 Masato Kinboshi
Masato Kinboshi Akio Ikeda
Akio Ikeda Yukihiro Ohno
Yukihiro Ohno- 1Department of Pharmacology, Osaka University of Pharmaceutical Sciences, Takatsuki, Japan
- 2Department of Epilepsy, Movement Disorders and Physiology, Graduate School of Medicine, Kyoto University, Kyoto, Japan
Astrocytes regulate potassium and glutamate homeostasis via inwardly rectifying potassium (Kir) 4.1 channels in synapses, maintaining normal neural excitability. Numerous studies have shown that dysfunction of astrocytic Kir4.1 channels is involved in epileptogenesis in humans and animal models of epilepsy. Specifically, Kir4.1 channel inhibition by KCNJ10 gene mutation or expressional down-regulation increases the extracellular levels of potassium ions and glutamate in synapses and causes hyperexcitation of neurons. Moreover, recent investigations demonstrated that inhibition of Kir4.1 channels facilitates the expression of brain-derived neurotrophic factor (BDNF), an important modulator of epileptogenesis, in astrocytes. In this review, we summarize the current understanding on the role of astrocytic Kir4.1 channels in epileptogenesis, with a focus on functional and expressional changes in Kir4.1 channels and their regulation of BDNF secretion. We also discuss the potential of Kir4.1 channels as a therapeutic target for the prevention of epilepsy.
Introduction
Epilepsy is a common neurological disease that is characterized by recurrent seizures caused by neuronal hyperexcitation. The current antiepileptic agents predominantly act on neuronal ion channels (e.g., blockers of voltage-gated sodium channels and calcium channels), glutamate receptors [e.g., antagonists of a-amino-3-hydroxy-5-methyl-isoxazolopropionic acid (AMPA) receptors], or GABAergic inhibitory systems (e.g., modulators of the GABAA receptor/chloride channel complex and inhibitors of GABA transaminase), which aim to suppress excessive neural excitation (1, 2). Therapy with these antiepileptic drugs is effective in about 70% of epilepsy patients, whereas seizure control is not achieved for the remaining 30% of patients (3, 4). Thus, there is a high unmet need for novel therapeutic targets or agents to treat refractory epilepsy.
Numerous findings show that astrocytes, the major cell component of glial cells in the central nervous system (CNS), actively regulate the excitability and plasticity of neurons by forming tripartite synapses in conjunction with presynaptic and postsynaptic neural components (5–10). Specifically, astrocytes regulate ion homeostasis and extracellular space volume, metabolize neurotransmitters (e.g., glutamate, GABA, and glycine), and secrete various neuroactive molecules including gliotransmitters [e.g., glutamate, D-serine, adenosine 5′-triphosphate (ATP)], neurotrophic factors [e.g., brain-derived neurotrophic factor (BDNF) and glia-derived neurotrophic factor (GDNF)], and cytokines [e.g., tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β)] (11–13). Among these functions of astrocytes, a spatial buffering system for potassium ions (K+) plays an important role in the maintenance of neuronal excitability, which transports excessive extracellular K+ secreted from excited neurons to sites with lower K+ concentrations (e.g., microcapillaries) (14–18). This potassium clearance mechanism is primarily mediated by astrocytic inwardly rectifying potassium (Kir) channels containing Kir4.1 subunits (Kir4.1 channels) (16–21).
In this review, we introduce the current understanding regarding the pathophysiological role of astrocytic Kir4.1 channels in the development of epilepsy (epileptogenesis). Further, we discuss the potential of Kir4.1 channels as a therapeutic target for the prevention of epilepsy.
Spatial Potassium Buffering and Astrocytic Kir4.1 Channels
Extracellular K+ levels are critical for determining the resting membrane potential of neurons and are normally maintained at ca. 3–5 mM (22). Physiological neural activity leads to an elevation of <1 mM in extracellular K+ concentration (23). Increases of 10–12 mM K+ in ceiling levels are induced during excessive neural activity due to electrical stimulation (24). Astrocytes rapidly transport K+ from synapses, where K+ is secreted from neurons during the repolarization phase, to regions with lower K+ levels (e.g., microcapillaries) by coupling into a syncytium through gap junctions (Figure 1) (14–21, 25). This astrocytic K+ clearance mechanism, known as “spatial potassium buffering,” is vital for maintaining K+ homeostasis and preventing neural hyperexcitability during normal brain function. In addition, spatial potassium buffering is known to be linked to glutamate uptake via glutamate transporters [e.g., excitatory amino acid transporters 1 (EAAT1) and EAAT2] and water transport via aquaporin-4 (AQP4) by astrocytes (26–31). Moreover, both connexin30 and connexin43 in astrocytic gap junctions were shown to play a critical role in normal K+ redistribution, using double knockout techniques in mice (32, 33).
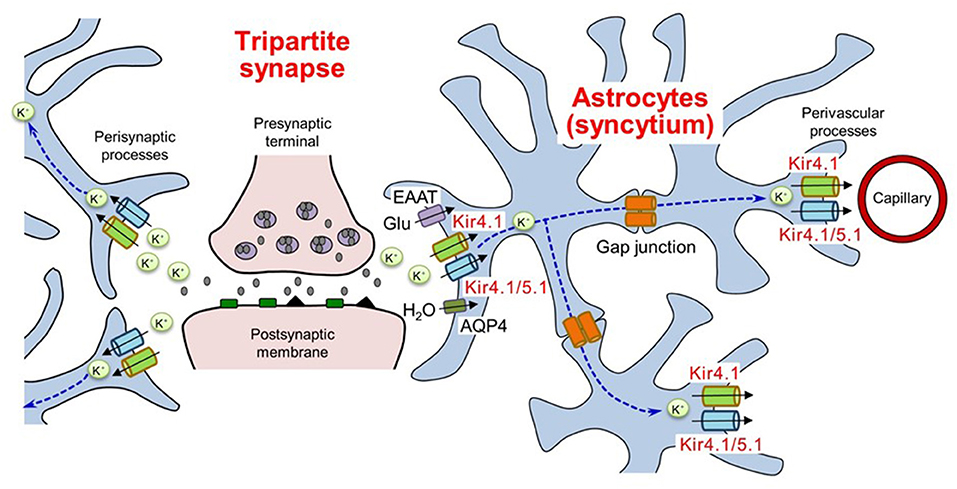
Figure 1. Spatial K+ buffering of astrocytes in tripartite synapses. Astrocytes uptake extracellular K+ secreted from neurons and release K+ in regions with lower K+ levels by coupling into a syncytium through gap junctions. The K+ buffering mechanism is corelated with glutamate uptake and water transport by astrocytes. EAAT, excitatory amino acid transporters; AQP4, aquaporin 4. Modified from Ohno et al. (25).
The influx of K+ into astrocytes is mainly mediated by Kir channels containing Kir4.1 and Kir5.1 subunits, which are highly expressed in astrocytes and retinal Müller cells (16–21, 34–39). Kir4.1 subunits have two transmembrane (TM) domains with an extracellular ion selectivity filter, including the GYG signature sequence, which construct Kir channels by forming tetramers (Figure 2) (25, 39). Two types of Kir4.1-containing Kir channels (Kir4.1 channels), the homo-tetramer of Kir4.1 and the hetero-tetramer of Kir4.1 and Kir5.1, conduct large inward K+ currents at potentials negative to K+ equilibrium potential (EK) and moderate outward K+ currents at those positive to EK (Figure 2) (25, 39, 40).
