 Ibitayo Abigail Ademuwagun1,2
Ibitayo Abigail Ademuwagun1,2 Solomon Oladapo Rotimi1,2
Solomon Oladapo Rotimi1,2 Steffen Syrbe3Yvonne Ukamaka Ajamma1
Steffen Syrbe3Yvonne Ukamaka Ajamma1 Ezekiel Adebiyi1,4,5*
Ezekiel Adebiyi1,4,5*- 1Covenant University Bioinformatics Research, Covenant University, Ota, Nigeria
- 2Department of Biochemistry, Covenant University, Ota, Nigeria
- 3Clinic for Pediatric and Adolescent Medicine, Heidelberg University, Heidelberg, Germany
- 4Department of Computer and Information Sciences, Covenant University, Ota, Nigeria
- 5Division of Applied Bioinformatics, German Cancer Research Center (DKFZ), Heidelberg, Germany
Genetic epilepsy occurs as a result of mutations in either a single gene or an interplay of different genes. These mutations have been detected in ion channel and non-ion channel genes. A noteworthy class of ion channel genes are the voltage gated sodium channels (VGSCs) that play key roles in the depolarization phase of action potentials in neurons. Of huge significance are SCN1A, SCN1B, SCN2A, SCN3A, and SCN8A genes that are highly expressed in the brain. Genomic studies have revealed inherited and de novo mutations in sodium channels that are linked to different forms of epilepsies. Due to the high frequency of sodium channel mutations in epilepsy, this review discusses the pathogenic mutations in the sodium channel genes that lead to epilepsy. In addition, it explores the functional studies on some known mutations and the clinical significance of VGSC mutations in the medical management of epilepsy. The understanding of these channel mutations may serve as a strong guide in making effective treatment decisions in patient management.
Introduction
Epilepsy is a neurological disorder with spontaneous and re-occurring seizures that are borne from a population of cortical neurons, exhibiting hyper-synchronized discharge of high current action potentials in the brain (1, 2). Etiologically, epilepsy may have genetic, infectious, metabolic, structural, immunological, or unknown origin (3). Structural epilepsies are acquired following brain trauma, stroke, injuries, inflammations or infections, unlike genetic (formerly idiopathic) epilepsies which are primarily due to underlying genetic mutations (3). Studies in this field have shown that epilepsy may have a combination of two or more etiologies (4). For instance, many disorders involving the central nervous system (CNS) malformations such as tubulinopathies, tuberous sclerosis and polymicrogyria are comorbid with epilepsy and have genetic causes (5). The discovery of several ion channel gene mutations in epilepsy has led to categorizing epilepsy as a channelopathy. Epilepsy associated channel proteins fall in the class of ionotropic ligand receptors for major neurotransmitters (such as acetylcholine {muscarinic} receptors, glutamatergic receptors and GABAergic receptors) and the voltage gated ion channel proteins including sodium (Na+), calcium (Ca2+), potassium (K+), and chloride (Cl−) channels (6) are involved in bio-signaling in the CNS.
Voltage gated sodium ion channels (VGSCs) are heteromeric proteins involved in the generation and propagation of action potentials in brain neural cells (7). They are membrane-associated proteins and are responsible for conducting currents of sodium ions down their concentration gradient into the cell (8). Mutations associated with these channel genes are the most common cause of genetic epilepsies, with thousands of them reported in diverse populations (9). To understand the role of genetic variants in epilepsy, many studies have focused on the possible functional implications of a vast array of VGSC mutations. Often, these studies involved attempts to understand the mechanism by which induced mutations alter neuron physiology, using model systems. Some functional studies involved heterologous expression of mutated channel proteins in human embryonic cell lines or human induced pluripotent stem cells (hiPSCs) and the study of the aberrant network conduction in these transfected cells using voltage patch clamp techniques. Other methods involve the use of genetically modified rodent models (10) [e.g., Q54 mice, bearing the 879GAL881QQQ mutation in SCN2A (11)]. Several strategies for functional studies have been developed, which has helped to unravel the underlying molecular mechanisms by which many mutations trigger seizures. These have exposed other knowledge areas like the impact of mutations in modifier genes and the understanding of differences in patients' response to drugs. Therefore, this review aims to discuss VGSCs mutations in the alpha and beta subunit genes of sodium channels that lead to epilepsy, some functional studies of these mutations and their significance to epilepsy management.
Search Criteria
A review of recent relevant literature was done on PubMed and GoogleScholar. Advanced search was done using the following keyword combinations: SCN1A mutations in epilepsy, SCN1B mutations in epilepsy, SCN2A mutations in epilepsy, SCN3A mutations in epilepsy, SCN8A mutations in epilepsy and functional studies of VGSCs in epilepsy. Search results (articles) containing the following word combination were excluded: Anticonvulsant properties of plant extracts, bioorganic compounds synthesized as antiepileptic drugs (AEDs), mechanism of action of new AEDs, clinical trials of drugs against epilepsy, heterocyclic compounds, imidazoles sulfonamides with anticonvulsant properties, quantitative structure activity relationship (QSAR) models for VGSC functions, biomarker studies in epilepsy syndromes and clinical trials of AEDs.
VGSC Genes and Their Tissue Distributions
There are nine different sodium channel α-subunit genes that code for Nav1.1 to Nav1.9 (12) channel proteins. SCN1A to SCN5A encode Nav1.1-Nav1.5 proteins, while SCN8A to SCN11A encode Nav1.6-Nav1.9 proteins, respectively (13). On the other hand, five different ß-subunit proteins have been reported, namely: ß1 (product of SCN1B), ß1B (product of SCN1B splice variant), ß2 (SCN2B), ß3 (SCN3B), and ß4 (SCN4B) (14). In humans, the expression of VGSCs follows a tissue dependent pattern and each sodium channel possesses cognate kinetic properties that are both native to the tissue and relate to its function. SCN1A, SCN2A, and SCN3A have their locus clustered within a 600 kb region on the long arm of the Homo sapiens chromosome 2q24. The following are the VGSC α subunit genes highly expressed in the human brain: SCN1A, SCN2A, SCN3A, and SCN8A (Table 1). Hence, these are the genes involved in epilepsy pathogenesis. Other sodium channels α subunit genes that have high expression in muscles are involved in other pathologies. For example, SCN4A mutants cause hypokalemic periodic paralysis (type 2) (15), myotonia congenita and myasthenic syndrome (16) while SCN5A variants result in atrial fibrillation (type 10) (17), brugada syndrome (type 1) (18), and cardiac death (19) (Table 2). As for ß subunit genes, they display a wide distribution pattern in the brain. The proteins encoded by SCN1B (i.e., β1 and β1B are developmentally regulated). The β subunits occur as transmembrane polypeptides having their amino terminus protruding extracellularly and their carboxyl terminus protruding intracellularly, interacting with the proteins of the cytoskeleton. A conserved immunoglobulin (Ig) region is located at the extracellular portion, just like cell adhesion proteins (37). These proteins are found in close interactions with cellular adhesion proteins (38). In fact, some studies have shown the adhesive property of β subunits in the brain cells (39). Apart from seizure disorders, SCN1B variants also cause artrial fibrillation (type 13) and brugada syndrome (type 5) (27, 30).
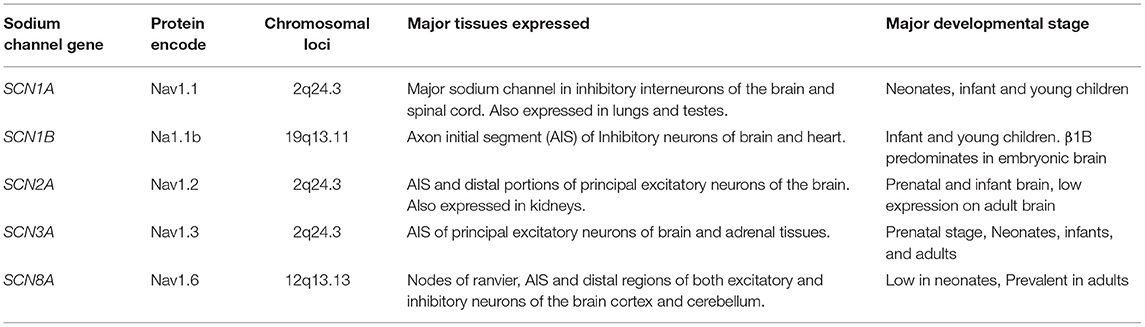
Table 1. Sodium channel genes involved in epilepsy, proteins encoded, genetic loci, and distribution in human tissues.
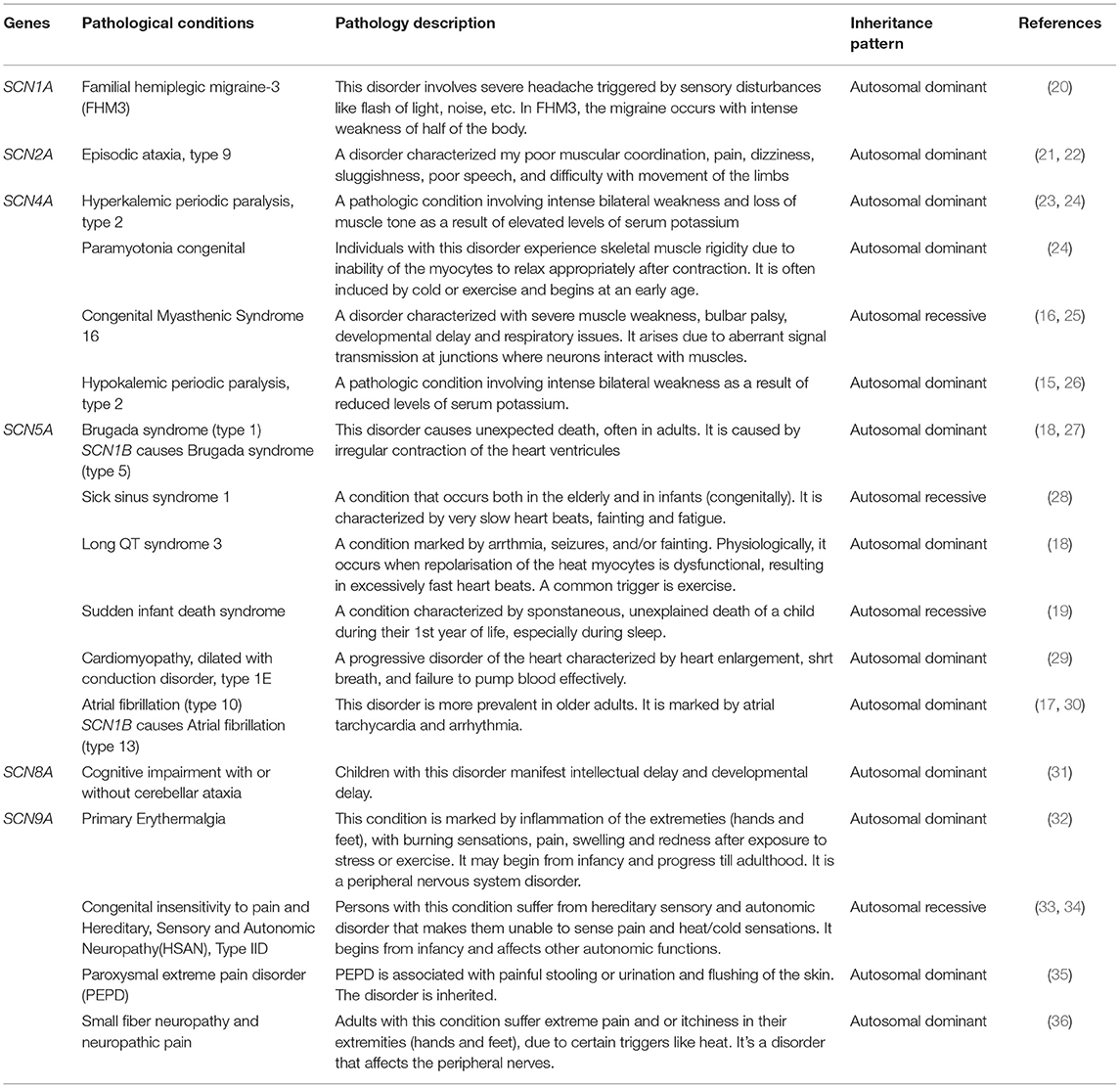
Table 2. Other pathologies associated with sodium channels.
SCN2A, SCN3A, and SCN8A are three main α subunit genes that are mostly expressed on excitatory neurons. On inhibitory interneurons, SCN1A has high expression while SCN8A is also expressed to lesser extent (40, 41). The expression of these channels is usually concentrated on the AIS of neurons. However, SCN2A are mildly expressed on the soma and dendrites since they may promote back propagation of action potentials to the cell body (42). Nav1.6 is present mainly in the nodes of ranvier and distal portions of the AIS, but shows lower distribution on the dendrites and soma (43). Nav1.6 plays a higher role in action potential generation than Nav1.2 because of its relatively lower threshold for activation. Both channels differ in their functions despite the strong similarities they exhibit. Due to the differential expression patterns of sodium channels on inhibitory and excitatory neurons, polymorphisms in them tend to result in different seizure patterns and syndromes.
