
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol., 21 January 2021
Sec. Pediatric Neurology
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.625428
This article is part of the Research TopicCerebral Palsy: New Developments View all 17 articles
 Sara A. Lewis1,2
Sara A. Lewis1,2 Sheetal Shetty1,2Bryce A. Wilson1,2
Sheetal Shetty1,2Bryce A. Wilson1,2 Aris J. Huang3
Aris J. Huang3 Sheng Chih Jin4Hayley Smithers-Sheedy5
Sheng Chih Jin4Hayley Smithers-Sheedy5 Michael C. Fahey6
Michael C. Fahey6 Michael C. Kruer1,2,3*
Michael C. Kruer1,2,3*Cohort-based whole exome and whole genome sequencing and copy number variant (CNV) studies have identified genetic etiologies for a sizable proportion of patients with cerebral palsy (CP). These findings indicate that genetic mutations collectively comprise an important cause of CP. We review findings in CP genomics and propose criteria for CP-associated genes at the level of gene discovery, research study, and clinical application. We review the published literature and report 18 genes and 5 CNVs from genomics studies with strong evidence of for the pathophysiology of CP. CP-associated genes often disrupt early brain developmental programming or predispose individuals to known environmental risk factors. We discuss the overlap of CP-associated genes with other neurodevelopmental disorders and related movement disorders. We revisit diagnostic criteria for CP and discuss how identification of genetic etiologies does not preclude CP as an appropriate diagnosis. The identification of genetic etiologies improves our understanding of the neurobiology of CP, providing opportunities to study CP pathogenesis and develop mechanism-based interventions.
Cerebral palsy (CP) describes a disorder of motor function resulting from maldevelopment or injury to the developing brain. The motor disorders of CP are frequently accompanied by other associated impairments including intellectual disability, epilepsy and sensory impairments (1). CP is relatively common with an estimated prevalence of 1.3–1.9 cases per 1,000 live births in high income countries (2, 3). Whilst often described as a childhood disorder, CP is a lifelong condition. Although many risk factors have been recognized, for many individuals identifying the etiology of their CP can be difficult.
One example of the complexity of CP etiology is preterm birth. Babies born preterm have an increased risk of CP. However, not all babies born preterm go on to develop CP. Being born preterm puts an infant at-risk for additional complications, including respiratory injury (hypoxia, hypercarbia, or hyperoxia), infectious/inflammatory insults (sepsis or necrotizing enterocolitis), hemodynamic compromise (hypotension, intraventricular hemorrhage, or thrombosis). These factors can contribute to cell death in the brain or alter the maturation of neurons and glia, resulting in abnormal white matter tracts (4) diminished cerebral volumes (5), or cerebellar hypoplasia (6). Further, genetic factors contribute to risk for preterm birth (7). Together this indicates the pathophysiology of CP may result from a combination of genetic, environmental, and genetic factors. Other CP risk factors, such as congenital anomalies or intrauterine growth restriction, may also reflect underlying genetic etiologies (8–11). Recognizing how genetic changes contribute to CP is a rapidly developing field with important themes.
Recent studies have implicated genetic factors as contributors or causes of CP, including single nucleotide variants (SNVs) and genomic copy number variants (CNVs). Putatively deleterious CNVs have been found in several CP cohorts, although estimates of molecular etiologic yield have varied considerably depending on study criteria (12–16). The initial study of 50 unselected CP cases using microarrays implicated novel variants in CP (12). Segel et al. (13) used microarrays to study an enriched cohort of 52 individuals with cryptogenic CP (i.e., no known cause). By applying American College of Medical Genetics & Genomics (ACMG) criteria to interpret their results, they identified 31% with pathogenic or likely pathogenic CNVs. Oskoui et al. (14) studied an unselected cohort of 147 individuals with CP using microarrays and evaluated detected CNVs using rigorous criteria based on ACMG guidelines. They found evidence for pathogenic or likely pathogenic CNVs in 9.6% of their cases. Zarrei et al. (15) assessed pathogenic or likely pathogenic CNVs in 97 individuals with otherwise unselected hemiplegic CP. When focusing on rare, de novo CNVs, they identified a molecular diagnostic yield of 7.2%. When they expanded their criteria to include inherited CNVs, known DECIPHER or ClinGen loci or CNVs that encompass genes known to play a role in NDD or brain/muscle disease, the potential yield rose to 23.7%. Finally, Corbett et al. (16) assessed CNV determination from whole exome data, finding an additional 3.7% of cases could be considered “solved” when CNVs are considered alongside SNVs.
In these studies, recurrent likely pathogenic CNVs were identified, such as a 2p25.3 deletion, 22q11.2 deletions and duplications, and Xp monosomy (Table 2). The relationship between a genetic deletion/duplication and CP can be complex. For example, 22q11.2 deletions can be associated with major congenital heart defects and polymicrogyria, with the cardiac abnormalities predisposing individuals to stroke and perioperative complications. Alternatively, 22q11.2 deletions are associated with dystonia in the absence of cardiac anomalies (17, 18). In addition, efforts to identify pathogenic CNVs are limited by clinical variability and microdeletion/microduplication boundaries that only partially overlap. Continued analysis and reporting of CNV findings alongside massively parallel SNV analyses are needed to expand knowledge of CNVs that can lead to CP.
The foundational study conducted by McMichael et al. (10) sequenced 98 unselected CP trios and found that six (6%) had a de novo (4 cases) or inherited (2 cases) predicted deleterious variant in a known disease-associated gene. Eight (8%) harbored a potentially pathogenic variant in a novel candidate CP gene. Six of these were de novo and two were inherited. A small study conducted by Schnekenberg et al. (19) found de novo mutations by WES in 3 out of 4 cases with ataxic CP. Subsequently, Takezawa et al. (20) whole exome sequenced 17 CP trios. These cases represented full-term births without diagnostic MRI findings. Nine of the 17 (53%) cases had pathogenic or likely pathogenic variants in known disease-associated genes by ACMG criteria.
Although these initial studies laid the foundation for the field, there were challenges in reconciling findings in candidate genes with known Online Mendelian Inheritance in Man (OMIM; https://www.omim.org) genes. Limitations of these studies included small sample sizes and a lack of robust statistical methods and well-matched controls. An international collaborative publication (21) addressed several of these limitations, reporting the largest trio-based cohort to date using a rigorously vetted statistical approach supported by mechanistic and biological validations.
Advanced paternal age had previously been associated with risk for CP (22) and is known to be associated with de novo mutations. De novo mutations were found to be enriched 1.2-fold in CP cases compared to the expected value estimated from background germline mutation rate (21, 23). Models predict between 27 and 124 genes likely contribute to CP through a de novo mechanism (21), with the number of identified genes therefore anticipated to rise exponentially with the number of trios sequenced. Gene discovery for CP is a growing area of interest with a recent literature search reporting 57 published genetic studies on individuals described as having CP. However, many of these reports are case studies and some included cases which do not meet current consensus criteria for CP (24), complicating interpretation.
