 Jacky Ganguly
Jacky Ganguly Mandar Jog
Mandar Jog- Movement Disorder Centre, London Health Sciences Centre, University of Western Ontario, London, ON, Canada
The spectrum of tauopathy encompasses heterogenous group of neurodegenerative disorders characterized by neural or glial deposition of pathological protein tau. Clinically they can present as cognitive syndromes, movement disorders, motor neuron disease, or mixed. The heterogeneity in clinical presentation, genetic background, and underlying pathology make it difficult to classify and clinically approach tauopathy. In the literature, tauopathies are thus mostly highlighted from pathological perspective. From clinical standpoint, cognitive syndromes are often been focussed while reviewing tauopathies. However, the spectrum of tauopathy has also evolved significantly in the domain of movement disorders and has transgressed beyond the domain of primary tauopathies. Secondary tauopathies from neuroinflammation or autoimmune insults and some other “novel” tauopathies are increasingly being reported in the current literature, while some of them are geographically isolated. Because of the overlapping clinical phenotypes, it often becomes difficult for the clinician to diagnose them clinically and have to wait for the pathological confirmation by autopsy. However, each of these tauopathies has some clinical and radiological signatures those can help in clinical diagnosis and targeted genetic testing. In this review, we have exposed the heterogeneity of tauopathy from a movement disorder perspective and have provided a clinical approach to diagnose them ante mortem before confirmatory autopsy. Additionally, phenotypic variability of these disorders (chameleons) and the look-alikes (mimics) have been discussed with potential clinical pointers for each of them. The review provides a framework within which new and as yet undiscovered entities can be classified in the future.
Introduction
Tauopathies are a heterogeneous group of neurodegenerative disorders, pathologically characterized by neuronal and/or glial inclusions of the microtubule-binding protein, tau. Heterogeneity spans many domains from the clinical presentation, anatomical localization, genetic variations, and radiological and pathological signs. Neuroanatomical vulnerability may be a key to the heterogeneity (the concept of “molecular nexopathies”) (1). Many factors can be implicated including “strain” specificity of the tau protein, biochemical property of the abnormal protein according to its post-translational modification, “prion-like” propagation capacity, interaction with other co-existent proteins like alpha-synuclein or TDP43, seeding or “permissive templating” property, intrinsic vulnerability of the affected structure, genetic, and epigenetic factors and environmental influences (1, 2).
The spectrum of tauopathy is still unfolding and transcending beyond the domain of primary tauopathies. While secondary tauopathy from autoimmune insult like in anti-IgLON5 disease brings up the topic of complex interaction between autoimmunity and neurodegeneration (3), geographically isolated tauopathies highlight the environmental impact. Apart from this, some novel tauopathies are also increasingly being described in the literature (4).
Clinically, tauopathies present as movement disorders, dementia, and motor neuron disease, either in isolation or in varied combinations (5), based on the vulnerable anatomical structures being affected by the pathological protein accumulation. In terms of genetics, MAPT gene containing N terminal domain (N1, N2) and microtubule binding domain (R1, R2, R3, R4), on chromosome 17q21 encodes the protein tau. Due to alternative splicing of the MAPT gene, three repeat (2N3R, 1N3R, 0N3R) or four repeat (2N4R, 1N4R, 0N4R) tau isoforms are formed (6). On the other hand, depending upon numerous single nucleotide polymorphisms (SNPs) and a 900kb inversion, H2 and H1 haplotypes of MAPT gene are formed and have impact on the phenotypic presentation (7).
In the literature, tauopathy has been discussed mostly as a pathological entity with its detailed pathological intricacies. Pathological confirmation of the diagnosis of tauopathy mostly depends on autopsy findings. However, pathological diagnosis is often confounded by the presence of multiple other proteins and thus it becomes difficult to determine whether the accumulated tau is pathological or an innocent bystander. In vivo biomarkers like CSF tau and tau-PET imaging are still research-based tools. Additionally, each of these tauopathies has some clinical and radiological signature that can predict the underlying genetics and pathology.
In order to clarify this complexity of heterogeneity of tauopathies, in this review, we have approached tauopathy from a clinical standpoint highlighting mainly the movement disorder perspective, focused on the clinical presentations (chameleons) and their phenotypic look-alikes (mimics). A critical review of the current status of the classification of tauopathies will be followed by the clinical spectrum of primary, secondary, and geographically isolated tauopathies to understand the heterogeneity. Potential clinical and radiological clues will be discussed for each of them. Finally, a practical approach is presented to guide the clinician in day to day practice. Specifically, the phenotype of familial frontotemporal dementia with parkinsonism has been clinically dissected further at the end, because it is one of the commonest overlapping phenotypes of tauopathies.
Classification of Tauopathies—Current Status and Pitfalls
Tauopathies have been conventionally classified from a pathological perspective into two groups—(A) Primary tauopathies where tau is the predominant pathology including three repeat (3R-) and four repeat (4R-) tauopathies, (B) Secondary tauopathies where additional etiologies (e.g., amyloid, trauma, and autoimmune) are involved for tau deposition (Figure 1) (8). However, some tauopathies are geographically isolated like Guadeloupean parkinsonism (9), Western pacific amyotrophic lateral sclerosis and parkinsonism-dementia complex (ALS/PDC) (10), and Nodding syndrome of northern Uganda (11). While exact etiopathogenesis of these geographically isolated tauopathies are still unknown, environmental impact (discussed later) has been highlighted in many studies. Thus, whether to include them in the group of secondary tauopathies or not, is still a matter of debate.
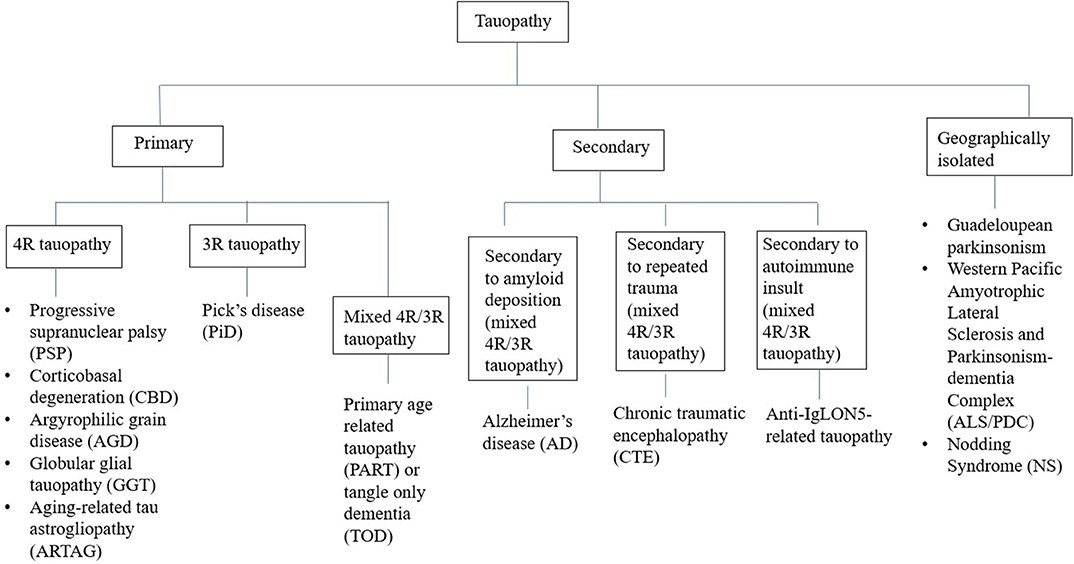
Figure 1. Classification of tauopathies.
Recently, Hõglinger et al. (12) have highlighted syndromic classification of tauopathy based on the predominant domain affected (cognitive or motor):
1. Cognitive syndromes: Behavioral variant of frontotemporal dementia (bvFTD), non-fluent agrammatic variant of primary progressive aphasia (nfavPPA), semantic variant of primary-progressive aphasia (svPPA), and amnestic syndrome of hippocampal type (AS).
2. Motor syndromes: Richardson syndrome (RS), Parkinson syndrome (P), corticobasal syndrome (CBS), primary gait freezing (PGF), cerebellar syndrome (C), and primary lateral sclerosis (PLS).
However, a primary tauopathy like progressive supranuclear palsy (PSP) or corticobasal degeneration (CBD) can present with different cognitive and motor syndromes (chameleons) and many a times there is phenotypic overlap of cognitive and motor syndrome like in familial FTD with parkinsonism linked to MAPT (FTDP-17).
Apart from these, tauopathies can be classified based on the etiology like genetic (e.g., MAPT related), autoimmune (e.g., anti IgLON5 related), traumatic (e.g., chronic traumatic encephalopathy), etc. It can also be classified based on the area of brain predominantly involved like frontal cortex (e.g., behavioral variant frontotemporal dementia/bvFTD, progressive supranuclear palsy-frontal variant/PSP-F), parietal cortex (e.g., corticobasal syndrome/CBS), peri-sylvian (e.g., progressive nonfluent aphasia/PNFA), limbic (e.g., argyrophilic grain disease/AGD), brainstem (e.g., progressive supranuclear palsy-Richardson's type/PSP-RS, anti IgLON5 related), or cerebellum (e.g., PSP-C).
Primary 4R- and 3R-Tauopathies
In this large group of primary tauopathies, CBD, GGT, AGD, and PiD are primarily pathological diagnosis where as corticobasal syndrome (CBS) is a clinical term. PSP can be described as both, a pathological or a clinical entity. In this review, PSP and CBS have been discussed with their phenotypic presentations (chameleons) and look-alikes (mimics). PNFA/PPA-G has also been discussed because it is primarily a clinical diagnosis and its pathology is mostly FTLD-tau. In the literature, GGT, AGD, and PiD have been traditionally discussed from a pathological standpoint. We have highlighted the clinical and radiological clues for suspecting GGT, AGD, and PiD clinically before confirmatory autopsy. We have sub-classified primary tauopathies according to the predominant clinical presentation like, movement disorder (PSP, CBS), language dysfunction (PNFA) and cognitive dysfunction or mixed (GGT, AGD, and PiD). However, movement disorders can be associated with the second and the third subtype in varied proportions.