SCN1A: Role in Epilepsy and Epilepsy Management
SCN1A mutations are the most prevalent amongst all VGSCs mutations in epilepsy and over 1,250 pathogenic variants are responsible for various epilepsies (44). In fact, amongst all epilepsy genes, SCN1A mutations are the most implicated (6, 45). Most Dravet syndrome (DS) and Generalized Epilepsy with Febrile Seizures Plus (GEFS+) cases have mutations in SCN1A. Both inherited and de novo mutations in SCN1A genes cause epilepsy and epileptic encephalopathies. For example, over three-fourth of DS cases are due to de novo mutations in SCN1A genes, while SCN1A mutations causing GEFS+ are frequently inherited (46). Many SCN1A mutations cause loss-of-function [e.g., de novo SCN1A mutations leading to DS occur as loss-of-function mutations on inhibitory interneurons (6)]. These mutations decrease the activity of GABAergic inhibitory interneurons (6). SCN1A variants in seizure disorders may occur as various mutation types such as missense, nonsense, protein-truncating variants, etc. For instance, about 50% of all DS cases arise from missense mutation while many others arise from non-sense mutations, deletions, frameshifts and splice-site variants (47, 48). Although DS and GEFS+ can be used as prototypes to study SCN1A variants, these mutations are also involved in other sub-types of epilepsy (13, 49).
Initially, research done using heterologous expression systems proposed that SCN1A mutations cause gain-of-function arising from dysfunctional inactivation of channel (50). Later, this suggestion was challenged after a host of loss-of-function mutations were discovered in patients with GEFS+. The effect of the deleterious missense mutations often results in a change in the biophysical properties of the Nav1.1 channel, causing a positive shift in the voltage dependence of activation with or without very slow inactivation of channel (51). Using bacterial artificial chromosome (BAC) transgenic mouse model, Tang et al. (52) reported that the effect of the R1648 mutation of SCN1A channel was a distorted resilience from inactivation, thereby implying loss-of-function. More recently, functional studies affirming loss-of-function were reported by Kluckova et al. (53) who investigated nine known and five novel variants, and observed clear partial and total loss-of-functions in different SCN1A variants. Four of them, E78K, E1587K, W384X, and R1596C resulted in mutant channels that failed to produce any measurable sodium currents (total function loss) while two, E788K and M909K resulted in partial loss. Three other variants (D249E, E78D, and T1934I) prevented the channel from recovering from the active state as required. Since SCN1A channels are majorly expressed on inhibitory interneurons, such loss-of-function mutations are expected to result in hyperexcitability.
Seizure manifestations and neuro-behavioral alterations observed in different epilepsies depend on the nature of mutations identified in sodium channels. Hence, VGSC gene screening is vital in epilepsy management. The total loss-of-function mutations in SCN1A results in more severe disorders. Meanwhile, the amino-acid-substituting point mutations causing change in function of sodium channels contribute to less severe epileptic disorders (6). This is exemplified in DS that arises from the loss-of-function mutation in SCN1A gene which is a more severe epileptic disorder than the GEFS+ (6, 9, 54). Zhang et al. (55) showed the relationship between clinical presentations of 13 children with SMEI having multiple seizure types and the incidence of having SCN1A mutations. Out of the 13 cases examined, 10 individuals with mutations in SCN1A were identified. They include 1 frameshift and 9 non-synonymous (seven missense and two non-sense) mutations. The authors concluded that despite identifying a well-known electroclinical pattern in these patients, a definitive genetic diagnostic approach involving SCN1A screening must be carried out for proper patient management. In another study involving 59 Italians and one Spanish patient having cryptogenic epilepsies, Zucca et al. (56) reported 12 SCN1A mutations. The mutational screening of SCN1A in 13 infants showed the presence of missense, non-sense, and frameshift mutations in patients with DS. Only 15% of the patients had abnormal brain imaging results, while most of them had normal electroencephalographic (EEG) activity (55). The consideration for genetic diagnosis in patients with epilepsy should not be teased out despite the absence of definite EEG results.
Microduplications in the 2q24.3 chromosomal region spanning through the genetic loci for SCN1A, SCN2A, and SCN3A that might influence expression levels of SCN2A were reported (57). The screened family seemed to manifest a seizure pattern that was different from the typical KCNQ2 or SCN2A associated syndromes. SCN1A mutations showed lesser degree of electrophysiological alterations than SCN3A mutations. Hence, more intense phenotypes were observed in the former (58). Also, frameshift mutations have been identified as one of the major underlying mutations in SCN1A responsible for DS (59). Tuncer et al. (59) reported a new SCN1A mutation in a 10-year-old Turkish boy who had seizure onset at 8 months old. Mutational screening of SCN1A revealed the presence of a deletion c.4018delC that resulted in a shift in the open reading frame, causing the premature introduction of a stop codon. The patient had a history of refractory seizures, which could not be effectively managed using monotherapy of phenobarbital, valproic acid, or other AEDs. Upon genetic screening, patient management took the course of a combined therapy using valproic acid (VPA) and stiripentol that proved effective. The authors reported the presence of a borderline phenotype rather than the regular DS (59). Genetic investigation improves diagnosis and may help in selecting better effective combination therapies in epilepsy management.
Studies by Shi et al. (60) showed the association between single nucleotide polymorphisms (SNPs) in SCN1A in epileptic Chinese patients and their recovery rates when valproic acid was administered. A common SCN1Aallele, rs3812718G>A, was linked with a positive drug response to valproic acid in the studied Chinese population (60). This was inconsistent with reports from previous studies (61) suggesting the discontinuation of sodium channel blockers as effective therapy against seizures in SCN1A-positive patients. This indicates that some population-based variants may alter patients' response to drugs, but this may be absent in other populations.
Fang et al. (62) reported a study of 24 epileptic patients of Chinese origin who had previously tested positive on SCN1A mutation screening. About 67% of the patients had missense mutations and about 17% had non-sense mutations. One patient had splice-site and frameshift mutations while large deletions were identified in only two of the patients. The patients' genetic background with respect to SCN1A mutations had impact on the medications that proved effective. For instance, ~33% of the patients were refractory to treatment with antiepileptic drugs (AEDs) while ~46% were drug responsive. The dietary intervention using ketogenic diet proved successful in three patients, who were rather non-responsive to AEDs. The use of sodium channel blockers proved unsuccessful in over 41% of the patients (62). Sodium channel blockers are being discontinued as a choice of treatment for the management of SCN1A-related seizure disorders due to their proven exacerbating effects in the presence of such mutations. In epilepsy management, the importance of mutational screening cannot be overemphasized because it gives a road map to effective diagnosis and treatment, while avoiding therapies that may aggravate the condition.
The discovery of pathogenic haploinsufficiency in DS has contributed to the clinical management of patients. Sodium channel antagonists like lamotrigine, phenytoin and carbamazepine are being discontinued in patients with DS and other epilepsy syndromes associated with SCN1A mutations (61). This was not the case with earlier studies on the management of SCN1A disorders. For instance, Krikova et al. (63) studied the effective therapeutic dose of lamotrigine (a sodium channel antagonist) in heterozygous and homozygous SCN1A mutations and observed the highest effective dose amongst those with homozygous SCN1A mutation. Today, the application of adjunctive therapies is highly sought after. However, their efficacy in the absence of adverse drug response is a matter of concern. An example is Cannabidiol (CBD) oil which has resulted in effective seizure management in patients with DS but several adverse events ranging from moderate to severe have been observed (64). New AEDs are already being considered for the treatment of DS. Two important drugs amongst these are stiripentol and fenfluramine. Although it originated in the 1970's, stiripentol is increasingly gaining attention for its minimal adverse reactions and its capacity to effectively manage seizures in DS, epilepsy of infancy with migrating focal seizures (EIMFS) and other intractable seizures (65). The initial uproar regarding the safety of humans using fenfluramine in weight management has gradually reduced with the release of new findings showing its efficacy in seizure management for patients with DS. Ceulemans et al. (66) recorded 70% success in a study involving the treatment of patients with DS with fenfluramine. On an average, the patients stayed seizure-free for 6 years. The result of a 5-year follow-up of the same cohort, revealed the safety of fenfluramine in patients with DS with effective seizure management, and only two patients recorded cardiovascular thickening (67). Another study involving nine patients revealed that over 70% of patients on fenfluramine had reduction in motor seizures (68, 69). These studies applied fenfluramine at low doses. Lagae et al. (70) reported the outcome of a Phase III clinical trial which suggests that the drug is safe for use as a combination drug at low doses (<30 mg per day) for an extended treatment period over several years with no adverse cardiovascular effects in young patients. These results suggest a breakthrough for the treatment of SCN1A-seizure disorders, with DS as a prototype. More clinical trials for fenfluramine are still ongoing and interesting findings are being reported.
Using a combination of immunochemistry and immunofluorescence, Guo et al. (71) reported an increased level of expression of sodium channel alpha subunits 1 and 3, and β1 subunits in the hippocampal region of spontaneous epileptic rats. This study revealed that cellular changes that result in the overexpression of some channel proteins may have critical roles to play in seizure generation. Also, the impact of epigenetic modifications is still gaining attention in epilepsy studies. Drugs that can specifically inhibit the activities of DNA methyl transferases (DNMTs) and Histone deacetylases (HDACs) are already being applied, especially in patients having temporal lobe epilepsy (TLE) (72). Therefore, proper understanding of epigenetic mechanisms of epileptogenesis would lead to the development of more adjunctive therapies in the management of epilepsy.
Functional studies (Table 3) can help detect gain or loss-of-function variants that may guide drug design and understanding of the relationships between variants and drug response. An example of the impact of genetic variant on drug response was recently reported by Zhao et al. (77) who examined the association between the SCN1A SNPs; rs3812718 and rs2298771 and the response to carbamazepine in a Chinese population. Resistance to drugs was observed with rs3812718 but not with rs2298771.
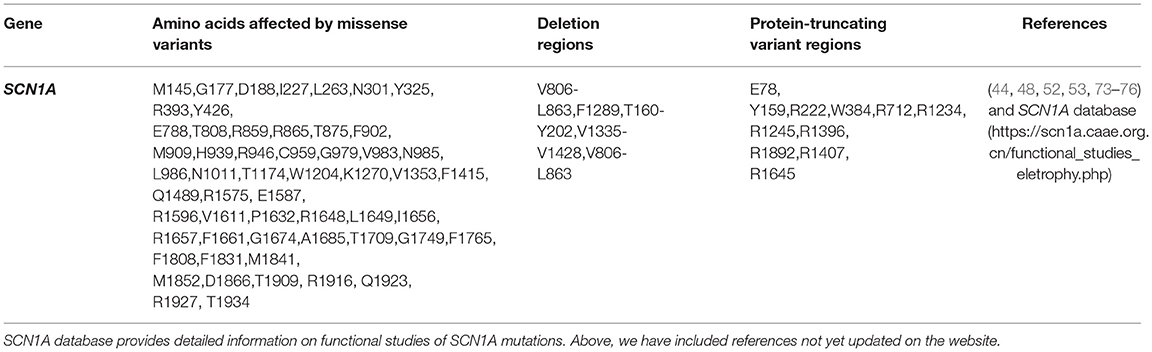
Table 3. Regions of SCN1A with functionally validated epilepsy variants with references.
Progress is being recorded in functional studies of channel mutations. Liu et al. (48) was the first to apply neurons derived from human induced pluripotent skin fibroblasts to study functional effects of SCN1A mutation in a neuron-related disorder. Studies of splice-site mutation of SCN1A revealed clear loss-of-function using induced iPSCs. The authors compared results of using heterologous expression systems like tsA201 and xenopus oocyte cells with results of patient- and control-derived pyramidal and bipolar neurons studies. While transfected heterologous system had little or no detectable current, the hiPSC-neurons delivered hyperexcitation as in the case with typical DS (48). They observed that hiPSCs neurons utilized the actual genetic architecture of the patients' neurons to study functional impacts rather than using other mammalian cells that may introduce some bias into the results due to inter-specie differences. Also, hiPSCs permit the studying of mutations in the presence of the patient's own genetic modifiers hereby providing more effective tailor-made therapies. Furthermore, neurons are more suitable for expressing channel biophysical properties than other mammalian cells and they serve as better models. Xie et al. (44) recently demonstrated the high potential of using hiPSC-neurons for functional studies by applying the K1270 SCN1A mutation. They observed a reduced depolarization signal in the mutant channel which suggested loss-of-function. Likewise, they realized that this approach improved the understanding of molecular mechanism by which mutations exert their effects on neurons.
Genetic screening may reveal population-based variants responsible for differences in response to drugs in diverse populations. One advantage of identifying population-related variants with their functional studies is that it will pave way for the development or determination of drugs that are more suitable for that population.
We have collated the SCN1A regions having variants whose functional studies have been reported in Figure 1, while Table 3 shows the affected regions with references.
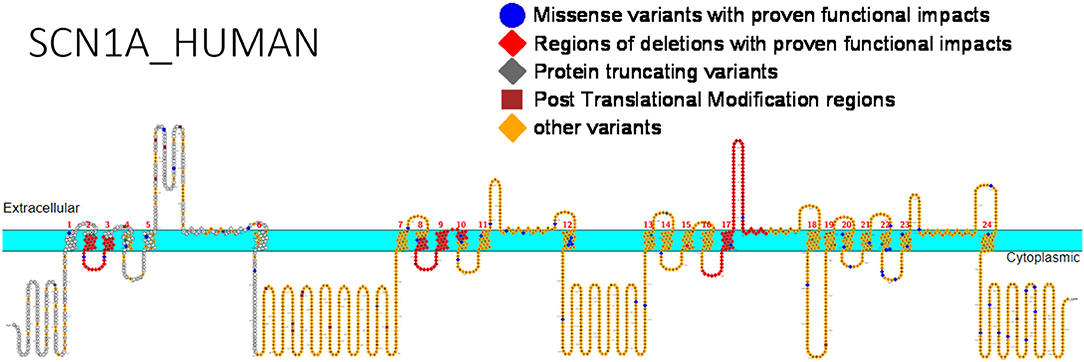
Figure 1. Primary structure of SCN1A protein showing regions with functionally validated epilepsy-variants from literature. Protein-truncating variant region (black), regions of deletions with proven functional impacts (red), regions of missense variants with proven functional impacts (blue), regions of post-translational modifications (brown), other variants (orange). [Created using Protter (78). Visualization link: https://bit.ly/2NWfncp].