With the increase in our knowledge of the role that genetics play in CP, distinctions between genetic and environmental/acquired etiologies are blurring and mixed and multiple insults are increasingly recognized. In some cases, genetic risk factors predispose individuals to environmental insults. Such a predisposition has been described for hemorrhagic stroke due to COL4A1 and COL4A2 mutations (25) and hypotonia before birth causing delivery complications and perinatal asphyxia, exemplified by 1p36 deletion syndrome (26). Differences in genetic backgrounds, such as single nucleotide polymorphisms in glutamate transport (27) and COX-1/2 receptors (28), might also underly susceptibility to CP after exposure to environmental risk factors (29).
The long-recognized increased risk of CP for males (30) also provides evidence for complex interactions between genetic contributions, predisposition, and responses to brain injury. For instance, female rats have more dystonia-like outcomes, and males have more spasticity-like outcomes after the same hypoxic-ischemic injury (31). Together, this suggests differences in genetics and subsequent physiology likely play a role in determining the development of CP in individuals exposed to environmental risk factors. Despite the considerable advances that have been made, the relative contribution of environmental vs. genetic causal factors remains unclear.
Using careful case ascertainment to ensure sequenced individuals have CP is crucial for interpretation of genetic findings as either consistent with CP or not (24). Some cohorts may include individuals with progressive neurological impairments who do not fulfill the criteria for CP (32). Although these findings are important in their own right, genetic findings from individuals with rare pediatric movement disorders that do not meet the diagnostic criteria for CP should not be included in lists of CP-associated genes. Full reporting of the age of onset, movement disorder subtype, the presence or absence of neurodegeneration (including if there was follow-up over time and age of the last contact) and comorbidities will be necessary for the interpretation of the pathogenicity of variants in future cases presenting with mutations in those genes.
Determining whether a genetic variant or group of variants accounts for an individual's phenotype can be difficult without subsequent laboratory-based validation studies delineating the variant's effect on RNA or protein function. In some instances, the detection of multiple individuals in a CP cohort with predicted damaging variants in the same gene can provide strong statistical evidence for a bona fide association with CP if enrichment is present and consistent inheritance patterns are evident. A few CP-associated genes have clear loss-of-function variants, such as stop gain and frameshift variants in CTNNB1 (21). Splice site variants can be challenging to interpret, but have been confirmed to affect mRNA sequence for AMPD2 and CTNNB1 as well as protein function in the case of AMPD2 (20). However, most variants are missense mutations, and when bioinformatically predicted deleterious de novo missense mutations are systematically tested, many do not change protein function (33), highlighting the importance of experimental validation. De novo variants also can also contribute through gain of function or even change of function effects. For example, FBXO31 and RHOB variants did not demonstrate loss of function deficits. However, when studied in more detail, gain/change of function mechanisms were identified (21). Given that many genes can have multiple functions, diverse localizations and myriad interactions, defects can be hard to detect in a single assay, making it more challenging to rule out candidate genes as well. TUBA1A missense variants, for example, can have subtle effects on microtubule shape and protein interactions, which may contribute to considerable phenotype heterogeneity (34). Even in some cases where genes have been shown to have an essential role in movement in model organism studies using zebrafish or Drosophila, the human variants were not tested to confirm variant-specific effects (21, 35).
The sheer number of genes that are being implicated in CP translates into a small but growing number of recurrent genes with a limited number of affected patients harboring variants in these genes. Larger cohorts with cases and controls utilizing methods to detect statistical enrichment of genes with deleterious variants are needed to overcome the recurrence gap. Gene matching platforms such as GeneMatcher (36) are going to be crucial for finding other patients with these rare genetic variants with clinical concordance. Current approaches to overcome limitations in identifying recurrent genes include using pathway enrichment and disease gene overlap to identify genes with a higher probability of contributing to CP pathology.
The criteria for identifying “a CP-associated gene” may vary based on the context prompting the question. Determining whether genetic variants explain an individual's clinical condition requires more stringent criteria than may be required for studying genetic networks contributing to the disease through multifactorial mechanisms. We propose a list of criteria for evaluating the level of evidence of CP genetic findings through the process of gene discovery, laboratory study, and finally, clinical application (Table 1). We hope that these proposed criteria will facilitate ongoing dialogue in the field amongst clinicians, basic scientists, and genomic researchers.
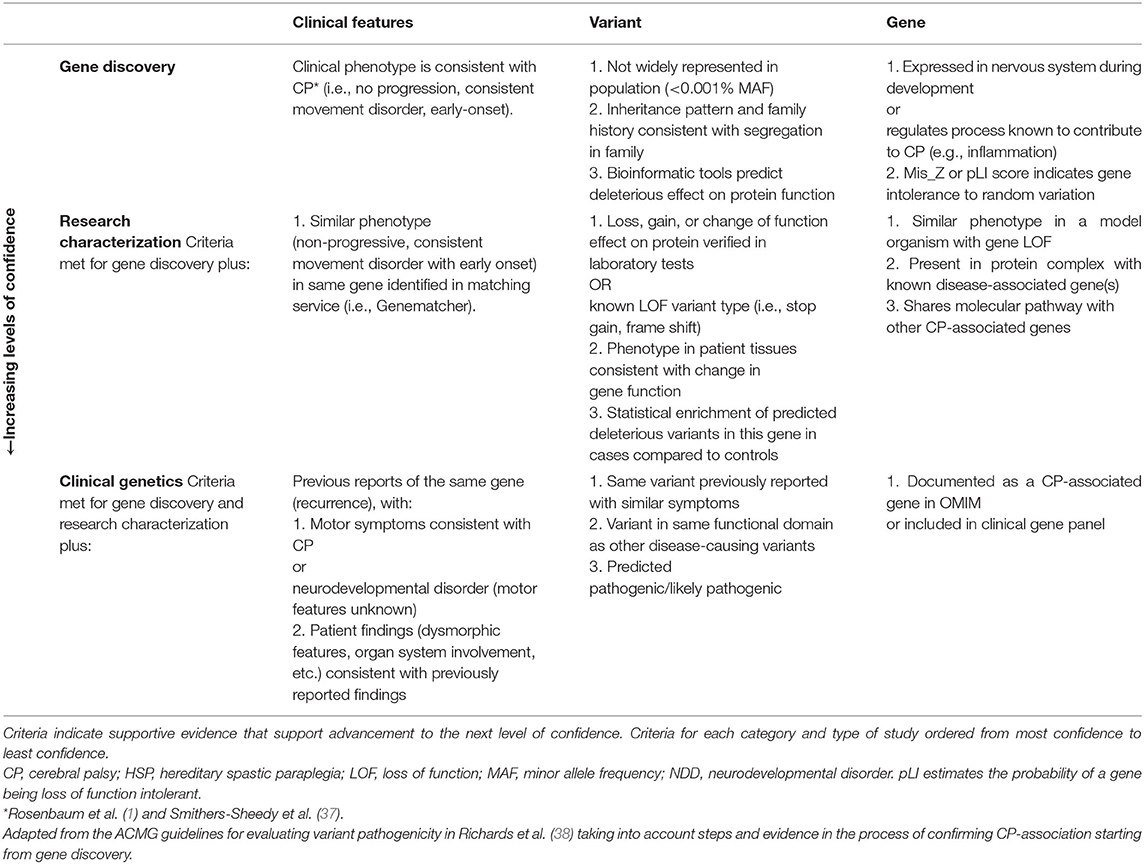
Table 1. Proposed criteria for prioritizing potential CP-associated genes.