Predominant Movement Disorder Presentation
Progressive Supranuclear Palsy (PSP)
Axial rigidity, facial dystonia, retrocollis, vertical supranuclear gaze palsy (VSGP), early postural instability, and pseudobulbar palsy are clinical pointers for classic PSP or Richardson's phenotype (PSP-RS). Apart from this, PSP can have varied phenotypic presentations (chameleons) like parkinsonian type (PSP-P), progressive gait freezing (PSP-PGF), etc. (Table 1) (13–15). According to the latest MDS criteria, four core clinical features should be assessed for varying levels of certainty for PSP pathology: (i) oculomotor dysfunction (e.g., VSGP, slow vertical saccades, square wave jerks, and eyelid apraxia), (ii) postural instability within 3 years (e.g., spontaneous loss of balance, unprovoked falls, tendency to fall on pull-test), (iii) akinesia (e.g., progressive gait freezing, akinetic rigid, predominantly axial parkinsonism), and (iv) cognitive dysfunction (e.g., non-fluent aphasia, apraxia of speech, frontal cognitive/behavioral presentation) (16). Levodopa resistance (<30% improvement of the MDS UPDRS III score on a levodopa challenge), dysphagia, hypokinetic spastic dysarthria and photophobia are useful suggestive features of PSP (16). Dorsal midbrain atrophy is the characteristic radiological finding leading to “Morning glory,” “Mickey Mouse” signs on axial MR images and “Hummingbird,” “Penguin silhouette” signs on sagittal MR images (17). Midbrain/Pons (M/P) ratio < 0.52, midbrain AP diameter measurement < 9.35 mm (18), MR Parkinsonism Index (MRPI) > 13.55 (19), and MRPI 2.0 > 2.18 for PSP-P, > 2.50 for PSP-RS (20) are other helpful radiological signs. MAPT H1 haplotype, specially H1c sub-haplotype and recently described H1d, H1g, and H1o sub-haplotypes of MAPT are associated with increased risk of PSP. Classic PSP pathology is characterized by “tufted astrocytes” and “globose” neurofibrillary tangles. Predominantly PSP pathology is seen in the progressive gait freezing phenotype (PSP-PGF) and in Richardson's phenotype (PSP-RS), whereas in the other variants of PSP, the pathology is often mixed or of non-PSP pathology (21). Alzheimer's disease (AD) pathology and argyrophilic grains (AG) are commonly associated co-pathologies (22). Overall, the phenotypic presentation of PSP depends on the brain area that is more vulnerable to the pathological protein accumulation like frontal lobe in PSP-F, parietal lobe in PSP-CBS, temporal lobe in PSP-SL, midbrain in PSP-RS, basal ganglia (post-synaptic striatal) in PSP-P, pons in PSP-PGF and cerebellum in PSP-C (Table 1) (23, 24).

Table 1. Chameleons of PSP and CBS.
Corticobasal Syndrome (CBS)
Asymmetric parkinsonism, limb dystonia, myoclonus, saccadic apraxia, ideomotor apraxia, cortical sensory deficits, and alien limb phenomena are classic clinical clues for CBS. Like PSP, CBS also has varied phenotypic presentations apart from this classic phenotype. Though in the latest criteria for CBD by Armstrong et al. (30) four phenotypic presentations have been described, certain other presentations have been reported in the literature (Table 1). Cognitive presentation of CBD (CBD-Cog) mimicking bvFTD or AD is an increasingly recognized phenotype with apathy, executive dysfunction, language, and visuospatial problems (37). Asymmetric cortical atrophy predominantly affecting peri-rolandic region, posterior frontal, and parietal lobes (38) is the radiological hallmark of CBS, but the predominant area of atrophy varies with underlying pathology. CBS-CBD and CBS-PSP pathology: focal atrophy involving premotor cortex, posterior superior frontal lobe and supplementary motor area (SMA), CBS-TDP-43, and CBS-AD pathology: more widespread gray matter loss, CBS-TDP43 pathology: fronto-temporal involvement (particularly prefrontal cortex) and CBS-AD pathology: temporo-parietal involvement (particularly parietal cortex) (39). In 25–56% of cases, clinical diagnosis of CBS correlates with classic CBD pathology (CBD–CBS) (40). Astrocytic plaques, ballooned, or achromatic neurons and argyrophilic threads are pathological hallmarks of classic CBD pathology. However, CBS phenotype can be associated with various other pathological entities apart from classic CBD pathology like, AD pathology, PSP pathology, and FTLD-TDP43 pathology (40).
Because of the overlapping phenotypes, whether PSP and CBD are two different disorders or are part of a spectrum, is a matter of debate (41, 42). Phenotypically, PSP and CBD pathology both can present as speech/language (SL) dysfunction (agrammatic non-fluent aphasia/speech apraxia), frontal cognitive/behavioral presentation (F), Richardson's syndrome, and corticobasal syndrome (43). Interestingly, if a patient presents with VSGP or slowing of vertical saccade, axial or symmetric limb rigidity or akinesia, limb apraxia, and postural instability, the patient can be classified as PSP-CBS or CBD-PSP. To get rid of this conundrum, Movement Disorder Society (MDS) criteria (2017) for PSP have introduced the novel diagnostic category “probable 4R-tauopathy” for joint clinical recognition of the patients with PSP and CBD pathology and to facilitate the research on 4R-tau targeted therapeutic strategies (16, 44). Probable 4R-tauopathy includes “possible PSP with SL” and “possible PSP with CBS” apart from all “probable PSP.”
On top of this, vertical gaze palsy can be seen in a lot of other disorders apart from PSP-RS and asymmetric dystonic stiff limb presentation can be seen in other disorders besides CBS. Thus, clinicians should always be aware of these look-alikes (“mimics”) of PSP and CBS (45, 46) (Table 2).

Table 2. Mimics of PSP and CBS.
Predominant Language Dysfunction Presentation
Progressive Nonfluent Aphasia (PNFA) or Agrammatic Variant of Primary Progressive Aphasia (PPA-G)
PNFA/PPA-G can clinically present with apraxia of speech (AOS), agrammatism, or mixed. AOS manifests as slow, labored, effortful, hesitant speech with inconsistent speech sound error and aprosody. “Groping after the target sound” is characteristic. Patients have difficulty to utter polysyllabic words and sequences of syllables (e.g., “puh-tuh-kuh”) (109). Phonemic speech sound errors are more common (110). Errors in grammar mainly affects syntax, function words, use of conjunction and verb (109). Clinically, PPA-G must be differentiated from the semantic variant PPA-S (single word comprehension and object knowledge is affected with intact repetition, commonly TDP43 pathology) and logopenic variant PPA-L (word finding difficulty with “tip-of-the-tongue” hesitation and impaired repetition, commonly AD pathology) (111). Orofacial apraxia is a common association with PPA-G. Signs of PSP or CBS may arise as the disease evolves (109). Predominately left peri-sylvian atrophy involving left posterior fronto-insular region (inferior frontal gyrus and insula) is seen in MRI brain (111). Around 30% of the cases are genetic and association with MAPT, PGRN and C9orf72 have been reported (112). PSP pathology is common in AOS variant with more dysarthric presentation and CBD pathology with more sentence comprehension deficit (113). Sometimes PiD pathology, TDP43-A pathology if there is associated ALS (nfvPPA-ALS) or AD pathology is also seen (112–114).
Predominant Cognitive or Mixed Presentation
Three other primary tauopathies namely globular glial tauopathy (GGT), argyrophilic grain disease (AGD) and Pick's disease (PiD) present as cognitive or mixed (cognitive and movement disorder overlap). Clinical, radiological, and pathological features of these entities have been described in Table 3.
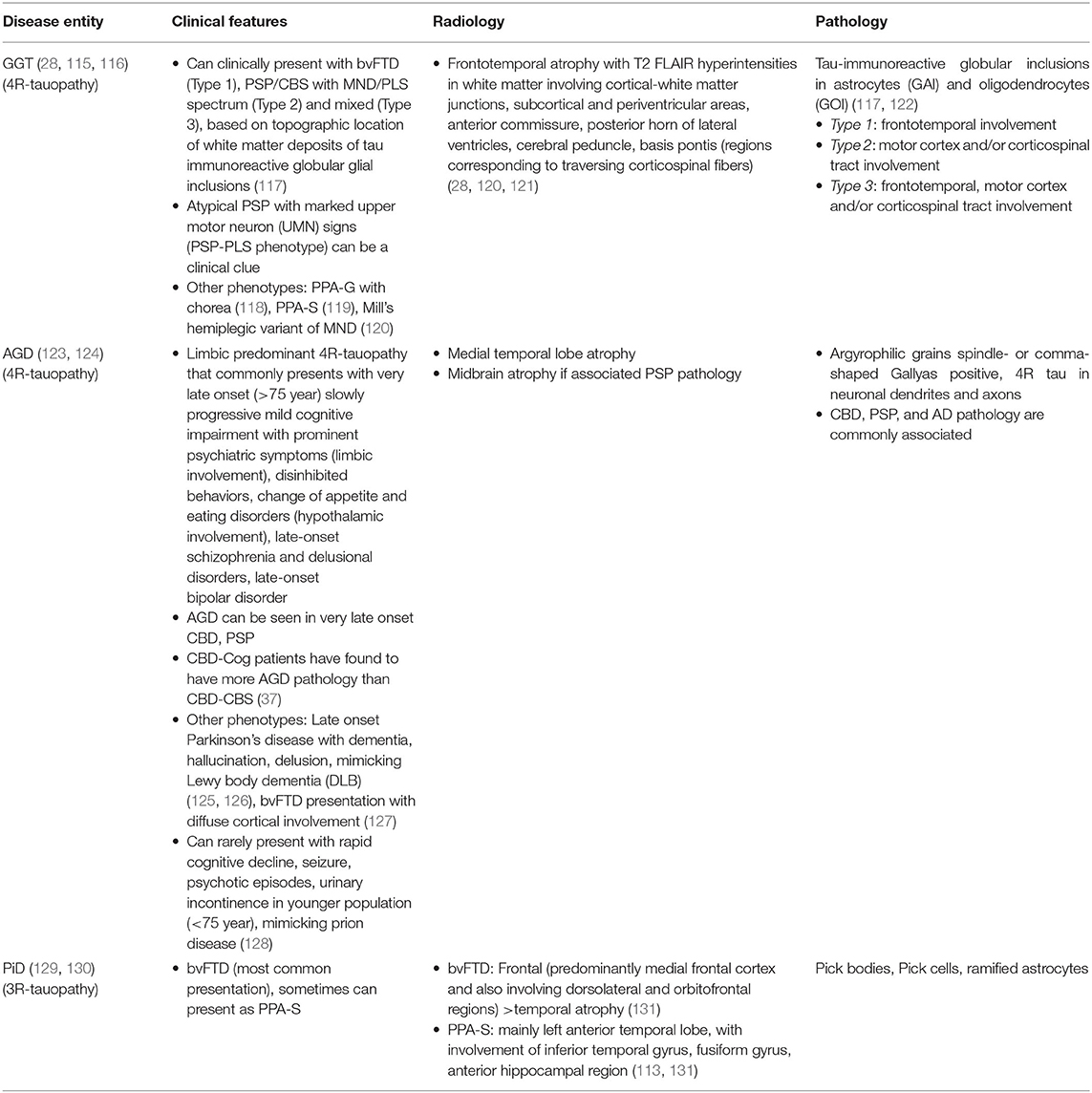
Table 3. Clinical, radiological, and pathological clues for GGT, AGD, and PiD.