SCN1B: Role in Epilepsy and Epilepsy Management
Mutations in the modulating ß subunits of the type 1 sodium channel genes have equally been confirmed as another cause of epilepsy. Wallace et al. (79) was the first to report a pathogenic missense mutation, p.(C121W), in SCN1B gene inherited in an autosomal dominant pattern in an Australian GEFS+ patient. Studies using heterologous expression systems revealed that this mutation hindered the function of β subunits both as an ancillary protein and adhesive protein in addition to altering gating characteristics of the channel. It also disrupts the Ig loop of the subunit, hereby inhibiting glycosylation (38, 80–82). Many mutations have been identified in this gene since then. One homozygous mutation was reported in a case of epileptic encephalopathy (83). Another study identified four mutations in SCN1B having linkage to epileptic seizures. All these mutations were found on the extracellular portion of the protein (84). This portion on the modulating ß-subunit was essential in channel gating. The four mutations were identified in patients having GEFS+. The most occurring missense substitution was the point mutation replacing cysteine with tryptophan p.(C121W) (84). More insights into the roles of sodium channel β subunits have shown that they do more than simply modify the functions of the α-subunits. In the absence of α subunits, they can function as indicator molecules in electrically activated cells. In addition, they play key roles in brain morphogenesis, modification of potassium channels, regulation of channel expression and cell to cell interactions (10).
Mutations in SCN1B have been reported in patients having GEFS+ phenotype, DS and Temporal lobe epilepsy (TLE) (79, 84, 85). However, many of these mutations are localized in the extracellular region of the auxiliary channel protein. Most functional studies on SCN1B revealed loss-of-function mutations, especially interfering with their ability to augment VGSC gating (79). The functional implication of these β subunit mutations is that they may impede the role of the principal pore forming α-subunits (86). Meanwhile, molecular studies to unravel definite pathogenicity of mutations in SCN1B are still incomplete.
Kruger et al. (87) reported the functional studies of the missense mutation, p.(C121W) that was initially reported by Wallace et al. (79). Using an experimental model of childhood febrile seizures, the authors compared the response of heterozygous SCN1B-C121W (SCN1B+/w) mice with SCN1B-null allele. The SCN1B+/w mice exhibited higher predisposition to heat-induced paroxysms, suggesting a gain-of-function mechanism (87). The results were contrary to the initial conclusions from co-expression studies using Xenopus oocytes and mammalian cell lines that suggested a loss-of-function of channel protein as a mechanism of action for this mutation (38, 79, 80). The response of the channels to the sodium ion channel blocker; phenytoin, decreased in the presence of the SCN1B mutation p.(C121W) (88).
Patino et al. (89) examined the functional implications of the mutation, p.G257R, earlier identified by Patino et al. (83) in β1B subunit which is mainly expressed in embryonic brains. They demonstrated that unlike β1, the splice-variant, β1B, does not function as a transmembrane polypeptide. Rather, it served as a secreted protein acting as a ligand in neuronal growth. Furthermore, they revealed that the mutation might alter sodium ion currents in the brain, using Chinese Hamster Ovary cells co-expressing Nav1.3 and β1B subunits. Similarly, their results suggested that p.(G257R) mutation acted in a similar manner to a previously reported missense mutation p.(R125C) which inhibited the release of β1B polypeptide into the extracellular environment, hence annulling its function in neuronite outgrowth (89).
Studies involving the mutant SCN1B gene in heterologous expression systems require co-expression with the α-subunit gene. Most mutations in SCN1B are found on the extracellular immunoglobulin fold (whose primary role is in cell to cell adhesion and interaction with the α-subunits) with few exceptions (e.g., one mutation on ß1B subunit was identified in its C-terminal domain). Phenotypic expression of these mutations and functional characterization suggested a loss-of-function mechanism. Additionally, the ß1 subunit had interactions with some types of potassium channels, thus aiding repolarization of these channels (90), by accelerating the activation of the potassium channel and suppressing its inactivation. In GEFS+ patients, where variants p.(C121W) and p.(R85C) of SCN1B have been described, there was a loss of effects of ß1 function on the Kv1.3 potassium channel (91). Identification and characterization of the roles of ß1 and ß1B variants in other forms of epilepsy, especially their effects on channel gating are of crucial importance. The role of SCN1B was recently demonstrated in developmental and epileptic encephalopathy. The mutation, R85C of SCN1B was identified as a loss-of-function variant (92). This was verified by Dang et al. (93) who studied the missense mutations: p.(R85C), p.(R85H), and p.(R89H). Dang et al. (93) confirmed a loss-of-function mechanism for the R85 variants. Clinical management of a child with refractory seizures proved ineffective with several combination therapies including levetiracetam, clobazam, Valproate and topiramate. However, the administration of Phenytoin resulted in effective management of the seizures (93). Despite this finding, the treatment of SCN1B-related epilepsy with sodium channel blockers remains inconclusive. There is still sparse information on the successful treatment in SCN1B-related epilepsy. Hence, more work is required, including functional studies using induced hIPSCs. Figure 2 shows SCN1B primary structure with regions having seizure-prone variants that have been functionally validated from literature while Table 4 shows affected regions with references.
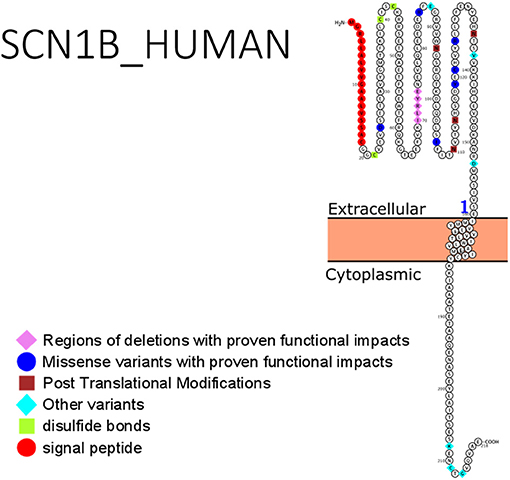
Figure 2. Primary structure of SCN1B protein showing regions with functionally validated epilepsy-variants from literature. Region of signal peptide (red), missense variant regions with proven functional impacts (blue), post-translational modifications (brown), other variants (aqua), regions of disulfide bonds (Lemon). [Created using Protter (78). Visualization link: https://bit.ly/366Lz6C].

Table 4. Regions of SCN1B with functionally validated epilepsy variants with references.
SCN2A: Role in Epilepsy and Epilepsy Management
There are two isoforms of SCN2A: one is expressed in infancy while the other is expressed in adulthood. The adult isoform has lower threshold for excitation than those of a neonate, thus, it is more excitable (10). Reid et al. (98) reported about 20 pathogenic variants of SCN2A leading to benign familiar neonatal infantile seizure (BFNIS) and GEFS+. The pathogenic mutations in this gene have been reported in several epilepsies of different severities. There is an age dependent seizure resolution in BFNIS that relates to the physiological reorganization of the axon initial segment during development. This rearrangement leads to the substitution of SCN2A with SCN8A in the distal regions of the AIS. Thus, reducing the impact of the mutant SCN2A on neuronal function (99). SCN2A and SCN8A mutations have been reported in some patients with severe epileptic encephalopathies (100). Ogiwara et al. (101) reported another more severe epileptic encephalopathy arising from de novo SCN2A mutations with clinical manifestations that resemble those of DS. Whole exome sequencing is continuously having more relevance in epilepsy genetics, especially in its potential to reveal genes whose mutations may have epistatic interactions by acting as genetic modifiers of disease. Studies by Shi et al. (102) to examine the impacts of SCN2A, GABRG2, and SCN1A in DS amongst Japanese population showed 29 mutations in SCN1A and 3 in SCN2A. The missense mutations, F328V and D322N were inherited while R1312T was de novo (102). Many SCN2A mutations leading to refractory epilepsies are often de novo rather than inherited (103).
Frameshift mutations have been noted in SCN2A in children having intellectual disability (ID) without seizure history (104). The first non-sense, protein-truncating mutation p.(R102X) in SCN2A was reported in patients having refractory seizure and ID by Kamiya et al. (105). This mutation was heterozygous and absent in parents of the reported patients. Electrophysiologic analysis using HEK 293 cells and voltage patch clamp studies showed that the mutation caused a hyperpolarization shift. Liao et al. (99) examined the clinical presentations in one child with neonatal seizures accompanied by pain and episodic ataxia. The authors reported a missense mutation p.(A263V) whose functional consequence is a rise in sodium currents in the mutant channel (99). Despite these promising results, mutation studies of SCN2A in patients with seizure disorders have not shown clearly defined phenotypes unlike SCN1A and SCN8A (106).
The role of SCN2A mutations in Early Onset Epileptic Encephalopathy (EOEE) was reported by Nakamura et al. (107) in a study involving 328 individuals with various types of EE. Fifteen novel amino acid replacing mutations were reported with ~86% (12) occurring de novo. Amongst the cases studied, all mutations in patients having Ohtahara syndrome occurred in the polypeptide loop that connects two transmembrane regions of Nav1.2. A country-wide study of patients with acute encephalopathy and fever-induced status epilepticus was recorded. Two amino acid substituting mutations in SCN2A were identified, one was previously identified in patients with DS p.(F328V) and the other was novel p.(I172V) (108).
Missense mutations in SCN2A have shown varying effects on the mutant protein. Cell culture studies using two BFNIS gene mutations revealed a depolarizing shift in voltage dependence of inactivation and an elevated/persistent current generation depicting gain-of-function as one mechanism in which SCN2A mutations cause excitability (109). Gain-of-function mutations in Nav1.2 cause seizures since these sodium channels are highly expressed on excitatory neurons. In a study conducted by Xu, Thomas (110) it was evident that gain-of-function mutations of the neonatal Nav1.2 predisposes to a higher seizure frequency than those of adult. The mutation in neonatal Nav1.2 increased excitability to a level that was similar to that expressed by the adult phenotype (110). This explains the self-limiting effect of BFNIS with advancement in age (10). Further studies are required to identify and characterize more SCN2A mutations and their involvements in epilepsies.
SCN2A mutations have been shown to be the second most prevalent mutation following KCNT1 (111) in patients having EIMFS. Treatment with phenytoin at high dosage proved effective in 75% of the cases having multiple epileptic syndromes including EIMFS and OS (112). The authors reported 11 missense mutations in SCN2A including a deletion mutation, that resulted in an amino acid substitution (112). Reports from Matalon et al. (113) supported the claim that SCN2A mutations are involved in early infantile epilepsies. They presented three children having refractory seizures with de novo SCN2A mutations. The three patients reported were found to be negative for mutational screening in the following epilepsy genes: FOXG1, CDKL5, MECP2, ARX, SCN1A, STXBP1, SLC25A22, PCDH19, SPTAN1, and ARHGEF9. The study showed that SCN2A does not merely act as a disease modifying gene but can independently trigger seizures. Three amino acid substituting de novo mutations were reported (113). Exome studies by Baasch et al. (103) revealed one new de novo missense mutation p.(R1882L) in SCN2A in patients with epilepsy and intellectual disability. The authors presented another supporting claim that pathogenic SCN2Amutationswere often de novo.
Zeng et al. (114) reported the clinical phenotypes of 21 children having mutations in SCN2A. Approximately 48% of the cases had inherited the mutations from their parents while the remaining had de novo mutations in SCN2A. Majority (about 86%) of the children had partial seizures and most of the patients (about 90%) manifested more than one seizure type. Less than one-third of the examined patients had infantile spasms. De novo mutations in this gene were found to be associated with delayed development.
Seizures arising from SCN2A mutations are well-controlled by high therapeutic doses of the sodium channel blocker, phenytoin (112). Two other sodium channel antagonistic seizure medications (lidocaine and mexiletine) were reported to be effective anecdotally in infants having intractable epilepsy due to de novo mutations in SCN2A (115). This was after a combination therapy including topiramate, levetiracetam, lacosamide, oxcarbazepine, and phenytoin proved abortive. Ketogenic diet including adjunctive therapies involving treatments with pyridoxine, folinic acid, pyridoxyl-5-phosphate was also applied to no avail. This medication was replicated in another patient carrying a de novo SCN2A missense mutation p.(G999L) and a successful seizure management was recorded (115). Conflicting reports in the management of SCN2A-related seizures were recorded until 2017 when Wolff et al. (116) identified a trend in the functional studies of SCN2A-variants. They reported that loss-of-function in SCN2A variants were more associated with seizures beginning over 3 months of age and were not properly managed using sodium channel antagonists. However, in infants <3 months a gain-of-function trend was observed and the administration of sodium channel blockers proved very effective (116). This trend requires further studies for clear validation and clinical applications.