We have also reviewed the extant literature for cohort-based next-generation sequencing of CP. Our search strategies were expanded to include patients from cohorts of pediatric movement disorders with non-progressive spasticity, dystonia, ataxia, or chorea that meet CP diagnostic criteria (1) with WES sequencing since 2015. We curated 127 gene variants from WES cohort studies and 32 CNVs from array and WES studies (Supplemental Table 1). To determine which genes have strong evidence for causing CP, we identified genes that meet ≥1 criteria from all sections of Table 1. We found 18 genes and 5 CNVs meeting these criteria that have been described in 2 or more patients (Table 2).
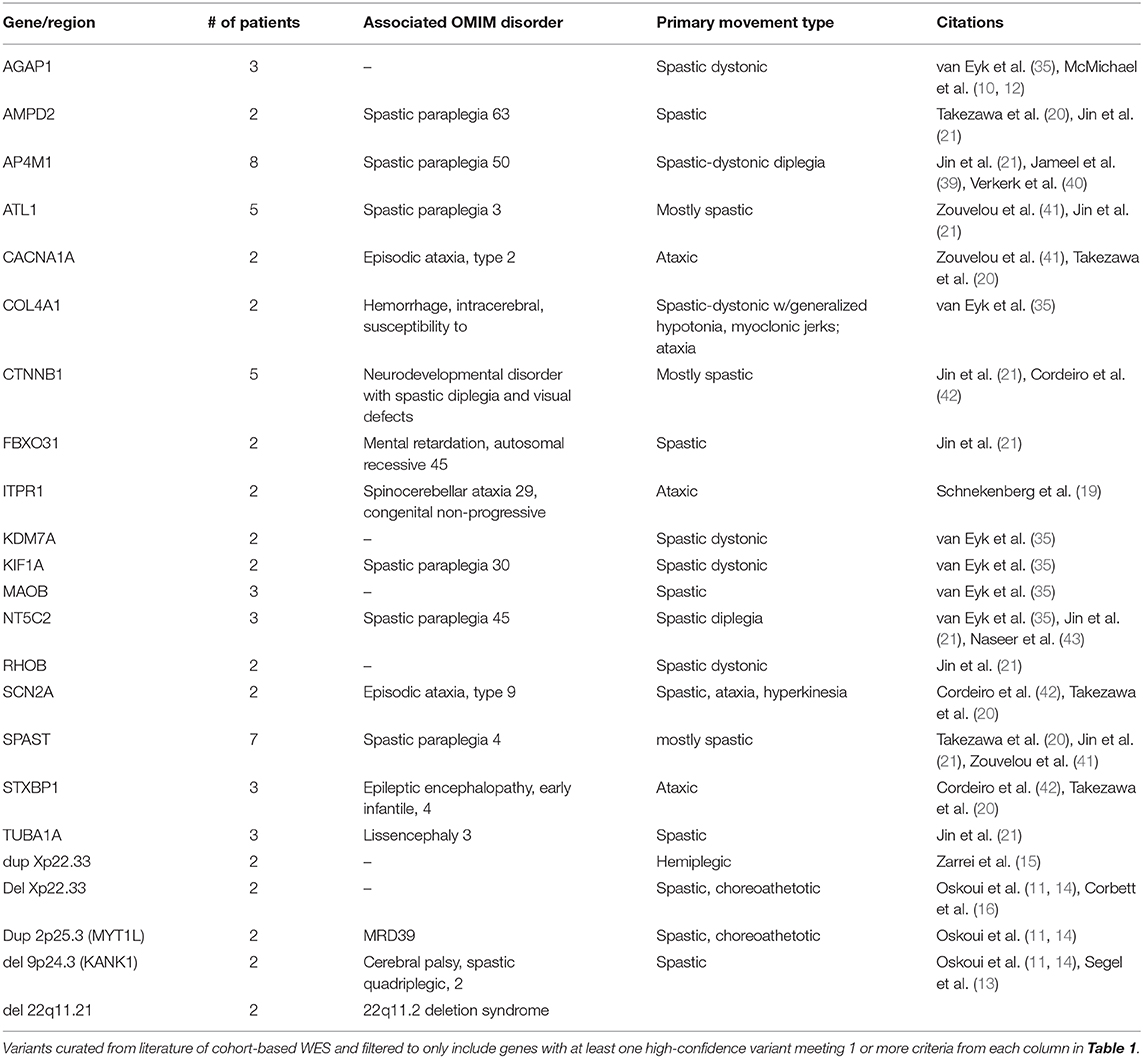
Table 2. Recurrent CP genes and copy number variants.
Identifying genes responsible for “pure” movement disorder phenotypes has proved quite challenging, but themes are emerging. Genes causing the canonically progressive hereditary spastic paraplegia and the early onset, stable course of spastic CP overlap. Several of these genes, including ATL1, SPAST, and AP4 complex members, converge on intracellular membrane trafficking and distribution, regulating the shape of organelles such as the endoplasmic reticulum (44). Genes causing mixed spastic-dystonic CP include AGAP1, CTNNB1, FBXO31, KDM7A, KIF1A, and RHOB, which indicates diverse biological processes can contribute to motor dysfunction. Genes crucial for basal ganglia development, NKX2-1, and cyclic nucleotide regulation in the striatum, including GNAO1, ADCY5, and PDE10A (45) have been identified in choreic CP. Finally, mutations in several ion channel genes have been found in ataxic CP patients including KCNC3, ITPR1 (19) and CACNA1A (20, 41). Neurotransmission might be more broadly involved as mutations in STXBP1, a regulator of syntaxin, have also been found in ataxic CP (19, 20). Taken together, some CP motor subtypes and other motor disorders appear to share not only overlapping phenotypic features, but also overlapping genes and genetic pathways (46).
