report of the quality standards subcommittee of the American academy of neurology. Neurology. (2006) 67:203–10. doi: 10.1212/01.wnl.0000227183.21314.cd
46. Stammet P, Dankiewicz J, Nielsen N, Fays F, Collignon O, Hassager C, et al. Protein S100 as outcome predictor after out-of-hospital cardiac arrest and targeted temperature management at 33 C and 36 C. Crit Care. (2017) 21:153. doi: 10.1186/s13054-017-1729-7
47. Sandroni C, Cavallaro F, Callaway CW, D'Arrigo S, Sanna T, Kuiper MA, et al. Predictors of poor neurological outcome in adult comatose survivors of cardiac arrest: a systematic review and meta-analysis. Part 2: patients treated with therapeutic hypothermia. Resuscitation. (2013) 84:1324–38. doi: 10.1016/j.resuscitation.2013.06.020
48. Welsh FA, Sims RE, Harris VA. Mild hypothermia prevents ischemic injury in gerbil hippocampus. J Cereb Blood Flow Metab. (1990) 10:557–63. doi: 10.1038/jcbfm.1990.98
49. Bart RD, Takaoka S, Pearlstein RD, Dexter F, Warner DS. Interactions between hypothermia and the latency to ischemic depolarization: implications for neuroprotection. Anesthesiology. (1998) 88:1266–73. doi: 10.1097/00000542-199805000-00018
50. Patel PM, Drummond JC, Cole DJ, Yaksh TL. Differential temperature sensitivity of ischemia-induced glutamate release and eicosanoid production in rats. Brain Res. (1994) 650:205–11. doi: 10.1016/0006-8993(94)91783-3
51. Winfree CJ, Baker CJ, Connolly ES, Fiore AJ, Solomon RA. Mild hypothermia reduces penumbral glutamate levels in the rat permanent focal cerebral ischemia model. Neurosurgery. (1996) 38:1216–22. doi: 10.1097/00006123-199606000-00034
52. Kawai N, Okauchi M, Morisaki K, Nagao S. Effects of delayed intraischemic and postischemic hypothermia on a focal model of transient cerebral ischemia in rats. Stroke. (2000) 31:1982–9. doi: 10.1161/01.str.31.8.1982
53. Wang G., Deng H., Maier C., Sun G., Yenari M. Mild hypothermia reduces ICAM-1 expression, neutrophil infiltration and microglia/monocyte accumulation following experimental stroke. Neuroscience. (2002) 114:1081–90. doi: 10.1016/S0306-4522(02)00350-0
54. Van Hemelrijck A, Vermijlen D, Hachimi-Idrissi S, Sarre S, Ebinger G, Michotte Y. Effect of resuscitative mild hypothermia on glutamate and dopamine release, apoptosis and ischaemic brain damage in the endothelin-1 rat model for focal cerebral ischaemia. J Neurochem. (2003) 87:66–75. doi: 10.1046/j.1471-4159.2003.01977.x
55. Van Hemelrijck A, Hachimi-Idrissi S, Sarre S, Ebinger G, Michotte Y. Post-ischaemic mild hypothermia inhibits apoptosis in the penumbral region by reducing neuronal nitric oxide synthase activity and thereby preventing endothelin-1-induced hydroxyl radical formation. Eur J Neurosci. (2005) 22:1327–37. doi: 10.1111/j.1460-9568.2005.04331.x
56. Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. (2002) 346:557–63. doi: 10.1056/NEJMoa003289
57. Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. (2002) 346:549–56. doi: 10.1056/NEJMoa012689
58. Zeiner A, Holzer M, Sterz F, Schörkhuber W, Eisenburger P, Havel C, et al. Hyperthermia after cardiac arrest is associated with an unfavorable neurologic outcome. Arch Intern Med. (2001) 161:2007–12. doi: 10.1001/archinte.161.16.2007
59. Look X, Li H, Ng M, Lim ETS, Pothiawala S, Tan KBK, et al. Randomized controlled trial of internal and external targeted temperature management methods in post- cardiac arrest patients. Am J Emerg Med. (2018) 36:66–72. doi: 10.1016/j.ajem.2017.07.017
60. Arrich J, Holzer M, Havel C, Warenits A-M, Herkner H. Pre-hospital versus in-hospital initiation of cooling for survival and neuroprotection after out-of-hospital cardiac arrest. Cochrane Database Syst Rev. (2016). doi: 10.1002/14651858.CD010570.pub2
61. Scales DC, Cheskes S, Verbeek PR, Pinto R, Austin D, Brooks SC, et al. Prehospital cooling to improve successful targeted temperature management after cardiac arrest: a randomized controlled trial. Resuscitation. (2017) 121:187–94. doi: 10.1016/j.resuscitation.2017.10.002