As in the case with SCN1A mutations, dietary interventions are sometimes effective in managing SCN2A-related seizures. The use of modified Atkins diet in one Chinese child was found to reduce seizures that presented with refractory seizures, infantile spasms and delayed development including autism-related characteristics. An SCN2A mutation p.(E1211K) which had been earlier identified in an infantile spasm case was detected (117). In spite of past failed attempts in the use of ketogenic diets in managing SCN2A-seizures (115), ketogenic diet has proved successful in managing SCN2A-epilepsy when adopted early enough (118, 119).
Apart from determining the treatment route, genetic variants may also determine drug response, and this has been reported for SCN2A. An association between SCN2A variants and selective drug response to valproic acid (VPA) was recently reported by Shi et al. (60) in Chinese patients. The allele, rs2304016A>G was negatively associated with response to VPA. Epistatic interactions amongst genes may equally determine how patients respond to drugs. The impact of epistatic interactions affecting response to valproic acid was studied in Chinese Han population (120). The following SCN2A SNPs had epistatic interactions influencing VPA response: rs10197716 and rs2119068, rs10197716, and rs11889342. Furthermore, interactions between SCN2A (rs7598931) and UDP-Glucuronosyltransferase-2B7 gene (UGT2B7) (rs12233719) were reported (120). Functional studies can help detect gain- or loss-of-function variants that can guide drug design, identify high risk population-related variants that are important for genetic testing and understand relationships between variants and drug response. Figure 3 shows SCN2A primary structure with regions having variants have been functionally validated from literature while Table 5 shows the affected regions with references.
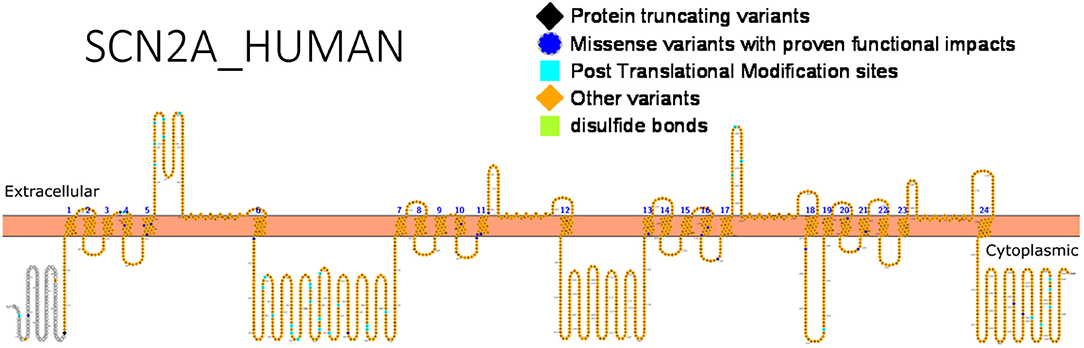
Figure 3. Primary structure of SCN2A protein showing regions with functionally validated epilepsy-variants from literature. Protein-truncating variant regions (black), region of missense variants with proven functional impacts (blue), post-translational modifications (aqua), other variants (orange) and regions of disulfide bonds (lemon) [Created using Protter (78). Visualization link: https://bit.ly/2HLNnbW].

Table 5. Regions of SCN2A with functionally validated epilepsy variants with references.
SCN3A: Role in Epilepsy and Epilepsy Management
The SCN3A gene is highly expressed in brains of neonates and adults. It is similarly expressed both in principal excitatory and inhibitory interneurons. Point mutations in SCN3A genes have been reported in patients with focal epilepsies (58, 123). Holland et al. (12) presented the first SCN3A mutations in cryptogenic focal seizures. In managing the patient's seizures, previous treatments that involved the administration of sodium channel blockers (carbamazepine or oxcarbazepine) had been futile. After mutational screening, the authors reported 1 new missense mutation (K354Q) in the polypeptide link between S5-S6 of domain I, and 3 novel synonymous variants in SCN3A. Electrophysiological studies of the missense mutation using SCN5A gene show a rise in sodium ion currents causing defects in inactivation in excitatory neurons. Although this suggests that the mutation acts through a gain-of function mechanism, the use of an isoform rather than SCN3A itself, may introduce a bias in the accurate measurement of the effect of this mutation. The first mechanistic study of SCN3A mutation using transfected neurons of the hippocampus was reported in 2010. Estacion et al. (124) provided the functional studies of the p.(K354Q) mutation (earlier reported by Holland et al. in 2008) in Nav1.3 channels using hippocampal cells from rats. The authors validated that this mutation increased ionic currents across the channel and decreased the threshold for Na+, resulting in hyperexcitation. A more improved approach now involves the use of neurons derived from patients' fibroblasts to express mutant channels and better results are being recorded in terms of studying the channels' biophysical properties. Functional studies of even more SCN3A mutations have shown an elevated ramp current in mutant channels, further suggesting gain-of-function (123).
Apart from gain-of-functions mutations, loss-of-functions mutations are implicated in focal epilepsies. Chen et al. (58) reported an SCN3A mutation in some patients with GEFS+. The identified mutation was consequential in creating a depolarizing shift in voltage-dependent activation and inactivation of the sodium channel, including delayed recovery from slow inactivation. Hence resulting in channel inactivity. Furthermore, Lamar et al. (125) identified a novel mutation in SCN3A yielding the mutant Nav1.3 protein p.(L247P) which is associated with focal epilepsy in childhood and delayed development. The authors demonstrated the role of a novel SCN3A mutation p. (L247P) in seizure pathogenesis. They reported that loss-of-function mutation in SCN3A, reduced trafficking of channel proteins or down regulated expression may result in epilepsy. Despite this, more studies have shown a prevalence of gain-of-function mutations for SCN3A. Zaman et al. (126) studied some SCN3A missense variants including p. (I875T), p. (P1333L), p. (V1769A), p. (R1642C), and p. (L1799Q). The result of biophysical studies on channel mutation showed a prevalence of gain-of-function with higher amplitude of inactivation and a lowered threshold of activating the channel compared to the wildtype channel. This effect was impeded by the application of 2 specific sodium channel blockers (lacosamide and phenytoin). Thus, implying their efficacy in managing SCN3A disorders. Mixed results of gain and loss-of-function are recorded because SCN3A is known to be expressed also on inhibitory neurons and this may explain why loss-of-function in this gene may also result in seizures.
Microduplication mutations have likewise been implicated as epilepsy-related SCN3A mutations. Yoshitomi et al. (127) reported 3 patients manifesting focal seizures and infantile spasms having microduplication mutation on the 2q24.3 chromosomal loci. Eight additional duplications were reported by Thuresson et al. (128). The region duplicated was overlapped and the spanned range approximately between 0.05 and 7.63 Mb in size. This resulted in an extra SCN2A copy which caused benign familial infantile seizures (BFIS), but had no link with cognitive decline (128). Recently, the role of deletion mutations has been expounded. A causative de novo deletion mutations in the 2q24.3 region was reported in a patient with West syndrome who also manifested symptoms of autism. This deletion (~1.1 Mb) covers the region encoding Nav1.2 and Nav1.3 (129).
Apart from de novo mutations, inherited mutations are known to cause SCN3A-seizure disorders. Using targeted exome sequencing including 412 epilepsy genes in 63 patients and their families, an inherited missense mutation p.(R621C) was identified in SCN3A but no functional studies was done (130). Zaman et al. (126) asserted that since many reported SCN3A variants occur de novo, they might have roles in modifying the effects of other pathogenic variants from different genes, which exhibit high penetrance. Functional analysis of these channel variants will assist in unraveling the molecular mechanism underlying phenotypic expression and shed more light on research to develop more effective therapies. Figure 4 shows SCN3A primary structure with regions having variants that have been functionally validated from literature while Table 6 shows the affected regions with references.
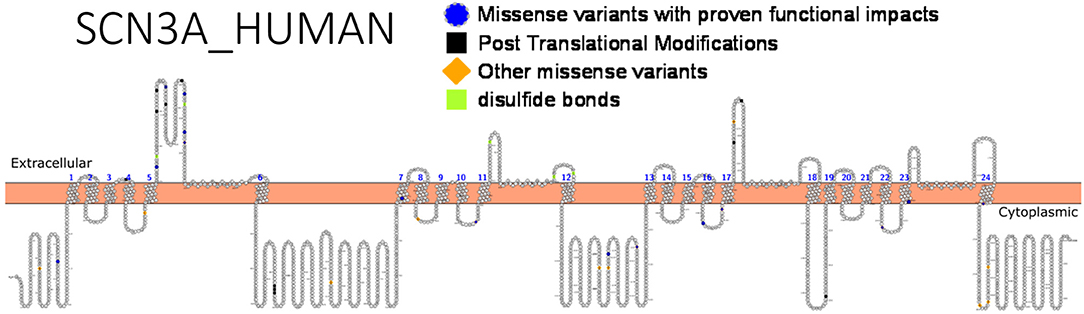
Figure 4. Primary structure of SCN3A protein showing regions with functionally validated epilepsy-variants from literature. Regions of missense variants with proven functional impacts (blue), regions of post-translational modifications (black), other missense variants (orange) [Created using Protter (78). Visualization link: https://bit.ly/2V8ZeDL].

Table 6. Regions of SCN3A with functionally validated epilepsy variants with references.
SCN8A: Role in Epilepsy and Epilepsy Management
Mutations in SCN8A gene is the major underlying factor responsible for pathogenesis of SCN8A encephalopathies; a seizure disorder marked with early onset paroxysmal manifestations characterized by hypersynchronous electrical discharges originating in the brain. This usually occurs within the first 18 months after birth, particularly after the 3rd month (131). Clinical manifestations include cognitive disability and developmental delay. About half of the children involved did not walk or sit properly (or not at all), and often manifest defects with the peripheral limbic functions. One-tenth of them have experienced sudden unexpected death in epilepsy (SUDEP). The antiepileptic drugs in the class of sodium channel blockers often guarantee transient improvement in few of the affected patients. However, the control of the paroxysmal discharge is far from sufficient (132).
Recent works in genomics have identified SCN8A as an epilepsy gene. Nav1.6 are widely distributed on the AIS of principal excitatory neurons and inhibitory interneurons (133). Expression of Nav1.6 is not limited to the CNS but is largely expressed in PNS neural cells. The expression of SCN8A is highly concentrated at the AIS of both excitatory and inhibitory neurons (48). In both neuron types, Nav1.6 coordinates excitation of neurons (43, 134). Studies using SCN8A knockout mice revealed that this gene is primarily involved in neuronal excitability in excitable cells such as purkinje cells of the cerebellum, ganglion cells of the retina and hippocampal cells (135). Hence, these mice exhibit early (juvenile) death (136). Furthermore, Nav1.6 is the main VGSC at nodes of Ranvier of mature neurons (43), and its under-expression inhibited nerve conduction (137).
Initial genetic studies using rodents revealed that the SCN8A gene is also mutated in epilepsy with absence seizures, a genetic generalized epilepsy (GGE) (138). Papale et al. (138) was the first to demonstrate that SCN8A is also a candidate gene in epilepsy using a mouse model with absence epilepsy. Heterozygous SCN8A-knock out mice manifested unprovoked spike-wave discharges which is typical of absence epileptic seizures.
Early infantile epileptic encephalopathy Type 13 (also called SCN8A encephalopathy) is caused by mutations of the SCN8A gene encoding the alpha subunit of Nav1.6 channel protein (132). Family studies have repeatedly shown de novo mutation in affected individuals, often arising from gain-of-function missense non-synonymous mutations. The mutant sodium channel exhibit enhanced activity (139). In a report by Blanchard et al., the physiological change observed in a mutant Nav1.6 was dysfunctional channel inactivation and a hyperpolarizing shift in voltage dependence of excitation resulting in an untimely or early channel re-opening (140). This implies that such mutations are localized on the Nav1.6 channels located on the principal excitatory neurons.
Veeramah et al. (141) reported a case of de novo heterozygous missense mutation in a child with severe infantile encephalopathy which started at 6 months. It was characterized with epileptic convulsions during the 4th year and was associated with SUDEP at age 15. This was a p.(N1768D) mutation in the Nav1.6 sodium channel expressed in the brain. Upon the expression of this mutant gene in ND7 cells, an enhanced ramp current with a corresponding shift in quick inactivation that was in favor of depolarization was observed. This led to hyperexcitability in pyramidal neurons of the hippocampus. In addition to this, incomplete inactivation of the channel was detected and there was a depolarizing shift in the voltage dependence of inactivation of the mutant channel (141). Complete identification and characterization of this mutation was typical of a gain-of-function, associated with enhanced ramp current. Hence, they suggested that a gain-of-function mutation in SCN8A can cause epileptic encephalopathy. The implication of this new mutation was a dysfunctional channel with an impaired inactivation potential leading to neuronal hyperactivity. Several large exome sequencing projects have equally identified 11 de novo mutations of SCN8A in singleton patients with epilepsy with or without intellectual disability (135, 142). In addition, another de novo mutation of SCN8A was discovered in a child having epileptic encephalopathy with congenital abnormalities (143).
Prior to this, a loss-of-function frameshift mutation (also identified in parents) caused by a 2 bp deletion p.(Pro1719ArgfsX6) was detected as a possible underlying factor for cerebellar ataxia associated with cognitive problems (144). Another study also identified seven potentially pathogenic variants in SCN8A, in patients manifesting seizures and intellectual disorder (142). Four of them were de novo and uninherited from any of their parents, another was paternally inherited (father was asymptomatic), while the last two were unknown. Studies on SCN8A null mice revealed reduced excitation in the hippocampal neurons (145) while experimental works to overexpress SCN8A in excitatory neurons were associated with seizures (146).