Several case-control studies have used high-throughput-omics approaches to discover dysfunctional pathways and networks associated with CP. In other areas of disease research integrated-omics approaches have been used to consolidate genomic, epigenomic, and transcriptomic findings, but this approach has yet to be systematically applied to CP. Nonetheless, early findings are proving interesting.
Genes with mutations detected in CP cluster into pathways governing neurite extension including the extracellular matrix, cell-matrix interactions, cytoskeletal dynamics, and Rho GTPase function. Several genes in these pathways cause locomotor impairments in Drosophila loss of function models (21), indicating that CP genes may regulate connectivity of central nervous system circuits regulating movement.
The concept that CP-associated genes regulate nervous system connectivity is further supported by the clinical and genetic overlap of CP with other neurodevelopmental disorders. Patients with CP often have intellectual disability (~45%), epilepsy (~40%), and autism (~7%) (47, 48). Jin et al. (21) found a 1.7–2.0-fold enrichment of NDD genes among CP candidate genes detected by WES. Genetic pleiotropy has been described for several CP candidate genes. For example, SCN8A encodes a sodium channel with well-characterized mutations causing epileptic encephalopathies (49). Additionally, de novo mutations in SCN8A have been identified in CP (10) and intellectual disability without seizures (50). KCNMA1 encodes a voltage and calcium gated potassium channel with different mutations associated with a spectrum of neurodevelopmental phenotypes including intellectual disability, developmental delay, axial and ataxic hypotonia, epilepsy, and dyskinesia (51, 52). The association of CP with known NDD genes deserves special mention as potential phenotypic expansions as in some cases, movement disorders have not been well-described previously (53). In most cases, the literature has not indicated the absence of a movement disorder but rather has remained silent on the issue, focusing instead on dysmorphic features, intellectual disability, etc. Thus, genetic disruption of brain development can result in CP, other neurodevelopmental disorders, or a combination.
Epigenomic analyses reveal alterations in axon guidance, actin cytoskeleton, and cell signaling with parallels to genomic findings. In monozygotic twins discordant for CP, pathway enrichment analysis for alterations in genome-wide DNA methylation at birth (54) shows changes in MAPK signaling, hypoxia-associated signaling, inflammation, cell adhesion, cytokine–cytokine receptor interaction, and Ras signaling. In unrelated individuals with CP, methylation differences have been identified in genes involved in axonal guidance, the actin cytoskeleton, insulin and ephrin receptors, crosstalk between dendritic cells and natural killer cells, TGF-β, Wnt, neuregulin, PI3K/AKT, and tight junction signaling (55). These findings support the notion that perinatal hypoxic-ischemia and inflammatory responses are associated with an eventual CP outcome. However, it is not clear whether a causal relationship exists, as these studies did not control for environmental risk factors, which could themselves account for the epigenetic changes. Additional cohorts studying many more epigenomes with careful attention to age, risk factors, and statistical methods are needed to determine if the changes are consistent, and how that relates to patient outcomes. Further studies linking genomic and epigenomic findings are also warranted.
Transcriptomic analysis of lymphoblastoid cell lines derived from 182 CP patients with a mix of environmental, genetic, and indeterminate etiologies revealed 387 significantly differentially expressed genes (56). Pathway enrichment analysis using this gene set demonstrated downregulated signal transduction and cell signaling pathways including brain-derived neurotrophic factor, upregulated immune function genes, and altered amyloid precursor protein A (APP) processing. Despite differing etiologies and age at the time of collection, the authors found overlap in dysregulation of MAPK signaling that aligned with findings from epigenetic studies. Many of the patients in this cohort have been reported in McMichael et al. (10) or Jin et al. (21) suggesting defects in genes that govern cell signaling (MAPK/PI3K/AKT) may lead to neuronal wiring defects and CP. One fascinating question regarding these findings is whether the observed difference in expression is primary (i.e. fundamentally related to the cause of CP) or secondary (i.e. a consequence of chronic spasticity/dystonia, muscle contracture, etc.) in nature.
Given these findings converge on neurotrophic and stress-response signaling pathways as well as cell adhesion, cytoskeletal maintenance, and actin dynamics, it will be important to replicate and further integrate these results. In the future, stratification of findings by etiology may reveal either consistent or disparate mechanisms. For both epigenetic and transcriptomic studies, the stage of development, time post-injury, and tissue type sampled will impact the resulting profiles. Harmonized study designs across—omics platforms will facilitate cross comparisons between epigenomics, transcriptomics, and proteomics, among other techniques. A serial study of individuals assessed using multiple techniques might be most revealing. Detailed catalogs of patient phenotypes will further facilitate interpretation of these complex findings and may help discern pathways which drives pathophysiology vs. those representing compensatory mechanisms.
The international consensus definition for CP defines the disorder as “a group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to non-progressive disturbances that occurred in the developing fetal or infant brain” (1). In 2019 the International Cerebral Palsy Genomics Consortium (ICPGC) released a consensus statement reaffirming that CP is defined by clinical phenotype rather than etiology (57). Therefore, a CP diagnosis applies if the definitional criteria are met, regardless if there is a genetic etiology (37). However, a recent survey of physicians who treat people with CP across different specialties revealed variability in current clinical practice. When presented with a hypothetical scenario, only 67% would make a diagnosis of CP if there was a consistent clinical CP phenotype with a genetic cause identified (58). There is thus a need for more clinician awareness and training on genetic etiologies of CP and implications for clinical practice in order to avoid revisionist diagnosis for children who fit clinical criteria for CP.
It is also important for clinicians to be able to distinguish between CP and other disorders to plan treatment and care. Inborn errors of metabolism (IEM) appear early in life with different types of disordered movements, including hypotonia, dystonia, and chorea (59). The stakes are high: 67 of the 110 inborn errors of metabolism that can resemble CP are treatable (59). Therefore, there is a need to quickly and accurately identify the underlying etiology. We posit that CP is not currently, and should not be defined as an “untreatable” disorder. This begs the question: When is it CP and when is it an inborn error of metabolism?
We suggest considering IEM as a potential etiology of CP and using the following classifications based on whether the condition is stable or degenerative:
1. If after treating the metabolic disturbance, the disability resolves without permanent brain injury, the patient should be described as having a treatable IEM.
2. If after treating the metabolic disturbance, the injury to the brain and disability remain but does not worsen, the patient should be described as having CP with a neurometabolic etiology.
3. If the disability progresses, with or without treatment, the patient can be described as having a non-CP rare pediatric movement disorder caused by a degenerative IEM.
A diagnostic classification of both CP and an IEM would be similar to indicating that a person has epilepsy due to an underlying neurometabolic disorder. In many cases, this would be beneficial for access to treatments and services needed for the optimal patient care (37).