Secondary Tauopathies
Anti IgLON5 Disease
Anti IgLON5 mediated secondary tauopathy stands at a critical juncture of autoimmunity and neurodegeneration, where deposition of hyperphosphorylated tau (both 3R and 4R) occurs mainly in the hypothalamus, brainstem, and hippocampus. Initially, it was described as an antibody mediated sleep disorder (132, 133). Subsequently, many other phenotypes (chameleons) have emerged and most of the times they overlap (51, 134–137) (Table 4). MRI brain is mostly normal or may show cerebellar, brainstem atrophy (138). Recognition of these clinical phenotypes of anti IgLON5 are necessary because of its treatability with immunomodulators can prevent further neurodegeneration.
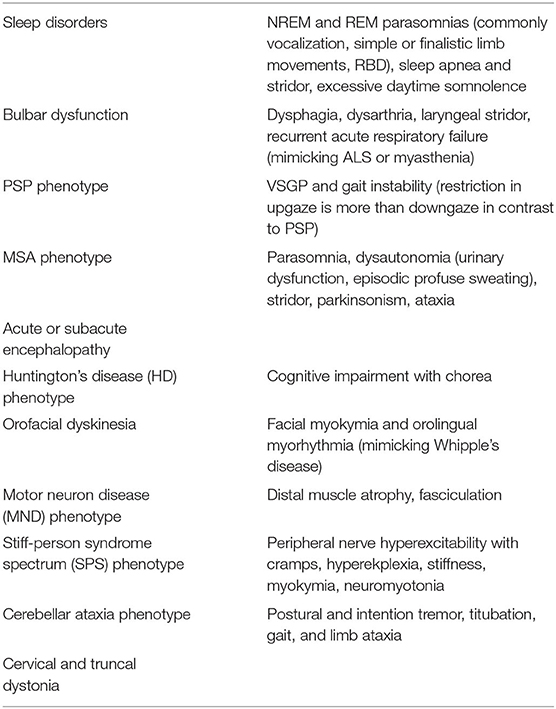
Table 4. Phenotypic presentations (chameleons) of IgLON5 disease.
Chronic Traumatic Encephalopathy (CTE)
CTE is mainly a neurocognitive syndrome related to repeated traumatic brain injury (TBI) where both 3R- and 4R- tau deposition is seen (like AD). TBI likely ignites a vicious cycle of neuroinflammation and tau phosphorylation, deposition (139). Susceptibility depends on multiple factors like carrying ApoE4 allele, cognitive reserve, etc. It was initially described in boxers and named as “punch-drunk syndrome” or “dementia puglistica.” Subsequently, the disease got noticed among athletes like football players and war veterans. Gardner et al. (140) has classified the older ones as “classic CTE” (parkinsonism followed by cognitive symptoms) and the recent ones as “modern CTE” (behavioral symptoms affecting mood/affect followed by cognitive symptoms). From clinical perspective, Jordan et al. (141) have divided CTE into three phenotypes: (1) Behavioral and psychiatric (aggression, impulsivity, delusions, depression, suicidality) that can mimic bvFTD; (2) Cognitive (affecting attention, executive, memory, visuospatial domains) that can mimic FTD or AD; (3) Motor (parkinsonism, ataxia, dysarthria, spasticity). Chronic post-concussive syndrome (CPCS) comes as differential but its temporal relation with the acute concussive event and the presence of headache are the helpful differentiating features (139). Pathologically CTE differs from AD, though both are secondary tauopathy with mixed 3R- and 4R-tau deposition. Perivascular deposition of tau positive NFTs along the depth of cortical sulci is the pathological hallmark of CTE. TDP-43 inclusions are more common in CTE, while Aß amyloid deposition is more in AD (142). Additionally, tau filaments in CTE have a unique ß-helix region with a hydrophobic cavity, containing cofactors necessary for tau aggregation and propagation (143).
Alzheimer's Disease (AD)
AD is the most common cause of dementia worldwide. Pathologically, extracellular Aß amyloid plaques and intracellular tau (mixed 3R and 4R) positive neurofibrillary tangles are seen (144). Apart from classic amnestic presentation, non-amnestic phenotypes of AD are increasingly being recognized like, language variant (e.g., logopenic aphasia with word finding difficulty), visuospatial variant (posterior cortical atrophy/PCA with impaired spatial cognition), behavioral variant (executive dysfunction with impaired reasoning, problem solving), and mixed cognitive-motor presentation (atypical parkinsonism) (145, 146). In the latest National Institute on Aging and Alzheimer's Association (NIA-AA) Research Framework criteria (2018), AD has been re-defined based on the underlying pathology (amyloid pathology/A, tau pathology/T and neurodegeneration/N) that can be documented in vivo by biomarkers or by post-mortem examination (147). From a movement disorder perspective, many studies have reported extrapyramidal signs including parkinsonism in AD with widely varied prevalence (20–100%) (148). Parkinsonism in AD is mostly unresponsive to levodopa and the patients with AD-parkinsonism phenotype usually show relatively rapid progression, severe deficits on neuropsychological testing and high frequency of major depression and dysthymia (149). However, in these scenarios of cognitive-motor overlap, the clinicians must differentiate cortical “pseudo-parkinsonian” features like ideomotor apraxia, paratonic rigidity, and frontal/higher level gait disorders from true parkinsonian (nigrostriatal) features like bradykinesia, lead-pipe rigidity, and parkinsonian/middle-level gait disorders (148). Dementia with Lewy bodies/DLB (occurrence of dementia prior to or within a year of onset of motor symptoms, cognitive fluctuation, well-formed visual hallucination, neuroleptic sensitivity, and autonomic dysfunction), Parkinson's disease dementia/PDD (onset of dementia after 1 year of parkinsonian motor symptoms) and Creutzfeldt–Jakob disease/CJD (rapid progression, cerebellar ataxia, seizure, and chorea) can mimic AD-parkinsonism phenotype with dementia, rigidity, and myoclonus (149). Lastly, genes responsible for early onset AD (EOAD) like presenilin 1 (PSEN1) and amyloid precursor protein (APP) can give rise to atypical parkinsonism like corticobasal syndrome (CBS) (95, 96). Besides the CBS phenotype, dystonia in AD can also be drug-induced (e.g., rivastigmine, mirtazapine, neuroleptics) (150–152). Choline esterase inhibitors (ChEIs) can induce truncal dystonia in the form of Pisa syndrome (tonic lateral flexion of the trunk) in patients of AD (153–155). Apart from parkinsonism, cortical reflex myoclonus is common in advanced AD (in around 50%). Myoclonus can appear early in the disease course in early onset familial AD and in AD with faster progression (156). Small amplitude postural jerky tremor (minipolymyoclonus) has also been reported in AD (157).
Geographically Isolated Tauopathies—Sociocultural and Environmental Impact
Guadeloupean Parkinsonism
High frequency of atypical parkinsonism with PSP like presentation is seen in French-Caribbean islands of Guadeloupe and Martinique (158). Two phenotypes have been described: (1) Guadeloupean PSP-like syndrome (Gd-PSP) with levodopa-resistant parkinsonism, early postural instability, and supranuclear gaze palsy (differs from classic PSP phenotype because of the high frequency of tremor, dysautonomia, and hallucination); (2) Guadeloupean Parkinsonism-dementia complex (Gd-PDC) with levodopa-resistant parkinsonism, subcortical dementia, and hallucination (9). Eating the fruits and infusions of the leaves of Annona muricata (soursop), containing Annonacin (toxic inhibitors of the mitochondrial respiratory chain complex I) has been proposed as a risk factor. Apart from supratentorial atrophy, 3rd ventricular dilatation (in both subgroups) and midbrain atrophy in Gd-PSP (like classic PSP), hypointense signals noted in T2 FLAIR, T2* sequences over substantia nigra, red nucleus, globus pallidus, and putamen in both subgroups (an important radiological clue) (9). Pathologically, they can mimic PSP or may have some atypical features like absence of tufted astrocytes and more tau positive neurons than true NFT (159).
Western Pacific Amyotrophic Lateral Sclerosis and Parkinsonism-Dementia Complex (ALS/PDC)
Historically, epidemiologist Kurland and neurologist Mulder described the high incidence of atypical parkinsonism and familial ALS in Guam (southernmost of the Mariana islands) in native Chamorro tribe. Subsequently, Hirano et al. termed it as “Parkinsonism-dementia complex of Guam (PDC)” because of the common association of dementia (160). Three high-incidence foci have been described so far in the literature: (1) Guam, USA (“Lytico-bodig” disease in Chamorro tribe), (2) PapuaNew Guinea, Indonesia (Auyu and Jakai tribe) (161), and (3) Hohara and Kozagawa regions of Kii Peninsula, Honshu Island, Japan (“Muro” disease) (10). Though the clinical description varied in literature, most common presentation described was rapidly progressive, familial, symmetric akinetic-rigid parkinsonism (PSP or “Bodig” phenotype) along with distal muscle atrophy (ALS or “Lytico” phenotype), hyperreflexia, vertical gaze palsy, and dementia (160). While overall the incidence of ALS/PDC has decreased, in Kii peninsula it is still being reported because of the use of traditional medicines (10). Ophthalmomyiasis-like pigmentary retinopathy (“criss-crossed tracks of depigmentation” of the retinal pigment epithelium) has been reported in these patients of Kii peninsula (162). MRI brain shows rapidly progressive frontotemporal atrophy mainly in PDC subtype (163). Typical “Hummingbird sign” (164) has been reported too. Pathologically, ALS/PDC can be called a “multiple proteinopathy” because apart from widespread 3R- and 4R-tau positive neuronal and glial inclusions, NFTs throughout the gray and white matter, including in the lower motor neurons in the spinal cord, TDP-43 deposits and accumulation of alpha synuclein deposition as Lewy bodies and Lewy neurites were also noted in the amygdala, substantia nigra and locus coeruleus (165). Several etiological hypotheses exist in the literature. Ingestion of toxic chemicals in the flour from seed of cycad plants containing toxic β-methylamino-l-alanine (l-BMAA) and cycasin has been proposed in “cycad hypothesis.” Japanese folk medicine (Kampo) also contains Sotetsu seed (cycad) (10). Cyanobacteria (Blue-green algae) containing toxic BMAA reaches the cycad seeds via the roots and these native people cook bats who eat these cycad seeds (160). Interestingly, C9orf mutation has recently been reported in some of these people of Kii peninsula (166), but not in Guam (167).