The understanding of the role of SCN8A in seizure initiation and epilepsy became well-known following the detection of de novo mutations in patients having epileptic encephalopathies with varying seizure severities. Another heterozygous mutation c.2300C>T in SCN8A was identified in a patient possessing delayed development, cognitive disability, and unmanageable seizures. This produced a mutated Nav1.6 polypeptide with a substituted amino acid p.(T767I) (100). Functional characterization of this mutation further revealed a gain-of-function, with a hyperpolarizing shift (10 mV) in voltage dependence of activation, including a rise in ramp current. These works enhanced the understanding of the gain-of-function roles of SCN8A mutations in epileptic encephalopathies, and more importantly, that SCN8A is a causative gene in children epilepsies. The works of Pan and Cummins (147) corroborate the gain-of-function theory, thereby suggesting a hyperpolarization-shift as a mechanism of action for the R850Q mutation. They further applied and recommended the use of computational simulation to predict the impact of variants on channel function.
However, loss-of-function mutations had been experimentally implicated in SCN8A. de Kovel et al. (148) demonstrated that the loss-of-function missense mutation in Nav1.6 p.(R223G) on inhibitory interneurons is another cause of infantile epileptic encephalopathies. Functional characterization studies showed a loss-of-function depicted by a significant decrease in the amplitude of current. Both gain-of-function mutations affecting Nav1.6 channels on principal excitatory neurons and loss-of-function mutations that affect Nav1.6 channels on inhibitory interneurons may lead to seizure generation.
Sprissler et al. (149) identified a new drug target and expanded the pathogenesis of SCN8A encephalopathies by comparing gene expression patterns before and after seizure onset and afterwards in a mouse model of early infantile epileptic encephalopathy Type 13 (149). The mutation, p.(N1768D) was introduced into the genome of the mouse by TALEN targeting. These knock-in mice exactly demonstrated the seizure disorder around the 3rd and 4th months of life (139). In addition, developmental disabilities were expressed in the mice, characterized by skewed postures, impeded movements, and tremors, which are typical of SCN8A encephalopathies. This further establish that either gain or loss-of-function mutations in SCN8A cause epilepsy. Recent studies show that loss-of-function mutations predispose to cognitive decline and intellectual disability without seizures while gain-of-function mutations result in more severe phenotypes associated with intractable seizures (150). More functional studies (using neurons) that will corroborate the findings of Liu et al. (150) are required to establish these genotype-phenotype relationships.
The pharmacological response of patients with gain-of-function mutations in SCN8A has been studied (151). Using a study involving four patients with SCN8A mutations, the authors discovered that sodium channel antagonists such as phenytoin at high therapeutic doses effectively manage SCN8A seizures. This is true in the case of a Chinese girl bearing the SCN8A mutation, p.(N1318S), in whom the administration of sodium channel antagonists proved successful (152).
A difference in drug response can occur in patients with different sodium channel mutations as observed in the cases treatment of SCN1A and SCN8A related seizures with sodium channel blockers. The former resulted in exacerbated seizures while the latter led to seizure mitigation. Hence, genetic testing is indispensable. Functional studies can help detect gain or loss-of-function variants that can guide drug design, identify high risk, population-related variants that are important for genetic testing and understand relationships between variants and specific drug response. Functional validation has been done for almost 20 different SCN8A variants (147). Figure 5 shows SCN8A primary structure with regions having variants have been functionally validated from literature while Table 7 shows the affected regions with references.
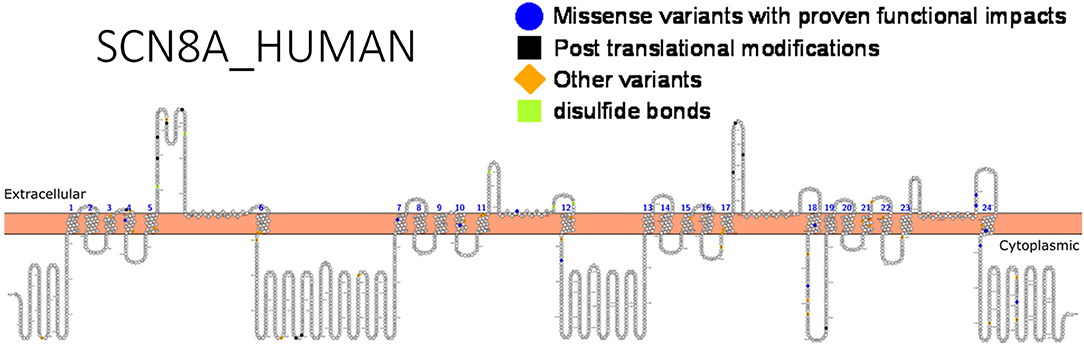
Figure 5. Primary structure of SCN8A protein showing regions with functionally validated epilepsy-variants from literature. Regions of missense variants with proven functional impacts (blue), regions of post-translational modifications (PTMs) (black), other variants (orange) and regions of disulfide bonds (lemon). [Created using Protter (78). Visualization link: https://bit.ly/2Vz6AB6].

Table 7. Regions of SCN8A with Functionally validated epilepsy variants with references.
SCN9A: An Epilepsy Gene or Not?
The sodium channel; Nav1.7 is expressed in the brain stem, cerebral cortex and spinal cord, but its major expression is in the peripheral nervous system (PNS) (155). Some SCN9A variants have been found in epilepsy and autism spectrum disorders. Also, many mutations in this gene are related to inherited chronic pain syndromes, erythermalgia, familial rectal pain (156, 157). Some SCN9A missense mutations have also been reported in patients with DS. Because the first SCN9A mutations identified occurred concurrently with SCN1A mutations, they were initially categorized as “genes with modifying roles to SCN1A” on inhibitory interneurons. But later on, six missense mutations were discovered in patients with febrile seizures not having SCN1A mutation, suggesting that SCN9A could be a major participant in cryptogenic epilepsies (158). Recently, Zhang et al. (159) discovered a possibly pathogenic missense variant; Y1958C causing GEFS+ in a Chinese family. In silico prediction algorithms validated this variant as likely deleterious but functional validation is still required. Due to the sparsity of SCN9A mutations in epilepsy, reports classifying it as a true epilepsy gene are inconclusive.
Genome Wide Association Studies (GWAS) of Sodium Channel Genes Involved in Epilepsy
Some reports from GWAS of epilepsy in different populations has shown some high-risk susceptibility single nucleotide polymorphisms (SNPs) within sodium channel genes (Table 8). These include studies of European, Asian, African-American origin.
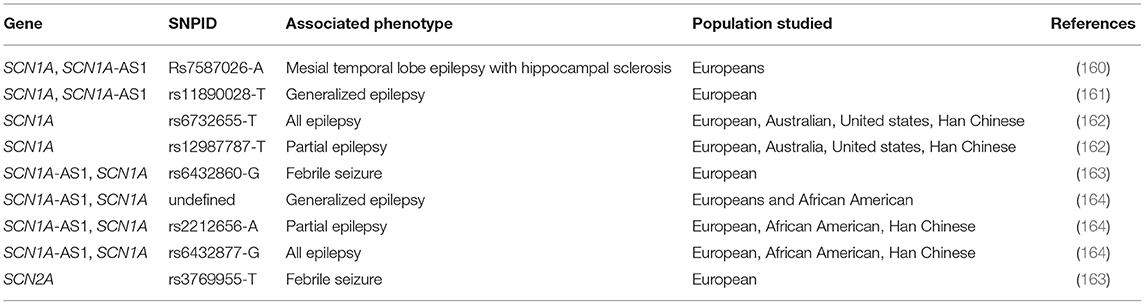
Table 8. GWAS reports of sodium channel genes involved in epilepsy.
In spite of the abundance of studies pointing out the roles of sodium channel genetic mutations in epilepsy, results published on the GWAS catalog (https://www.ebi.ac.uk/gwas/) are still relatively few. Feenstra et al. (163) conducted a genome wide association studies on the genetic causality of febrile seizures in individuals presenting with vaccine-associated and vaccine-unassociated seizures (163). Genetic loci associated with epilepsy (2q24.3) included two different sodium channel genes (rs6432860 in SCN1A and rs3769955 in SCN2A). More GWA studies in diverse population have confirmed the involvement of the genetic loci, 2q24.3 in both monogenic and polygenic epilepsies. In a cohort study of 8696 cases, the SNP, rs6732655-T was associated with all epilepsy phenotypes while rs12987787-T was associated with focal epilepsies (162). Both SNPs mapped to SCN1A within the 2q24.3 genomic loci.
A larger cohort of 15,212 cases identified 16 genomic loci associated with different epilepsy phenotypes (164). Amongst these was the 2q24.3 locus harboring SCN1A-SCN3A. The lead SNPs in this region were rs6432877-G for all epilepsies and rs11890028 (G) for genetic generalized epilepsies. The same locus was previously identified as causality for common generalized epilepsies (161). Very large sample sizes are required to increase power and identify more loci in GWAS of epilepsy. As a complex and genetically heterogenous disorder, it is also difficult to measure heritability in polygenic epilepsies. Hence, small cohort sizes (e.g., <3,500 cases) may be unable to establish heritability of rare variants with small effect sizes (165). This warrants the adoption of large cohort sizes. It would be advantageous if many countries would adopt the establishment of biobanks with detailed sampling information, as this would minimize sampling time in large cohorts studies (166).
Many SNPs are expected to have mitigated or aggravated effects when they occur in the presence of other gene mutations (i.e., epistatic interactions). Although GWAS reports may give a clue to the presence of pleiotropic effects amongst genes (164), they do not account for the actual damage-potential of the variants under the influence of mutations from other genes, Hence, necessitating functional studies.
Despite the drawbacks of GWAS, its innate capacity still needs to be harnessed. More research collaborations and funding are required to promote GWAS in regions with high epilepsy burden. GWAS of epilepsy in epilepsy endemic regions are expected to discover ethnically related polymorphisms and genetic diversity of epilepsy amongst different populations (167).
Conclusion and Future Considerations
Since epilepsy is a polygenic and heterogenous disorder, the increasingly cheap cost of whole genome and exome sequencing is rapidly advancing variant identification in candidate genes, including those acting as disease modifiers. The exponential increase in detected variants has created a huge need for more functional studies to validate the underlying mechanisms. Even though the evolutionary conservation amongst the different α-subunits has prevented the designing of drugs specifically for different VGSC channels, some progress has been recorded in determining effective therapies based on the knowledge of molecular mechanisms of mutations. Meanwhile, the outcome bias introduced in the functional analyses done using heterologous mammalian cells lines should be rapidly eliminated using patient-derived, genetically engineered neurons from hiPSCs or transgenic animals. Moreover, computational methods should be considered to guide decision-making for appropriate variants for functional studies. Above all, the field of epilepsy genetics has experienced a rapid improvement and much understanding has been gained over the past decades.
Limitations of This Study
Reports reviewed in this article were majorly from European, Asian, and American populations. We observed the absence of genetic studies on epilepsy from African populations, despite the high prevalence of this disorder in Africa. Fodjo et al. (168) reported the prevalence of epilepsy in West Africa as 13.14 per 1,000 persons, Akinsulore and Adewuya (169) reported an incidence rate of 5.3–37 per 100 persons in Nigeria, Esterhuizen et al. (167) reported a median occurrence of 14.2 per 1,000 in Sub-Saharan Africa while Mukuku et al. (170) reported 5.2–74 per 100 persons with epilepsy. Although variances occur depending on the studied region, there is remarkably higher prevalence of epilepsy in low- and middle-income countries (LMICs) than in developed nations (171). Over three-fourths of persons having epilepsy live in the LMICs, and about two-thirds of these cases have unknown etiology (167, 170). Although some reports suggest a relationship between epilepsy and CNS infections in Africa (168, 172), the etiology of most epilepsy remains unknown. This emphasizes the role of genetics and an urgent need for genetic investigations.
Author Contributions
AI, RS, and AE developed the concept of the manuscript. AI wrote the first draft. RS, AE, AY, and SS reviewed and modified the draft. All authors approved the final version of the work.
Funding
This work was funded by the National Institutes of Health (NIH) Fogarty Common Fund grant number 1U2RTW010679.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate Prof. Dr. Johannes Lemke and Dr. Julia Hentschel for reviewing and making corrections to this manuscript.
Abbreviations
GABRG2, Gamma-aminobutyric acid receptor, gamma-2; KCNT1, Potassium channel, subfamily T, member 1; KCNQ2, Potassium channel, voltage-gated, KQT-like subfamily, member 2; FOXG1, Forkhead box G1; CDKL5, Cyclin-dependent kinase-like 5; MECP2, Methyl-CpG-binding protein 2; ARX, Aristaless-related homeobox, x-linked; SCN1A, Sodium channel, neuronal type I, alpha subunit; SCN2A, Sodium channel, voltage-gated, type II, alpha subunit; SCN3A, Sodium channel, voltage-gated, type III, alpha subunit; SCN1B, Sodium channel, voltage-gated, type I, beta subunit; SCN8A, Sodium channel, voltage-gated, type VIII, alpha subunit; SCN9A, Sodium channel, voltage-gated, type IX, alpha subunit; STXBP1, Syntaxin-binding protein 1; SLC25A22, Solute carrier family 25 (mitochondrial carrier, glutamate), member 22; PCDH19, Protocadherin 19; SPTAN1, Spectrin, alpha, nonerythrocytic 1; ARHGEF9, Rho guanine nucleotide exchange factor 9.