Genetic findings previously thought to singularly lead to neurodegenerative disorders have now been identified to lead to non-degenerative motor disorders that meet CP criteria. There is increasing evidence that different mutations in the same gene can lead to either degenerative or developmental phenotypes that probably reflect different effects on cellular biology. Careful phenotyping and follow up of patients with SPAST and ATL1 mutations have revealed that some have phenotypes that are stable over decades (60, 61). KCNC3 variants from infant-onset disease change channel gating properties and increase neuron excitability compared to variants from adult-onset disease, which were associated with reduced channel activity and high-frequency neuronal firing. Further, these effects can manifest with different temporal patterns, with the infant-associated variant disrupting dendrite and axon branching as a developmental feature absent from the adult-associated variant (62). Mutations in TRIO were recently shown to lead to distinct, domain-specific effects on development (63). This evidence suggests that manifestations may vary depending on the nature of a given mutation and potentially other moderating factors. A finding in a gene linked to a potentially progressive disorder should trigger a careful re-appraisal of the patient's phenotype and may influence testing, monitoring, and follow up, but should not reflexively prompt a revision of diagnosis unless clinically appropriate based on the patient's course.
New genetic findings are raising new questions. For instance, do genetic mutations disrupt brain development in consistent ways? What imaging findings indicate a genetic cause of CP? There have been several studies on brain malformations in movement disorders (64). Periventricular leukomalacia (PVL) detected by MRI has classically been interpreted as caused by perinatal stress (65). However, PVL was also detected in patients with ATL1 and RHOB variants (21) and 5/12 patients with a neuroimaging finding of PVL had no pre- or perinatal risk factors or complications (66). This finding suggests that rather than representing a single etiology, PVL reflects white matter abnormalities where the timing can be either pre- or perinatal (67). In addition, individuals with CP often have structurally normal or mild, non-specific anatomic MRI findings (68). Functional MRI may add additional dimensionality to the understanding of brain connectivity (69). Compared to those with abnormal MRI findings, CP patients with normal MRIs are more likely to have been born at term and experienced an uneventful perinatal period. They are also more likely to have dyskinetic and diplegic CP types but show no differences in the severity of impairment or presence of comorbidities (70). More study is currently needed to determine how MRI could predict genetic contributions to CP.
Genetic findings can and will inform personalized medicine over time. For instance, in a randomized case-control clinical trial, polymorphisms in immune genes (IL1β, PAI1, IL6R) were associated with improved neurodevelopmental outcomes, including reduced rates of CP, after magnesium sulfate intervention in women at imminent risk of premature delivery (71). Genetic etiologies may also inform interventions such as deep brain stimulation (DBS). In some cases, DBS outcomes can be anticipated based on prior genetic findings (72, 73). Other genes may inform preventative or monitoring efforts to ensure optimal outcomes, such as avoiding head injury and managing cardiovascular risk factors in COL4A1 patients. Due to increasingly compelling evidence that CP can have a genetic component, future studies should test whether using WES and CNV analysis can guide clinical care for CP. Surveys of patients and their families with disorders of unknown etiology support the use of WES as part of the diagnostic process, with the greatest benefit reported from those who obtained positive findings (74). This underscores the need for additional gene discovery and translational research to improve the interpretation of such tests and establish clinical guidelines for utilization in diagnostic assessment.
In summary, genetic etiology is a notable contributor to the development of CP, potentially through both disrupted brain development and dysregulated responses to risk factors. A small number of recurrent genes with strong evidence of pathogenicity have been described. However, there are likely hundreds more genes that await discovery and/or validation with large-cohort sequencing and follow up studies on the molecular consequences of patient-associated variants. Future studies will also be needed to provide a more in-depth analysis of the function of genes in individuals with overlapping phenotypes using platforms like Genematcher (36). A comprehensive list of all genes that have been identified for CP, not just those from cohort WES studies reviewed here, will be an essential development for the field, particularly as clinical CP sequencing panels are already being offered. Since CP-associated genes have not been systematically curated in OMIM, this represents an opportunity for the field. In the interim, the inclusion of NDD genes with or without previously described movement disorder phenotypes compatible with CP can serve as a surrogate.
Efforts to find genetic etiologies by CP motor type have been limited, in part due to genetic and phenotypic heterogeneity. Identifying CP-subtypes may become more feasible with meta-analysis with high numbers of patients with detailed clinical information provided for every individual in the cohort, facilitated by the International Cerebral Palsy Genomics Consortium (www.icpgc.org). Identifying genes that can cause specific motor types will be crucial for studying the mechanisms underlying those disorders as well as identifying pathways and other features to guide further gene discovery and the development of personalized treatments.
Progress in understanding CP pathology and developing new treatments has been impeded by the limited availability of animal models for functional studies. As monogenic forms of CP are increasingly identified, this will allow development of important genetic models to better understand changes to neurobiology and development in CP. Studies to date have identified relatively few genes as sequenced cohorts have been comparatively small, compounded by a relative lack of deep phenotyping. As more genetic studies are conducted, there will be an increased need for validation analyses to definitively link variants and genes with cerebral palsy. This improved characterization of individual genes will also facilitate subsequent gene discovery, pathways contributing to CP pathogenesis, and the development of targeted interventions. A better understanding of CP genomics will also enable the identification of risk genes contributing through a multifactorial, rather than strictly monogenic way. Together, the discovery of genetic factors relevant to CP provides new opportunities for detailed study driving development of treatments and interventions to improve the lives of people living with CP. Updating diagnostic criteria and practice parameters to incorporate indications for genetic testing and interpretation should be considered to provide clarity and guidance of how to classify CP considering the evolving genetic landscape.
SL conceptualized and wrote the manuscript. SS, BW, and AH contributed to literature review, variant curation, and drafting of the manuscript. SJ, HS-S, MF, and MK made intellectual contributions and edits. All authors approved the final manuscript.
This work was funded in part by NIH award 1R01NS106298 to MK, the Australian American Fulbright Foundation to MF, and salary support through a National Health and Medical Research Council of Australia Early Career Fellowship (1144566) and from the Australasian Cerebral Palsy Clinical Trials Network to HS-S, and the K99/R00 Pathway to Independence Award (K99HL143036 and R00HL143036-02) to SJ.
MK serves as a consultant to PTC Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the many children and adults with CP and their families who have contributed to our studies. Without their commitment to improving the state of the science in CP, this work would not have been possible.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.625428/full#supplementary-material
Supplemental Table 1. Curated sequence variants from whole exome sequencing studies of cerebral palsy cohorts.
Supplemental Table 2. Curated copy number variants from whole exome and microarray studies of cerebral palsy cohorts.
1. Rosenbaum P, Paneth N, Leviton A, Goldstein M, Bax M, Damiano D, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl. (2007) 109:8–14.