Nodding Syndrome (NS)
Children in the East Africa, mostly the Acholi tribe in northern Uganda (“lucluc”), Wapogoro tribe of Tanzania (“kifafa cha kusinzia”) and South Sudan suffer from a deleterious syndrome initially presenting as stereotypical head dropping movements (triggered by food, cold weather) that gradually leads to cognitive impairment, malnutrition, impaired growth, seizures, epileptic encephalopathy, and parkinsonism in late stage (168, 169). Often the affected children die by accidental drowning and burns. Clinically, NS overlaps with sub-Saharan Nakalanga syndrome (NLS) with pituitary dwarfism (169). Several etiological hypotheses exist in the literature, autoimmune reaction toleiomodin-1 epitope of Onchocerca volvulus (nematode causing river blindness) has been mostly mentioned. Recently, widespread tau-immunoreactive NFTs and pre-tangles have been noted, mostly in the gyral crests of the frontal and temporal cortex, brainstem, substantia nigra, and locus coeruleus (11). MRI shows varying degree of cortical atrophy mainly involving fronto-temporal regions (170). Presence of tau pathology in NS is a pathological factor for the disease or just an effect of repeated seizure, is still to be determined.
Cluster of PSP in Northern France
Caparros-Lefebvre et al. (171) have reported a cluster of older onset (mean age 74 years) PSP cases (53% PSP-P, 33% PSP-RS) from suburban towns centered on Wattrelos and Leers, northern France. Etiopathogenesis has been linked to the environmental toxic exposure from the industrial dumping of phosphate and chromate ores in the territory.
Being familiar with these disease phenotypes are needed because many a times patients migrate and a so-called “geographically isolated” tauopathy can present to a clinician practicing far away.
Novel Tauopathies—Are They?
Mulroy et al. (4) in their recent paper on “novel tauopathies,” have highlighted how pathological tau deposition is interestingly being noted in some other movement disorders like ADCY5 related dyskinesia, Beta-propeller protein associated neurodegeneration (BPAN), Benign hereditary chorea (BHC) Type 2, Huntington's disease (HD), Progressive ataxia, and palatal tremor (PAPT) and Spinocerebellar ataxia (SCA 11, 31). These are mostly isolated case reports and whether there is any pathological significance of tau in these disorders or tau is just an innocent bystander, is largely unknown. Presence of tau may be because of co-existing other known tauopathy, as a part of “mixed proteinopathy” or be just because of old age. Similarly, tau pathology is also seen in post encephalitic parkinsonism (172), Niemann-Pick type C disease (173) subacute sclerosing panencephalitis (SSPE) (174) and in prion disease like Gerstmann-Straussler-Scheinker disease (GSS) (175) although the clinico-pathological significance is still unknown.
A Practical Clinical Approach to Tauopathies and Movement Disorders
Considering tauopathies from a movement disorder perspective, interlacing clinical phenotypes and pathological, genetic heterogeneity often make it difficult to diagnose them clinically. Some clinical pointers like VSGP, frontal disinhibited behavior, amyotrophy, prominent language involvement, chorea, and cerebellar ataxia can be helpful clues for the clinician on this regard (Figure 2).
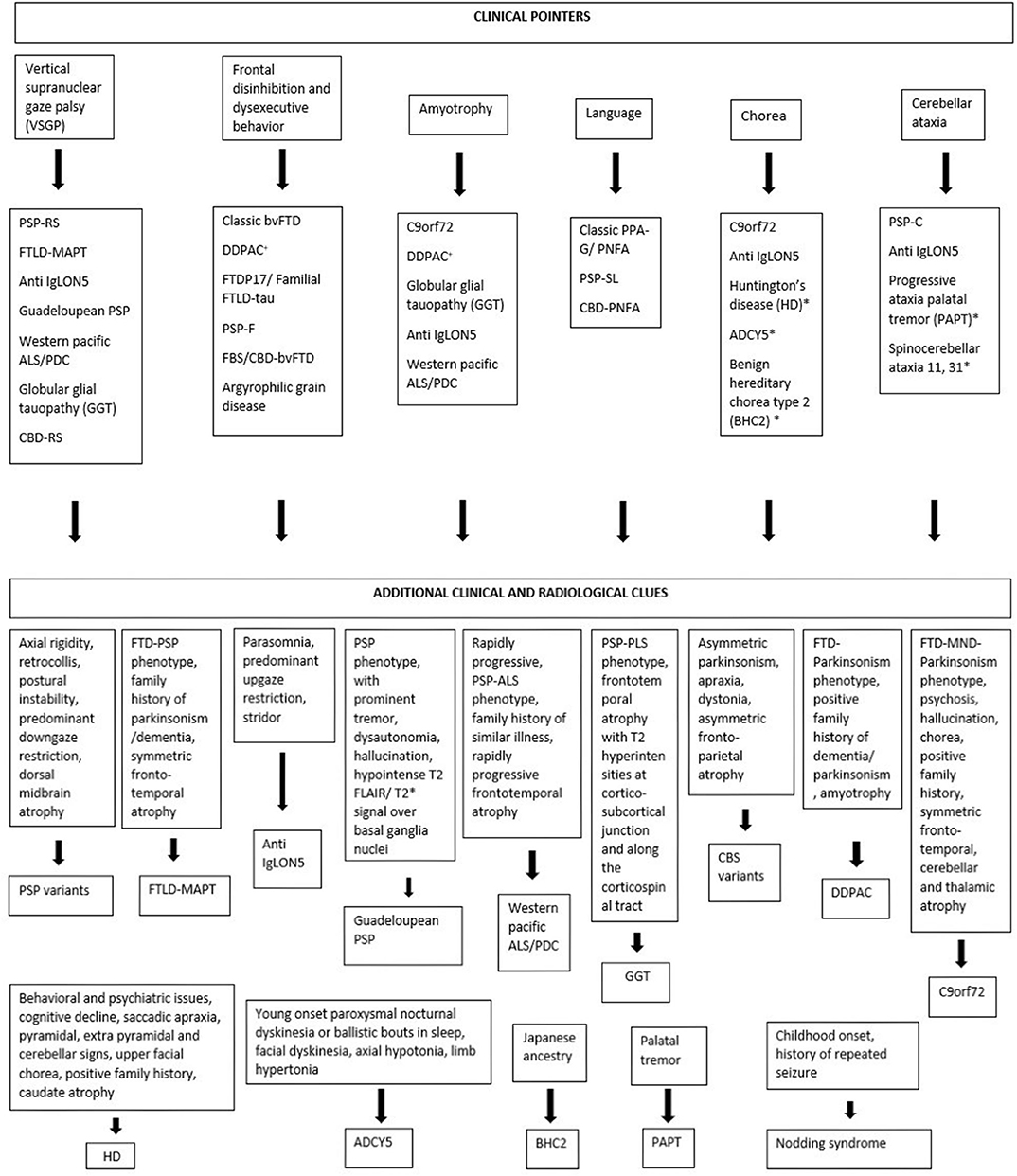
Figure 2. Clinical pointers for diagnosing tauopathies. *Novel tauopathies, +DDPAC, disinhibition–dementia–parkinsonism–amyotrophy complex related to MAPT mutation (intron 10 + 14).
Familial FTD With Parkinsonism—A Phenotypic Overlap
The overlap of familial frontotemporal dementia and parkinsonism needs special attention because it is one of the commonest presenting phenotypes baffling the movement disorder specialists. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) has recently been described as “familial FTLD-tau” because of the similarity of neuropathological features and disease progression between patients of familial FTLD-tau with MAPT mutations and sporadic FTLD-tau subtypes (PiD, PSP, CBD, and GGT) (176). Parkinsonism associated with familial FTD and MAPT mutation varies from mild to the aggressive form in severity and can occur early or late in this spectrum (57). Chromosome 17 carries another gene named progranulin (PGRN), that is also linked with the spectrum of frontotemporal dementia-parkinsonism, but with TAR DNA binding protein 43 (TDP43) inclusions instead of tau. Apart from these two common genetic associations (MAPT and PGRN), parkinsonism in familial FTD can also be liked with chromosome 9 open reading frame 72 (C9orf72) gene where overlap with motor neuron disease (FTD-MND) is commonly seen (177, 178) (Table 6). Four other less common genetic links reported in familial FTD with parkinsonism cases are—chromatin modifying protein 2B (CHMP2B), transactive response DNA-binding protein (TARDBP), valosin-containing protein (VCP), and fused-in-sarcoma (FUS) genes (179–181).
Phenotypically, MAPT and PGRN both can present with akinetic-rigid variant of parkinsonism. But MAPT commonly presents with PSP like phenotype with symmetric motor involvement while PGRN presents with corticobasal syndrome (CBS) like phenotype with asymmetric involvement and parietal lobe signs like apraxia, dyscalculia, visuoperceptual, and visuospatial dysfunction (57, 58, 182). Penetrance is 100% in MAPT, while it is age dependent in PGRN and reaches about 90% at the age of 70 (58). So, if there is no family history, MAPT is unlikely but PGRN can still be a possibility. Progression of disease is relatively faster in PGRN (131) and hallucinations (183) are more common. Radiologically, symmetric fronto-temporal atrophy is seen in MAPT involving anteromedial temporal lobe and orbitofrontal region (184) while caudate atrophy (185) (also common in FUS) (186) can also be seen. However, PGRN commonly presents with asymmetric fronto-temporal atrophy and more prominent posterior atrophy involving temporo-parietal, parieto-occipital regions (58, 182). White matter hyper-intensities are more frequent in PGRN (187). Additional cerebellar and thalamic atrophy can be seen in C9orf72 along with symmetric frontotemporal atrophy (188, 189) (Table 5).