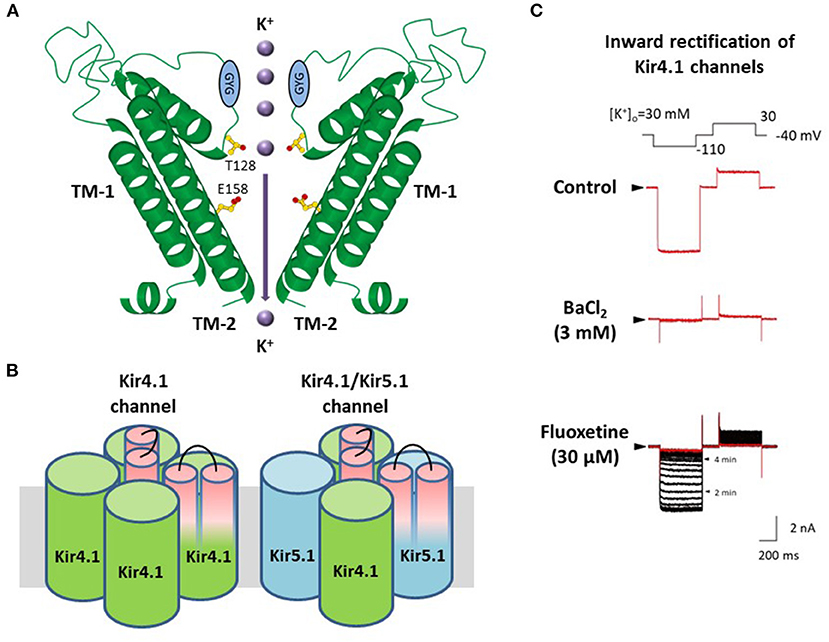
Figure 2. Molecular structure and properties of Kir4.1 channels. (A) Kir4.1 subunits have two transmembrane (TM) helices with one extracellular loop, including the GYG signature sequence of the K+ selectivity filter. (B) Kir4.1 subunits construct two types of channels, the homo-tetramer of Kir4.1 and the hetero-tetramer of Kir4.1 and Kir5.1. (C) Kir4.1 channels (homo-tetramer of Kir4.1) conduct large inward and relatively small outward K+ currents. Selective serotonin reuptake inhibitors, fluoxetine, inhibit Kir4.1 channel currents. Modified from Ohno et al. (25).
Pharmacological studies have shown that among CNS agents, several antidepressants reversibly inhibited K+ currents via Kir4.1 channels in a subunit-dependent manner. Tricyclic antidepressants (TCAs) such as nortriptyline, amitriptyline, desipramine, and imipramine, blocked Kir4.1 channels in a voltage-dependent manner, while selective serotonin reuptake inhibitors (SSRIs) including fluoxetine and sertraline inhibited Kir4.1 channels in a voltage-independent manner (Figure 2) (41–44). The inhibitory effects of antidepressants for Kir4.1 channels were achieved at concentrations considered to be within a range of brain concentrations for clinical treatment of depression. Antidepressant treatment is reported to increase the risk of seizure incidence (45, 46), which may be due to antidepressant drug actions on Kir4.1 channels.
Alanine-scanning mutagenesis studies on the antidepressant-Kir4.1 channel interaction demonstrated that these antidepressant agents specifically blocked the Kir4.1 channel pore (47). Two amino acids, T128 and E158, on pore and TM-2 helices respectively, can bind to antidepressants. Recently, anti-malarial agents such as quinacrine and chloroquine, and the anti-protozoal agent, pentamidine, have also been shown to inhibit Kir4.1 channels by binding to T128 and E158, similar to antidepressant agents (48–50). Although few reports are available on drug-Kir4.1 interaction, information about structure-based action on Kir4.1 channels is important for designing novel treatment compounds for epilepsy and reducing the potential of seizure side effects.
Kir4.1 Channels in Epilepsy Patients
Mutations in the human KCNJ10 gene encoding Kir4.1 were reported to cause the epileptic disorders known as “EAST” (Epilepsy, Ataxia, Sensorineural deafness and Tubulopathy) and “SeSAME” (Seizures, Sensorineural deafness, Ataxia, Mental retardation, and Electrolyte imbalance) syndrome (OMIM 612780) (51–53). Patients with EAST/SeSAME syndrome initially manifest generalized tonic-clonic seizures (GTCSs) within a few months after birth and are treated with anticonvulsant agents such as valproate and phenobarbital. The KCNJ10 mutations responsible for EAST/SeSAME syndrome have been shown to be T57I, R65P, R65C (cytoplasmic end of TM-1), F75L, F75C, G77R (TM-1), V91Gfs*197, F119Gfs*25, C140R (extracellular loop between TM-1 and TM-2), T164I, A167V (TM-2), R175Q, R199X, R240H, V259*, G275Vfs*7, and R297C (C-terminal domain) (51–64). These homozygous or compound heterozygous mutations disrupted Kir4.1 channel function to varying degrees from completely to moderately. Moreover, novel loss-of-function mutations (I60T, I60M, G163D, R171Q, A201T, I209T, and T290A) in KCNJ10 were identified in patients with atypical EAST/SeSAME syndrome lacking one or more core clinical manifestations (65–69). In contrast, heterozygous gain-of-function mutations (R18Q, V84M, and R348H) in KCNJ10 caused autism spectrum disorders with spastic seizures and intellectual disability (70–72).
Electrophysiological investigations demonstrated that Kir currents were significantly reduced in hippocampal specimens from refractory temporal lobe epilepsy (TLE) patients, using patch-clamp techniques (73–75). The impairment of glial K+ uptake sensitive to Ba2+, a blocker of Kir channels, was also in sclerotic hippocampal slices from patients with epilepsy (76, 77). Furthermore, astrocytic Kir4.1 expression has been shown to decrease in the sclerotic hippocampus of TLE patients (78–80). Additionally, flavoprotein fluorescence imaging, visualizing neuronal activities without exogenous dyes in living tissues taken from epilepsy patients, showed that epileptiform activities propagated from the subiculum of the hippocampus with sclerosis, where the Kir4.1 expression of astrocytes was markedly down-regulated (81). In refractory partial epilepsy pathologically diagnosed as “focal cortical dysplasia type 1,” Kir4.1 expression was decreased in the epileptogenic regions where direct current (DC) shifts were detected using wide-band electroencephalography (EEG) recordings (82). These DC shifts preceding conventional ictal pattern and high frequency oscillations (HFOs), known as “active DC shifts,” were suggested to reflect the extracellular K+ accumulation caused by the dysfunction of astrocytic potassium buffering, which can be EEG biomarkers for the epileptic zone (82). Therefore, Kir4.1 channel dysfunction affected by gene mutations or expressional down-regulation is likely to be involved in the pathogenesis of human epileptic disorders.
Kir4.1 Channels in Animal Epilepsy Models
Kir4.1 homozygous deletion in mice reduced body weight gain and caused progressive weakness by postnatal day (P) 8–10, although heterozygous mice showed no pathological behavior (83, 84). Subsequently, Kir4.1 knockout mice exhibited jerky movements and severe deficits in controlling voluntary movements, posture, and balance, and consequently died by P24. In addition, studies using conditional knockout techniques have reported that mice with conditional knockout of astrocytic Kir4.1 developed pronounced body tremor, ataxia, and stress-induced GTCSs, which were suggested to be involved in astrocytic membrane depolarization and impaired uptake of extracellular K+ following neural activity (Table 1) (30, 85, 86). Moreover, Kir4.1 conditional knockout also reduced glutamate uptake by astrocytes (30). This impairment of glutamate clearance resulted from the dysfunction of EAATs due to membrane depolarization in astrocytes (29–31, 87, 88).