2. Report of the Australian Cerebral Palsy Register Birth years 1995–2012 (2018) Available online at: https://cpregister.com/wp-content/uploads/2019/02/Report-of-the-Australian-Cerebral-Palsy-Register-Birth-Years-1995-2012.pdf (accessed October 7, 2020).
3. Sellier E, Platt MJ, Andersen GL, Krägeloh-Mann I, De La Cruz J, Cans C, et al. (2016) Decreasing prevalence in cerebral palsy: a multi-site European population-based study, 1980 to 2003. Dev Med Child Neurol. 58:85–92. doi: 10.1111/dmcn.12865
4. Hagen MW, Riddle A, McClendon E, Gong X, Shaver D, Srivastava T, et al. Role of recurrent hypoxia-ischemia in preterm white matter injury severity. PLoS ONE. (2014) 9:e112800. doi: 10.1371/journal.pone.0112800
5. Chau CMY, Ranger M, Bichin M, Park MTM, Amaral RSC, Chakravarty M, et al. Hippocampus, Amygdala, and Thalamus Volumes in Very Preterm Children at 8 Years: Neonatal Pain and Genetic Variation. Front Behav Neurosci. (2019) 13:51. doi: 10.3389/fnbeh.2019.00051
6. Gano D, Barkovich AJ. Cerebellar hypoplasia of prematurity: causes and consequences. Handb Clin Neurol. (2019) 162:201–16. doi: 10.1016/B978-0-444-64029-1.00009-6
7. Strauss JF, Romero R, Gomez-Lopez N, Haymond-Thornburg H, Modi BP, Teves ME, et al. Spontaneous preterm birth: advances toward the discovery of genetic predisposition. Am J Obstet Gynecol. (2018) 218:294–314. doi: 10.1016/j.ajog.2017.12.009
8. Goldsmith S, Mcintyre S, Andersen GL, Gibson C, Himmelmann K, Blair E, et al. Congenital anomalies in children with pre- or perinatally acquired cerebral palsy: an international data linkage study. Dev Med Child Neurol. (2020). doi: 10.1111/dmcn.14602. [Epub ahead of print].
9. Odding E, Roebroeck ME, Stam HJ. The epidemiology of cerebral palsy: incidence, impairments and risk factors. Disabil Rehabil. (2006) 28:183–191. doi: 10.1080/09638280500158422
10. McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatry. (2015) 20:176–82. doi: 10.1038/mp.2014.189
11. Oskoui M, Coutinho F, Dykeman J, Jetté N, Pringsheim T. An update on the prevalence of cerebral palsy: a systematic review and meta-analysis. Dev Med Child Neurol. (2013) 55:509–19. doi: 10.1111/dmcn.12080
12. McMichael G, Girirajan S, Moreno-De-Luca A, Gecz J, Shard C, Nguyen LS, et al. Rare copy number variation in cerebral palsy. Eur J Hum Genet. (2014) 22:40–5. doi: 10.1038/ejhg.2013.93
13. Segel R, Ben-Pazi H, Zeligson S, Fatal-Valevski A, Aran A, Gross-Tsur V, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. (2015) 84:1660–8. doi: 10.1212/WNL.0000000000001494
14. Oskoui M, Gazzellone MJ, Thiruvahindrapuram B, Zarrei M, Andersen J, Wei J, et al. Clinically relevant copy number variations detected in cerebral palsy. Nat Commun. (2015) 6:7949. doi: 10.1038/ncomms8949
15. Zarrei M, Fehlings DL, Mawjee K, Switzer L, Thiruvahindrapuram B, Walker S, et al. De novo and rare inherited copy-number variations in the hemiplegic form of cerebral palsy. Genet Med. (2018) 20:172–80. doi: 10.1038/gim.2017.83
16. Corbett MA, van Eyk CL, Webber DL, Bent SJ, Newman M, Harper K, et al. Pathogenic copy number variants that affect gene expression contribute to genomic burden in cerebral palsy. NPJ Genom Med. (2018) 3:33. doi: 10.1038/s41525-018-0073-4
17. Van Iseghem V, McGovern E, Apartis E, Keren B, Vidailhet M, Roze E, et al. Subcortical myoclonus and associated dystonia in 22q11.2 deletion syndrome. Tremor Other Hyperkinet Mov (N Y). (2020) 10:729. doi: 10.7916/tohm.v0.729
18. Kontoangelos K, Maillis A, Maltezou M, Tsiori S, Papageorgiou CC. Acute dystonia in a patient with 22q11.2 deletion syndrome. Ment Illn. (2015) 7:5902. doi: 10.4081/mi.2015.5902
19. Schnekenberg PR, Perkins EM, Miller JW, Davies WIL, D'Adamo MC, Pessia M, et al. De novo mutations in patients diagnosed with ataxic cerebral palsy. Brain. (2015) 138:1817–32. doi: 10.1093/brain/awv117
20. Takezawa Y, Kikuchi A, Haginoya K, Niihori T, Numata-Uematsu Y, Inui T, et al. Genomic analysis identifies masqueraders of full-term cerebral palsy. Ann Clin Transl Neurol. (2018) 5:538–51. doi: 10.1002/acn3.551
21. Jin SC, Lewis SA, Bakhtiari S, Zeng X, Sierant MC, Shetty S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. (2020) 52:1046–56. doi: 10.1038/s41588-020-0695-1
22. Fletcher NA, Foley J. Parental age, genetic mutation, and cerebral palsy. J Med Genet. (1993) 30:44–6. doi: 10.1136/jmg.30.1.44
23. Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM. A framework for the interpretation of de novo mutation in human disease. Nat Genet. (2014) 46:944–0. doi: 10.1038/ng.3050
24. Pham R, Mol BW, Gecz J, MacLennan AH, MacLennan SC, Corbett MA, et al. Definition and diagnosis of cerebral palsy in genetic studies: a systematic review. Dev Med Child Neurol. (2020) 62:1024–30. doi: 10.1111/dmcn.14585
25. Kinoshita K, Ishizaki Y, Yamamoto H, Sonoda M, Yonemoto K, Kira R, et al. De novo p.G696S mutation in COL4A1 causes intracranial calcification and late-onset cerebral hemorrhage: a case report and review of the literature. Eur J Med Genet. (2020) 63:103825. doi: 10.1016/j.ejmg.2019.103825
26. Carter LB, Battaglia A, Cherry A, Manning MA, Ruzhnikov MR, Bird LM, et al. Perinatal distress in 1p36 deletion syndrome can mimic hypoxic ischemic encephalopathy. Am J Med Genet A. (2019) 179:1543–6. doi: 10.1002/ajmg.a.61266
27. Rajatileka S, Odd D, Robinson MT, Spittle AC, Dwomoh L, Williams M, et al. (2018) Variants of the EAAT2 glutamate transporter gene promoter are associated with cerebral palsy in preterm infants. Mol Neurobiol. 55:2013–24. doi: 10.1007/s12035-017-0462-1