Table 5. Clinical and radiological clues for familial FTD with Parkinsonism.
In addition to this, specific mutations in the MAPT gene can present with subtle phenotypic differences (57) (Table 6). On the other hand, Forrest et al. have noted pathological variability with specific mutations in MAPT like PSP pathology in S305S, CBD pathology in S305S, IVS10+16 and R406W, PiD pathology in K257T and GGT pathology in P301L, IVS10+16 mutation (176).
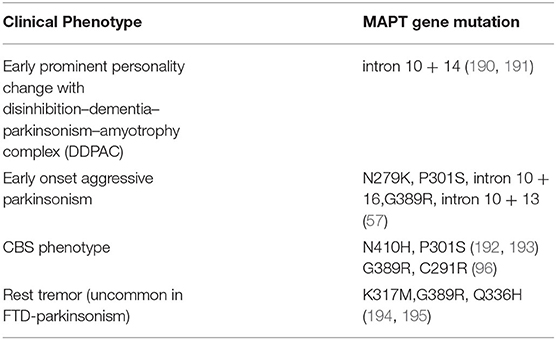
Table 6. Clinical phenotypes associated with MAPT gene mutations.
Conclusion
Tauopathy is a complex clinico-pathological hub encompassing multiple facets of movement disorders, dementia, and motor neuron disease. Topographic localization of tau accumulation shapes the clinical phenotype with varied combination of these domains. They can present with diverse overlapping phenotypes and their presentation can be mimicked by a lot of other diseases. Recognizing these “chameleons” and “mimics” are necessary from clinical and therapeutic standpoints. Emerging secondary tauopathies and geographically isolated tauopathies, many a time having relation with secondary environmental factors, are continuously invoking more and more research on the pathogenesis of tauopathy. Obviously, we can't stamp a disorder as “tauopathy” by mere presence of tau pathology in the brain. But is there a reliable clinical criterion for tagging a disorder as “tauopathy” ? MDS-PSP criteria has introduced the diagnostic category of “probable 4R-tauopathy” for ante mortem diagnosis of patients with PSP or CBD pathology (16). However, the spectrum of tauopathy is expanding far beyond these two pathological subtypes and tau deposition is being seen in different disease entities. Confirming the role of tau as a pathogenic factor for these disorders is the unmet need of the hour. The crosstalk between autoimmunity and neurodegeneration or neuroinflammation and tau aggregation also demands further research on this regard (196).
Traditionally, tauopathy has been depicted in the literature either as a pathological construct or from a cognitive perspective, while the evolving movement disorder domain of tauopathy is often neglected. Intertwining of the clinical, radiological, genetic, and pathological domains of movement disorder makes the spectrum intriguing but creates diagnostic confusion. Subtle clinical and radiological clues are the keys here to navigate through this conundrum. They are not only helpful for targeted genetic testing and predicting the pathology before autopsy, but also can open the door for utilizing newer biomarkers like ligand gated imaging or CSF biochemistry more efficiently and encourage further research on protein based therapeutic strategies.
Author Contributions
JG: organization and execution of the research project and writing of the first draft of the manuscript. MJ: conception of the research project and review and critique of the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
No authors have received any funding from any institution, including personal relationships, interests, grants, employment, affiliations, patents, inventions, honoraria, consultancies, royalties, stock options/ownership, or expert testimony for the last 12 months.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Warren JD, Rohrer JD, Schott JM, Fox NC, Hardy J, Rossor MN. Molecular nexopathies: a new paradigm of neurodegenerative disease. Trends Neurosci. (2013) 36:561–9. doi: 10.1016/j.tins.2013.06.007
2. Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. (2014) 82:1271–88. doi: 10.1016/j.neuron.2014.04.047
3. Landa J, Gaig C, Planagumà J, Saiz A, Antonell A, Sanchez-Valle R, et al. Effects of IgLON5 antibodies on neuronal cytoskeleton: a link between autoimmunity and neurodegeneration. Ann Neurol. (2020). doi: 10.1002/ana.25857. [Epub ahead of print].
4. Mulroy E, Jaunmuktane Z, Balint B, Erro R, Latorre A, Bhatia KP. Some new and unexpected tauopathies in movement disorders. Mov Disord Clin Pract. (2020) 7:616–26. doi: 10.1002/mdc3.12995
5. Murley AG, Coyle-Gilchrist I, Rouse MA, Jones PS, Li W, Wiggins J, et al. Redefining the multidimensional clinical phenotypes of frontotemporal lobar degeneration syndromes. Brain. (2020) 143:1555–71. doi: 10.1093/brain/awaa097
6. Ling H. Untangling the tauopathies: Current concepts of tau pathology and neurodegeneration. Park Relat Disord. (2018) 46:S34–S8. doi: 10.1016/j.parkreldis.2017.07.031
7. Caffrey TM, Wade-Martins R. Functional MAPT haplotypes: bridging the gap between genotype and neuropathology. Neurobiol Dis. (2007) 27:1–0. doi: 10.1016/j.nbd.2007.04.006
8. Rösler TW, Tayaranian Marvian A, Brendel M, Nykänen NP, Höllerhage M, Schwarz SC, et al. Four-repeat tauopathies. Prog Neurobiol. (2019) 180:101644. doi: 10.1016/j.pneurobio.2019.101644
9. Lannuzel A, Höglinger GU, Verhaeghe S, Gire L, Belson S, Escobar-Khondiker M, et al. Atypical parkinsonism in Guadeloupe: a common risk factor for two closely related phenotypes? Brain. (2007) 130:816–27. doi: 10.1093/brain/awl347
10. Spencer PS, Palmer VS, Kihira T, Yoshida S, Reis J, Yabushita M, et al. Kampo medicine and Muro disease (Amyotrophic Lateral Sclerosis and Parkinsonism-Dementia Complex). eNeurologicalSci. (2020) 18:100230. doi: 10.1016/j.ensci.2020.100230
11. Pollanen MS, Onzivua S, Robertson J, McKeever PM, Olawa F, Kitara DL, et al. Nodding syndrome in Uganda is a tauopathy. Acta Neuropathol. (2018) 136:691–7. doi: 10.1007/s00401-018-1909-9
12. Höglinger GU, Respondek G, Kovacs GG. New classification of tauopathies. Rev Neurol (Paris). (2018) 174:664–8. doi: 10.1016/j.neurol.2018.07.001
13. Armstrong MJ. Progressive supranuclear palsy: an update. Curr Neurol Neurosci Rep. (2018) 18:1–9. doi: 10.1007/s11910-018-0819-5
14. Picillo M, Erro R, Cuoco S, Tepedino MF, Manara R, Pellecchia MT, et al. MDS PSP criteria in real-life clinical setting: motor and cognitive characterization of subtypes. Mov Disord. (2018) 33:1361–5. doi: 10.1002/mds.27408
15. Iankova V, Respondek G, Saranza G, Painous C, Cámara A, Compta Y, et al. Video-tutorial for the Movement Disorder Society criteria for progressive supranuclear palsy. Parkinsonism Relat Disord. (2020). doi: 10.1016/j.parkreldis.2020.06.030. [Epub ahead of print].
16. Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. (2017) 32:853–64.
17. Jalal MJ, Menon M. “Humming bird sign”, “Mickey Mouse sign”, and “morning glory sign” in progressive supranuclear palsy. Menoufia Med J. (2017) 30:325. doi: 10.4103/mmj.mmj_204_16
18. Massey LA, Jager HR, Paviour DC, O'Sullivan SS, Ling H, Williams DR, et al. The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology. (2013) 80:1856–61. doi: 10.1212/WNL.0b013e318292a2d2
19. Morelli M, Arabia G, Novellino F, Salsone M, Giofre L, Condino F, et al. MRI measurements predict PSP in unclassifiable parkinsonisms: a cohort study. Neurology. (2011) 77:1042–7. doi: 10.1212/WNL.0b013e31822e55d0
20. Quattrone A, Morelli M, Nigro S, Quattrone A, Vescio B, Arabia G, et al. A new MR imaging index for differentiation of progressive supranuclear palsy-parkinsonism from Parkinson's disease. Park Relat Disord. (2018) 54:3–8. doi: 10.1016/j.parkreldis.2018.07.016
21. Boxer AL, Yu J-T, Golbe LI, Litvan I, Lang AE, Höglinger GU. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. (2017) 16:552–63. doi: 10.1016/S1474-4422(17)30157-6
22. Jecmenica Lukic M, Kurz C, Respondek G, Grau-Rivera O, Compta Y, Gelpi E, et al. Copathology in progressive supranuclear palsy: does it matter? Mov Disord. (2020) 35:984–93. doi: 10.1002/mds.28011
23. Coughlin DG, Litvan I. Progressive supranuclear palsy: advances in diagnosis and management. Parkinsonism Relat Disord. (2020) 73:105–16. doi: 10.1016/j.parkreldis.2020.04.014
24. Kovacs GG, Lukic MJ, Irwin DJ, Arzberger T, Respondek G, Lee EB, et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. (2020) 140:99–119. doi: 10.1007/s00401-020-02158-2