62. Nordberg P, Taccone FS, Truhlar A, Forsberg S, Hollenberg J, Jonsson M, et al. Effect of trans-nasal evaporative intra-arrest cooling on functional neurologic outcome in out-of-hospital cardiac arrest: the PRINCESS randomized clinical trial. JAMA. (2019) 321:1677–85. doi: 10.1001/jama.2019.4149
63. Bernard SA, Smith K, Finn J, Hein C, Grantham H, Bray JE, et al. Induction of therapeutic hypothermia during out-of-hospital cardiac arrest using a rapid infusion of cold saline: the RINSE trial (rapid infusion of cold normal saline). Circulation. (2016) 134:797–805. doi: 10.1161/CIRCULATIONAHA.116.021989
64. Kirkegaard H, Soreide E, de Haas I, Pettila V, Taccone FS, Arus U, et al. Targeted temperature management for 48 vs 24 hours and neurologic outcome after out-of-hospital cardiac arrest: a randomized clinical trial. JAMA. (2017) 318:341–50. doi: 10.1001/jama.2017.8978
65. Jeppesen AN, Hvas A-M, Duez CHV, Grejs AM, Ilkjær S, Kirkegaard H. Prolonged targeted temperature management compromises thrombin generation: A randomised clinical trial. Resuscitation. (2017) 118:126–32. doi: 10.1016/j.resuscitation.2017.06.004
66. NIH. Targeted Hypothermia Versus Targeted Normothermia After Out-of-hospital Cardiac Arrest-Full Text View-ClinicalTrials.gov. (2020). Available online at: https://clinicaltrials.gov/ct2/show/NCT02908308 (accessed November 11, 2019).
67. May TL, Riker RR, Fraser GL, Hirsch KG, Agarwal S, Duarte C, et al. Variation in sedation and neuromuscular blockade regimens on outcome after cardiac arrest. Crit Care Med. (2018) 46:e975–80. doi: 10.1097/CCM.0000000000003301
68. Salciccioli JD, Cocchi MN, Rittenberger JC, Peberdy MA, Ornato JP, Abella BS, et al. Continuous neuromuscular blockade is associated with decreased mortality in post-cardiac arrest patients. Resuscitation. (2013) 84:1728–33. doi: 10.1016/j.resuscitation.2013.06.008
69. Lascarrou JB, Le Gouge A, Dimet J, Lacherade JC, Martin-Lefèvre L, Fiancette M, et al. Neuromuscular blockade during therapeutic hypothermia after cardiac arrest: observational study of neurological and infectious outcomes. Resuscitation. (2014) 85:1257–62. doi: 10.1016/j.resuscitation.2014.05.017
70. Stöckl M, Testori C, Sterz F, Holzer M, Weiser C, Schober A, et al. Continuous versus intermittent neuromuscular blockade in patients during targeted temperature management after resuscitation from cardiac arrest-A randomized, double blinded, double dummy, clinical trial. Resuscitation. (2017) 120:14–9. doi: 10.1016/j.resuscitation.2017.08.238
71. Lee BK Cho IS Oh JS Choi WJ Wee JH Kim CS . Continuous neuromuscular blockade infusion for out-of-hospital cardiac arrest patients treated with targeted temperature management: A multicenter randomized controlled trial. PLoS ONE. (2018) 13:e0209327. doi: 10.1371/journal.pone.0209327
72. Breivik H, Safar P, Sands P, Fabritius R, Lind B, Lust P, et al. Clinical feasibility trials of barbiturate therapy after cardiac arrest. Crit Care Med. (1978) 6:228–44. doi: 10.1097/00003246-197807000-00004
73. Erickson KM, Pasternak JJ, Weglinski MR, Lanier WL. Temperature management in studies of barbiturate protection from focal cerebral ischemia: systematic review and speculative synthesis. J Neurosurg Anesthesiol. (2009) 21:307–17. doi: 10.1097/ANA.0b013e3181aa03eb
74. Harman F, Hasturk AE, Yaman M, Arca T, Kilinc K, Sargon MF, et al. Neuroprotective effects of propofol, thiopental, etomidate, and midazolam in fetal rat brain in ischemia-reperfusion model. Child's Nerv Syst. (2012) 28:1055–62. doi: 10.1007/s00381-012-1782-0
75. Smith DS, Rehncrona S, Siesjö BK. Barbiturates as protective agents in brain ischemia and as free radical scavengers in vitro. Acta Physiol Scand Suppl. (1980) 492:129–34.
