 Fujun Liu
Fujun Liu Shucai Fan
Shucai Fan Xin Tang
Xin Tang Liangxue Zhou
Liangxue Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 15 December 2020
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.604612
Background: Adult sellar region atypical teratoid/rhabdoid tumor (AT/RT) is a rare lesion. We aimed to elucidate clinical, radiologic, and pathological characteristics, treatment strategies, and outcomes of this disease.
Methods: Five adult sellar AT/RT patients were retrospectively analyzed between January 2015 and December 2018. In addition, we performed a review of the reported data on adult sellar AT/RT.
Results: Patients (n = 5) were female with a median age of 50 years. The mean duration of symptoms, of which headache was the most frequent, was 1.6 months (range, 2 weeks−8 months). The average tumor size was 2.82 cm (range, 1.9–4.5 cm). All lesions were irregularly shaped. MRI showed heterogeneous enhancement in three of five lesions. Four of five patients underwent subtotal resection (STR) and one gross total resection (GTR). Whereas, one patient received post-operative adjuvant radiotherapy, one patient received post-operative combination of radio- and chemotherapy. The review of the reported data showed that 39 cases of adult sellar AT/RT had been reported. The estimated median overall survival (OS) was 23 months with a 1-year survival estimate of 59.7%. The median OS for patients with GTR was 28 months and 17 months for patients with STR. Kaplan–Meier analysis showed that patients with high (≥35%) MIB-1/Ki67 index value had a significantly shorter OS compared with those with low (<35%) index value (p = 0.033), and that patients who received post-operative combination radio- and chemotherapy had longer OS than that of those who did not (p < 0.001).
Conclusion: Adult sellar region AT/RT is a rapidly growing tumor with a poor prognosis. High levels of MIB1/Ki-67 on histology may indicate aggressive feature of the tumor. Maximal safe resection followed by adjuvant radiotherapy combined with chemotherapy may be the optimal therapeutic strategy for adult sellar region AT/RT.
Atypical teratoid/rhabdoid tumor (AT/RT) of the central nervous system (CNS), which occurs chiefly in children below the age of 3 years, is an aggressive malignant tumor (1, 2). According to the 2016 WHO Classification of Tumors of the CNS, AT/RT is defined as having specific genetic alterations in the INI1/SMARCB1/hSNF5, or rarely, in the SMARCA4/BRG1 genes (3). Initially described in the early 1990s by Horn et al. (4), adult AT/RT is an extremely rare tumor, commonly located in the sellar region (5), with a lifetime risk estimated at <1 per million individuals (6). A recent study of AT/RT showed hemangiopericytoma-like stag-horn vasculature and lack of INI1 expression in the adult sellar region featured by rhabdoid tumors, which was identified as a clinicopathologically and genetically distinct variant of AT/RT (7). However, the clinical and radiological features, pathological characteristics, treatment strategies, and clinical outcomes of adult sellar region AT/RT remain ill-defined due to its rarity. In the present study, we report the clinical features, radiologic findings, pathological characteristics, treatment, and outcomes of a series of five adult sellar region AT/RT diagnosed in our hospital and also review the literature related to adult sellar region AT/RT found on the PubMed database and Google Scholar.
Between January 2015 and December 2018, five adult patients with sellar region AT/RT received surgical resection in the neurosurgery department of West China Hospital of Sichuan University. The diagnosis of the adult sellar region AT/RT was based on the 2016 WHO Classification of CNS Tumors (3). This study was approved by the West China Hospital Ethics Committee. Written informed consent had been obtained from all patients' families previously.
Relevant data were extracted retrospectively from patients' medical and surgical records. Clinical data including patient age, sex, symptoms, duration of symptoms before diagnosis, tumor size, extent of surgical resection (subtotal resection or gross total resection), endocrine hormone levels, histological findings, surgical outcomes, and clinical follow-up were analyzed.
All patients had pre-operative gadolinium-enhanced cranial MRI scans, including T1-weighted, T2-weighted, and gadolinium-enhanced T1-weighted sequences. The size of the tumors, cystic lesions, characteristics of contrast enhancement, peri-tumor edema, tumor shape, and hydrocephalus were obtained from the pre-operative post-contrast imaging. The tumor size in MRI was defined as the largest diameter of the lesion. The characteristics of contrast enhancement of MRI were classified into homogeneous and heterogeneous contrast enhancement. CT was performed to help with pre-operative differential diagnosis and CT density, calcifications, and intratumoral hemorrhage were also recorded. Tumor specimens were fixed in 10% buffered neutral formalin, paraffin-embedded, sectioned, and processed for H&E staining for routine histological evaluation and immunohistochemical staining.
The extent of surgical resection was classified into gross total resection (GTR; no distinct residual tumor) and subtotal resection (STR; >90% of tumor removal), which was evaluated according to the surgeon's operative notes and <72 h post-operative imaging analyses. Patients were assessed by an oncologist for adjuvant therapy after surgical resection. The post-operative adjuvant radiotherapy, chemotherapy, and overall survival (OS), defined as the time from date of surgical resection until death or last follow-up, were reviewed. The neurologic functional outcomes were also assessed using the Karnofsky performance scale (KPS). The endpoint of this study was the OS and recurrence-free survival (RFS); RFS was defined as the time between surgical resection and tumor recurrence on neuroimaging studies.
IBM SPSS Statistics (version 24, IBM Corp.) were applied for data analysis. For quantitative data, means ± SD or median are presented. Categorical data are presented as frequencies and percentages. OS was analyzed using Kaplan–Meier estimates and the log-rank test for comparisons. A p-value ≤0.05 was considered as statistically significant.
PubMed and Google Scholar were searched for case reports and series relevant to adult sellar AT/RT reported up to October 12, 2020. “Sellar,” “Suprasellar,” “Adult,” and “Atypical Teratoid/Rhabdoid Tumor” were used as either key words or medical subject headings. In addition, references from articles previously identified were used for further cases. The results were manually screened for studies focusing on clinical experience in diagnosis and/or treatment of adult sellar AT/RT in human subjects.