References
1. Boison D. The biochemistry and epigenetics of epilepsy: focus on adenosine and glycine. Front Mol Neurosci. (2016) 9:1–7. doi: 10.3389/fnmol.2016.00026
2. Chen T, Giri M, Xia Z, Subedi YN, Li Y. Genetic and epigenetic mechanisms of epilepsy: a review. Neuropsychiatric Dis Treatment. (2017) 13:1841–59. doi: 10.2147/NDT.S142032
3. Falco-walter JJ, Scheffer IE, Fisher RS. The new definition and classification of seizures and epilepsy. Epilepsy Res. (2018) 139:73–9. doi: 10.1016/j.eplepsyres.2017.11.015
4. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE POSITION PAPER ILAE classification of the epilepsies : position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
5. Sweet KM, Shaw DW, Chapman T. Cerebral palsy and seizures in a child with tubulinopathy pattern dysgenesis and focal cortical dysplasia. Radiol Case Rep. (2017) 12:396–400. doi: 10.1016/j.radcr.2016.12.008
6. Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia. (2010) 51:1650–8. doi: 10.1111/j.1528-1167.2010.02640.x
7. Bechi G, Rusconi R, Cestèle S, Striano P, Franceschetti S, Mantegazza M. Rescuable folding defective NaV1.1 (SCN1A) mutants in epilepsy: properties, occurrence, and novel rescuing strategy with peptides targeted to the endoplasmic reticulum. Neurobiol Dis. (2015) 75:100–14. doi: 10.1016/j.nbd.2014.12.028
9. Claes LR, Deprez L, Suls A, Baets J, Smets K, Van Dyck T, et al. The SCN1A variant database: a novel research and diagnostic tool. Hum Mutat. (2009) 30:E904–20. doi: 10.1002/humu.21083
10. Kaplan ID, Isom L, Petrou S. Role of sodium channels in epilepsy. Cold Spring Harb Perspect Med. (2016) 6:a022814. doi: 10.1101/cshperspect.a022814
11. Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, et al. A gain-of-function mutation in the sodium channel gene SCN2A results in seizures and behavioral abnormalities. Neurosicnce. (2001) 102:307–17. doi: 10.1016/S0306-4522(00)00479-6
12. Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, et al. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci Lett. (2008) 433:65–70. doi: 10.1016/j.neulet.2007.12.064
13. Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol. (2010) 588:1849–59. doi: 10.1113/jphysiol.2010.187484
14. Brackenbury WJ, Isom LL. Na channel beta subunits: overachievers of the ion channel family. Front Pharmacol. (2011) 2:53. doi: 10.3389/fphar.2011.00053
15. Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. (2009) 72:1544–7. doi: 10.1212/01.wnl.0000342387.65477.46
16. Tsujino A, Maertens C, Ohno K, Shen X-M, Fukuda T, Harper CM, et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Nat Acad Sci USA. (2003) 100:7377–82. doi: 10.1073/pnas.1230273100
17. Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. (2008) 117:1927–35. doi: 10.1161/CIRCULATIONAHA.107.757955
18. Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. Clin Invest. (2008) 118:2219–29. doi: 10.1172/JCI34057
19. Albert CM, Nam EG, Rimm EB, Jin HW, Hajjar RJ, Hunter DJ, et al. Cardiac sodium channel gene variants and sudden cardiac death in women. Circulation. (2008) 117:16–23. doi: 10.1161/CIRCULATIONAHA.107.736330
20. Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet Neurol. (2005) 366:371–6. doi: 10.1016/S0140-6736(05)66786-4
21. Schwarz N, Bast T, Gaily E, Golla G, Gorman KM, Griffiths LR, et al. Clinical and genetic spectrum of SCN2A-associated episodic ataxia. Eur J Pediat Neurol. 2019:438–47. doi: 10.1016/j.ejpn.2019.03.001
22. Fazeli W, Becker K, Herkenrath P, Duchting C, Korber F, Landgraf P, et al. Dominant SCN2A mutation causes familial episodic ataxia and impairment of speech development. Neuropediatrics. (2018) 49:379–84. doi: 10.1055/s-0038-1668141
23. Jurkat-Rott K, Mitrovic N, Hang C, Kouzmekine A, Iaizzo P, Herzog J, et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Nat Acad Sci USA. (2000) 97:9549–54. doi: 10.1073/pnas.97.17.9549
24. Gay S, Dupuis D, Faivre L, Masurel-Paulet A, Labenne M, Colombani M, et al. Severe neonatal non-dystrophic myotonia secondary to a novel mutation of the voltage-gated sodium channel (SCN4A) gene. Am J Med Genet. (2008) 146A:380–3. doi: 10.1002/ajmg.a.32141
25. Arnold WD, Feldman DH, Ramirez S, He L, Kassar D, Quick A, et al. Defective fast inactivation recovery of Na(v)1.4 in congenital myasthenic syndrome. Ann Neurol. (2015) 77:840–50. doi: 10.1002/ana.24389
26. Francis DG, Rybalchenko V, Struyk A, Cannon SC. Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia. Neurology. (2011) 76:1635–41. doi: 10.1212/WNL.0b013e318219fb57
27. Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott J-J, et al. Sodium channel beta-1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. (2008) 118:2260–8. doi: 10.1172/JCI33891
28. Makita N, Sasaki K, Groenewegen WA, Yokota T, Yokoshiki H, Murakami T, et al. Congenital atrial standstill associated with coinheritance of a novel SCN5A mutation and connexin 40 polymorphisms. Heart Rhythm. (2005) 2:1128–34. doi: 10.1016/j.hrthm.2005.06.032
29. Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KI, et al. Sodium channel mutations and susceptibility of heart failure and atrial fibrillation. JAMA. (2005) 293:447–54. doi: 10.1001/jama.293.4.447
30. Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, et al. Mutations in sodium channel beta-1- and beta-2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. (2009) 2:268–78. doi: 10.1161/CIRCEP.108.779181
31. Wagnon JL, Barker BS, Ottolini M, Park Y, Volkheimer A, Valdez P, et al. Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol Genet. (2017) 3:1–6. doi: 10.1212/NXG.0000000000000170
32. Michiels JJ, te Morsche RHM, Jansen JBMJ, Drenth JPH. Autosomal dominant erythermalgia associated with a novel mutation in the voltage-gated sodium channel alpha-subunit Na(v)1.7. Arch Neurol. (2005) 62:1587–90. doi: 10.1001/archneur.62.10.1587
33. Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. (2006) 444:894–8. doi: 10.1038/nature05413
34. Yuan J, Matsuura E, Higuchi Y, Hashiguchi A, Nakamura T, Nozuma S, et al. Hereditary sensory and autonomic neuropathy type IID caused by an SCN9A mutation. Neurology. (2013) 80:1641–9. doi: 10.1212/WNL.0b013e3182904fdd
35. Meglic A, Perkovic-Benedik M, Trebusak Podkrajsek K, Bertok S. Painful micturition in a small child: an unusual clinical picture of paroxysmal extreme pain disorder. Pediat Nephrol. (2014) 29:1643–6. doi: 10.1007/s00467-014-2819-2
36. Devigili G, Eleopra R, Pierro T, Lombardi R, Rinaldo S, Lettieri C, et al. Paroxysmal itch caused by gain-of-function Na(v)1.7 mutation. Pain. (2014) 155:1702–7. doi: 10.1016/j.pain.2014.05.006
37. Lombardi R, Dong J, Rodriguez G, Al. E. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. (2009) 104:1076–84. doi: 10.1161/CIRCRESAHA.109.196899
38. Meadows L, Isom L. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. (2005) 67:448–58. doi: 10.1016/j.cardiores.2005.04.003
39. Chen S, Gurha P, Lombardi R. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ Res. (2014) 114:454–68. doi: 10.1161/CIRCRESAHA.114.302810
40. Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, et al. Preferential inactivation of SCN1A in parvalbumin interneurons increases seizure susceptibility. Neurobiol Dis. (2012) 49C:211–20. doi: 10.1016/j.nbd.2012.08.012
41. Du J, Simmons S, Brunklaus A, Adiconis X, Hession CC, Fu Z, et al. Differential excitatory vs inhibitory SCN expression at single cell level regulates brain sodium channel function in neurodevelopmental disorders. Eur J Paediatric Neurol. (2020) 24:129–33. doi: 10.1016/j.ejpn.2019.12.019
42. Oliva MK, McGarr TC, Beyer BJ, Gazina E, Kaplan DI, Cordeiro L, et al. Physiological and genetic analysis of multiple sodium channel variants in a model of genetic absence epilepsy. Neurobiol Dis. (2014) 67:180–90. doi: 10.1016/j.nbd.2014.03.007
43. Lorincz A, Nusser Z. Molecular identity of dendritic voltage gated sodium channels. Science. (2010) 328:906–9. doi: 10.1126/science.1187958
44. Xie Y, Ng NN, Safrina OS, Ramos CM, Ess KC, Schwartz PH, et al. Comparisons of dual isogenic human iPSC pairs identify functional alterations directly caused by an epilepsy associated SCN1A mutation. Neurobiol Dis. (2020) 134:104627. doi: 10.1016/j.nbd.2019.104627
45. Surovy M, Soltysova A, Kolnikova M, Sykora P, Ilencikova D, Ficek A, et al. Novel SCN1A variants in Dravet syndrome and evaluating a wide ap- proach of patient selection. Gen Physiol Biophys. (2016) 35:333–42. doi: 10.4149/gpb_2016002
46. Hirose S, Scheffer IE, Marini C, De Jonghe PAE, Goldman AM, Kauffman M, et al. SCN1A testing for epilepsy: application in clinical practice. Epilepsia. (2013) 54:946–52. doi: 10.1111/epi.12168
47. Marini C, Scheffer IE, Nabbout R, Suls A, De Jonghe P, Zara F, et al. The genetics of Dravet syndrome. Epilepsia. (2011) 52:24–9. doi: 10.1111/j.1528-1167.2011.02997.x
48. Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, O'Malley HA, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol. (2013) 74:128–39. doi: 10.1002/ana.23897
49. Meisler MH, O'Brien JE. Gene interactions and modifiers in epilepsy. In: (US) NCfBI, editor. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. 4th ed. Oxford: Oxford University Press (2012). p. 1–12. doi: 10.1093/med/9780199746545.003.0059
50. Lossin C, Wang DW, Rhodes TH, Vanoye CG, George ALJ. Molecular basis of an inherited epilepsy. Neuron. (2002) 34:877–84. doi: 10.1016/S0896-6273(02)00714-6
51. Lossin C, Rhodes TH, Desai RR, Vanoye CG, Wang D, Carniciu S, et al. Epilepsy-associated dysfunction in the voltagegated neuronal sodium channel SCN1A. J Neurosci. (2003) 23:11289–95. doi: 10.1523/JNEUROSCI.23-36-11289.2003
52. Tang B, Dutt K, Papale L, Rusconi R, Shankar A, Hunter J, et al. A BAC transgenic mouse model reveals neuron subtype-specific effects of a Generalized Epilepsy with Febrile Seizures Plus (GEFS+) mutation. Neurobiol Dis. (2009) 35:91–102. doi: 10.1016/j.nbd.2009.04.007
53. Kluckova D, Kolnikova M, Lacinova L, Jurkovicova-Tarabova B, Foltan T, Demko V, et al. A Study among the genotype, functional alternations, and phenotype of 9 SCN1A mutations in epilepsy patients. Sci Rep. (2020) 10:1–13. doi: 10.1038/s41598-020-67215-y
54. Lossin C. A catalog of SCN1A variants. Brain Dev. (2009) 31:114–30. doi: 10.1016/j.braindev.2008.07.011
55. Zhang YH, Sun HH, Liu XY, Ma XW, Yang ZX, Xiong H, et al. Clinical features and SCN1A gene mutation analysis of severe myoclonic epilepsy of infancy. Zhonghua er ke za zhi. (2008) 46:769–73.
56. Zucca C, Redaelli F, Epifanio R, Zanotta N, Romeo A, Lodi M, et al. Cryptogenic epileptic syndromes related to SCN1A. Twelve novel mutations identified. Arch Neurol. (2008) 65:489–94. doi: 10.1001/archneur.65.4.489
57. Heron SE, Scheffer IE, Grinton BE, Eyre H, Oliver KL, Bain S, et al. Familial neonatal seizures with intellectual disability caused by a microduplication of chromosome 2q24. Epilepsia. (2010) 51:1865–9. doi: 10.1111/j.1528-1167.2010.02558.x
58. Chen YJ, Shi YW, Xu HQ, Chen ML, Gao MM, Sun WW, et al. Electrophysiological differences between the same pore region mutation in SCN1A and SCN3A. Mol Neurobiol. (2015) 51:1263–70. doi: 10.1007/s12035-014-8802-x
59. Tunçer GÖ, Teber S, Albayrak P, Kutluk MG, Deda G. A case of Dravet Syndrome with a newly defined mutation in the SCN1A gene. Turkish Archiv Pediatr. (2018) 53:259. doi: 10.5152/TurkPediatriArs.2018.4197
60. Shi L, Zhu M, Li H, Wen Z, Chen X, Luo J, et al. SCN1A and SCN2A polymorphisms are associated with response to valproic acid in Chinese epilepsy patients. Eur J Clin Pharmacol. (2019) 2019:1–9. doi: 10.1007/s00228-019-02633-0
61. Ottman R, Hirose S, Jain S, Lerche H, Lopes-Cendes I, Noebels Jea JL, et al. Genetic testing in the epilepsies - report of the ILAE Genetics Commission. Epilepsia. (2010) 51:655–70. doi: 10.1111/j.1528-1167.2009.02429.x
62. Fang ZX, Hong SQ, Li TS, Wang J, Xie LL, Han W, et al. Genetic and phenotypic characteristics of SCN1A-related epilepsy in Chinese children. NeuroReport. (2019) 2019:1259. doi: 10.1097/WNR.0000000000001259
63. Krikova E, Val'dman E, Avakian GN, AndreevIaA EV, Denisov FK, Rider RR, et al. Association study of the SCN1 gene polymorphism and effective dose of lamotrigine. Zhurnal Nevrologii i Psikhiatrii Imeni SS Korsakova. (2009) 109:57–62.