76. Brain Resuscitation Clinical Trial I Study Group. Randomized clinical study of thiopental loading in comatose survivors of cardiac arrest. N Engl J Med. (1986) 314:397–403. doi: 10.1056/NEJM198602133140701
77. Monsalve F, Rucabado L, Ruano M, Cuñat J, Lacueva V, Viñuales A. The neurologic effects of thiopental therapy after cardiac arrest. Intensive Care Med. (1987) 13:244–8.
78. Ma D, Wilhelm S, Maze M, Franks NP. Neuroprotective and neurotoxic properties of the “inert” gas, xenon. Br J Anaesth. (2002) 89:739–46.
79. Jawad N, Rizvi M, Gu J, Adeyi O, Tao G, Maze M, et al. Neuroprotection (and lack of neuroprotection) afforded by a series of noble gases in an in vitro model of neuronal injury. Neurosci Lett. (2009) 460:232–6. doi: 10.1016/j.neulet.2009.05.069
80. Arola OJ, Laitio RM, Roine RO, Grönlund J, Saraste A, Pietilä M, et al. Feasibility and cardiac safety of inhaled xenon in combination with therapeutic hypothermia following out-of-hospital cardiac arrest. Crit Care Med. (2013) 41:2116–24. doi: 10.1097/CCM.0b013e31828a4337
81. Brevoord D, Beurskens CJP, van den Bergh WM, Lagrand WK, Juffermans NP, Binnekade JM, et al. Helium ventilation for treatment of post-cardiac arrest syndrome: a safety and feasibility study. Resuscitation. (2016) 107:145–9. doi: 10.1016/j.resuscitation.2016.07.004
82. Tamura T, Hayashida K, Sano M, Suzuki M, Shibusawa T, Yoshizawa J, et al. Feasibility and safety of hydrogen gas inhalation for post-cardiac arrest syndrome– first-in-human pilot study. Circ J. (2016) 80:1870–3. doi: 10.1253/circj.CJ-16-0127
83. Laitio R, Hynninen M, Arola O, Virtanen S, Parkkola R, Saunavaara J, et al. Effect of inhaled xenon on cerebral white matter damage in comatose survivors of out-of-hospital cardiac arrest. JAMA. (2016) 315:1120. doi: 10.1001/jama.2016.1933
84. Tamura T, Hayashida K, Sano M, Onuki S, Suzuki M. Efficacy of inhaled HYdrogen on neurological outcome following BRain ischemia during post-cardiac arrest care (HYBRID II trial): study protocol for a randomized controlled trial. Trials. (2017) 18:488. doi: 10.1186/s13063-017-2246-3
85. Hölscher C. Potential role of glucagon-like Peptide-1 (GLP-1) in neuroprotection. CNS Drugs. (2012) 26:871–82. doi: 10.2165/11635890-000000000-00000
86. Combs CK. Are GLP-1 receptor agonists useful against traumatic brain injury? J Neurochem. (2015) 135:1059–61. doi: 10.1111/jnc.13224
87. Holst JJ, Burcelin R, Nathanson E. Neuroprotective properties of GLP-1: theoretical and practical applications. Curr Med Res Opin. (2011) 27:547–58. doi: 10.1185/03007995.2010.549466
88. Wiberg S, Hassager C, Schmidt H, Thomsen JH, Frydland M, Lindholm MG, et al. Neuroprotective effects of the glucagon-like peptide-1 analog exenatide after out-of-hospital cardiac arrest. Circulation. (2016) 134:2115–24. doi: 10.1161/CIRCULATIONAHA.116.024088
89. Wang D, Jiang Q, Du X. Protective effects of scopolamine and penehyclidine hydrochloride on acute cerebral ischemia-reperfusion injury after cardiopulmonary resuscitation and effects on cytokines. Exp Ther Med. (2018) 15:2027–31. doi: 10.3892/etm.2017.5646
90. Hassan TB, Jagger C, Barnett DB. A randomised trial to investigate the efficacy of magnesium sulphate for refractory ventricular fibrillation. Emerg Med J. (2002) 19:57–62. doi: 10.1136/emj.19.1.57