Of the five patients in our institution, all were female, and median age at diagnosis was 50 years (range, 29–80 years). The most common symptom was headache (n = 5; 100%), which was usually severe and unbearable, followed by visual disturbance (n = 3; 60%), ptosis (n = 2; 40%), numbness in limbs (n = 2; 40%), and galactorrhea (n = 1; 20%). The average duration of symptoms before surgery was 1.6 months (range, 2 weeks−8 months). The examination of cerebrospinal fluid for five patients was normal on admission to hospital. The pre-operative hormone profiles including luteinizing hormone (LH), prolactin, growth hormone (GH), cortisol, plasma adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), thyroid-stimulating hormone (TSH), and free thyroxine 4 (FT4) are listed in Table 1. Four patients had hypopituitarism and one patient had a normal anterior pituitary function.
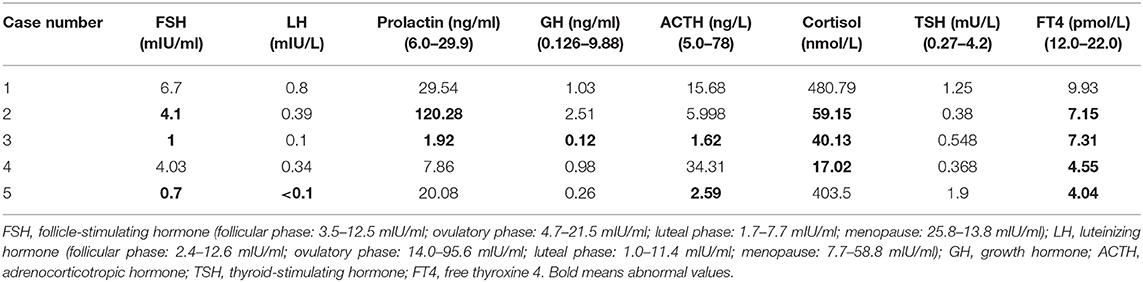
Table 1. Pre-operative levels of various hormones in 5 adult patients with sellar region AT/RT.
All patients underwent both MRI and CT scan pre-operatively. The radiological features are listed in Table 2. Five lesions were located in the sellar region. The median tumor size was 2.7 cm (range, 1.9–4.5 cm). On CT, of the five lesions, one was high dense and four were isodense, and none of the lesions had calcification. On MRI, T1-weighted imaging showed all five lesions to be isointense, and on T2-weighted imaging, three lesions were hyperintense, one isointense, and one isointense with hyperintense. Whereas, one patient experienced intratumoral hemorrhage, none showed hydrocephalus. Four of the five lesions appeared solid and one was solid and cystic. Most lesions exhibited heterogeneous enhancement and had irregular shapes (Figure 1). Peritumoral edema of the lesions that frequently extended into the cavernous sinus and also involving the surrounding brain tissue was noted.

Table 2. Radiological features of five adult patients with sellar region AT/RT.
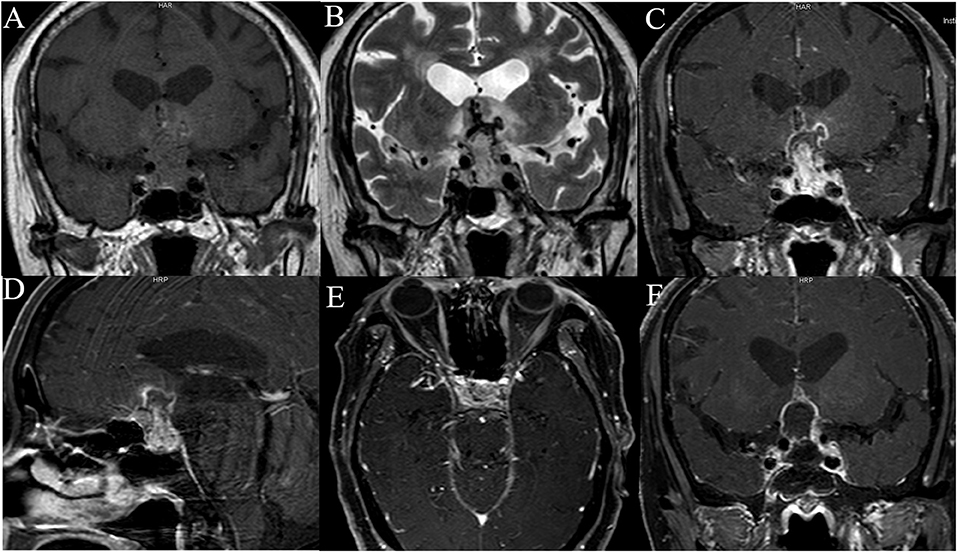
Figure 1. The neuroimaging of case 5. Pre-operative T1-weighted image (A), T2-weighted image (B), coronal (C), sagittal (D), axial (E), T1-weighted contrast-enhanced MRI of case 5 show intrasellar heterogeneously enhanced solid and cystic mass with suprasellar extension compressing and encasing the optic chiasm, invasion of bilateral cavernous sinus. Post-operative MRI scan of the brain shows subtotal resection of the tumor (F).
The pathological data are summarized in Table 3. Histologically, adult sellar region AT/RT consisted of medium-sized cells showing identifiable rhabdoid morphology and some atypical epithelioid-like spindle cells. Primitive neuroepithelial or interstitial components were also observed. Notably, three of the five lesions had hemangiopericytoma-like stag-horn vasculature (Figure 2). Immunohistochemistry showed loss of INI1, S-100, GFAP, and desmin expression in all five lesions, which were immunopositive for CD99 (66.6%; 2 of 3), CK (60.0%; 3 of 5), CD34 (33.3%; 1 of 3), EMA (50%; 2 of 4), and Syn (10%; 1 of 5). Staining for MIB-1/Ki-67 index test showed median MIB1/Ki-67 levels of 40% in all five lesions (range, 20–50%).
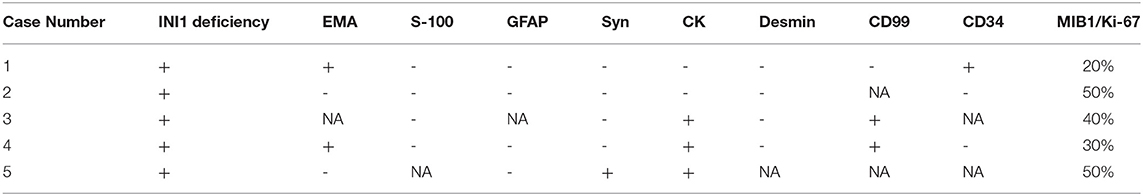
Table 3. Immunohistochemical characteristics of five adult patients with sellar region AT/RT.