64. Cross JH, Devinsky O, Marsh E, Miller I, Nabbout R, Scheffer IE, et al. Cannabidiol (CBD) reduces convulsive seizure frequency in Dravet syndrome: results of a multi-center, randomized, controlled trial. Epilepsia. (2017) 58:S12. doi: 10.1111/epi.13944
65. Eschbach K, Knupp KG. Stiripentol for the treatment of seizures in Dravet syndrome. Expert Rev Clin Pharmacol. (2019) 12:379–88. doi: 10.1080/17512433.2019.1605904
66. Ceulemans B, Boel M, Leyssens K, Van Rossem C, Neels P, Jorens PG, et al. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia. (2012) 53:1131–9. doi: 10.1111/j.1528-1167.2012.03495.x
67. Ceulemans B, Schoonjans AS, Marchau F, Paelinck BP, Lagae L. Five-year extended follow-up status of 10 patients with Dravet syndrome treated with fenfluramine. Epilepsia. (2016) 57:e129–34. doi: 10.1111/epi.13407
68. Schoonjans A, Paelinck BP, Marchau F, Gunning B, Gammaitoni A, Galer BS, et al. Low-dose fenfluramine significantly reduces seizure frequency in Dravet syndrome: a prospective study of a new cohort of patients. Eur J Neurol. (2017) 24:309–14. doi: 10.1111/ene.13195
69. Schoonjans AS, Marchau F, Paelinck BP, Lagae L, Gammaitoni A, Pringsheim M, et al. Cardiovascular safety of low-dose fenfluramine in Dravet syndrome: a review of its benefit-risk profile in a new patient population. Curr Med Res Opin. (2017) 33:1773–81. doi: 10.1080/03007995.2017.1355781
70. Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. (2019) 394:2243–54. doi: 10.1016/S0140-6736(19)32500-0
71. Guo F, Yu N, Cai J-q, Quinn T, Zong Z-h, Zeng Y-j, et al. Voltage-gated sodium channel NAv1.1, Nav1.3 and β1 subunit were up-regulated in the hippocampus of spontaneously epileptic rat. Brain Res Bulletin. (2008) 75:179–87. doi: 10.1016/j.brainresbull.2007.10.005
72. Younus I, Reddy DS. Epigenetic interventions for epileptogenesis: a new frontier for curing epilepsy. Pharmacol Therapeut. (2017) 177:108–22. doi: 10.1016/j.pharmthera.2017.03.002
73. Jiao J, Yuanyuan Y, Yiwu S, Jiayu C, Ru G, Yong F, et al. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum Mol Genet. (2013) 22:4241–52. doi: 10.1093/hmg/ddt275
74. Higurashi N, Uchida T, Lossin C, Misumi Y, Okada Y, Akamatsu W, et al. A human Dravet syndrome model from patient induced pluripotent stem cells. Mol Brain. (2013) 6:19. doi: 10.1186/1756-6606-6-19
75. Sun Y, Paşca SP, Portmann T, Goold C, Worringer KA, Guan W, et al. A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet syndrome patients. Elife. (2016) 5:e13073. doi: 10.7554/eLife.13073
76. Liu J, Gao C, Chen W, Ma W, Li X, Shi Y, et al. CRISPR/Cas9 facilitates investigation of neural circuit disease using human iPSCs: mechanism of epilepsy caused by an SCN1A loss-of-function mutation. Transl Psychiatry. (2016) 6:e703. doi: 10.1038/tp.2015.203
77. Zhao GX, Zhang Z, Cai WK, Shen ML, Wang P, He GH. Associations between CYP3A4, CYP3A5 and SCN1A polymorphisms and carbamazepine metabolism in epilepsy: a meta-analysis. medRxiv. (2020). doi: 10.1101/2020.03.03.20030783
78. Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. (2013) 30:884–6. doi: 10.1093/bioinformatics/btt607
79. Wallace RH, Wang DW, Singh R, Scheffer IE, George ALJ, Phillips HA, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Naþ-channel beta1 subunit gene SCN1B. Nat Genetics. (1998) 19:366e70. doi: 10.1038/1252
80. Baroni D, Barbieri R, Picco C, Moran O. Functional modulation of voltage-dependent sodium channel expression by wild type and mutated C121W-β1 subunit. J Bioenerg Biomembr. (2013) 45:45:353–68. doi: 10.1007/s10863-013-9510-3
81. O'Malley HA, Isom LL. Sodium channel β subunits : emerging targets in channelopathies. Annu Rev Physiol. (2015) 77:481–504. doi: 10.1146/annurev-physiol-021014-071846
82. Shimizu H, Miyazaki H, Ohsawa N, Shoji S, Ishizuka-Katsura Y, Tosaki A, et al. Structure-based site-directed photo-crosslinking analyses of multimeric cell-adhesive interactions of voltage-gated sodium channel β subunits. Sci Rep. (2016) 6:26618. doi: 10.1038/srep26618
83. Patino GA, Claes LRF, Lopez-Santiago LF, Slat EA, Dondeti RSR, Chen C, et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci. (2009) 29:10764–78. doi: 10.1523/JNEUROSCI.2475-09.2009
84. Scheffer IE, Harkin LA, Grinton BE, Dibbens LM, Turner SJ, Zielinski MA, et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain. (2007) 130:100–9. doi: 10.1093/brain/awl272
85. Ogiwara I, Nakayama T, Yamagata T, Ohtani H, Mazaki E, Tsuchiya S, et al. A homozygous mutation of voltage-gated sodium channel beta(I) gene SCN1B in a patient with Dravet syndrome. Epilepsy. (2012) 53:e200e3. doi: 10.1111/epi.12040
86. Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL, et al. Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J Neurosci. (2009) 29:2027e42. doi: 10.1523/JNEUROSCI.4531-08.2009
87. Kruger LC, O'Malley HA, Hull JM, Kleeman A, Patino GA, Isom LL. β1-C121W is down but not out: epilepsy-associated SCN1B-C121W results in a deleterious gain-of-function. J Neurosci. (2016) 36:6213–24. doi: 10.1523/JNEUROSCI.0405-16.2016
88. Lucas PT, Meadows LS, Nicholls J, Ragsdale DS. An epilepsy mutation in the beta1 subunit of the voltage-gated sodium channel results in reduced channel sensitivity to phenytoin. Epilepsy Res. (2005) 64:77–84. doi: 10.1016/j.eplepsyres.2005.03.003
89. Patino GA, Brackenbury WJ, Bao Y, Lopez-santiago LF, Malley HAO, Chen C, et al. Voltage-gated Na channel β1B : a secreted cell adhesion molecule involved in human epilepsy. J Neurosci. (2011) 31:14577–91. doi: 10.1523/JNEUROSCI.0361-11.2011
90. Marionneau C, Carrasquillo Y, Norris AJ, Townsend RR, Isom LL, Link AJ, et al. The sodium channel accessory subunit Navb1 regulates neuronal excitability through modulation of repolarizing voltage-gated Kþ channels. J Neurosci. (2012) 32:5716e27. doi: 10.1523/JNEUROSCI.6450-11.2012
91. Nguyen HM, Miyazaki H, Hoshi N, Smith BJ, Nukina N, Goldin AL, et al. Modulation of voltage-gated K+ channels by the sodium channel beta1 subunit. Proc Natl Acad Sci USA. (2012) 109:18577–82. doi: 10.1073/pnas.1209142109
92. Aeby A, Sculier C, Bouza AA, Askar B, Lederer D, Schoonjans AS, et al. SCN1B-linked early infantile developmental and epileptic encephalopathy. Ann Clin Transl Neurol. (2019) 6:2354–67. doi: 10.1002/acn3.50921
93. Dang LT, Quinonez SC, Becka BR, Isom LL, Joshi SM. Dramatic improvement in seizures with phenytoin treatment in an individual with refractory epilepsy and a SCN1B variant. Pediatric Neurol. (2020) 108:121–2. doi: 10.1016/j.pediatrneurol.2020.03.012
94. Audenaert D, Claes L, Ceulemans B, Lofgren A, Van Broeckhoven C, De Jonghe P. A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology. (2003) 61:854–6. doi: 10.1212/01.WNL.0000080362.55784.1C
95. Baroni D, Picco C, Moran O. A mutation of SCN1B associated with GEFS+ causes functional and maturation defects of the voltage-dependent sodium channel. Hum Mutat. (2018) 39:1402-15. doi: 10.1002/humu.23589
96. Ramadan W, Patel N, Anazi S, Kentab AY, Bashiri FA, Hamad MH, et al. Confirming the recessive inheritance of SCN1B mutations in developmental epileptic encephalopathy. Clin Genet. (2017) 92:327–31. doi: 10.1111/cge.12999
97. Tammaro P, Conti F, Moran O. Modulation of sodium current in mammalian cells by an epilepsy-correlated beta-1-subunit mutation. Biochem Biophys Res Commun. (2002) 291:1095–101. doi: 10.1006/bbrc.2002.6570
98. Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Progr Neurobiol. (2009) 87:41–57. doi: 10.1016/j.pneurobio.2008.09.016
99. Liao Y, Anttonen A, Liukkonen E, Gaily E, Maljevic S, Schubert S, et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology. (2010) 75:1454–8. doi: 10.1212/WNL.0b013e3181f8812e
100. Estacion M, O'Brien JE, Conravey A, Hammer MF, Waxman SG, Dib-Hajj SD, et al. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol Dis. (2014) 69:117–23. doi: 10.1016/j.nbd.2014.05.017
101. Ogiwara I, Ito K, Sawaishi Y, Osaka H, Mazaki E, Inoue I, et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. (2009) 73:1046–53. doi: 10.1212/WNL.0b013e3181b9cebc
102. Shi X, Yasumoto S, Nakagawa E, Fukasawa T, Uchiya S, Hirose S. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev. (2009) 31:758–62. doi: 10.1016/j.braindev.2009.08.009
103. Baasch AL, Hüning I, Gilissen C, Klepper J, Veltman JA, Gillessen-Kaesbach G, et al. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia. (2014) 55:25–9. doi: 10.1111/epi.12554
104. Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. (2012) 380:1674–82. doi: 10.1016/S0140-6736(12)61480-9
105. Kamiya K, Kaneda M, Sugawara T, Mazaki E, Okamura N, Montal M, et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci. (2004) 24:2690–8. doi: 10.1523/JNEUROSCI.3089-03.2004
106. Shi X, Yasumoto S, Kurahashi H, Nakagawa E, Fukasawa T, Uchiya S, et al. Clinical spectrum of SCN2A mutations. Brain Dev. (2012) 34:541–5. doi: 10.1016/j.braindev.2011.09.016
107. Nakamura K, Kato M, Osaka H, Yamashita S, Nakagawa E, Haginoya K, et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. (2013) 81:1–8. doi: 10.1212/WNL.0b013e3182a43e57
108. Saitoh M, Ishii A, Ihara Y, Hoshino A, Terashima H, Kubotac M, et al. Missense mutations in sodium channel SCN1A and SCN2A predispose children to encephalopathy with severe febrile seizures. Epilepsy Res. (2015) 117:1–6. doi: 10.1016/j.eplepsyres.2015.08.001
109. Liao Y, Deprez L, Maljevic S, Pitsch J, Claes L, Hristova D, et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain. (2010) 133:1403–14. doi: 10.1093/brain/awq057
110. Xu R, Thomas EA, Jenkins M, Gazina EV, Chiu C, Heron SE, et al. A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Mol Cell Neurosci. (2007) 35:292–301. doi: 10.1016/j.mcn.2007.03.003
111. Barcia G, Fleming M, Deligniere A, Gazula V-R, Brown MR, Langouet M, et al. De novo gainof- function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. (2012) 44:1255–9. doi: 10.1038/ng.2441
112. Howell KB, Mcmahon JM, Mackay MT, Rodriguez- V, Clark D, Freeman JL, et al. SCN2A encephalopathy A major cause of epilepsy of infancy with migrating focal seizures. Neurology. (2015) 85:1–10. doi: 10.1212/WNL.0000000000001926
113. Matalon D, Goldberg E, Medne L, Marsh ED. Confirming an expanded spectrum of SCN2A mutations : a case series. Epileptic Disord. (2014) 16:13–8. doi: 10.1684/epd.2014.0641
114. Zeng Q, Zhang Y, Yang X, Zhang J, Liu A, Liu X, et al. Phenotype study of SCN2A gene related epilepsy. Zhonghua Er Ke Za Zhi. (2018) 56:518–23. doi: 10.3760/cma.j.issn.0578-1310.2018.07.009
115. Foster LA, Johnson MR, Macdonald JT, Karachunski PI, Henry TR, Nascene DR, et al. Infantile epileptic encephalopathy associated with SCN2A mutation responsive to oral mexiletine. Pediatric Neurol. (2016) 16:1–11. doi: 10.1016/j.pediatrneurol.2016.10.008
116. Wolff M, Johannesen K, Hedrich U, Masnada S, Rubboli G, Gardella Eea. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. (2017) 2017:awx054. doi: 10.1093/brain/awx054
117. Wong VCN, Fung CW, Kwong AKY. SCN2A mutation in a Chinese boy with infantile spasm - response to Modified Atkins Diet. Brain Dev. (2015) 37:729–32. doi: 10.1016/j.braindev.2014.10.008