91. Longstreth WT, Fahrenbruch CE, Olsufka M, Walsh TR, Copass MK, Cobb LA. Randomized clinical trial of magnesium, diazepam, or both after out-of-hospital cardiac arrest. Neurology. (2002) 59:506–14. doi: 10.1212/wnl.59.4.506
92. Roine RO, Kaste M, Kinnunen A, Nikki P. Safety and efficacy of nimodipine in resuscitation of patients outside hospital. Br Med J. (1987) 294:20. doi: 10.1136/bmj.294.6563.20
93. Roine RO, Kaste M, Kinnunen A, Nikki P, Sarna S, Kajaste S. Nimodipine after resuscitation from out-of-hospital ventricular fibrillation. A placebo-controlled, double-blind, randomized trial. JAMA. (1990) 264:3171–7.
94. Forsman M, Aarseth HP, Nordby HK, Skulberg A, Steen PA. Effects of nimodipine on cerebral blood flow and cerebrospinal fluid pressure after cardiac arrest: correlation with neurologic outcome. Anesth Analg. (1989) 68:436–43.
95. Gueugniaud PY, Gaussorgues P, Garcia-Darennes F, Bancalari G, Roux H, Robert D, et al. Early effects of nimodipine on intracranial and cerebral perfusion pressures in cerebral anoxia after out-of-hospital cardiac arrest. Resuscitation. (1990) 20:203–12.
96. Brain Resuscitation Clinical Trial II Study Group. A randomized clinical study of a calcium-entry blocker (lidoflazine) in the treatment of comatose survivors of cardiac arrest. N Engl J Med. (1991) 324:1225–31. doi: 10.1056/NEJM199105023241801
97. Bishop RL, Weisfeldt ML. Sodium bicarbonate administration during cardiac arrest. Effect on arterial pH PCO2, and osmolality. JAMA. (1976) 235:506–9.
98. Marsh JD, Margolis TI, Kim D. Mechanism of diminished contractile response to catecholamines during acidosis. Am J Physiol. (1988) 254:H20–7. doi: 10.1152/ajpheart.1988.254.1.H20
99. Vukmir RB, Katz L, Group SBS. Sodium bicarbonate improves outcome in prolonged prehospital cardiac arrest. Am J Emerg Med. (2006) 24:156–61. doi: 10.1016/J.AJEM.2005.08.016
100. Ahn S, Kim YJ, Sohn CH, Seo DW, Lim KS, Donnino MW, et al. Sodium bicarbonate on severe metabolic acidosis during prolonged cardiopulmonary resuscitation: a double-blind, randomized, placebo-controlled pilot study. J Thorac Dis. (2018) 10:2295–02. doi: 10.21037/jtd.2018.03.124
101. Pilcher J, Weatherall M, Shirtcliffe P, Bellomo R, Young P, Beasley R. The effect of hyperoxia following cardiac arrest –a systematic review and meta-analysis of animal trials. Resuscitation. (2012) 83:417–22. doi: 10.1016/j.resuscitation.2011.12.021
102. Roberts BW, Kilgannon JH, Hunter BR, Puskarich MA, Pierce L, Donnino M, et al. Association between early hyperoxia exposure after resuscitation from cardiac arrest and neurological disability. Circulation. (2018) 137:2114–24. doi: 10.1161/CIRCULATIONAHA.117.032054
103. Thomas M, Voss S, Benger J, Kirby K, Nolan JP. Cluster randomised comparison of the effectiveness of 100% oxygen versus titrated oxygen in patients with a sustained return of spontaneous circulation following out of hospital cardiac arrest: a feasibility study. PROXY: post ROSC OXYgenation study. BMC Emerg Med. (2019) 19:16. doi: 10.1186/s12873-018-0214-1