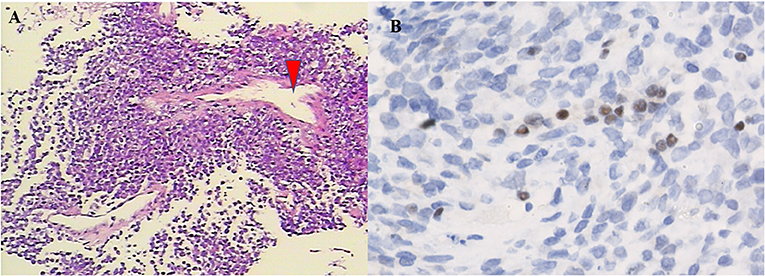
Figure 2. Pathological features of the tumor. On H&E staining (A, ×100), the tumor was composed of atypical cells with pink cytoplasm and prominent nuclei, which arranged in fascicular pattern forming a thin-walled stag-horn hemangiopericytoma-like vasculature (arrowhead). Immunohistochemistry. The tumor cells lack expression of the INI1 protein (B, ×400).
All five patients underwent surgical resection via endoscopic transnasal sphenoidal approach. Since the tumor exhibited blurred boundaries with the surrounding structure and invaded into the cavernous sinus and brain tissue, only one patient underwent GTR whereas the other four underwent STR. No surgery-related complications were observed during the early post-operative period. On 1-month post-operation, two patients (case 4 and case 1) had an excellent KPS score of 100 and 90, respectively. However, one patient (case 2) experienced disease progression (KPS score, 50) and two patients (case 3 and case 5) died (KPS score, 0). One patient (case 4) who survived 8 months received post-operative adjuvant radiotherapy with a total dose of 60 Gy applied in 30 fractions and local gamma knife radiation therapy once. One patient (case 1), with an OS of 4 months, received 52 Gy radiation in 28 fractions followed by six cycles of chemotherapy (cisplatin and dacarbazine). Because of the advanced age and poor medical condition of case 5, the family refused post-operative adjuvant radiotherapy and chemotherapy. Two patients (case 2 and case 3) showed rapid disease progression after resection, leading to deterioration of their condition, and therefore did not complete post-operative RT and chemotherapy course. Clinical follow-up after surgery was available for all five patients. At the last follow-up, the median RFS was 1 month (range, 0.5–2 months) and the median OS was 2 months (range, 1–8 months).
All searches generated 235 articles in total. After carefully reading of the titles and abstracts, 188 irrelevant articles, 3 non-English, and 10 repeated articles were excluded. Thirty-four relevant reports were selected for a full-text review. Finally, the remaining 26 reports are summarized in Table 4 (7–32).
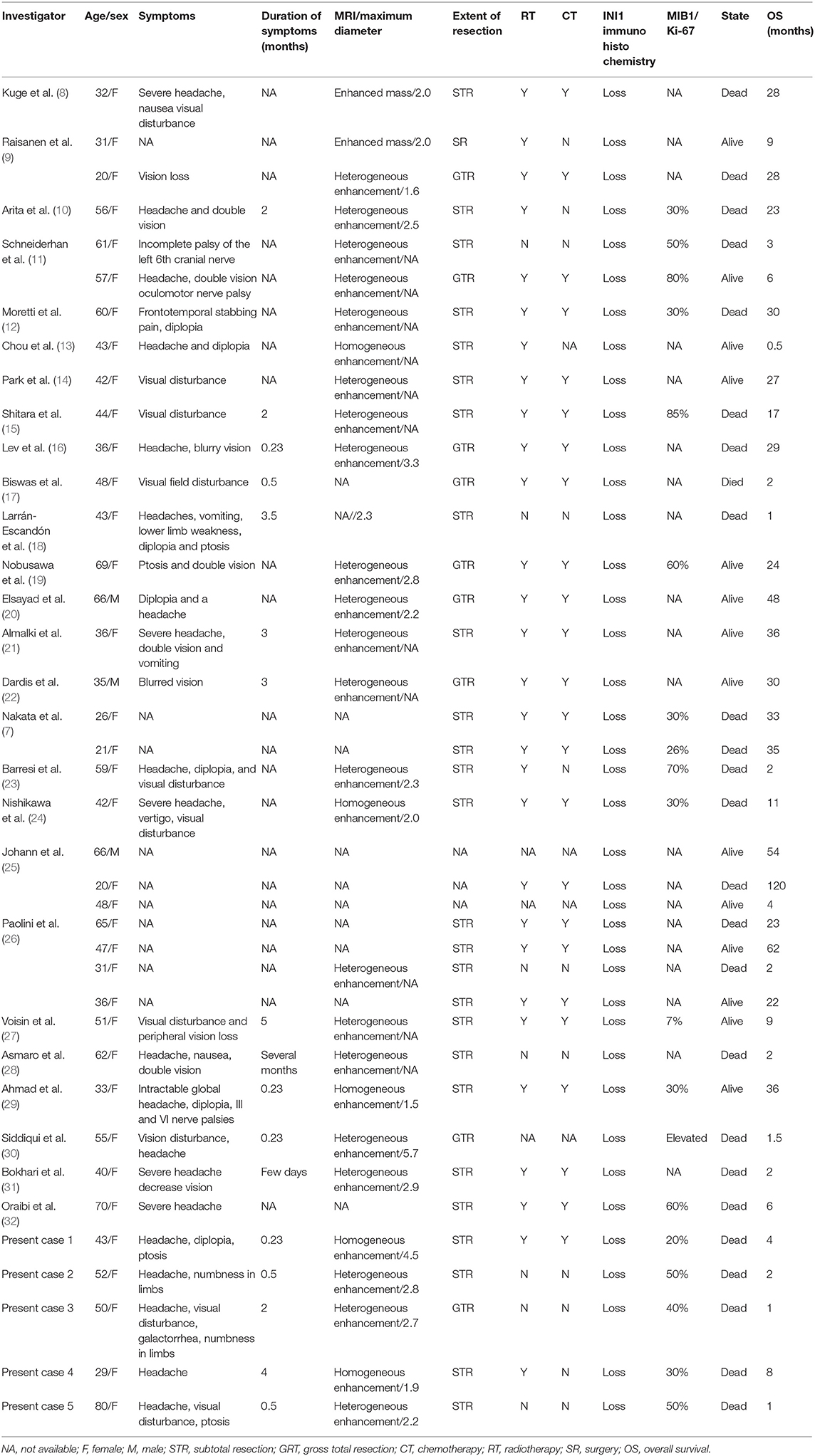
Table 4. Key characteristics of reported cases of adult sellar region AT/RT.