Table 1. Pathophysiological changes in Kir4.1 channels in animal epilepsy models.
Numerous studies using animal models of epilepsy showed that astrocytic Kir4.1 expressional changes were involved in seizure induction and susceptibility. Specifically, Kir4.1 expression was significantly reduced in Noda epileptic rats (NER), a hereditary epilepsy model (Table 1) (89). NER exhibited frequent spontaneous GTCSs associated with two genetic loci, chromosome (Chr) 1q32-33 and Chr5q22, including cholecystokinin B receptor (Cckbr), suppressor of tumorigenicity 5 (St5), and PHD finger protein 24 (Phf24) (90–95). In NER, Kir4.1 expression was region-specifically reduced in the amygdala, where the expression of Fos protein, a biological marker of neural excitation, significantly elevated (89). Moreover, Leucine-Rich Glioma-Inactivated 1 (Lgi1) mutant rats, a model of human autosomal dominant lateral temporal lobe epilepsy (ADLTE), showed reduced astrocytic Kir4.1 expression in specific regions, including both the lateral and medial temporal lobes, after the acquisition of audiogenic seizure susceptibility (Table 1) (96). In these regions, neural hyperexcitation during seizures was confirmed using Fos immunohistochemical techniques (97). Auditory stimuli for seizure induction consisted of sound stimulation twice, priming stimulation at P16 and test stimulation at 8 weeks. Priming stimulation induces epileptogenesis caused by Lgi1 mutation without spontaneous seizure phenotypes (96, 98). Interestingly, the Kir4.1 expression in astrocytes was reduced during the time-course of epileptogenesis before application of test stimulation at the age of 8 weeks in Lgi1 mutant rats (96). These findings indicate that the dysfunction of Kir4.1 channels is involved not only in evoking seizure generation, but also in chronic development of epilepsy (epileptogenesis).
Furthermore, the Kcnj10 single nucleotide polymorphism (SNP) with T262S variation that disrupts Kir4.1 channel activity has been identified as the mutation responsible for seizure susceptibility of DBA/2 mice (Table 1) (99, 100). A rodent epilepsy model induced by fluid percussion injury or albumin injection also exhibited down-regulation of Kir4.1 expression in regions related to seizure foci (Table 1) (101, 102). Kir4.1 expression was transiently reduced after status epilepticus (SE) in temporal lobe epilepsy (TLE) models induced by electrical stimulation, although the expression of Kir4.1 returned to the normal level 1 week after SE (Table 1) (103). In contrast to epilepsy models with convulsive seizures, no changes in Kir4.1 expression were detected in Groggy rats, an absence epilepsy model (Table 1) (104). In addition, hyperglycemia has been reported to reduce Kir4.1 expression and disrupt the clearance of both K+ and glutamate using astrocyte primary cultures (105). Type 2 diabetic mice (db/db) also showed down-regulation of Kir4.1 expression and dysfunction in K+ intake that were associated with hippocampal neural hyperexcitability (Table 1) (106). These studies may explain the epileptic predisposition of type 2 diabetes patients (107, 108).
Astrocytic Kir4.1-BDNF System in Epileptogenesis
BDNF is a member of the neurotrophin family essential for the normal development and function of the CNS. Specifically, BDNF regulates cell survival, neurogenesis, neuronal sprouting, synaptic plasticity, and reactive gliosis by binding to tropomyosin-related kinase (Trk) receptors, especially TrkB receptors (109–112). The neurotrophic properties of BDNF potentially produce therapeutic effects for neurodegenerative diseases (e.g., Alzheimer's disease, Parkinson's disease, and Huntington's disease) and neuropsychiatric diseases (e.g., depression and bipolar disorder) (113–119). However, elevated expression of BDNF is known to be involved in the pathogenesis of epilepsy in various animal models and human brains (111, 120, 121). In addition, inhibition of BDNF/TrkB signaling has been shown to suppress the development of epilepsy in animal models (122–126).
While the expressional levels of BDNF were higher in neurons, astrocytic BDNF expression and BDNF/TrkB signaling have also been shown to contribute to brain functions under physiological and pathophysiological conditions (127–130). A recent study demonstrated that BDNF overexpression in astrocytes caused neuronal hyperexcitability and cell death, and deteriorated the phenotypes in lithium pilocarpine-induced TLE models, which were suggested to be mediated by astrocytic TrkB receptors, rather than neural TrkB receptors (131).
Astrocytic Kir4.1 channels have been shown to modulate BDNF expression using astrocyte primary cultures (25, 44, 132). Several antidepressant agents (e.g., imipramine and amitriptyline), which reportedly inhibited Kir4.1 channels in a subunit-specific manner (41, 43, 47), facilitated the expression of BDNF in astrocytes (133–135). Furthermore, the relative potencies of antidepressant agents for BDNF induction were consistent with those for the blockade of Kir4.1 channels, but not for the inhibition of 5-HT reuptake (43, 44). In addition, Kir4.1 knockdown by small interfering RNA (siRNA) transfection significantly elevated BDNF expression in astrocytes, which was suppressed by a MEK1/2 inhibitor, but not by a p38 MAPK inhibitor or a JNK inhibitor (44). These results suggest that the reduced function of Kir4.1 channels facilitates BDNF expression in astrocytes by activating the Ras/Raf/MEK/ERK pathway (Figure 3) (25, 44, 132). This hypothesis was supported by previous studies showing that the Ras/Raf/MEK/ERK signaling pathway regulates the transcription of BDNF and other survival/plasticity genes through interaction with cyclic AMP response element binding protein (CREB) (136, 137). It is therefore likely that Kir4.1 channels play a key role in modulating epileptogenesis by controlling not only the extracellular K+ and glutamate levels in synapses, but also the BDNF expression in astrocytes. The astrocytic Kir4.1-BDNF system is expected to serve as a novel target for the treatment of epilepsy, especially epileptogenesis.
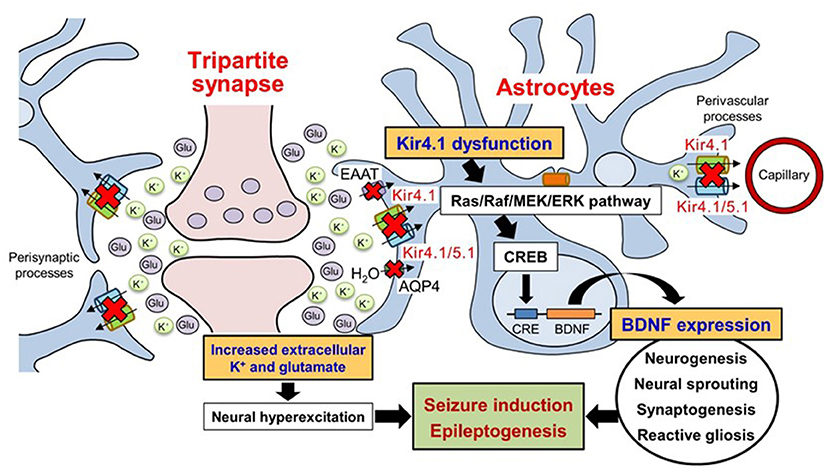
Figure 3. Schematic drawing illustrating the effects of Kir4.1 dysfunction on neural hyperexcitation and astrocytic BDNF expression. Dysfunction (genetic mutation, down-regulated expression, and pharmacological blockade) of Kir4.1 channels increases extracellular K+ and glutamate at synapses and causes neural hyperexcitability. The dysfunction of Kir4.1 channels activates the Ras/Raf/MEK/ERK signaling pathway and facilitates BDNF expression in astrocytes. Based on these changes, astrocytic Kir4.1 channels play important roles in modulating seizure induction and epileptogenesis. Modified from Ohno et al. (25).