104. Kuisma M, Boyd J, Voipio V, Alaspää A, Roine RO, Rosenberg P. Comparison of 30 and the 100% inspired oxygen concentrations during early post-resuscitation period: a randomised controlled pilot study. Resuscitation. (2006) 69:199–206. doi: 10.1016/j.resuscitation.2005.08.010
105. Young P, Bailey M, Bellomo R, Bernard S, Dicker B, Freebairn R, et al. HyperOxic therapy OR NormOxic therapy after out-of-hospital cardiac arrest (HOT OR NOT): a randomised controlled feasibility trial. Resuscitation. (2014) 85:1686–91. doi: 10.1016/j.resuscitation.2014.09.011
106. Bray JE, Hein C, Smith K, Stephenson M, Grantham H, Finn J, et al. Oxygen titration after resuscitation from out-of-hospital cardiac arrest: a multi-centre, randomised controlled pilot study (the EXACT pilot trial). Resuscitation. (2018) 128:211–5. doi: 10.1016/j.resuscitation.2018.04.019
107. Bray JE, Smith K, Hein C, Finn J, Stephenson M, Cameron P, et al. The EXACT protocol: a multi-centre, single-blind, randomised, parallel-group, controlled trial to determine whether early oxygen titration improves survival to hospital discharge in adult OHCA patients. Resuscitation. (2019) 139:208–13. doi: 10.1016/j.resuscitation.2019.04.023
108. Jakkula P, Reinikainen M, Hästbacka J, Loisa P, Tiainen M, Pettilä V, et al. Targeting two different levels of both arterial carbon dioxide and arterial oxygen after cardiac arrest and resuscitation: a randomised pilot trial. Intensive Care Med. (2018) 44:2112–21. doi: 10.1007/s00134-018-5453-9
109. Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. (2008) 7:156–67. doi: 10.1038/nrd2466
110. Jung K-H, Chu K, Ko S-Y, Lee S-T, Sinn D-I, Park D-K, et al. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke. (2006) 37:2744–50. doi: 10.1161/01.STR.0000245116.40163.1c
111. Dezfulian C, Alekseyenko A, Dave KR, Raval AP, Do R, Kim F, et al. Nitrite therapy is neuroprotective and safe in cardiac arrest survivors. Nitric Oxide. (2012) 26:241–50. doi: 10.1016/j.niox.2012.03.007
112. Kim F, Dezfulian C, Empey PE, Morrell M, Olsufka M, Scruggs S, et al. Usefulness of intravenous sodium nitrite during resuscitation for the treatment of out-of-hospital cardiac arrest. Am J Cardiol. (2018) 122:554–9. doi: 10.1016/j.amjcard.2018.04.060
113. Sirén AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA. (2001) 98:4044–9. doi: 10.1073/pnas.051606598
114. Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A, et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med. (2003) 198:971–5. doi: 10.1084/jem.20021067
115. Cariou A, Claessens Y-E, Pène F, Marx J-S, Spaulding C, Hababou C, et al. Early high-dose erythropoietin therapy and hypothermia after out-of-hospital cardiac arrest: a matched control study. Resuscitation. (2008) 76:397–404. doi: 10.1016/j.resuscitation.2007.10.003
116. Grmec Š, Strnad M, Kupnik D, Sinkovič A, Gazmuri RJ. Erythropoietin facilitates the return of spontaneous circulation and survival in victims of out-of-hospital cardiac arrest. Resuscitation. (2009) 80:631–7. doi: 10.1016/j.resuscitation.2009.03.010
117. Cariou A, Deye N, Vivien B, Richard O, Pichon N, Bourg A, et al. Early high-dose erythropoietin therapy after out-of-hospital cardiac arrest. J Am Coll Cardiol. (2016) 68:40–9. doi: 10.1016/j.jacc.2016.04.040