Since the initial description of adult sellar AT/RT, 39 cases, including our case series, constitute the current main body of reported data on adult sellar AT/RT. The estimated median OS was 23 months (range, 1–120 months) with a 1-year survival estimate of 59.7%. The mean age was 46.3 ± 15.1 years. Of the 39 patients, 36 (94.9%) were female and 3 (7.7%) were male. The symptoms were reported in 29 patients, and 20 patients (92.3%) suffered from headache and visual disturbance. The characteristics of contrast enhancement data were available for 26 patients, and the heterogeneous enhancement was observed in 21 of these patients (72.4%). The characteristics of contrast enhancement (p = 0.621) did not have a significant effect on the OS in Kaplan–Meier analysis. In addition, the median value of MIB1/Ki-67 was 35% (range, 7–85%) in 18 patients, and those patients were classified into two groups by the median value of MIB1/Ki-67. Kaplan–Meier analysis showed that patients with high (≥35%) MIB-1/Ki67 index value had a significantly shorter OS compared with those with low (<35%) index value (p = 0.033; Figure 3A). Furthermore, of the 39 patients, 36 underwent surgical resection including 26 STR, 9 GTR, and one with an unknown extent of resection, while the treatment information for the remaining 3 patients were unavailable. The median OS for patients with GTR was 28 and 17 months for patients with STR. However, the Kaplan–Meier analysis results did not show any significant difference between extent of surgical resection and OS (p = 0.501). The data of post-operative adjuvant therapy were available in 35 patients. Twenty-four received combination radio- and chemotherapy; four received radiation therapy alone after resection and none of the cases received chemotherapy alone. The Kaplan–Meier analysis demonstrated that patients who received post-operative combination radio- and chemotherapy had a significantly longer OS than that of those who did not (p < 0.001; Figure 3B). In addition, the role of radiotherapy toward adult sellar AT/RT could not be affirmatively concluded because of the small sample size.
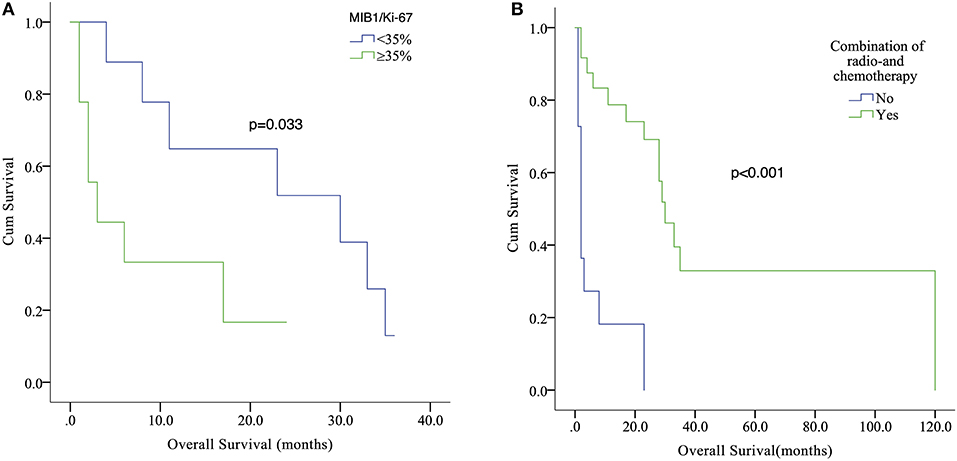
Figure 3. Kaplan–Meier analysis showed that patients with high (≥35%) MIB-1/Ki67 index value had a significantly shorter OS compared with those with low (<35%) index value (p = 0.033) (A). Relationship between post-operative combination radio- and chemotherapy (Yes or No) and survival outcome (p < 0.001) (B).
Adult sellar region AT/RT is an extremely rare and fast-growing neoplasm of the CNS. To the best of our knowledge, only 34 cases of adult sellar region AT/RT were previously reported in English-language journals in the form of case reports or retrospective case series. A literature search of all previous reports of adult sellar region AT/RT are summarized in Table 4 (7–32).
Thus far, only three cases of adult male patients with sellar region AT/RT have been described (20, 22, 25), indicating a female gender preponderance, which was also reflected in our case series. This suggests that women are susceptible to sellar region AT/RT, but further studies are warranted to reach a definitive conclusion. Interestingly, in the pediatric AT/RT population, there is a higher incidence in males (5, 33). Furthermore, pediatric AT/RT occurs chiefly <3 years old (34). The mean age was 46.3 ± 15.1 years in the 39 cases discussed here. The average symptom duration of adult sellar region AT/RT in our case series was 1.6 months (range, 2 weeks−8 months), which was consistent with previously reported studies (range, 1 week−5 months) (10, 15–18, 21, 22, 27). Because other sellar region diseases may exhibit similar symptoms, the clinical manifestations of adult sellar region AT/RT are likely non-specific, and mainly depend on the tumor size. Indeed, the most frequently reported symptoms were headache and visual disturbance (1–3, 7, 9, 14, 15, 21–26, 28–31, 33–38). Remarkably, headache in the five cases was usually severe and unbearable, characterized by whole-brain distending pain that was not mitigated by mild analgesics or weak opioids.