28. Kapitanović Vidak H, Catela Ivković T, Vidak Z, Kapitanović S. COX-1 and COX-2 polymorphisms in susceptibility to cerebral palsy in very preterm infants. Mol Neurobiol. (2017) 54:930–8. doi: 10.1007/s12035-016-9713-9
29. Pregnolato S, Chakkarapani E, Isles AR, Luyt K. Glutamate transport and preterm brain injury. Front Physiol. (2019) 10:417. doi: 10.3389/fphys.2019.00417
30. SCPE. Prevalence and characteristics of children with cerebral palsy in Europe. Dev Med Child Neurol. (2002) 4:633–40. doi: 10.1111/j.1469-8749.2002.tb00848.x
31. Aravamuthan BR, Gandham S, Young AB, Rutkove SB. Sex may influence motor phenotype in a novel rodent model of cerebral palsy. Neurobiol Dis. (2020) 134:104711. doi: 10.1016/j.nbd.2019.104711
32. Matthews AM, Blydt-Hansen I, Al-Jabri B, Andersen J, Tarailo-Graovac M, Price M, et al. Atypical cerebral palsy: genomics analysis enables precision medicine. Genet Med. (2019) 21:1621–8. doi: 10.1038/s41436-018-0376-y
33. Miosge LA, Field MA, Sontani Y, Cho V, Johnson S, Palkova A, et al. Comparison of predicted and actual consequences of missense mutations. Proc Natl Acad Sci USA. (2015) 112:E5189–98. doi: 10.1073/pnas.1511585112
34. Belvindrah R, Natarajan K, Shabajee P, Bruel-Jungerman E, Bernard J, Goutierre M, et al. Mutation of the α-tubulin Tuba1a leads to straighter microtubules and perturbs neuronal migration. J Cell Biol. (2017) 216:2443–61. doi: 10.1083/jcb.201607074
35. van Eyk CL, Corbett MA, Frank MSB, Webber DL, Newman M, Berry JG, et al. Targeted resequencing identifies genes with recurrent variation in cerebral palsy. NPJ Genom Med. (2019) 4:27. doi: 10.1038/s41525-019-0101-z
36. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. (2015) 36:928–30. doi: 10.1002/humu.22844
37. Smithers-Sheedy H, Badawi N, Blair E, Cans C, Himmelmann K, Krägeloh-Mann I, et al. What constitutes cerebral palsy in the twenty-first century? Dev Med Child Neurol. (2014) 56:323–8. doi: 10.1111/dmcn.12262
38. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Medical Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
39. Jameel M, Klar J, Tariq M, Moawia A, Altaf Malik N, Seema Waseem S, et al. A novel AP4M1 mutation in autosomal recessive cerebral palsy syndrome and clinical expansion of AP-4 deficiency. BMC Med Genet. (2014) 15:133. doi: 10.1186/s12881-014-0133-2
40. Verkerk AJ, Schot R, Dumee B, Schellekens K, Swagemakers S, Bertoli-Avella AM, et al. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet. (2009) 85:40–52. doi: 10.1016/j.ajhg.2009.06.004
41. Zouvelou V, Yubero D, Apostolakopoulou L, Kokkinou E, Bilanakis M, Dalivigka Z, et al. The genetic etiology in cerebral palsy mimics: the results from a Greek tertiary care center. Eur J Paediatr Neurol. (2019) 23:427–37. doi: 10.1016/j.ejpn.2019.02.001
42. Cordeiro D, Bullivant G, Siriwardena K, Evans A, Kobayashi J, Cohn RD, et al. Genetic landscape of pediatric movement disorders and management implications. Neurol Genet. (2018) 4:e265. doi: 10.1212/NXG.0000000000000265
43. Naseer MI, Abdulkareem AA, Pushparaj PN, Bibi F, Chaudhary AG. Exome analysis identified novel homozygous splice site donor alteration in NT5C2 gene in a Saudi Family Associated With Spastic Diplegia Cerebral Palsy, Developmental Delay, and Intellectual Disability. Front Genet. (2020) 11:14. doi: 10.3389/fgene.2020.00014
44. Blackstone C. Converging cellular themes for the hereditary spastic paraplegias. Curr Opin Neurobiol. (2018) 51:139–46. doi: 10.1016/j.conb.2018.04
45. Diggle CP, Sukoff Rizzo SJ, Popiolek M, Hinttala R, Schülke JP, Kurian MA, et al. Biallelic mutations in PDE10A lead to loss of striatal PDE10A and a hyperkinetic movement disorder with onset in infancy. Am J Hum Genet. 98:735–43. doi: 10.1016/j.ajhg.2016.03.015
46. Synofzik M, Schüle R. Overcoming the Divide between ataxias and spastic paraplegias: shared phenotypes, genes, and pathways. Mov Disord. (2017) 32:332–45. doi: 10.1002/mds.26944
47. Reid SM, Meehan EM, Arnup SJ, Reddihough DS. Intellectual disability in cerebral palsy: a population-based retrospective study. Dev Med Child Neurol. (2018) 60:687–94. doi: 10.1111/dmcn.13773
48. Christensen D, Van Naarden Braun K, Doernberg NS, Maenner MJ, Arneson CL, Durkin MS, et al. Prevalence of cerebral palsy, co-occurring autism spectrum disorders, and motor functioning—Autism and Developmental Disabilities Monitoring Network, USA, 2008. Dev Med Child Neurol. (2014) 56:59–65. doi: 10.1111/dmcn.12268
49. Wagnon JL, Meisler MH. Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy. Front Neurol. (2015) 6:104. doi: 10.3389/fneur.2015.00104
50. Wagnon JL, Barker BS, Ottolini M, Park Y, Volkheimer A, Valdez P, et al. Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol Genet. (2017) 3:e170. doi: 10.1212/NXG.0000000000000170
51. Liang L, Li X, Moutton S, Cogné B, Saint-Martin A, Hurst ACE, et al. De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum Mol Genet. (2019) 28:2937–51. doi: 10.1093/hmg/ddz117
52. Yeşil G, Aralaşmak A, Akyüz E, Içagasioglu D, Uygur Sahin T, Bayram Y. Expanding the phenotype of homozygous KCNMA1 mutations; dyskinesia, epilepsy, intellectual disability, cerebellar and corticospinal tract atrophy. Balkan Med J. (2018) 35:336–9. doi: 10.4274/balkanmedj.2017.0986