118. Tsai MS, Huang CH, Chang WT, Chen WJ, Hsu CY, Hsieh CC, et al. The effect of hydrocortisone on the outcome of out-of-hospital cardiac arrest patients: a pilot study. Am J Emerg Med. (2007) 25:318–25. doi: 10.1016/j.ajem.2006.12.007
119. Mentzelopoulos SD, Malachias S, Chamos C, Konstantopoulos D, Ntaidou T, Papastylianou A, et al. Vasopressin, steroids, and epinephrine and neurologically favorable survival after in-hospital cardiac arrest. JAMA. (2013) 310:270–9. doi: 10.1001/jama.2013.7832
120. Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol. (2015) 78:129–41. doi: 10.1016/j.yjmcc.2014.08.018
121. Argaud L, Cour M, Dubien P-Y, Giraud F, Jossan C, Riche B, et al. Effect of cyclosporine in nonshockable out-of-ospital cardiac arrest: the CYRUS randomized clinical trial. JAMA Cardiol. (2016) 1:557–65. doi: 10.1001/jamacardio.2016.1701
122. Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, et al. Effects of coenzyme Q10 in early parkinson disease. Arch Neurol. (2002) 59:1541. doi: 10.1001/archneur.59.10.1541
123. Cocchi MN, Giberson B, Berg K, Salciccioli JD, Naini A, Buettner C, et al. Coenzyme Q10 levels are low and associated with increased mortality in post-cardiac arrest patients. Resuscitation. (2012) 83:991–5. doi: 10.1016/j.resuscitation.2012.03.023
124. Damian MS, Ellenberg D, Gildemeister R, Lauermann J, Simonis G, Sauter W, et al. Coenzyme Q10 combined with mild hypothermia after cardiac arrest: a preliminary study. Circulation. (2004) 110:3011–6. doi: 10.1161/01.CIR.0000146894.45533.C2
125. Callaway CW, Soar J, Aibiki M, Böttiger BW, Brooks SC, Deakin CD, et al. Part 4: advanced life support. Circulation. (2015) 132:S84–145. doi: 10.1161/CIR.0000000000000273
126. Perkins GD, Cottrell P, Gates S. Is adrenaline safe and effective as a treatment for out of hospital cardiac arrest? BMJ. (2014) 348:g2435. doi: 10.1136/bmj.g2435
127. Ristagno G, Tang W, Huang L, Fymat A, Chang Y-T, Sun S, et al. Epinephrine reduces cerebral perfusion during cardiopulmonary resuscitation*. Crit Care Med. (2009) 37:1408–15. doi: 10.1097/CCM.0b013e31819cedc9
128. Perkins GD, Ji C, Deakin CD, Quinn T, Nolan JP, Scomparin C, et al. A randomized trial of epinephrine in out-of-hospital cardiac arrest. N Engl J Med. (2018) 379:711–21. doi: 10.1056/NEJMoa1806842
129. Maria V, Pasquale B, Carmine I, Giuseppe S. Epinephrine for out of hospital cardiac arrest: A systematic review and meta-analysis of randomized controlled trials. Resuscitation. (2019) 136:54–60. doi: 10.1016/j.resuscitation.2019.01.016
130. Curley G, Laffey JG, Kavanagh BP. Bench-to-bedside review: Carbon dioxide. Crit Care. (2010) 14:220. doi: 10.1186/cc8926
131. Godoy DA, Seifi A, Garza D, Lubillo-Montenegro S, Murillo-Cabezas F. Hyperventilation therapy for control of posttraumatic intracranial hypertension. Front Neurol. (2017) 8:250. doi: 10.3389/fneur.2017.00250
132. Eastwood GM, Schneider AG, Suzuki S, Peck L, Young H, Tanaka A, et al. Targeted therapeutic mild hypercapnia after cardiac arrest: a phase II multi-centre randomised controlled trial (the CCC trial). Resuscitation. (2016) 104:83–90. doi: 10.1016/j.resuscitation.2016.03.023
133. Eastwood G. Targeted Therapeutic Mild Hypercapnia After Resuscitated Cardiac Arrest (TAME). Clin Identifier NCT03114033 (2018).