Pre-surgery levels of serum pituitary hormone levels are non-specific for differential diagnosis of adult sellar region AT/RT because pituitary hormone deficiency is also seen in large non-functioning pituitary adenoma (35). There are contradicting reports of serum pituitary hormone levels within the normal range before surgery (12, 23, 24) or diminished levels in adult sellar region AT/RT patients (16, 21). Accordingly, in our study, pituitary hormone deficiency was found in four patients, and anterior pituitary function was normal in one patient. The cause of hypopituitarism may be related to the lesion compression or invasion of the normal pituitary gland.
Radiologic features are crucial for pre-operative diagnosis of adult sellar region AT/RT and CT and MRI are the common modalities of choice for diagnosing these tumors. CT scan of the adult AT/RT usually show either isodense or, rarely, hyperdense lesions (12, 18, 28), and accordingly, 80% of adult sellar region AT/RTs in our case series were isodense, and 20% hyperdense. Rare hyperdense lesions of sellar region AT/RT usually indicate intratumor hemorrhage, which was confirmed in the present study and Asmaro et al. (28) Hyperdense sellar and suprasellar mass also should be differentiated from craniopharyngioma or pituitary macroadenoma by CT imaging. It was isointense on T1-weighted, hyperintense or isointense on T2-weighted images (14, 16, 21). The lack of calcification in our five adult sellar region AT/RT cases and the accompanying features of the lesions, namely, irregular shapes, and involving brain tissue, have not been reported in the literature. On MRI, the lesions extended into the cavernous sinus, compressing and displacing the optic chiasm, and involved the surrounding brain tissue. In addition, the majority of sellar region AT/RT showed heterogeneous enhancements, and characteristics of contrast enhancement did not have a significant effect on the OS. Noticeably, in the sellar region, pituitary tumors that are relatively regular and hourglass in shape often exhibit homogeneous enhancements. Furthermore, the slow growth of pituitary tumors relative to the sellar region AT/RT. Thus far, none of pediatric sellar AT/RT was reported (27). Therefore, the radiological features of the pediatric sellar AT/RT were unclear.
The morphological and immunohistochemical features of the tumor predominantly dictate diagnosis of AT/RT. Intraoperatively, soft-textured tumors colored gray-white or gray-red with abundant blood supply and blurred boundaries with surrounding structure that had invaded the cavernous sinus and clivus were observed in our case series. Histologically, the AT/RT tumors were densely packed with rhabdoid, neuroepithelial, and epithelial cells that were teratoid in nature (36). In our study, most lesions in the adult sellar region AT/RT had hemangiopericytoma-like stag-horn vasculature on histopathologic examination, which may differ from that of non-sellar region AT/RT (7, 19). Immunohistochemical findings of cellular markers in the adult sellar region AT/RT of our cases were in concordance to that observed previously (9, 37). The lack of expression of S-100, GFAP, and desmin in adult sellar region AT/RT in our cases is neither universal nor specific because these markers can be positive in some adult sellar region AT/RTs (11, 12, 16). Noticeably, the loss of INI1/SMARCB1/SMARCA4/BRG1 protein expression has been reported as a main feature of AT/RT in 34 cases examined in the literature (3, 39), which was similar to the five cases in our study.
The robust expression of MIB1/Ki-67 may imply the highly proliferative nature of an aggressive tumor. The median value of MIB1/Ki-67 was 35% in our pooled analysis (range, 7–85%). The Kaplan–Meier analysis showed that patients with high (≥35%) MIB-1/Ki67 index value had a significantly shorter OS compared with those with low (<35%) index value (p = 0.033). It was our novel finding that high levels (≥35%) of MIB1/Ki-67 in the adult sellar region AT/RT may indicate aggressive feature.
Because of the rareness of adult sellar region AT/RT, a consistent treatment protocol has not yet been established. However, a multimodal approach including surgical resection, radiation, and chemotherapy has been applied for the treatment of adult sellar region AT/RT. Although surgical resection is the main treatment, the goal of gross total resection is difficult to achieve because of the unclear boundaries, abundant blood supply, and invasion of the tumor into the cavernous sinus and clivus. Of the 39 cases, 36 underwent surgical resection including 26 STR, 9 GTR, and one with an unknown extent of resection (9), whereas the treatment information for the remaining three patients were unavailable (25). Furthermore, the extent of resection on OS remains unclear. Whereas, some studies noted a favorable outcome with GTR (15, 38, 40), others found no difference in survival between those who received GTR and STR (5, 41). We found that the median OS for patients with GTR was 28 months and 17 months for patients with STR. However, the Kaplan–Meier analysis results did not show any significant difference between extent of surgical resection and OS. Nevertheless, considering that adult sellar region AT/RT is aggressive in nature and fast-growing, maximal safe resection may be the optimal strategy, although the prognostic impact of extent of resection on sellar region AT/RT remains controversial. For pediatric patients with AT/RT, some studies have reported that GTR was likely beneficial when compared with STR (33, 42). However, other studies found no significant difference in the survival based on the extent of resection (35, 43).
Although post-operative chemotherapy using ifosfamide, carboplatin, and etoposide is an important supplementary therapy for AT/RT, and doxorubicin, methotrexate, and temozolomide have been explored for adult sellar region AT/RT (7, 16, 17, 19), the best chemotherapeutic regimen for adult AT/RT remains unclear (44, 45). Some of these agents can favorably penetrate into the intraparenchymal tumor and cerebrospinal fluid (44, 45). Because none of the 34 previous cases and in our case series received chemotherapy alone after resection, the responsiveness of adult sellar region AT/RT to chemotherapy cannot be assessed and thus, remains to be proven. However, chemotherapy has shown benefits; Hilden et al. (46) showed that post-operative chemotherapy achieved complete (6 patients) or partial (6 patients) response in 12 of 22 pediatric AT/RT cases.