25. Ling H. Clinical Approach to Progressive Supranuclear Palsy. J Mov Disord. (2016) 9:3–13. doi: 10.14802/jmd.15060
26. Ling H, Massey LA, Lees AJ, Brown P, Day BL. Hypokinesia without decrement distinguishes progressive supranuclear palsy from Parkinson's disease. Brain. (2012) 135:1141–53. doi: 10.1093/brain/aws038
27. Maetzler W, Rattay TW, Hobert MA, Synofzik M, Bader A, Berg D, et al. Freezing of Swallowing. Mov Disord Clin Pract. (2016) 3:490–3. doi: 10.1002/mdc3.12314
28. Liu AJ, Chang JE, Naasan G, Boxer AL, Miller BL, Spina S. Progressive supranuclear palsy and primary lateral sclerosis secondary to globular glial tauopathy: a case report and a practical theoretical framework for the clinical prediction of this rare pathological entity. Neurocase. (2020) 26:91–7. doi: 10.1080/13554794.2020.1732427
29. Ando S, Kanazawa M, Onodera O. Progressive supranuclear palsy with predominant cerebellar ataxia. J Mov Disord. (2020) 13:20–6. doi: 10.14802/jmd.19061
30. Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. (2013) 80:496–503. doi: 10.1212/WNL.0b013e31827f0fd1
31. Day GS, Sung Lim T, Hassenstab J, Goate AM, Grant EA, Roe CM, et al. Differentiating cognitive impairment due to corticobasal degeneration and Alzheimer disease. Neurology. (2017) 88:1273–81. doi: 10.1212/WNL.0000000000003770
32. Abbate C, Trimarchi PD, Manzoni L, Quarenghi AM, Salvi G, Pietro, Inglese S, et al. A posterior variant of corticobasal syndrome: evidence from a longitudinal study of cognitive and functional status in a single case. Cogent Psychol. (2018) 5. doi: 10.1080/23311908.2018.1452868
33. Lang AE. Cortical basal ganglionic degeneration presenting with “progressive loss of speech output and orofacial dyspraxia.” J Neurol Neurosurg Psychiatry. (1992) 55:1101. doi: 10.1136/jnnp.55.11.1101
34. Thümler BH, Urban PP, Davids E, Siessmeier M, Schreckenberger T, Benz P, et al. Dysarthria and pathological laughter/crying as presenting symptoms of corticobasal- ganglionic degeneration syndrome. J Neurol. (2003) 250:1107–8. doi: 10.1007/s00415-003-0075-7
35. Kimura N, Kumamoto T, Hanaoka T, Hazama Y, Nakamura K, Arakawa R. Corticobasal degeneration presenting with progressive conduction aphasia. J Neurol Sci. (2008) 269:163–8. doi: 10.1016/j.jns.2007.12.017
36. Rossor MN, Tyrrell PJ, Warrington EK, Thompson PD, Marsden CD, Lantos P. Progressive frontal gait disturbance with atypical Alzheimer's disease and corticobasal degeneration. J Neurol Neurosurg Psychiatry. (1999) 67:345–52. doi: 10.1136/jnnp.67.3.345
37. Sakae N, Santos OA, Pedraza O, Litvan I, Murray ME, Duara R, et al. Clinical and pathologic features of cognitive-predominant corticobasal degeneration. Neurology. (2020) 95:e35–e45. doi: 10.1212/WNL.0000000000009734
38. Constantinides VC, Paraskevas GP, Paraskevas PG, Stefanis L, Kapaki E. Clinical Parkinsonism & Related Disorders Corticobasal degeneration and corticobasal syndrome : a review. Clin Park Relat Disord. (2019) 1:66–71. doi: 10.1016/j.prdoa.2019.08.005
39. Whitwell JL, Jack CR, Boeve BF, Parisi JE, Ahlskog JE, Drubach DA, et al. Imaging correlates of pathology in corticobasal syndrome. Neurology. (2010) 75:1879–87. doi: 10.1212/WNL.0b013e3181feb2e8
40. Ling H, O'Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. (2010) 133:2045–57. doi: 10.1093/brain/awq123
41. Höglinger GU. Is it useful to classify progressive supranuclear palsy and corticobasal degeneration as different disorders? No. Mov Disord Clin Pract. (2018) 5:141–4. doi: 10.1002/mdc3.12582
42. Ling H, Macerollo A. Is it useful to classify PSP and CBD as different disorders? Yes. Mov Disord Clin Pract. (2018) 5:145–8. doi: 10.1002/mdc3.12581
43. Respondek G, Grimm MJ, Piot I, Arzberger T, Compta Y, Englund E, et al. Validation of the movement disorder society criteria for the diagnosis of 4-repeat tauopathies. Mov Disord. (2020) 35:171–6. doi: 10.1002/mds.27872
44. Lang AE. Comment on “Is it Useful to Classify PSP and CBD as Different Disorders?” Mov Disord Clin Pract. (2018) 5:564–5. doi: 10.1002/mdc3.12676
45. Giagkou N, Bhatia KP, Höglinger GU, Stamelou M. Genetic mimics of the non-genetic atypical parkinsonian disorders - the ‘atypical’ atypical. Int Rev Neurobiol. (2019) 149:327–51. doi: 10.1016/bs.irn.2019.10.008
46. Stamelou M, Quinn NP, Bhatia KP. “Atypical” atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy-a diagnostic guide. Mov Disord. (2013) 28:1184–99. doi: 10.1002/mds.25509
47. Eggink H, Brandsma R, van der Hoeven JH, Lange F, de Koning TJ, Tijssen MAJ. Teaching video neuro images : the “round the houses” sign as a clinical clue for Niemann-Pick disease type C. Neurology. (2016) 86:e202. doi: 10.1212/WNL.0000000000002660
48. Kresojević N, Mandić-Stojmenović G, Dobričić V, Petrović I, Brajković L, Stefanova E, et al. Very late-onset niemann pick type c disease: example of progressive supranuclear palsy look-alike disorder. Mov Disord Clin Pract. (2020) 7:211–4. doi: 10.1002/mdc3.12892
49. Dalmau J. Clinical analysis of anti-Ma2-associated encephalitis. Brain. (2004) 127:1831–44. doi: 10.1093/brain/awh203
50. Gövert F, Leypoldt F, Junker R, Wandinger K-P, Deuschl G, Bhatia KP, et al. Antibody-related movement disorders - a comprehensive review of phenotype-autoantibody correlations and a guide to testing. Neurol Res Pract. (2020) 2:6. doi: 10.1186/s42466-020-0053-x
51. Gaig C, Graus F, Compta Y, Högl B, Bataller L, Brüggemann N, et al. Clinical manifestations of the anti-IgLON5 disease. Neurology. (2017) 88:1736–43. doi: 10.1212/WNL.0000000000003887
52. Chen H, Wu J, Irani SR. Distinctive magnetic resonance imaging findings in IgLON5 antibody disease. JAMA Neurol. (2020) 77:125. doi: 10.1001/jamaneurol.2019.3638
53. Merchán-del Hierro X, Rojas G, Aldinio V, Bres-Bullrich M, Da-Prat G, Ebner R, et al. Moaning phenomenon and rapidly progressive dementia in anti LGI-1 associated progressive supranuclear palsy syndrome. Tremor Other Hyperkinet Mov. (2020) 10:8. doi: 10.5334/tohm.65
54. van Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, de Bruijn MAAM, et al. Anti-LGI1 encephalitis. Neurology. (2016) 87:1449–456. doi: 10.1212/WNL.0000000000003173
55. Averbuch-Heller L, Paulson GW, Daroff RB, Leigh RJ. Whipple's disease mimicking progressive supranuclear palsy: the diagnostic value of eye movement recording. J Neurol Neurosurg Psychiatry. (1999) 66:532–5. doi: 10.1136/jnnp.66.4.532
56. Black DF, Aksamit AJ, Morris JM. MR imaging of central nervous system whipple disease: a 15-year review. Am J Neuroradiol. (2010) 31:1493–7. doi: 10.3174/ajnr.A2089
57. Baizabal-Carvallo JF, Jankovic J. Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat Rev Neurol. (2016) 12:175–85. doi: 10.1038/nrneurol.2016.14
58. Espay AJ, Litvan I. Parkinsonism and frontotemporal dementia: the clinical overlap. J Mol Neurosci. (2011) 45:343–9. doi: 10.1007/s12031-011-9632-1
59. Williams DR, Hadeed A, Najim AS, Wreikat A, Lees AJ. Kufor Rakeb Disease: autosomal recessive, levodopa- responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. (2005) 20:1264–71. doi: 10.1002/mds.20511
60. Yang X, Xu Y. Mutations in the ATP13A2 gene and Parkinsonism: a preliminary review. Biomed Res Int. (2014) 2014:1–9. doi: 10.1155/2014/371256
61. Henao AI, Pira S, Herrera DA, Vargas SA, Montoya J, Castillo M. Characteristic brain MRI findings in ataxia-neuropathy spectrum related to POLG mutation. Neuroradiol J. (2016) 29:46–8. doi: 10.1177/1971400915621324
62. Newsway V, Fish M, Rohrer JD, Majounie E, Williams N, Hack M, et al. Europe PMC Funders Group Perry syndrome due to the DCTN1 G71R mutation - a distinctive L-DOPA responsive disorder with behavioural syndrome, vertical gaze palsy and respiratory failure. (2015) 25:767–70. doi: 10.1002/mds.22950
63. Erro R, Stamelou M, Bhatia KP. editors. (Familial) PSP Look-Alike: Perry Syndrome. In: Case Studies in Movement Disorders. Cambridge: Cambridge University Press. (2017). p. 24–5.