134. Bhate TD, McDonald B, Sekhon MS, Griesdale DEG. Association between blood pressure and outcomes in patients after cardiac arrest: a systematic review. Resuscitation. (2015) 97:1–6. doi: 10.1016/j.resuscitation.2015.08.023
135. Bro-Jeppesen J, Annborn M, Hassager C, Wise MP, Pelosi P, Nielsen N, et al. Hemodynamics and vasopressor support during targeted temperature management at 33°C versus 36°C after out-of-hospital cardiac arrest. Crit Care Med. (2015) 43:318–27. doi: 10.1097/CCM.0000000000000691
136. Jakkula P, Pettilä V, Skrifvars MB, Hästbacka J, Loisa P, Tiainen M, et al. Targeting low-normal or high-normal mean arterial pressure after cardiac arrest and resuscitation: a randomised pilot trial. Intensive Care Med. (2018) 44:2091–101. doi: 10.1007/s00134-018-5446-8
137. Ameloot K, De Deyne C, Eertmans W, Ferdinande B, Dupont M, Palmers P-J, et al. Early goal-directed haemodynamic optimization of cerebral oxygenation in comatose survivors after cardiac arrest: the Neuroprotect post-cardiac arrest trial. Eur Heart J. (2019) 40:1804–14. doi: 10.1093/eurheartj/ehz120
138. Lederer W, Lichtenberger C, Pechlaner C, Kroesen G, Baubin M. Recombinant tissue plasminogen activator during cardiopulmonary resuscitation in 108 patients with out-of-hospital cardiac arrest. Resuscitation. (2001) 50:71–6. doi: 10.1016/s0300-9572(01)00317-3
139. Böttiger BW, Bode C, Kern S, Gries A, Gust R, Glätzer R, et al. Efficacy and safety of thrombolytic therapy after initially unsuccessful cardiopulmonary resuscitation: a prospective clinical trial. Lancet. (2001) 357:1583–5. doi: 10.1016/S0140-6736(00)04726-7
140. Lederer W, Lichtenberger C, Pechlaner C, Kinzl J, Kroesen G, Baubin M. Long-term survival and neurological outcome of patients who received recombinant tissue plasminogen activator during out-of-hospital cardiac arrest. Resuscitation. (2004) 61:123–9. doi: 10.1016/j.resuscitation.2003.12.016
141. Abu-Laban RB, Christenson JM, Innes GD, van Beek CA, Wanger KP, McKnight RD, et al. Tissue plasminogen activator in cardiac arrest with pulseless electrical activity. N Engl J Med. (2002) 346:1522–8. doi: 10.1056/NEJMoa012885
142. Böttiger BW, Arntz H-R, Chamberlain DA, Bluhmki E, Belmans A, Danays T, et al. Thrombolysis during resuscitation for out-of-hospital cardiac arrest. N Engl J Med. (2008) 359:2651–62. doi: 10.1056/NEJMoa070570
143. Bjelland TW, Dale O, Kaisen K, Haugen BO, Lydersen S, Strand K, et al. Propofol and remifentanil versus midazolam and fentanyl for sedation during therapeutic hypothermia after cardiac arrest: a randomised trial. Intensive Care Med. (2012) 38:959–67. doi: 10.1007/s00134-012-2540-1
144. Paul M, Bougouin W, Dumas F, Geri G, Champigneulle B, Guillemet L, et al. Comparison of two sedation regimens during targeted temperature management after cardiac arrest. Resuscitation. (2018) 128:204–10. doi: 10.1016/j.resuscitation.2018.03.025
145. Borgquist O, Wise MP, Nielsen N, Al-Subaie N, Cranshaw J, Cronberg T, et al. Dysglycemia, glycemic variability, and outcome after cardiac arrest and temperature management at 33°C and 36°C. Crit Care Med. (2017) 45:1337–43. doi: 10.1097/CCM.0000000000002367
146. Oksanen T, Skrifvars MB, Varpula T, Kuitunen A, Pettilä V, Nurmi J, et al. Strict versus moderate glucose control after resuscitation from ventricular fibrillation. Intensive Care Med. (2007) 33:2093–100. doi: 10.1007/s00134-007-0876-8
147. Ribaric SF, Turel M, Knafelj R, Gorjup V, Stanic R, Gradisek P, et al. Prophylactic versus clinically-driven antibiotics in comatose survivors of out-of-hospital cardiac arrest-A randomized pilot study. Resuscitation. (2017) 111:103–9. doi: 10.1016/j.resuscitation.2016.11.025