The benefits of radiotherapy have been explored to treat and control both primary AT/RT and micro-metastatic disease (47, 48). Buscariollo et al. (35) reported that the median survival of 144 patients with AT/RT who received radiotherapy improved from 6 to 34 months. Another study documented that post-operative local radiotherapy for AT/RT with 54 Gy in 1.8 Gy daily fractions increased potential survival (20). Besides, several factors such as dose, technique, and timing can affect the outcomes from radiotherapy. Elsayed et al. (20) indicated that focal radiation doses ranging from 30 to 60 Gy can improve local control of AT/RT. Chen et al. (49) showed an increased disease-free survival in AT/RT from a total dose of more than 50 Gy and found that any delay in initiation of radiation therapy had an adverse effect on prognosis. Similarly, Tekautz et al. (48) also revealed that radiotherapy delivered early or late after surgical resection was associated with survival. Three cases from our literature review of 34 cases of adult sellar region AT/RT, and one patient (case 4) in our case series, who had an OS of 8 months, received post-operative radiotherapy alone. Because of these small sample sizes, we cannot further evaluate the OS benefits of radiotherapy alone in adult sellar region AT/RT.
The role of combination of radio- and chemotherapies among adult sellar region AT/RT patients remains to be determined (14, 26). We attempted to explore the effect of post-operative combination of radio- and chemotherapy on outcomes in adult sellar region AT/RT patients. In our pooled analysis, the data of post-operative adjuvant therapy was available in 35 patients, and 24 received combination radio- and chemotherapy. The Kaplan–Meier analysis demonstrated that patients who received post-operative combination radio- and chemotherapy had a significantly longer OS than that of those who did not (p < 0.001). Our results showed that combination of radio- and chemotherapy may be beneficial for adult sellar region AT/RT patients after surgical resection. In addition, further clinical studies may be needed to verify its efficacy. When it comes to the combination of radio- and chemotherapy for pediatric AT/RT, some of studies have demonstrated that the post-operative combination of radio- and chemotherapy was associated with better outcomes (33, 48).
Despite efforts to develop multimodal treatments, the overall prognosis of adult sellar region AT/RT remains poor. We found that adult patients with sellar region AT/RT had a median OS of 23 months (range, 1–120 months) in the 39 cases discussed here with a 1-year survival estimate of 59.7%, which was significantly longer than the median OS of 11.1 months among children with AT/RT (50). To improve OS, the novel treatment approaches are urgently needed for adult and pediatric AT/RT.
This study is not without limitations. First of all, it was limited by the retrospective nature and small sample size. Some of the results were based on pooled data from various cases or case series; therefore, heterogeneities in patient populations and treatment regimens should be kept in mind when interpreting the results. In addition, the information about MRI spine was lacking. As a result, the rarity of the disease limits additional statistical analyses, and finally, data on immunohistochemical staining were incomplete. Even though our study was the largest retrospective study on adult sellar region AT/RT until now, which can be helpful in the comprehensive understanding and management of adult sellar region AT/RT. Further prospective multicenter studies with larger sample sizes are warranted to support our results and identify the best treatment strategy for adult patients with sellar region AT/RT.
Adult sellar region AT/RT is a rapidly growing rare tumor in the CNS with a poor prognosis. Headache, usually severe and unbearable, is the main symptom. The disease shows a striking female predominance, which is its most prominent feature. Adult sellar region AT/RT usually exhibits irregular shape and heterogeneous enhancement and frequently extends into the cavernous sinus, involving even the surrounding brain tissue on MRI. The CT scan usually shows an isodense mass and rarely calcification. It was our novel finding that high levels of MIB1/Ki-67 may indicate aggressive feature. When an adult female patient has these features, it is recommended to include adult AT/RT in differential diagnosis of unclear malignant sellar lesions. Maximal safe resection followed by adjuvant radiotherapy combined with chemotherapy may be the optimal therapeutic strategy for adult sellar region AT/RT, and further clinical studies may be needed to verify its efficacy.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Sichuan University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
FL and ShucF: data curation. FL and XT: formal analysis. ShuaF and ShucF: methodology. FL: writing—original draft. LZ: writing—review and editing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AT/RT, Atypical teratoid/rhabdoid tumor; CNS, Central nervous system; GTR, Gross total resection; KPS, Karnofsky performance scale; OS, Overall survival; RFS, Recurrence-free survival; STR, Subtotal resection.
1. Horiguchi H, Nakata S, Nobusawa S, Uyama S, Miyamoto T, Ueta H, et al. Adult-onset atypical teratoid/rhabdoid tumor featuring long spindle cells with nuclear palisading and perivascular pseudorosettes. Neuropathology. (2017) 37:52–7. doi: 10.1111/neup.12317
2. Wu WW, Bi WL, Kang YJ, Ramkissoon SH, Prasad S, Shih HA, et al. Adult atypical teratoid/rhabdoid tumors. World Neurosurg. (2016) 85:197–204. doi: 10.1016/j.wneu.2015.08.076
3. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
4. Horn M, Schlote W, Lerch KD, Steudel WI, Harms D, Thomas E. Malignant rhabdoid tumor: primary intracranial manifestation in an adult. Acta Neuropathol. (1992) 83:445–8. doi: 10.1007/BF00713540
5. Chan V, Marro A, Findlay JM, Schmitt LM, Das S. A systematic review of atypical teratoid rhabdoid tumor in adults. Front Oncol. (2018) 8:567. doi: 10.3389/fonc.2018.00567
6. Woehrer A, Slavc I, Waldhoer T, Heinzl H, Zielonke N, Czech T, et al. Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer. (2010) 116:5725–32. doi: 10.1002/cncr.25540
7. Nakata S, Nobusawa S, Hirose T, Ito S, Inoshita N, Ichi S, et al. Sellar atypical teratoid/rhabdoid tumor (AT/RT): a clinicopathologically and genetically distinct variant of AT/RT. Am J Surg Pathol. (2017) 41:932–40. doi: 10.1097/PAS.0000000000000845
8. Kuge A, Kayama T, Tsuchiya D, Kawakami K, Saito S, Nakazato Y, et al. Suprasellar primary. malignant rhabdoid tumor in an adult: a case report. No Shinkei Geka. (2000) 28:351–8.
9. Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A. Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol. (2005) 15:23–8. doi: 10.1111/j.1750-3639.2005.tb00096.x
10. Arita K, Sugiyama K, Sano T, Oka H. Atypical teratoid/rhabdoid tumour in sella turcica in an adult. Acta Neurochir (Wien). (2008) 150:491–5; discussion 96. doi: 10.1007/s00701-008-1500-y
11. Schneiderhan TM, Beseoglu K, Bergmann M, Neubauer U, Macht S, Hanggi D, et al. Sellar atypical teratoid/rhabdoid tumours in adults. Neuropathol Appl Neurobiol. (2011) 37:326–9. doi: 10.1111/j.1365-2990.2010.01111.x
12. Moretti C, Lupoi D, Spasaro F, Chioma L, Di Giacinto P, Colicchia M, et al. Sella turcica atypical. teratoid/rhabdoid tumor complicated with lung metastasis in an adult female. Clin Med Insights Case Rep. (2013) 6:177–82. doi: 10.4137/CCRep.S12834
13. Chou S, Lo S, Wong H, Lau P, Cheung Y. Atypical teratoid / rhabdoid tumour in the sella turcica of a female adult. Hong Kong J Radiol. (2013) 16:65–8. doi: 10.12809/hkjr1311034
14. Park HG, Yoon JH, Kim SH, Cho KH, Park HJ, Kim SH, et al. Adult-onset sellar and suprasellar. atypical teratoid rhabdoid tumor treated with a multimodal approach: a case report. Brain Tumor Res Treat. (2014) 2:108–13. doi: 10.14791/btrt.2014.2.2.108
15. Shitara S, Akiyama Y. Atypical teratoid/rhabdoid tumor in sellar turcica in an adult: a case report and review of the literature. Surg Neurol Int. (2014) 5:75. doi: 10.4103/2152-7806.133105
16. Lev I, Fan X, Yu R. Sellar atypical teratoid/rhabdoid tumor: any pre-operative diagnostic. Clues? AACE Clinical Case Rep. (2015) 1:2–7. doi: 10.4158/EP14337.CR
17. Biswas S, Wood M, Joshi A, Bown N, Strain L, Martinsson T, et al. Exome sequencing of an adult pituitary atypical teratoid rhabdoid tumor. Front Oncol. (2015) 5:236. doi: 10.3389/fonc.2015.00236
18. Larrán-Escandón L, Mateo-Gavira I, Vilchez-López FJ, Gómez Cárdenas E, Aguilar Diosdado. M. Pituitary apoplexy as presentation of atypical teratoid/rhabdoid tumor in an adult. Endocrinología y Nutrición. (2016) 63:364–5. doi: 10.1016/j.endoen.2016.08.007
19. Nobusawa S, Hirato J, Sugai T, Okura N, Yamazaki T, Yamada S, et al. Atypical. Teratoid/Rhabdoid Tumor (AT/RT) arising from ependymoma: a type of AT/RT secondarily developing from other primary central nervous system tumors. J Neuropathol Exp Neurol. (2016) 75:167–74. doi: 10.1093/jnen/nlv017
20. Kriz J, Samhouri L, Haverkamp U, Straeter R, Stummer W, et al. Long-term survival. following additive radiotherapy in patients with atypical teratoid rhabdoid tumors. Strahlenther Onkol. (2016) 192:569–81. doi: 10.1007/s00066-016-0978-8
21. Almalki MH, Alrogi A, Al-Rabie A, Al-Dandan S, Altwairgi A, Orz Y. Atypical teratoid/rhabdoid tumor of the sellar region in an adult with long survival: case report and review of the literature. J Clin Med Res. (2017) 9:216–20. doi: 10.14740/jocmr2922w
22. Dardis C, Yeo J, Milton K, Ashby LS, Smith KA, Mehta S, et al. Atypical teratoid rhabdoid. Tumor: two case reports and an analysis of adult cases with implications for pathophysiology and treatment. Front Neurol. (2017) 8:247. doi: 10.3389/fneur.2017.00247
23. Barresi V, Lionti S, Raso A, Esposito F, Cannavo S, Angileri FF. Pituitary atypical teratoid. rhabdoid tumor in a patient with prolactinoma: a unique description. Neuropathology. (2018) 38:260–67. doi: 10.1111/neup.12440
24. Nishikawa A, Ogiwara T, Nagm A, Sano K, Okada M, Chiba A, et al. Atypical teratoid/rhabdoid. tumor of the sellar region in adult women: is it a sex-related disease? J Clin Neurosci. (2018) 49:16–21. doi: 10.1016/j.jocn.2017.12.010
25. Johann PD, Bens S, Oyen F, Wagener R, Giannini C, Perry A, et al. Sellar region atypical. Teratoid/Rhabdoid Tumors (ATRT) in adults display DNA methylation profiles of the ATRT-MYC subgroup. Am J Surg Pathol. (2018) 42:506–11. doi: 10.1097/PAS.0000000000001023
26. Paolini MA, Kipp BR, Sukov WR, Jenkins SM, Barr Fritcher EG, Aranda D, et al. Sellar region. Atypical Teratoid/Rhabdoid Tumors in adults: clinicopathological characterization of five cases and review of the literature. J Neuropathol Exp Neurol. (2018) 77:1115–21. doi: 10.1093/jnen/nly091
27. Voisin MR, Ovenden C, Tsang DS, Gupta AA, Huang A, Gao AF, et al. Atypical. Teratoid/Rhabdoid sellar tumor in an adult with a familial history of a germline SMARCB1 mutation: case report and review of the literature. World Neurosurg. (2019) 127:336–45. doi: 10.1016/j.wneu.2019.04.083
28. Asmaro K, Arshad M, Massie L, Griffith B, Lee I. Sellar Atypical Teratoid/Rhabdoid Tumor. Presenting with subarachnoid and intraventricular hemorrhage. World Neurosurg. (2019) 123:e31–e38. doi: 10.1016/j.wneu.2018.10.198
29. Ahmad M, Sekar V, Healy E, Rennie I, Cooke S. Case report: the unusual case of a sellar/suprasellar Extrarenal Rhabdoid tumour in an adult. Interdiscip Neurosurg. (2019) 17:36–9. doi: 10.1016/j.inat.2019.01.014
30. Siddiqui M, Thoms D, Samples D, Caron J. Atypical teratoid/rhabdoid tumor presenting with. subarachnoid and intraventricular hemorrhage. Surg Neurol Int. (2019) 10:139. doi: 10.25259/SNI-59-2019
31. Bokhari RA, Bafaqeeh M, Al-Obaysi S, Al-Aman A, Alshakweer W. Atypical. Teratoid/Rhabdoid tumor of the sellar region: a case report and review of the literature. J Neurol Res. (2020) 10:13–6. doi: 10.14740/jnr556