Kir4.1 Channels as a Novel Therapeutic Target for Prevention of Epilepsy
Based on the potential role of astrocytic Kir4.1 channels in epileptogenesis, normalizing the down-regulation of astrocytic Kir4.1 channel expression during epileptogenesis can be a therapeutic strategy to prevent epilepsy. We recently showed that repeated treatments with antiepileptic drugs (valproate, phenytoin, and phenobarbital), which are effective for convulsive seizures, commonly elevated the astrocytic Kir4.1 expression in the limbic region (138). These antiepileptic drugs have previously showed inhibitory effects on kindling development in animal models (139–141), in which the elevated expression of Kir4.1 channels may contribute to the prophylactic effect of these drugs. Moreover, we have shown that valproate prevented audiogenic epileptogenesis in Lgi1 mutant rats by elevating the down-regulated Kir4.1 expression during epileptogenesis in a dose-dependent manner (96). Although further studies are required to reveal the mechanisms underlying the Kir4.1 pathogenic changes in epileptogenesis, these findings support the notion that astrocytic Kir4.1 channels can be therapeutic targets for prevention of epilepsy. Specifically, novel compounds positively modulating astrocytic Kir4.1 channels are expected to have potential for treatment of epilepsy and epileptogenesis. Although no information on the structure-activity relationship for Kir4.1 channel stimulators is available, gain-of-function mutations of the KCNJ10 gene (e.g., R18Q in N-terminus and V84M in TM1 region) reported in patients with autism spectrum disorders may give hints for new drug discovery (70, 72). In addition, retigabine (ezogabine), an antiepileptic drug for focal onset seizures, may also give information since it primarily acts on neural KCNQ2-5 (Kv7.2-7.5) ion channels as a positive allosteric modulator (142).
Conclusion
Astrocytic Kir4.1 channels play a critical role in the regulation of brain homeostasis and neural excitability. Evidence is accumulating that dysfunction of astrocytic Kir4.1 channels is involved in epileptogenesis in both epilepsy patients and animal epilepsy models. Moreover, the reduced activity of Kir4.1 channels elevates the levels of extracellular K+ and glutamate at tripartite synapses and facilitates astrocytic BDNF expression, which can promote the development of epilepsy. Although data are limited, the approach to restore Kir4.1 down-regulation during epileptogenesis was actually effective to prevent the development of epilepsy in an animal model of epilepsy. Thus, the Kir4.1-BDNF system in astrocytes is expected to serve as a novel therapeutic target for epilepsy. especially epileptogenesis.
Author Contributions
MK, AI, and YO designed and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (YO and AI: 19H03574, AI: 15H05874).
Conflict of Interest
AI and MK belong to Department of Epilepsy, Movement Disorders and Physiology at Kyoto University is Industry-Academia Collaboration Courses, supported by Eisai Co., Ltd., Nihon Kohden Corporation, Otsuka Pharmaceutical Co., and UCB Japan Co., Ltd.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. (2007) 4:18–61. doi: 10.1016/j.nurt.2006.11.010
2. Lasoń W, Chlebicka M, Rejdak K. Research advances in basic mechanisms of seizures and antiepileptic drug action. Pharmacol Rep. (2013) 65:787–801. doi: 10.1016/S1734-1140(13)71060-0
3. Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. (2000) 342:314–9. doi: 10.1056/NEJM200002033420503
4. Kalilani L, Sun X, Pelgrims B, Noack-Rink M, Villanueva V. The epidemiology of drug-resistant epilepsy: a systematic review and meta-analysis. Epilepsia. (2018) 59:2179–93. doi: 10.1111/epi.14596
5. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. (1999) 22:208–15. doi: 10.1016/S0166-2236(98)01349-6
6. Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. (2007) 13:54–63. doi: 10.1016/j.molmed.2006.12.005
7. Halassa MM, Fellin T, Haydon PG. Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology. (2009) 57:343–6. doi: 10.1016/j.neuropharm.2009.06.031
8. Dityatev A, Rusakov DA. Molecular signals of plasticity at the tetrapartite synapse. Curr Opin Neurobiol. (2011) 21:353–9. doi: 10.1016/j.conb.2010.12.006
9. Pérez-Alvarez A, Araque A. Astrocyte-neuron interaction at tripartite synapses. Curr Drug Targets. (2013) 14:1220–4. doi: 10.2174/13894501113149990203
10. Hasan U, Singh SK. The astrocyte-neuron interface: an overview on molecular and cellular dynamics controlling formation and maintenance of the tripartite synapse. Methods Mol Biol. (2019) 1938:3–18. doi: 10.1007/978-1-4939-9068-9_1
11. Parpura V, Zorec R. Gliotransmission: exocytotic release from astrocytes. Brain Res Rev. (2010) 63:83–92. doi: 10.1016/j.brainresrev.2009.11.008
12. Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. (2013) 36:174–84. doi: 10.1016/j.tins.2012.11.008
13. Harada K, Kamiya T, Tsuboi T. Gliotransmitter release from astrocytes: functional, developmental, and pathological implications in the brain. Front Neurosci. (2016) 9:499. doi: 10.3389/fnins.2015.00499
14. Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. (2000) 36:291–300. doi: 10.1016/S0197-0186(99)00137-0
15. Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. (2004) 129:877–96. doi: 10.1016/j.neuroscience.2004.09.053
16. Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. (2004) 129:1045–56. doi: 10.1016/j.neuroscience.2004.06.008
17. Ohno Y, Tokudome K, Kunisawa N, Iha HA, Kinboshi M, Mukai T, et al. Role of astroglial Kir4.1 channels in the pathogenesis and treatment of epilepsy. Ther Targets Neurol Dis. (2015) 2:e476. doi: 10.14800/ttnd.476
18. Bellot-Saez A, Kékesi O, Morley JW, Buskila Y. Astrocytic modulation of neuronal excitability through K+ spatial buffering. Neurosci Biobehav Rev. (2017) 77:87–97. doi: 10.1016/j.neubiorev.2017.03.002
19. Neusch C, Papadopoulos N, Müller M, Maletzki I, Winter SM, Hirrlinger J, et al. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J Neurophysiol. (2006) 95:1843–52. doi: 10.1152/jn.00996.2005
20. Larsen BR, MacAulay N. Kir4.1-mediated spatial buffering of K+: experimental challenges in determination of its temporal and quantitative contribution to K+ clearance in the brain. Channels. (2014) 8:544–50. doi: 10.4161/19336950.2014.970448
21. Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, Olsen ML. The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. (2016) 132:1–21. doi: 10.1007/s00401-016-1553-1
22. Somjen GG. Extracellular potassium in the mammalian central nervous system. Annu Rev Physiol. (1979) 41:159–77. doi: 10.1146/annurev.ph.41.030179.001111
23. Syková E, Rothenberg S, Krekule I. Changes of extracellular potassium concentration during spontaneous activity in the mesencephalic reticular formation of the rat. Brain Res. (1974) 79:333–7. doi: 10.1016/0006-8993(74)90428-4
24. Heinemann U, Lux HD. Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res. (1977) 120:231–49. doi: 10.1016/0006-8993(77)90903-9
25. Ohno Y, Kinboshi M, Shimizu S. Inwardly rectifying potassium channel Kir4.1 as a novel modulator of BDNF expression in astrocytes. Int J Mol Sci. (2018) 19:3313. doi: 10.3390/ijms19113313
26. Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, et al. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. (1999) 26:47–54. doi: 10.1002/(SICI)1098-1136(199903)26:1<47::AID-GLIA5>3.0.CO;2-5
27. Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, et al. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci USA. (2003) 100:2106–11. doi: 10.1073/pnas.0437946100
28. Puwarawuttipanit W, Bragg AD, Frydenlund DS, Mylonakou MN, Nagelhus EA, Peters MF, et al. Differential effect of alpha-syntrophin knockout on aquaporin-4 and Kir4.1 expression in retinal macroglial cells in mice. Neuroscience. (2006) 137:165–75. doi: 10.1016/j.neuroscience.2005.08.051
29. Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, et al. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. (2007) 55:274–81. doi: 10.1002/glia.20455
30. Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. (2007) 27:11354–65. doi: 10.1523/JNEUROSCI.0723-07.2007
31. Olsen ML, Sontheimer H. Functional implications for Kir4.1 channels in glia biology: from K+ buffering to cell differentiation. J Neurochem. (2008) 107:589–601. doi: 10.1111/j.1471-4159.2008.05615.x
32. Pannasch U, Vargová L, Reingruber J, Ezan P, Holcman D, Giaume C, et al. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci USA. (2011) 108:8467–72. doi: 10.1073/pnas.1016650108
33. Walrave L, Vinken M, Leybaert L, Smolders I. Astrocytic connexin43 channels as candidate targets in epilepsy treatment. Biomolecules. (2020) 10:1578. doi: 10.3390/biom10111578
34. Ishii M, Horio Y, Tada Y, Hibino H, Inanobe A, Ito M, et al. Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Müller cell membrane: their regulation by insulin and laminin signals. J Neurosci. (1997) 17:7725–35. doi: 10.1523/JNEUROSCI.17-20-07725.1997
35. Poopalasundaram S, Knott C, Shamotienko OG, Foran PG, Dolly JO, Ghiani CA, et al. Glial heterogeneity in expression of the inwardly rectifying K+ channel, Kir4.1, in adult rat CNS. Glia. (2000) 30:362–72. doi: 10.1002/(SICI)1098-1136(200006)30:4<362::AID-GLIA50>3.0.CO;2-4
36. Ishii M, Fujita A, Iwai K, Kusaka S, Higashi K, Inanobe A, et al. Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina. Am J Physiol Cell Physiol. (2003) 285:C260–7. doi: 10.1152/ajpcell.00560.2002
37. Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J Biol Chem. (2004) 279:44065–73. doi: 10.1074/jbc.M405985200
38. Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, et al. International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev. (2005) 57:509–26. doi: 10.1124/pr.57.4.11
39. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. (2010) 90:291–366. doi: 10.1152/physrev.00021.2009
40. Tanemoto M, Kittaka N, Inanobe A, Kurachi Y. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J Physiol. (2000) 525:587–92. doi: 10.1111/j.1469-7793.2000.00587.x
41. Su S, Ohno Y, Lossin C, Hibino H, Inanobe A, Kurachi Y. Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J Pharmacol Exp Ther. (2007) 320:573–80. doi: 10.1124/jpet.106.112094
42. Ohno Y, Kurachi Y. Astroglial inwardly rectifying potassium channel Kir4.1 as a potential target for the novel antidepressant agents. In: Kaplan SP, editor. Drug Design Research Perspectives. New York, NY: Nova Science Publishers, Inc. (2007). p. 1–8.
43. Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res. (2007) 1178:44–51. doi: 10.1016/j.brainres.2007.08.018
44. Kinboshi M, Mukai T, Nagao Y, Matsuba Y, Tsuji Y, Tanaka S, et al. Inhibition of inwardly rectifying potassium (Kir) 4.1 channels facilitates brain-derived neurotrophic factor (BDNF) expression in astrocytes. Front Mol Neurosci. (2017) 10:408. doi: 10.3389/fnmol.2017.00408
45. Hill T, Coupland C, Morriss R, Arthur A, Moore M, Hippisley-Cox J. Antidepressant use and risk of epilepsy and seizures in people aged 20 to 64 years: cohort study using a primary care database. BMC Psychiatry. (2015) 15:315. doi: 10.1186/s12888-015-0701-9
46. Wang SM, Han C, Bahk WM, Lee SJ, Patkar AA, Masand PS, et al. Addressing the side effects of contemporary antidepressant drugs: a comprehensive review. Chonnam Med J. (2018) 54:101–12. doi: 10.4068/cmj.2018.54.2.101
47. Furutani K, Ohno Y, Inanobe A, Hibino H, Kurachi Y. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol Pharmacol. (2009) 75:1287–95. doi: 10.1124/mol.108.052936
48. Marmolejo-Murillo LG, Aréchiga-Figueroa IA, Moreno-Galindo EG, Navarro-Polanco RA, Rodríguez-Menchaca AA, Cui M, et al. Chloroquine blocks the Kir4.1 channels by an open-pore blocking mechanism. Eur J Pharmacol. (2017) 800:40–7. doi: 10.1016/j.ejphar.2017.02.024
49. Marmolejo-Murillo LG, Aréchiga-Figueroa IA, Cui M, Moreno-Galindo EG, Navarro-Polanco RA, Sánchez-Chapula JA, et al. Inhibition of Kir4.1 potassium channels by quinacrine. Brain Res. (2017) 1663:87–94. doi: 10.1016/j.brainres.2017.03.009
50. Aréchiga-Figueroa IA, Marmolejo-Murillo LG, Cui M, Delgado-Ramírez M, van der Heyden MAG, Sánchez-Chapula JA, et al. High-potency block of Kir4.1 channels by pentamidine: Molecular basis. Eur J Pharmacol. (2017) 815:56–63. doi: 10.1016/j.ejphar.2017.10.009
51. Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. (2009) 360:1960–70. doi: 10.1056/NEJMoa0810276
52. Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA. (2009) 106:5842–7. doi: 10.1073/pnas.0901749106
53. Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, et al. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci USA. (2010) 107:14490–5. doi: 10.1073/pnas.1003072107
54. Sala-Rabanal M, Kucheryavykh LY, Skatchkov SN, Eaton MJ, Nichols CG. Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1 (KCNJ10). J Biol Chem. (2010) 285:36040–8. doi: 10.1074/jbc.M110.163170