148. NIH. Antibiotherapy during therapeutic hypothermia to prevent infectious complications (ANTHARCTIC). (2020). Available online at: https://clinicaltrials.gov/ct2/show/NCT02186951
149. de Jonge JC, Wallet J, van der Worp HB. Fever worsens outcomes in animal models of ischaemic stroke: a systematic review and meta-analysis. Eur Stroke J. (2019) 4:29–38. doi: 10.1177/2396987318776421
150. Arrich J, Holzer M, Havel C, Müllner M, Herkner H. Hypothermia for neuroprotection in adults after cardiopulmonary resuscitation. Cochrane Database Syst Rev. (2016) 2:CD004128. doi: 10.1002/14651858.CD004128.pub4
151. Callaway CW, Coppler PJ, Faro J, Puyana JS, Solanki P, Dezfulian C, et al. Association of initial illness severity and outcomes after cardiac arrest with targeted temperature management at 36°C or 33°C. JAMA Netw open. (2020) 3:e208215. doi: 10.1001/jamanetworkopen.2020.8215
152. Wood T, Osredkar D, Puchades M, Maes E, Falck M, Flatebø T, et al. Treatment temperature and insult severity influence the neuroprotective effects of therapeutic hypothermia. Sci Rep. (2016) 6:23430. doi: 10.1038/srep23430
153. Colbourne F, Corbett D. Delayed and prolonged post-ischemic hypothermia is neuroprotective in the gerbil. Brain Res. (1994) 654:265–72. doi: 10.1016/0006-8993(94)90488-X
154. Cloostermans MC, Van Meulen FB, Eertman CJ, Hom HW, Van Putten MJAM. Continuous electroencephalography monitoring for early prediction of neurological outcome in postanoxic patients after cardiac arrest: a prospective cohort study. Crit Care Med. (2012) 40:2867–75. doi: 10.1097/CCM.0b013e31825b94f0
155. Keijzer HM, Hoedemaekers CWE, Meijer FJA, Tonino BAR, Klijn CJM, Hofmeijer J. Brain imaging in comatose survivors of cardiac arrest: Pathophysiological correlates and prognostic properties. Resuscitation. (2018) 133:124–36. doi: 10.1016/j.resuscitation.2018.09.012
156. Wiberg S, Hassager C, Thomsen JH, Frydland M, Høfsten DE, Engstrøm T, et al. GLP-1 analogues for neuroprotection after out-of-hospital cardiac arrest: study protocol for a randomized controlled trial. Trial. (2016) 17:304. doi: 10.1186/s13063-016-1421-2
157. Dronne M-A, Grenier E, Chapuisat G, Hommel M, Boissel J-P. A modelling approach to explore some hypotheses of the failure of neuroprotective trials in ischemic stroke patients. Prog Biophys Mol Biol. (2008) 97:60–78. doi: 10.1016/j.pbiomolbio.2007.10.001
158. Sena ES, van der Worp HB, Bath PMW, Howells DW, Macleod MR. Publication bias in reports of animal stroke studies leads to major overstatement of efficacy. PLoS Biol. (2010) 8:e1000344. doi: 10.1371/journal.pbio.1000344
159. Cressey D. Poorly designed animal experiments in the spotlight. Nature. (2015). doi: 10.1038/nature.2015.18559
160. Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLOS Biol. (2020) 18:e3000410. doi: 10.1371/journal.pbio.3000410
Keywords: postanoxic coma, post-cardiac arrest syndrome, resuscitation, cerebral ischaemia, hypoxic ischaemic brain injury
Citation: Nutma S, le Feber J and Hofmeijer J (2021) Neuroprotective Treatment of Postanoxic Encephalopathy: A Review of Clinical Evidence. Front. Neurol. 12:614698. doi: 10.3389/fneur.2021.614698
Received: 06 October 2020; Accepted: 19 January 2021;
Published: 18 February 2021.
Edited by:
Michael Kuiper, Medisch Centrum Leeuwarden, NetherlandsReviewed by:
Jeanne Teitelbaum, McGill University, CanadaBenjamin Aaron Emanuel, University of Southern California, United States
Copyright © 2021 Nutma, le Feber and Hofmeijer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sjoukje Nutma, cy5udXRtYUBtc3Qubmw=