32. Oraibi O, Mau C, Daggubati L, Khormi Y, Manni A, Zacharia B. MON-262 a case of the. suprasellar atypical teratoid rhabdoid tumor (ATRT) presenting in an adult treated with intrathecal chemotherapy. J Endocr Soci. (2020) 4:1261. doi: 10.1210/jendso/bvaa046.1261
33. Ma XJ, Li D, Wang L, Hao SY, Zhang LW, Zhang JT, et al. Overall survival of primary. Intracranial atypical teratoid rhabdoid tumor following multimodal treatment: a pooled analysis of individual patient data. Neurosurg Rev. (2020) 43:281–92. doi: 10.1007/s10143-018-1055-9
34. Fischer-Valuck BW, Chen I, Srivastava AJ, Floberg JM, Rao YJ, King AA, et al. Assessment of the treatment approach and survival outcomes in a modern cohort of patients with atypical teratoid rhabdoid tumors using the National Cancer Database. Cancer. (2017) 123:682–87. doi: 10.1002/cncr.30405
35. Buscariollo DL, Park HS, Roberts KB, Yu JB. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer. (2012) 118:4212–9. doi: 10.1002/cncr.27373
36. Avci Z Kaya IS Dogukan AS Aydin O Ismailoglu O. Pathology teach and tell: central nervous system atypical teratoid/rhabdoid tumor. Pediatr Pathol Mol Med. (2003) 22:443–7. doi: 10.1080/pdp.22.5.443.447
37. Las Heras F, Pritzker KP. Adult variant of atypical teratoid/rhabdoid tumor: immunohistochemical and ultrastructural confirmation of a rare tumor in the sella tursica. Pathol Res Pract. (2010) 206:788–91. doi: 10.1016/j.prp.2010.07.004
38. Fruhwald MC, Biegel JA, Bourdeaut F, Roberts CW, Chi SN. Atypical teratoid/rhabdoid. Tumors-current concepts, advances in biology, and potential future therapies. Neuro-oncology. (2016) 18:764–78. doi: 10.1093/neuonc/nov264
39. Chhabda S, Carney O, D'Arco F, Jacques TS, Mankad K. The 2016 World Health Organization classification of tumours of the Central Nervous System: what the paediatric neuroradiologist needs to know. Quant Imaging Med Su. (2016) 6:486–89. doi: 10.21037/qims.2016.10.01
40. Athale UH, Duckworth J, Odame I, Barr R. Childhood atypical teratoid rhabdoid tumor of the. Central nervous system: a meta-analysis of observational studies. J Pediat Hematol Oncol. (2009) 31:651–63. doi: 10.1097/MPH.0b013e3181b258a9
41. Biswas A, Kashyap L, Kakkar A, Sarkar C, Julka PK. Atypical teratoid/rhabdoid tumors: challenges and search for solutions. Cancer Manag Res. (2016) 8:115–25. doi: 10.2147/CMAR.S83472
42. Fossey M, Li H, Afzal S, Carret AS, Eisenstat DD, Fleming A, et al. Atypical teratoid rhabdoid. Tumor in the first year of life: the Canadian ATRT registry experience and review of the literature. J Neurooncol. (2017) 132:155–62. doi: 10.1007/s11060-016-2353-0
43. Yang WC, Yen HJ, Liang ML, Chen HH, Lee YY, Wong TT, et al. Role of early and aggressive. post-operative radiation therapy in improving outcome for pediatric central nervous system atypical teratoid/rhabdoid tumor. Childs Nerv Syst. (2019) 35:1013–20. doi: 10.1007/s00381-019-04126-y
44. Jacobs S, McCully CL, Murphy RF, Bacher J, Balis FM, Fox E. Extracellular fluid. concentrations of cisplatin, carboplatin, and oxaliplatin in brain, muscle, and blood measured using microdialysis in nonhuman primates. Cancer Chemoth Pharm. (2010) 65:817–24. doi: 10.1007/s00280-009-1085-7
45. Muldoon LL, Soussain C, Jahnke K, Johanson C, Siegal T, Smith QR, et al. Chemotherapy. delivery issues in central nervous system malignancy: a reality check. J Clin Oncol. (2007) 25:2295–305. doi: 10.1200/JCO.2006.09.9861
46. Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, et al. Central nervous. System atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. (2004) 22:2877–84. doi: 10.1200/JCO.2004.07.073
47. Balaton AJ, Vaury P, Videgrain M. Paravertebral malignant rhabdoid tumor in an adult. A case report with immunocytochemical study. Pathol Res Pract. (1987) 182:713–8.
48. Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, et al. Atypical. teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol. (2005) 23:1491–9. doi: 10.1200/JCO.2005.05.187
49. Chen YW, Wong TT, Ho DM, Huang PI, Chang KP, Shiau CY, et al. Impact of radiotherapy for. pediatric CNS atypical teratoid/rhabdoid tumor (single institute experience). Int J Radiat Oncol Biol Phys. (2006) 64:1038–43. doi: 10.1016/j.ijrobp.2005.10.001
50. Schrey D, Carceller Lechon F, Malietzis G, Moreno L, Dufour C, Chi S, et al. Multimodal. therapy in children and adolescents with newly diagnosed atypical teratoid rhabdoid tumor: individual pooled data analysis and review of the literature. J Neurooncol. (2016) 126:81–90. doi: 10.1007/s11060-015-1904-0
Keywords: atypical teratoid/rhabdoid tumor, clinicopathologic and radiologic features, adult female, sella region, treatment
Citation: Liu F, Fan S, Tang X, Fan S and Zhou L (2020) Adult Sellar Region Atypical Teratoid/Rhabdoid Tumor: A Retrospective Study and Literature Review. Front. Neurol. 11:604612. doi: 10.3389/fneur.2020.604612
Received: 10 September 2020; Accepted: 16 November 2020;
Published: 15 December 2020.
Edited by:
Soumen Khatua, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Sumit Gupta, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2020 Liu, Fan, Tang, Fan and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liangxue Zhou, emhseGxsbGxAMTYzLmNvbQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.