53. Kulkarni N, Tang S, Bhardwaj R, Bernes S, Grebe TA. Progressive movement disorder in brothers carrying a GNAO1 mutation responsive to deep brain stimulation. J Child Neurol. (2016) 31:211–4. doi: 10.1177/0883073815587945
54. Mohandas N, Bass-Stringer S, Maksimovic J, Crompton K, Loke YJ, Walstab J, et al. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin Epigenet. (2018) 10:25. doi: 10.1186/s13148-018-0457-4
55. Bahado-Singh RO, Vishweswaraiah S, Aydas B, Mishra NK, Guda C, Radhakrishna U. Deep learning/artificial intelligence and blood-based DNA epigenomic prediction of cerebral palsy. Int J Mol Sci. (2019). 20:2075. doi: 10.3390/ijms20092075
56. van Eyk CL, Corbett MA, Gardner A, van Bon BW, Broadbent JL, Harper K, et al. Analysis of 182 cerebral palsy transcriptomes points to dysregulation of trophic signaling pathways and overlap with autism. Transl Psychiatry. (2018) 8:88. doi: 10.1038/s41398-018-0136-4
57. MacLennan AH, Lewis S, Moreno-De-Luca A, Fahey M, Leventer RJ, McIntyre S, et al. Genetic or other causation should not change the clinical diagnosis of cerebral palsy. J Child Neurol. (2019) 34:472–6. doi: 10.1177/0883073819840449
58. Aravamuthan BR, Fehlings D, Shetty S, Fahey M, Gilbert L, Tilton A, et al. Variability in cerebral palsy diagnosis. Pediatrics. (2021) e2020010066. doi: 10.1542/peds.2020-010066
59. Leach EL, Shevell M, Bowden K, Stockler-Ipsiroglu S, van Karnebeek CDM. Treatable inborn errors of metabolism presenting as cerebral palsy mimics: systematic literature review. Orphanet J Rare Dis. (2014) 9:197. doi: 10.1186/s13023-014-0197-2
60. Schüle R, Wiethoff S, Martus P, Karle KN, Otto S, Klebe S, et al. Hereditary spastic paraplegia: clinicogenetic lessons from 608 patients. Ann Neurol. (2016) 79:646–58. doi: 10.1002/ana.24611
61. Parodi L, Fenu S, Barbier M, Duyckaerts C, Tezenas du Montcel S, Monin ML, et al. Spastic paraplegia due to SPAST mutations is modified by the underlying mutation and sex. Brain. (2018) 141:3331–42. doi: 10.1093/brain/awy285
62. Hsieh JY, Ulrich BN, Issa FA, Lin MCA, Brown B, Papazian DM. Infant and adult SCA13 mutations differentially affect Purkinje cell excitability, maturation, and viability in vivo. Elife. (2020) 9:e57358. doi: 10.7554/eLife.57358
63. Barbosa S, Greville-Heygate S, Bonnet M, Godwin A, Fagotto-Kaufmann C, Kajava AV, et al. Opposite modulation of RAC1 by mutations in TRIO is associated with distinct, domain-specific neurodevelopmental disorders. Am J Hum Genet. (2020) 106:338–55. doi: 10.1016/j.ajhg.2020.01.018
64. Korzeniewski SJ, Slaughter J, Lenski M, Haak P, Paneth N. The complex aetiology of cerebral palsy. Nat Rev Neurol. (2018) 14:528–43. doi: 10.1038/s41582-018-0043-6
65. Patel DR, Neelakantan M, Pandher K, Merrick J. Cerebral palsy in children: a clinical overview. Transl Pediatr. (2020) 9(Suppl. 1):S125–35. doi: 10.21037/tp.2020.01.01
66. Miller SP, Shevell MI, Patenaude Y, O'Gorman AM. Neuromotor spectrum of periventricular leukomalacia in children born at term. Pediatr Neurol. (2000) 23:155–9. doi: 10.1016/s0887-8994(00)00172-7
67. Lasry O, Shevell MI, Dagenais L, REPACQ Consortium. Cross-sectional comparison of periventricular leukomalacia in preterm and term children. Neurology. (2010) 74:1386–91. doi: 10.1212/WNL.0b013e3181dad62d
68. Numata Y, Onuma A, Kobayashi Y, Sato-Shirai I, Tanaka S, Kobayashi S, et al. Brain magnetic resonance imaging and motor and intellectual functioning in 86 patients born at term with spastic diplegia. Dev Med Child Neurol. (2013) 55:167–72. doi: 10.1111/dmcn.12013
69. Reid LB, Cunnington R, Boyd RN, Rose SE. Surface-based fMRI-driven diffusion tractography in the presence of significant brain pathology: a study linking structure and function in cerebral palsy. PLoS ONE. (2016) 11:e0159540. doi: 10.1371/journal.pone.0159540
70. Springer A, Dyck Holzinger S, Andersen J, Buckley D, Fehlings D, Kirton A, et al. Profile of children with cerebral palsy spectrum disorder and a normal MRI study. Neurology. (2019) 93:e88–96. doi: 10.1212/WNL.0000000000007726
71. Clark EAS, Weiner SJ, Rouse DJ, Mercer BM, Reddy UM, Iams JD, et al. Genetic variation, magnesium sulfate exposure, and adverse neurodevelopmental outcomes following preterm birth. Am J Perinatol. (2018) 35:1012–22. doi: 10.1055/s-0038-1635109
72. Brüggemann N, Kühn A, Schneider SA, Kamm C, Wolters A, Krause P, et al. Short- and long-term outcome of chronic pallidal neurostimulation in monogenic isolated dystonia. Neurology. (2015) 84:895–903. doi: 10.1212/WNL.000000000000131
73. Koy A, Cirak S, Gonzalez V, Becker K, Roujeau T, Milesi C, et al. Deep brain stimulation is effective in pediatric patients with GNAO1 associated severe hyperkinesia. J Neurol Sci. (2018) 391:31–9. doi: 10.1016/j.jns.2018.05.018
Keywords: cerebral palsy, genetics, genomics, neurodevelopmental disorders, neurogenetics
Citation: Lewis SA, Shetty S, Wilson BA, Huang AJ, Jin SC, Smithers-Sheedy H, Fahey MC and Kruer MC (2021) Insights From Genetic Studies of Cerebral Palsy. Front. Neurol. 11:625428. doi: 10.3389/fneur.2020.625428
Received: 03 November 2020; Accepted: 16 December 2020;
Published: 21 January 2021.
Edited by:
Sotiria D. Mastroyianni, Panagiotis & Aglaia Kyriakou Children's Hospital, GreeceReviewed by:
Atsuo Kikuchi, Tohoku University, JapanCopyright © 2021 Lewis, Shetty, Wilson, Huang, Jin, Smithers-Sheedy, Fahey and Kruer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael C. Kruer, a3J1ZXJtQGFyaXpvbmEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.