118. Su DJ, Lu JF, Lin LJ, Liang JS, Hung KL. SCN2A mutation in an infant presenting with migrating focal seizures and infantile spasm responsive to a ketogenic diet. Brain Dev. (2018) 40:8724–7. doi: 10.1016/j.braindev.2018.03.005
119. Turkdogan D, Thomas G, Demirel B. Ketogenic diet as a successful early treatment modality for SCN2A mutation. Brain Dev. (2019) 41:389–91. doi: 10.1016/j.braindev.2018.10.015
120. Lu Y, Su Q, Li M, Dayimu A, Dai X, Wang Z, et al. Association of SCN1A, SCN2A, and UGT2B7 polymorphisms with responsiveness to valproic acid in the treatment of epilepsy. BioMed Res Int. (2020) 2020:1–8. doi: 10.1155/2020/8096235
121. Misra SN, Kahlig KM, George ALJ. Impaired NaV1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia. (2008) 49:1535–45. doi: 10.1111/j.1528-1167.2008.01619.x
122. Kile KB, Tian N, Durand DM. SCN2A sodium channel mutation results in hyperexcitability in the hippocampus in vitro. Epilepsia. (2008) 49:488–99. doi: 10.1111/j.1528-1167.2007.01413.x
123. Vanoye CG, Gurnett CA, Holland KD, George AL, Kearney JA. Novel SCN3A variants associated with focal epilepsy in children. Neurobiol Dis. (2014) 62:313–22. doi: 10.1016/j.nbd.2013.10.015
124. Estacion M, Gasser A, Dib-Hajj SD, Waxman SG. A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Exp Neurol. (2010) 224:362–8. doi: 10.1016/j.expneurol.2010.04.012
125. Lamar T, Vanoye CG, Calhoun J, Wong JC, Dutton SBB, Jorge BS, et al. SCN3A deficiency associated with increased seizure susceptibility. Neurobiol Dis. (2017) 102:38–48. doi: 10.1016/j.nbd.2017.02.006
126. Zaman T, Helbig I, BoŽović IB, DeBrosse SD, Bergqvist AC, Wallis K, et al. Mutations in SCN3A cause early infantile epileptic encephalopathy. Ann Neurol. (2018) 83:703–17. doi: 10.1002/ana.25188
127. Yoshitomi S, Takahashi Y, Ishizuka M, Yamaguchi T. Three patients manifesting early infantile epileptic spasms associated with 2q24 microduplications. Brain Dev. (2015) 37:874–9. doi: 10.1016/j.braindev.2015.03.001
128. Thuresson A-C, Van Buggenhout G, Sheth F, Kamate M, Andrieux J, Clayton SJ, et al. Whole gene duplication of SCN2A and SCN3A is associated with neonatal seizures and a normal intellectual development. Clin Genet. (2016) 91:106–10. doi: 10.1111/cge.12797
129. Chong FP, Saitsu H, Sakai Y, Imagi T, Nakamura R, Matsukura M, et al. Deletions of SCN2A and SCN3A genes in a patient with West syndrome and autistic spectrum disorder. Seizure Eur J Epilepsy. (2018) 60:91–3. doi: 10.1016/j.seizure.2018.06.012
130. Wang Y, Du X, Bin R, Yu S, Xia Z, Zheng G. Genetic variants identified from epilepsy of unknown etiology in Chinese children by targeted exome sequencing. Sci Rep. (2017) 7:1–10. doi: 10.1038/srep46520
131. Larsen J, Carvill GL, Gardella E, Kluger G, Schmiedel G, Barisic N, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology. (2015) 84:480–9. doi: 10.1212/WNL.0000000000001211
132. Hammer MF, Wagnon JL, Mefford HC, Meisler MH. SCN8A-related epilepsy with encephalopathy. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. Gene Reviews. Seattle: University of Washington (2016) 1993–2021.
133. Sills GJ. Classical mechanisms of action of antiepileptic drugs. In: H Potschka, Lerche H, editors, Therapeutic Targets and Perspectives in the Pharmacological Treatment of Epilepsy. Bremen: UNI-MED Verlag. (2013). p. 62–5.
134. Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltagedependent ion channels in mammalian central neurons. Physiol Rev. (2008) 88:1407–47. doi: 10.1152/physrev.00002.2008
135. O'Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet. (2013) 4:213. doi: 10.3389/fgene.2013.00213
136. Sharkey LM, Cheng X, Drews V, Buchner DA, Jones JM, Justice MJ, et al. The ataxia3 mutation in the N-terminal cytoplasmic domain of sodium channel Nav1.6 disrupts intracellular trafficking. J Neurosci. (2009) 29:2733–41. doi: 10.1523/JNEUROSCI.6026-08.2009
137. Kearney JA, Buchner DA, de Haan G, Adamska M, Levin SI, Furay AR, et al. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel SCN8A (Nav1.6). Hum Mol Genet. (2002) 11:2765–75. doi: 10.1093/hmg/11.22.2765
138. Papale LA, Beyer B, Jones JM, Sharkey LM, Tufik S, Epstein M, et al. Heterozygous mutations of the voltage-gated sodium channel SCN8A are associated with spikewave discharges and absence epilepsy in mice. Hum Mol Genet. (2009) 18:1633–41. doi: 10.1093/hmg/ddp081
139. Wagnon JL, Barker BS, Hounshell JA, Haaxma CA, Shealy A, Moss T, et al. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol. (2016) 3:114–23. doi: 10.1002/acn3.276
140. Blanchard MG, Willemsen MH, Walker JB, Dib-Hajj SD, Waxman SG, Jongmans, et al. De novo gain-of-function and lossof- function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J Med Genet. (2015) 52:330–7. doi: 10.1136/jmedgenet-2014-102813
141. Veeramah KR, O'Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. de novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. (2012) 90:502–10. doi: 10.1016/j.ajhg.2012.01.006
142. Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. (2013) 45:825. doi: 10.1038/ng.2646
143. Vaher U, Nõukas M, Nikopensius T, Kals M, Annilo T, Nelis M, et al. De novo SCN8A mutation identified by whole-exome sequencing in a boy with neonatal epileptic encephalopathy, multiple congenital anomalies, and movement disorders. J Child Neurol. (2013) 29:NP202–6. doi: 10.1177/0883073813511300
144. Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Genet. (2006) 43:527–30. doi: 10.1136/jmg.2005.035667
145. Royeck M, Horstmann MT, Remy S, Reitze M, Yaari Y, Beck H. Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J Neurophysiol. (2008) 100:2361–80. doi: 10.1152/jn.90332.2008
146. Sun W, Wagnon JL, Mahaffey CL, Briese M, Ule J, Frankel WN. Aberrant sodium channel activity in the complex seizure disorder of Celf4 mutant mice. J Physiol. (2013) 591:241–55. doi: 10.1113/jphysiol.2012.240168
147. Pan Y, Cummins TR. Distinct functional alterations in SCN8A epilepsy mutant channels. J Physiol. (2020) 598:381–401. doi: 10.1113/JP278952
148. de Kovel CGF, Meisler MH, Brilstra EH, van Berkestijn FMC, Slot Rvt, van Lieshout S, et al. Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res. (2014) 108:1511–8. doi: 10.1016/j.eplepsyres.2014.08.020
149. Sprissler RS, Wagnon JL, Bunton-Stasyshyn RK, Meisler MH, Hammer MF. Altered gene expression profile in a mouse model of SCN8A encephalopathy. Exp Neurol. (2017) 288:134–41. doi: 10.1016/j.expneurol.2016.11.002
150. Liu Y, Schubert J, Sonnenberg L, Helbig KL, Hoei-Hansen CE, Koko M, et al. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain. (2019) 142:376–90. doi: 10.1093/brain/awy326
151. Boerma RS, Braun KP, van de Broek MP, van Berkestijn FM, Swinkels ME, Hagebeuk EO, et al. Remarkable phenytoin sensitivity in 4 children with SCN8A-related epilepsy: a molecular neuropharmacological approach. Neurotherapeutics. (2016) 13:192–7. doi: 10.1007/s13311-015-0372-8
152. Lin KM, Su G, Wang F, Zhang X, Wang Y, Ren J, et al. A de novo SCN8A heterozygous mutation in a child with epileptic encephalopathy: a case report. BMC Pediatrics. (2019) 19:1–6. doi: 10.1186/s12887-019-1796-9
153. Meisler MH. SCN8A encephalopathy: mechanisms and models. Epilepsia. (2019) 60:S86–91. doi: 10.1111/epi.14703
154. Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel SCN8A is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. (2007) 16:2892–9. doi: 10.1093/hmg/ddm248
155. Toledo-Aral J, Moss B, He Z, Koszowski A, Whisenand T, Levinson S, et al. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci USA. (1997) 94:1527–32. doi: 10.1073/pnas.94.4.1527
156. Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci. (2004) 24:8232–6. doi: 10.1523/JNEUROSCI.2695-04.2004
157. Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. (2004) 41:171–4. doi: 10.1136/jmg.2003.012153
158. Doty CN. SCN9A: another sodium channel excited to play a role in human epilepsies. Clin Genet. (2010) 77:326–8. doi: 10.1111/j.1399-0004.2009.01366_1.x
159. Zhang T, Chen M, Zhu A, Zhang X, Fang T. Novel mutation of SCN9A gene causing generalized epilepsy with febrile seizures plus in a Chinese family. Neurol Sci. (2020) 2020:1–5. doi: 10.1007/s10072-020-04284-x
160. Kasperaviciute D, Catarino C, Matarin M, Leu C, Novy J, Tostevin A, et al. Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain. (2013)136:3140–50. doi: 10.1093/brain/awt233
161. EPICURE Consortium, EMINet Consortium, Steffens M, Leu C, Ruppert A, Zara F, et al. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum Mol Genet. (2012) 21:5359–72. doi: 10.1093/hmg/dds373
162. International League Against Epilepsy Consortium on Complex Epilepsies. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol. (2014) 13:893–903. doi: 10.1016/S1474-4422(14)70171-1
163. Feenstra B, Pasternak B, Geller F, Carstensen L, Wang T, Huang F, et al. Common variants associated with general and MMR vaccine-related febrile seizures. Nat Genet. (2014) 46:1274–82. doi: 10.1038/ng.3129
164. International League Against Epilepsy Consortium on Complex Epilepsies. Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat Commun. (2018) 9:5269. doi: 10.1038/s41467-018-07524-z
165. Kasperaviciute D, Catarino C, Heinzen E, Depondt C, Cavalleri G, Caboclo L, et al. Common genetic variation and susceptibility to partial epilepsies: a genome-wide association study. Brain. (2010) 133:2136–47. doi: 10.1093/brain/awq130
166. Wijmenga C, Zhernakova A. The importance of cohort studies in the post-GWAS era. Nat Genet. (2018) 50:322–8 doi: 10.1038/s41588-018-0066-3
167. Esterhuizen AI, Carvill GL, Ramesar RS, Kariuki SM, Newton C, Poduri A, et al. Clinical application of epilepsy genetics in africa: is now the time? Front Neurol. (2018) 9:1–7. doi: 10.3389/fneur.2018.00276
168. Fodjo S, Nelson J, Remme JH, Preux PM, Colebunders R. Meta-analysis of epilepsy prevalence in West Africa and its relationship with onchocerciasis endemicity and control. Int Health. (2020) 12:192–202. doi: 10.1093/inthealth/ihaa012
169. Akinsulore A, Adewuya A. Psychosocial aspects of epilepsy in Nigeria: a review. African J Psychiatry. (2010) 13:351–6. doi: 10.4314/ajpsy.v13i5.63100
170. Mukuku O, Nawej P, Bugeme M, Nduu F, Mawaw PM, Luboya ON. Epidemiology of epilepsy in Lubumbashi, Democratic Republic of Congo. Neurol Res Int. (2020) 2020:5621461. doi: 10.1155/2020/5621461
171. Ba-Diop A, Marin B, Druet-Cabanac M, Ngoungou EB, Newton CR, Preux PM. Epidemiology, causes, and treatment of epilepsy in sub-Saharan Africa. Lancet Neurol. (2014) 13:1029–44. doi: 10.1016/S1474-4422(14)70114-0
Keywords: seizures, gain-of-functions, loss-of-functions, depolarization, VGSC
Citation: Ademuwagun IA, Rotimi SO, Syrbe S, Ajamma YU and Adebiyi E (2021) Voltage Gated Sodium Channel Genes in Epilepsy: Mutations, Functional Studies, and Treatment Dimensions. Front. Neurol. 12:600050. doi: 10.3389/fneur.2021.600050
Received: 28 August 2020; Accepted: 01 March 2021;
Published: 24 March 2021.
Edited by:
Mathias Toft, University of Oslo, NorwayReviewed by:
Manabu Funayama, Juntendo University, JapanFilippo M. Santorelli, Fondazione Stella Maris (IRCCS), Italy
Copyright © 2021 Ademuwagun, Rotimi, Syrbe, Ajamma and Adebiyi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ezekiel Adebiyi, ZXpla2llbC5hZGViaXlpQGNvdmVuYW50dW5pdmVyc2l0eS5lZHUubmc=