55. Tang X, Hang D, Sand A, Kofuji P. Variable loss of Kir4.1 channel function in SeSAME syndrome mutations. Biochem Biophys Res Commun. (2010) 399:537–41. doi: 10.1016/j.bbrc.2010.07.105
56. Williams DM, Lopes CM, Rosenhouse-Dantsker A, Connelly HL, Matavel A, O-Uchi J, et al. Molecular basis of decreased Kir4.1 function in SeSAME/EAST syndrome. J Am Soc Nephrol. (2010) 21:2117–29. doi: 10.1681/ASN.2009121227
57. Freudenthal B, Kulaveerasingam D, Lingappa L, Shah MA, Brueton L, Wassmer E, et al. KCNJ10 mutations disrupt function in patients with EAST syndrome. Nephron Physiol. (2011) 119:p40–8. doi: 10.1159/000330250
58. Scholl UI, Dave HB, Lu M, Farhi A, Nelson-Williams C, Listman JA, et al. SeSAME/EAST syndrome–phenotypic variability and delayed activity of the distal convoluted tubule. Pediatr Nephrol. (2012) 27:2081–90. doi: 10.1007/s00467-012-2219-4
59. Kara B, Ekici B, Ipekçi B, Aslanger AK, Scholl U. KCNJ10 gene mutation in an 8-year-old boy with seizures. Acta Neurol Belg. (2013) 113:75–7. doi: 10.1007/s13760-012-0113-2
60. Parrock S, Hussain S, Issler N, Differ AM, Lench N, Guarino S, et al. KCNJ10 mutations display differential sensitivity to heteromerisation with KCNJ16. Nephron Physiol. (2013) 123:7–14. doi: 10.1159/000356353
61. Abdelhadi O, Iancu D, Tekman M, Stanescu H, Bockenhauer D, Kleta R. Founder mutation in KCNJ10 in Pakistani patients with EAST syndrome. Mol Genet Genomic Med. (2016) 4:521–6. doi: 10.1002/mgg3.227
62. Papavasiliou A, Foska K, Ioannou J, Nagel M. Epilepsy, ataxia, sensorineural deafness, tubulopathy syndrome in a European child with KCNJ10 mutations: A case report. SAGE Open Med Case Rep. (2017) 5:1–6. doi: 10.1177/2050313X17723549
63. Severino M, Lualdi S, Fiorillo C, Striano P, De Toni T, Peluso S, et al. Unusual white matter involvement in EAST syndrome associated with novel KCNJ10 mutations. J Neurol. (2018) 265:1419–25. doi: 10.1007/s00415-018-8826-7
64. Celmina M, Micule I, Inashkina I, Audere M, Kuske S, Pereca J, et al. EAST/SeSAME syndrome: review of the literature and introduction of four new Latvian patients. Clin Genet. (2019) 95:63–78. doi: 10.1111/cge.13374
65. Al Dhaibani MA, El-Hattab AW, Holroyd KB, Orthmann-Murphy J, Larson VA, Siddiqui KA, et al. Novel mutation in the KCNJ10 gene in three siblings with seizures, ataxia and no electrolyte abnormalities. J Neurogenet. (2018) 32:1–5. doi: 10.1080/01677063.2017.1404057
66. Nicita F, Tasca G, Nardella M, Bellacchio E, Camponeschi I, Vasco G, et al. Novel homozygous KCNJ10 mutation in a patient with non-syndromic early-onset cerebellar ataxia. Cerebellum. (2018) 17:499–503. doi: 10.1007/s12311-018-0924-7
67. Nadella RK, Chellappa A, Subramaniam AG, More RP, Shetty S, Prakash S, et al. Identification and functional characterization of two novel mutations in KCNJ10 and PI4KB in SeSAME syndrome without electrolyte imbalance. Hum Genomics. (2019) 13:53. doi: 10.1186/s40246-019-0236-0
68. Zhang H, Zhu L, Wang F, Wang R, Hong Y, Chen Y, et al. Novel KCNJ10 compound heterozygous mutations causing EAST/SeSAME-like syndrome compromise potassium channel function. Front Genet. (2019) 10:912. doi: 10.3389/fgene.2019.00912
69. Morin M, Forst AL, Pérez-Torre P, Jiménez-Escrig A, Barca-Tierno V, García-Galloway E, et al. Novel mutations in the KCNJ10 gene associated to a distinctive ataxia, sensorineural hearing loss and spasticity clinical phenotype. Neurogenetics. (2020) 21:135–43. doi: 10.1007/s10048-020-00605-6
70. Sicca F, Imbrici P, D'Adamo MC, Moro F, Bonatti F, Brovedani P, et al. Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol Dis. (2011) 43:239–47. doi: 10.1016/j.nbd.2011.03.016
71. Guglielmi L, Servettini I, Caramia M, Catacuzzeno L, Franciolini F, D'Adamo MC, et al. Update on the implication of potassium channels in autism: K+ channelautism spectrum disorder. Front Cell Neurosci. (2015) 9:34. doi: 10.3389/fncel.2015.00034
72. Sicca F, Ambrosini E, Marchese M, Sforna L, Servettini I, Valvo G, et al. Gain-of-function defects of astrocytic Kir4.1 channels in children with autism spectrum disorders and epilepsy. Sci Rep. (2016) 6:34325. doi: 10.1038/srep34325
73. Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. (1998) 32:286–303. doi: 10.1016/S0920-1211(98)00059-X
74. Hinterkeuser S, Schröder W, Hager G, Seifert G, Blümcke I, Elger CE, et al. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. (2000) 12:2087–96. doi: 10.1046/j.1460-9568.2000.00104.x
75. Schröder W, Hinterkeuser S, Seifert G, Schramm J, Jabs R, Wilkin GP, et al. Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia. (2000) 41:S181–4. doi: 10.1111/j.1528-1157.2000.tb01578.x
76. Kivi A, Lehmann TN, Kovács R, Eilers A, Jauch R, Meencke HJ, et al. Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci. (2000) 12:2039–48. doi: 10.1046/j.1460-9568.2000.00103.x
77. Jauch R, Windmüller O, Lehmann TN, Heinemann U, Gabriel S. Effects of barium, furosemide, ouabaine and 4,4'-diisothiocyanatostilbene-2,2'-disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potassium concentration in hippocampal slices from rats and from patients with epilepsy. Brain Res. (2002) 925:18–27. doi: 10.1016/S0006-8993(01)03254-1
78. Das A, Wallace GC, Holmes C, McDowell ML, Smith JA, Marshall JD, et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience. (2012) 220:237–46. doi: 10.1016/j.neuroscience.2012.06.002
79. Heuser K, Eid T, Lauritzen F, Thoren AE, Vindedal GF, Taubøll E, et al. Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J Neuropathol Exp Neurol. (2012) 71:814–25. doi: 10.1097/NEN.0b013e318267b5af
80. Steinhäuser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia. (2012) 60:1192–202. doi: 10.1002/glia.22313
81. Kitaura H, Shirozu H, Masuda H, Fukuda M, Fujii Y, Kakita A. Pathophysiological characteristics associated with epileptogenesis in human hippocampal sclerosis. EBioMedicine. (2018) 29:38–46. doi: 10.1016/j.ebiom.2018.02.013
82. Ikeda A, Takeyama H, Bernard C, Nakatani M, Shimotake A, Daifu M, et al. Active direct current (DC) shifts and “Red slow”: two new concepts for seizure mechanisms and identification of the epileptogenic zone. Neurosci Res. (2020) 156:95–101. doi: 10.1016/j.neures.2020.01.014
83. Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. (2000) 20:5733–40. doi: 10.1523/JNEUROSCI.20-15-05733.2000
84. Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. (2001) 21:5429–38. doi: 10.1523/JNEUROSCI.21-15-05429.2001
85. Chever O, Djukic B, McCarthy KD, Amzica F. Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J Neurosci. (2010) 30:15769–77. doi: 10.1523/JNEUROSCI.2078-10.2010
86. Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, et al. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia. (2011) 59:1635–42. doi: 10.1002/glia.21205
87. Bay V, Butt AM. Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia. (2012) 60:651–60. doi: 10.1002/glia.22299
88. Frizzo ME. Can a selective serotonin reuptake inhibitor act as a glutamatergic modulator? Curr Ther Res Clin Exp. (2017) 87:9–12. doi: 10.1016/j.curtheres.2017.07.001
89. Harada Y, Nagao Y, Shimizu S, Serikawa T, Terada R, Fujimoto M, et al. Expressional analysis of inwardly rectifying Kir4.1 channels in Noda epileptic rat (NER). Brain Res. (2013) 1517:141–9. doi: 10.1016/j.brainres.2013.04.009
90. Noda A, Hashizume R, Maihara T, Tomizawa Y, Ito Y, Inoue M, et al. NER rat strain: a new type of genetic model in epilepsy research. Epilepsia. (1998) 39:99–107. doi: 10.1111/j.1528-1157.1998.tb01281.x
91. Maihara T, Noda A, Yamazoe H, Voigt B, Kitada K, Serikawa T. Chromosomal mapping of genes for epilepsy in NER: a rat strain with tonic-clonic seizures. Epilepsia. (2000) 41:941–9. doi: 10.1111/j.1528-1157.2000.tb00276.x
92. Hanaya R, Sasa M, Kiura Y, Ishihara K, Serikawa T, Kurisu K. Epileptiform burst discharges in hippocampal CA3 neurons of young but not mature Noda epileptic rats (NER). Brain Res. (2002) 950:317–20. doi: 10.1016/S0006-8993(02)03195-5
93. Jinde S, Masui A, Morinobu S, Noda A, Kato N. Differential changes in messenger RNA expressions and binding sites of neuropeptide Y Y1, Y2 and Y5 receptors in the hippocampus of an epileptic mutant rat: Noda epileptic rat. Neuroscience. (2002) 115:1035–45. doi: 10.1016/S0306-4522(02)00545-6
94. Kuramoto T, Voigt B, Nakanishi S, Kitada K, Nakamura T, Wakamatsu K, et al. Identification of candidate genes for generalized tonic-clonic seizures in Noda epileptic rat. Behav Genet. (2017) 47:609–19. doi: 10.1007/s10519-017-9870-2
95. Serikawa T, Kunisawa N, Shimizu S, Kato M, Iha HA, Kinboshi M, et al. Increased seizure sensitivity, emotional defects and cognitive impairment in PHD finger protein 24 (Phf24)-null rats. Behav Brain Res. (2019) 369:111922. doi: 10.1016/j.bbr.2019.111922
96. Kinboshi M, Shimizu S, Mashimo T, Serikawa T, Ito H, Ikeda A, et al. Down-regulation of astrocytic Kir4.1 channels during the audiogenic epileptogenesis in Leucine-Rich Glioma-Inactivated 1 (Lgi1) mutant rats. Int J Mol Sci. (2019) 20:1013. doi: 10.3390/ijms20051013
97. Fumoto N, Mashimo T, Masui A, Ishida S, Mizuguchi Y, Minamimoto S, et al. Evaluation of seizure foci and genes in the Lgi1L385R/+ mutant rat. Neurosci Res. (2014) 80:69–75. doi: 10.1016/j.neures.2013.12.008
98. Baulac S, Ishida S, Mashimo T, Boillot M, Fumoto N, Kuwamura M, et al. A rat model for LGI1-related epilepsies. Hum Mol Genet. (2012) 21:3546–57. doi: 10.1093/hmg/dds184
99. Ferraro TN, Golden GT, Smith GG, Martin JF, Lohoff FW, Gieringer TA, et al. Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm Genome. (2004) 15:239–51. doi: 10.1007/s00335-003-2270-3
100. Inyushin M, Kucheryavykh LY, Kucheryavykh YV, Nichols CG, Buono RJ, Ferraro TN, et al. Potassium channel activity and glutamate uptake are impaired in astrocytes of seizure-susceptible DBA/2 mice. Epilepsia. (2010) 51:1707–13. doi: 10.1111/j.1528-1167.2010.02592.x
101. Stewart TH, Eastman CL, Groblewski PA, Fender JS, Verley DR, Cook DG, et al. Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. J Neurophysiol. (2010) 104:3345–60. doi: 10.1152/jn.00398.2010
102. Frigerio F, Frasca A, Weissberg I, Parrella S, Friedman A, Vezzani A, et al. Long-lasting pro-ictogenic effects induced in vivo by rat brain exposure to serum albumin in the absence of concomitant pathology. Epilepsia. (2012) 53:1887–97. doi: 10.1111/j.1528-1167.2012.03666.x
103. Zurolo E, de Groot M, Iyer A, Anink J, van Vliet EA, Heimans JJ, et al. Regulation of Kir4.1 expression in astrocytes and astrocytic tumors: a role for interleukin-1 β. J Neuroinflammation. (2012) 9:280. doi: 10.1186/1742-2094-9-280
104. Harada Y, Nagao Y, Mukai T, Shimizu S, Tokudome K, Kunisawa N, et al. Expressional analysis of inwardly rectifying Kir4.1 channels in Groggy rats, a rat model of absence seizures. Arch Neurosci. (2014) 1:e18651. doi: 10.5812/archneurosci.18651
105. Rivera-Aponte DE, Méndez-González MP, Rivera-Pagán AF, Kucheryavykh YV, Kucheryavykh LY, Skatchkov SN, et al. Hyperglycemia reduces functional expression of astrocytic Kir4.1 channels and glial glutamate uptake. Neuroscience. (2015) 310:216–23. doi: 10.1016/j.neuroscience.2015.09.044
106. Méndez-González MP, Rivera-Aponte DE, Benedikt J, Maldonado-Martínez G, Tejeda-Bayron F, Skatchkov SN, et al. Downregulation of astrocytic Kir4.1 potassium channels is associated with hippocampal neuronal hyperexcitability in type 2 diabetic mice. Brain Sci. (2020) 10:72. doi: 10.3390/brainsci10020072
107. Yun C, Xuefeng W. Association between seizures and diabetes mellitus: a comprehensive review of literature. Curr Diabetes Rev. (2013) 9:350–54. doi: 10.2174/15733998113099990060
108. Lu CL, Chang YH, Sun Y, Li CY. A population-based study of epilepsy incidence in association with type 2 diabetes and severe hypoglycaemia. Diabetes Res Clin Pract. (2018) 140:97–106. doi: 10.1016/j.diabres.2018.03.020
109. Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. (2000) 23:639–45. doi: 10.1016/S0166-2236(00)01672-6
110. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. (2001) 24:677–736. doi: 10.1146/annurev.neuro.24.1.677
111. Jankowsky JL, Patterson PH. The role of cytokines and growth factors in seizures and their sequelae. Prog Neurobiol. (2001) 63:125–49. doi: 10.1016/S0301-0082(00)00022-8
112. Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. (2004) 22:123–31. doi: 10.1080/08977190410001723308
113. Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. (2001) 50:260–5. doi: 10.1016/S0006-3223(01)01083-6
114. Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol. (2001) 63:71–124. doi: 10.1016/S0301-0082(00)00014-9
115. Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. (2001) 293:493–8. doi: 10.1126/science.1059581
116. Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang DM. Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology. (2002) 43:1173–9. doi: 10.1016/S0028-3908(02)00217-4
117. Dias BG, Banerjee SB, Duman RS, Vaidya VA. Differential regulation of brain derived neurotrophic factor transcripts by antidepressant treatments in the adult rat brain. Neuropharmacology. (2003) 45:553–63. doi: 10.1016/S0028-3908(03)00198-9
118. Giralt A, Friedman HC, Caneda-Ferrón B, Urbán N, Moreno E, Rubio N, et al. BDNF regulation under GFAP promoter provides engineered astrocytes as a new approach for long-term protection in Huntington's disease. Gene Ther. (2010) 17:1294–308. doi: 10.1038/gt.2010.71
119. de Pins B, Cifuentes-Díaz C, Farah AT, López-Molina L, Montalban E, Sancho-Balsells A, et al. Conditional BDNF delivery from astrocytes rescues memory deficits, spine density, and synaptic properties in the 5xFAD mouse model of Alzheimer disease. J Neurosci. (2019) 39:2441–58. doi: 10.1523/JNEUROSCI.2121-18.2019
120. Hagihara H, Hara M, Tsunekawa K, Nakagawa Y, Sawada M, Nakano K. Tonic-clonic seizures induce division of neuronal progenitor cells with concomitant changes in expression of neurotrophic factors in the brain of pilocarpine-treated mice. Brain Res Mol Brain Res. (2005) 139:258–66. doi: 10.1016/j.molbrainres.2005.05.031
121. Jean YY, Lercher LD, Dreyfus CF. Glutamate elicits release of BDNF from basal forebrain astrocytes in a process dependent on metabotropic receptors and the PLC pathway. Neuron Glia Biol. (2008) 4:35–42. doi: 10.1017/S1740925X09000052
122. Binder DK, Routbort MJ, Ryan TE, Yancopoulos GD, McNamara JO. Selective inhibition of kindling development by intraventricular administration of TrkB receptor body. J Neurosci. (1999) 19:1424–36. doi: 10.1523/JNEUROSCI.19-04-01424.1999
123. Barton ME, Shannon HE. The seizure-related phenotype of brain-derived neurotrophic factor knockdown mice. Neuroscience. (2005) 136:563–9. doi: 10.1016/j.neuroscience.2005.08.008
124. Heinrich C, Lähteinen S, Suzuki F, Anne-Marie L, Huber S, Häussler U, et al. Increase in BDNF-mediated TrkB signaling promotes epileptogenesis in a mouse model of mesial temporal lobe epilepsy. Neurobiol Dis. (2011) 42:35–47. doi: 10.1016/j.nbd.2011.01.001
125. Grabenstatter HL, Russek SJ, Brooks-Kayal AR. Molecular pathways controlling inhibitory receptor expression. Epilepsia. (2012) 53:71–8. doi: 10.1111/epi.12036
126. Liu G, Gu B, He XP, Joshi RB, Wackerle HD, Rodriguiz RM, et al. Transient inhibition of TrkB kinase following status epilepticus prevents development of temporal lobe epilepsy. Neuron. (2013) 79:31–8. doi: 10.1016/j.neuron.2013.04.027
127. Zafra F, Lindholm D, Castrén E, Hartikka J, Thoenen H. Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J Neurosci. (1992) 12:4793–9. doi: 10.1523/JNEUROSCI.12-12-04793.1992
128. Miklic S, Juric DM, Carman-Krzan M. Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int J Dev Neurosci. (2004) 22:119–30. doi: 10.1016/j.ijdevneu.2004.03.001
129. Saha RN, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol. (2006) 1:212–22. doi: 10.1007/s11481-006-9020-8
130. Holt LM, Hernandez RD, Pacheco NL, Torres Ceja B, Hossain M, Olsen ML. Astrocyte morphogenesis is dependent on BDNF signaling via astrocytic TrkB.T1. Elife. (2019) 8:e44667. doi: 10.7554/eLife.44667
131. Fernández-García S, Sancho-Balsells A, Longueville S, Hervé D, Gruart A, Delgado-García JM, et al. Astrocytic BDNF and TrkB regulate severity and neuronal activity in mouse models of temporal lobe epilepsy. Cell Death Dis. (2020) 11:411. doi: 10.1038/s41419-020-2615-9
132. Ohno Y. Astrocytic Kir4.1 potassium channels as a novel therapeutic target for epilepsy and mood disorders. Neural Regen Res. (2018) 13:651–2. doi: 10.4103/1673-5374.230355
133. Takano K, Yamasaki H, Kawabe K, Moriyama M, Nakamura Y. Imipramine induces brain-derived neurotrophic factor mRNA expression in cultured astrocytes. J Pharmacol Sci. (2012) 120:176–86. doi: 10.1254/jphs.12039FP
134. Boku S, Hisaoka-Nakashima K, Nakagawa S, Kato A, Kajitani N, Inoue T, et al. Tricyclic antidepressant amitriptyline indirectly increases the proliferation of adult dentate gyrus-derived neural precursors: an involvement of astrocytes. PLoS ONE. (2013) 8:e79371. doi: 10.1371/journal.pone.0079371
135. Hisaoka-Nakashima K, Kajitani N, Kaneko M, Shigetou T, Kasai M, Matsumoto C, et al. Amitriptyline induces brain-derived neurotrophic factor (BDNF) mRNA expression through ERK-dependent modulation of multiple BDNF mRNA variants in primary cultured rat cortical astrocytes and microglia. Brain Res. (2016) 1634:57–67. doi: 10.1016/j.brainres.2015.12.057
136. Curtis J, Finkbeiner S. Sending signals from the synapse to the nucleus: possible roles for CaMK, Ras/ERK, and SAPK pathways in the regulation of synaptic plasticity and neuronal growth. J Neurosci Res. (1999) 58:88–95. doi: 10.1002/(SICI)1097-4547(19991001)58:1<88::AID-JNR9>3.0.CO;2-R
137. Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. (2012) 35:47–56. doi: 10.1016/j.tins.2011.11.004
138. Mukai T, Kinboshi M, Nagao Y, Shimizu S, Ono A, Sakagami Y, et al. Antiepileptic drugs elevate astrocytic Kir4.1 expression in the rat limbic region. Front Pharmacol. (2018) 9:845. doi: 10.3389/fphar.2018.00845
139. Silver JM, Shin C, McNamara JO. Antiepileptogenic effects of conventional anticonvulsants in the kindling model of epilepsy. Ann. Neurol. (1991) 29:356–63. doi: 10.1002/ana.410290404
140. Sasa M. A new frontier in epilepsy: novel antiepileptogenic drugs. J Pharmacol Sci. (2006) 100:487–94. doi: 10.1254/jphs.CPJ06010X
141. Löscher W, Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol Rev. (2010) 62:668–700. doi: 10.1124/pr.110.003046
Keywords: astrocytes, Kir4.1 channels, spatial potassium buffering, epilepsy, BDNF, glutamate, tripartite synapse
Citation: Kinboshi M, Ikeda A and Ohno Y (2020) Role of Astrocytic Inwardly Rectifying Potassium (Kir) 4.1 Channels in Epileptogenesis. Front. Neurol. 11:626658. doi: 10.3389/fneur.2020.626658
Received: 06 November 2020; Accepted: 08 December 2020;
Published: 23 December 2020.
Edited by:
Christian Steinhaeuser, Universität Bonn, GermanyReviewed by:
Devin K. Binder, University of California, Riverside, United StatesGerald Seifert, University Hospital Bonn, Germany
Copyright © 2020 Kinboshi, Ikeda and Ohno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukihiro Ohno, eW9obm9AZ2x5Lm91cHMuYWMuanA=