64. Konno T, Ross OA, Teive HAG, Sławek J, Dickson DW, Wszolek ZK. DCTN1-related neurodegeneration: perry syndrome and beyond. Parkinsonism Relat Disord. (2017) 41:14–24. doi: 10.1016/j.parkreldis.2017.06.004
65. Picillo M, Petrucci S, Valente EM, Pappatà S, Squame F, Ginevrino M, et al. Progressive supranuclear palsy-like phenotype in a GBA E326K mutation carrier. Mov Disord Clin Pract. (2017) 4:444–6. doi: 10.1002/mdc3.12406
66. Bertoni JM, Label LS, Sackelleres JC, Hicks SP. Supranuclear gaze palsy in familial Creutzfeldt-Jakob disease. Arch Neurol. (1983) 40:618–22. doi: 10.1001/archneur.1983.04050090054008
67. Fulbright RK, Hoffmann C, Lee H, Pozamantir A, Chapman J, Prohovnik I. MR imaging of familial creutzfeldt-jakob disease: a blinded and controlled study. Am J Neuroradiol. (2008) 29:1638–43. doi: 10.3174/ajnr.A1217
68. Ufkes NA, Woodard C, Dale ML. A case of Gerstmann-Straussler-Scheinker (GSS) disease with supranuclear gaze palsy. J Clin Mov Disord. (2019) 6:7. doi: 10.1186/s40734-019-0082-1
69. Ribosa-Nogué R, Pagonabarraga J, Gomez-Anson B, Granell-Moreno E, Sánchez-Valle R, Kulisevsky J. Gerstmann-Sträussler-Scheinker disease presenting with atypical parkinsonism, but typical magnetic resonance imaging findings of prion disease. Mov Disord Clin Pract. (2016) 3:93–95. doi: 10.1002/mdc3.12228
70. Erro R, Lees AJ, Moccia M, Picillo M, Penco S, Mosca L, et al. Progressive Parkinsonism, Balance Difficulties, and Supranuclear Gaze Palsy. JAMA Neurol. (2014) 71:104. doi: 10.1001/jamaneurol.2013.5149
71. Stojanov D, Vojinovic S, Aracki-Trenkic A, Tasic A, Benedeto-Stojanov D, Ljubisavljevic S, et al. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). Bosn J Basic Med Sci. (2015) 15:1–8. doi: 10.17305/bjbms.2015.247
72. Pfeffer G, Gorman GS, Griffin H, Kurzawa-Akanbi M, Blakely EL, Wilson I, et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain. (2014) 137:1323–36. doi: 10.1093/brain/awu060
73. De la Casa-Fages B, Fernández-Eulate G, Gamez J, Barahona-Hernando R, Morís G, García-Barcina M, et al. Parkinsonism and spastic paraplegia type 7: expanding the spectrum of mitochondrial Parkinsonism. Mov Disord. (2019) 34:1547–61. doi: 10.1002/mds.27812
74. Da Graça FF, De Rezende TJR, Vasconcellos LFR, Pedroso JL, Barsottini OGP, França MC. Neuroimaging in hereditary spastic paraplegias: current use and future perspectives. Front Neurol. (2019) 10:1117. doi: 10.3389/fneur.2018.01117
75. Gwinn-Hardy K, Chen JY, Liu H-C, Liu TY, Boss M, Seltzer W, et al. Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology. (2000) 55:800–5. doi: 10.1212/WNL.55.6.800
76. Nascimento FA, Marques Garcia BC, Teive HAG. Teaching Video NeuroImages: upward gaze palsy is a sign of spinocerebellar ataxia type 3. Neurology. (2018) 91:e494. doi: 10.1212/WNL.0000000000005913
77. Lin I-S, Wu R-M, Lee-Chen G-J, Shan D-E, Gwinn-Hardy K. The SCA17 phenotype can include features of MSA-C, PSP and cognitive impairment. Parkinsonism Relat Disord. (2007) 13:246–9. doi: 10.1016/j.parkreldis.2006.04.009
78. Park H, Kim H-J, Jeon BS. Parkinsonism in spinocerebellar ataxia. Biomed Res Int. (2015) 1–11. doi: 10.1155/2015/125273
79. Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset parkinsonism. Hum Mutat. (2013) 34:1208–15. doi: 10.1002/humu.22373
80. Huey ED, Ferrari R, Moreno JH, Jensen C, Morris CM, Potocnik F, et al. FUS and TDP43 genetic variability in FTD and CBS. Neurobiol Aging. (2012) 33:1016.e9–e17. doi: 10.1016/j.neurobiolaging.2011.08.004
81. Anor CJ, Xi Z, Zhang M, Moreno D, Sato C, Rogaeva E, et al. Mutation analysis of C9orf72 in patients with corticobasal syndrome. Neurobiol Aging. (2015) 36:2905.e1–e5. doi: 10.1016/j.neurobiolaging.2015.06.008
82. Souza PVS de, Pinto WBV de R, Oliveira ASB. C9orf72-related disorders: expanding the clinical and genetic spectrum of neurodegenerative diseases. Arq Neuropsiquiatr. (2015) 73:246–256. doi: 10.1590/0004-282X20140229
83. Lamb R, Rohrer JD, Real R, Lubbe SJ, Waite AJ, Blake DJ, et al. A novel TBK1 mutation in a family with diverse frontotemporal dementia spectrum disorders. Cold Spring Harb Mol Case Stud. (2019) 5:1–10. doi: 10.1101/mcs.a003913
84. Caroppo P, Camuzat A, De Septenville A, Couratier P, Lacomblez L, Auriacombe S, et al. Semantic and nonfluent aphasic variants, secondarily associated with amyotrophic lateral sclerosis, are predominant frontotemporal lobar degeneration phenotypes in TBK1 carriers. Alzheimers Dement. (2015) 1:481–6. doi: 10.1016/j.dadm.2015.10.002
85. Herrero Valverde A, Costa S, Timoteo Â, Ginestal R, Pimentel J. Rapidly progressive corticobasal degeneration syndrome. Case Rep Neurol. (2011) 3:185–90. doi: 10.1159/000329820
86. Zhang Y, Minoshima S, Vesselle H, Lewis DH. A case of Creutzfeldt-Jakob disease mimicking corticobasal degeneration. Clin Nucl Med. (2012) 37:e173–e5. doi: 10.1097/RLU.0b013e31824c5f0e
87. Necpál J, Stelzer M, Koščová S, Patarák M. A corticobasal syndrome variant of familial Creutzfeldt-Jakob disease with stroke-like onset. Case Rep Neurol Med. (2016) 2016:1–3. doi: 10.1155/2016/4167391
88. Lee W, Simpson M, Ling H, McLean C, Collins S, Williams DR. Characterising the uncommon corticobasal syndrome presentation of sporadic Creutzfeldt-Jakob disease. Park Relat Disord. (2013) 19:81–5. doi: 10.1016/j.parkreldis.2012.07.010
89. Kim Y-D, Kim J-S, Lee E-S, Yang D-W, Lee K-S, Kim Y-I. Progressive “vascular” corticobasal syndrome due to bilateral ischemic hemispheric lesions. Intern Med. (2009) 48:1699–702. doi: 10.2169/internalmedicine.48.2415
90. Engelen M, Westhoff D, de Gans J, Nederkoorn PJ. A 64-year old man presenting with carotid artery occlusion and corticobasal syndrome: a case report. J Med Case Rep. (2011) 5:357. doi: 10.1186/1752-1947-5-357
91. Koga S, Roemer SF, Kasanuki K, Dickson DW. Parkinsonism and related disorders cerebrovascular pathology presenting as corticobasal syndrome: an autopsy case series of “vascular CBS.” Park Relat Disord. (2019) 68:79–84. doi: 10.1016/j.parkreldis.2019.09.001
92. Morris HR, Lees AJ. Primary antiphospholipid syndrome presenting as a corticobasal degeneration syndrome. Mov Disord. (1999) 14:530–2. doi: 10.1002/1531-8257(199905)14:3<530::AID-MDS1030>3.0.CO;2-8
93. Martino D, Chew N-K, Mir P, Edwards MJ, Quinn NP, Bhatia KP. Atypical movement disorders in antiphospholipid syndrome. Mov Disord. (2006) 21:944–9. doi: 10.1002/mds.20842
94. Lee D-W, Eum S-W, Moon CO, Ma H-I, Kim YJ. Corticobasal syndrome associated with antiphospholipid syndrome without cerebral infarction. Neurology. (2014) 82:730–1. doi: 10.1212/WNL.0000000000000152
95. Navarro E, De Andrés C, Guerrero C, Giménez-Roldán S. Corticobasal syndrome in a family with early-onset Alzheimer's disease linked to a presenilin-1 gene mutation. Mov Disord Clin Pract. (2015) 2:388–94. doi: 10.1002/mdc3.12212
96. Abate F, Dati G, Ginevrino M, Valente EM, Barone P, Picillo M. APP-related corticobasal syndrome: expanding the list of corticobasal degeneration look alikes. Mov Disord Clin Pract. (2020) 7:849–51. doi: 10.1002/mdc3.13037
97. Scahill RI, Ridgway GR, Bartlett JW, Barnes J, Ryan NS, Mead S, et al. Genetic influences on atrophy patterns in familial Alzheimer's disease: a comparison of APP and PSEN1 mutations. J Alzheimers Dis. (2013) 35:199–212. doi: 10.3233/JAD-121255
98. Rubio-Agusti I, Kojovic M, Edwards MJ, Murphy E, Chandrashekar HS, Lachmann RH, et al. Atypical parkinsonism and cerebrotendinous xanthomatosis: report of a family with corticobasal syndrome and a literature review. Mov Disord. (2012) 27:1769–74. doi: 10.1002/mds.25229
99. Stelten BML, van de Warrenburg BPC, Wevers RA, Verrips A. Movement disorders in cerebrotendinous xanthomatosis. Park Relat Disord. (2019) 58:12–6. doi: 10.1016/j.parkreldis.2018.07.006
100. Warren JD, Mummery CJ, Al-Din AS, Brown P, Wood NW. Corticobasal degeneration syndrome with basal ganglia calcification: Fahr's disease as a corticobasal look-alike? Mov Disord. (2002) 17:563–7. doi: 10.1002/mds.10122
101. Balint B, Mahant N, Meinck HM, Fung V. Stiff limb syndrome mimicking corticobasal syndrome. Mov Disord Clin Pract. (2014) 1:354–6. doi: 10.1002/mdc3.12059
102. Graus F, Saiz A, Dalmau J. GAD antibodies in neurological disorders - insights and challenges. Nat Rev Neurol. (2020) 16:353–65. doi: 10.1038/s41582-020-0359-x
103. Muñoz González A, Contreras Chicote A, Vales Montero M, Velázquez Pérez JM, Dela Casa B, Luque Buzo E, et al. Antiglycine receptor antibodies and rapidly progressive corticobasal syndrome [abstract]. Mov Disord. (2018) 33(Suppl 2).
104. Carvajal-González A, Leite MI, Waters P, Woodhall M, Coutinho E, Balint B, et al. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain. (2014) 137:2178–92. doi: 10.1093/brain/awu142
105. Kasanuki K, Josephs KA, Ferman TJ, Murray ME, Koga S, Konno T, et al. Diffuse Lewy body disease manifesting as corticobasal syndrome a rare form of Lewy body disease. Neurology. (2018) 91:E268–E79. doi: 10.1212/WNL.0000000000005828
106. Mak E, Su L, Williams GB, O'Brien JT. Neuroimaging characteristics of dementia with Lewy bodies. Alzheimers Res Ther. (2014) 6:18. doi: 10.1186/alzrt248
107. Konno T, Yoshida K, Mizuno T, Kawarai T, Tada M, Nozaki H, et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol. (2017) 24:37–45. doi: 10.1111/ene.13125
108. Baba Y, Ghetti B, Baker MC, Uitti RJ, Hutton ML, Yamaguchi K, et al. Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol. (2006) 111:300–11. doi: 10.1007/s00401-006-0046-z
109. Marshall CR, Hardy CJD, Volkmer A, Russell LL, Bond RL, Fletcher PD, et al. Primary progressive aphasia: a clinical approach. J Neurol. (2018) 265:1474–90. doi: 10.1007/s00415-018-8762-6
110. Ash S, Mcmillan C, Gunawardena D, Avants B, Morgan B, Khan A, et al. Brain & Language Speech errors in progressive non-fluent aphasia. Brain Lang. (2010) 113:13–20. doi: 10.1016/j.bandl.2009.12.001
111. Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. (2011) 76:1006–14. doi: 10.1212/WNL.0b013e31821103e6
112. Grossman M. The non-fluent / agrammatic variant of primary progressive aphasia. Lancet Neurol. (2012) 11:545–55. doi: 10.1016/S1474-4422(12)70099-6
113. Montembeault M, Brambati SM, Gorno-Tempini ML, Migliaccio R. Clinical, anatomical, and pathological features in the three variants of primary progressive aphasia: a review. Front Neurol. (2018) 9:692. doi: 10.3389/fneur.2018.00692
114. Tan RH, Guennewig B, Dobson-Stone C, Kwok JBJ, Kril JJ, Kiernan MC, et al. The underacknowledged PPA-ALS. Neurology. (2019) 92:e1354 LP–e66. doi: 10.1212/WNL.0000000000007146
115. Josephs KA, Katsuse O, Beccano-Kelly DA, Lin W-L, Uitti RJ, Fujino Y, et al. Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol. (2006) 65:396–405. doi: 10.1097/01.jnen.0000218446.38158.61
116. Fu Y-J, Nishihira Y, Kuroda S, Toyoshima Y, Ishihara T, Shinozaki M, et al. Sporadic four-repeat tauopathy with frontotemporal lobar degeneration, Parkinsonism, and motor neuron disease: a distinct clinicopathological and biochemical disease entity. Acta Neuropathol. (2010) 120:21–32. doi: 10.1007/s00401-010-0649-2
117. Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol. (2013) 126:537–44. doi: 10.1007/s00401-013-1171-0
118. Kim E-J, Lee MJ, Lee J-H, Lee YM, Shin J-H, Shin M-J, et al. Globular glial tauopathy presenting as non-fluent/agrammatic variant primary progressive aphasia with chorea. Parkinsonism Relat Disord. (2017) 44:159–61. doi: 10.1016/j.parkreldis.2017.09.006
119. Graff-Radford J, Josephs KA, Parisi JE, Dickson DW, Giannini C, Boeve BF. Globular glial Tauopathy presenting as semantic variant primary progressive aphasia. JAMA Neurol. (2016) 73:123. doi: 10.1001/jamaneurol.2015.2711
120. Marsili L, Dickson DW, Espay AJ. Globular glial tauopathy may be mistaken for corticobasal syndrome-pointers for the clinician. Mov Disord Clin Pract. (2018) 5:439–41. doi: 10.1002/mdc3.12634
121. Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R, et al. White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol. (2008) 67:963–75. doi: 10.1097/NEN.0b013e318187a80f
122. Kovacs GG. Astroglia and tau: new perspectives. Front Aging Neurosci. (2020) 12:96. doi: 10.3389/fnagi.2020.00096
123. Rodriguez RD, Grinberg LT. Argyrophilic grain disease: an underestimated tauopathy. Dement Neuropsychol. (2015) 9:2–8. doi: 10.1590/S1980-57642015DN91000002
124. Yokota O, Miki T, Ikeda C, Nagao S, Takenoshita S, Ishizu H, et al. Neuropathological comorbidity associated with argyrophilic grain disease. Neuropathology. (2018) 38:82–97. doi: 10.1111/neup.12429
125. Sengoku R, Kaneda D, Kameyama M, Tokumaru A, Ishii K, Kanemaru K, et al. An atutopsy case of argyrophilic grain disease, clinically presenting with Parkinson dementia [abstract]. Mov Disord. (2018) 33.
126. Jicha GA, Nelson PT. Hippocampal sclerosis, argyrophilic grain disease, and primary age-related tauopathy. Contin Lifelong Learn Neurol. (2019) 25:208–33. doi: 10.1212/CON.0000000000000697
127. Gil MJ, Serrano S, Manzano MS, Cuadrado ML, Góméz E, Rábano A. Argyrophilic grain disease presenting as behavioral frontotemporal dementia. Clin Neuropathol. (2019) 38:8–13. doi: 10.5414/NP301122
128. Wurm R, Klotz S, Rahimi J, Katzenschlager R, Lindeck-Pozza E, Regelsberger G, et al. Argyrophilic grain disease in individuals younger than 75 years: clinical variability in an under-recognized limbic tauopathy. Eur J Neurol. (2020) 27:1856–66. doi: 10.1111/ene.14321
129. Rossor MN. Pick's disease: a clinical overview. Neurology. (2001) 56:S3–S5. doi: 10.1212/WNL.56.suppl_4.S3
130. Irwin DJ, Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE, et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol. (2016) 79:272–87. doi: 10.1002/ana.24559
131. Whitwell JL, Josephs KA. Recent advances in the imaging of frontotemporal dementia. Curr Neurol Neurosci Rep. (2012) 12:715–23. doi: 10.1007/s11910-012-0317-0
132. Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. (2014) 13:575–86. doi: 10.1016/S1474-4422(14)70051-1
133. Gaig C, Iranzo A, Santamaria J, Graus F. The sleep disorder in anti-lgLON5 disease. Curr Neurol Neurosci Rep. (2018) 18:41. doi: 10.1007/s11910-018-0848-0
134. Gaig C, Compta Y. Neurological profiles beyond the sleep disorder in patients with anti-IgLON5 disease. Curr Opin Neurol. (2019) 32:493–99. doi: 10.1097/WCO.0000000000000677
135. Tao Q, Wei Q, Song S, Yin X. Motor neuron disease-like phenotype associated with anti-IgLON5 disease. CNS Neurosci Ther. (2018) 24:1305–8. doi: 10.1111/cns.13038
136. Schöberl F, Levin J, Remi J, Goldschagg N, Eren O, Okamura N, et al. IgLON5: a case with predominant cerebellar tau deposits and leptomeningeal inflammation. Neurology. (2018) 91:180–2. doi: 10.1212/WNL.0000000000005859
137. Peeters I, Wiels W, De Raedt S, Flamez A. Unusual head movements in anti-IgLON5 disease. Mov Disord Clin Pract. (2020) 7:708–9. doi: 10.1002/mdc3.13016
138. Nissen MS, Blaabjerg M. Anti-IgLON5 disease: a case with 11-year clinical course and review of the literature. Front Neurol. (2019) 10:1056. doi: 10.3389/fneur.2019.01056
139. Fesharaki-Zadeh A. Chronic traumatic encephalopathy: a brief overview. Front Neurol. (2019) 10:713. doi: 10.3389/fneur.2019.00713
140. Gardner A, Iverson GL, McCrory P. Chronic traumatic encephalopathy in sport: a systematic review. Br J Sports Med. (2014) 48:84–90. doi: 10.1136/bjsports-2013-092646
141. Jordan BD. The clinical spectrum of sport-related traumatic brain injury. Nat Rev Neurol. (2013) 9:222–30. doi: 10.1038/nrneurol.2013.33
142. Katsumoto A, Takeuchi H, Tanaka F. Tau pathology in chronic traumatic encephalopathy and Alzheimer's disease: similarities and differences. Front Neurol. (2019) 10:980. doi: 10.3389/fneur.2019.00980
143. Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. (2019) 568:420–3. doi: 10.1038/s41586-019-1026-5
144. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. (2019) 14:32. doi: 10.1186/s13024-019-0333-5
145. Atri A. The Alzheimer's disease clinical spectrum. Med Clin North Am. (2019) 103:263–93. doi: 10.1016/j.mcna.2018.10.009
146. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:263–9. doi: 10.1016/j.jalz.2011.03.005
147. Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimers disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
148. Kurlan R, Richard IH, Papka M, Marshall F. Movement disorders in Alzheimers disease: more rigidity of definitions is needed. Mov Disord. (2000) 15:24–9. doi: 10.1002/1531-8257(200001)15:1<24::AID-MDS1006>3.0.CO;2-X
149. Starkstein SE, Merello M. Movement disorders in Alzheimer's disease. In: Merello M, Starkstein SE, editors. Movement Disorders in Dementias. London: Springer London (2014). pp. 129–40.
150. Pavlis CJ, Kutscher EC, Carnahan RM, Kennedy WK, Van Gerpen S, Schlenker E. Rivastigmine-induced dystonia. Am J Heal Pharm. (2007) 64:2468–70. doi: 10.2146/ajhp060399
151. Magnuson TM, Roccaforte WH, Wengel SP, Burke WJ. Medication-induced dystonias in nine patients with dementia. J Neuropsychiatry Clin Neurosci. (2000) 12:219–25. doi: 10.1176/jnp.12.2.219
152. van den Bosch S, Bouckaert F, Peuskens J. Mirtazapine-induced dystonia in a patient with Alzheimer's disease. Tijdschr Psychiatr. (2006) 48:153–7.
153. Vanacore N, Suzzareddu G, Maggini M, Casula A, Capelli P, Raschetti R. Pisa syndrome in a cohort of Alzheimer's disease patients. Acta Neurol Scand. (2005) 111:199–201. doi: 10.1111/j.1600-0404.2005.00388.x
154. Cossu G, Melis M, Melis G, Maccioni E, Putzu V, Catte O, et al. Reversible Pisa syndrome (pleurothotonus) due to the cholinesterase inhibitor galantamine: case report. Mov Disord. (2004) 19:1243–4. doi: 10.1002/mds.20164
155. Mimura Y, Kurose S, Takata T, Tabuchi H, Mimura M, Funayama M. Pisa syndrome induced by switching of a choline-esterase inhibitor treatment from donepezil to galantamine: a case report. BMC Neurol. (2020) 20:183. doi: 10.1186/s12883-020-01769-2
156. Caviness JN. Myoclonus and neurodegenerative disease-what's in a name? Parkinsonism Relat Disord. (2003) 9:185–92. doi: 10.1016/S1353-8020(02)00054-8
157. Wilkins DE, Hallett M, Erba G. Primary generalised epileptic myoclonus: a frequent manifestation of minipolymyoclonus of central origin. J Neurol Neurosurg Psychiatry. (1985) 48:506–16. doi: 10.1136/jnnp.48.6.506
158. Silveira-Moriyama L, Lees AJ. Endemic atypical parkinsonism. J Neurol Sci. (2018) 388:220–1. doi: 10.1016/j.jns.2018.02.018
159. Caparros-Lefebvre D, Sergeant N, Lees A, Camuzat A, Daniel S, Lannuzel A, et al. Guadeloupean Parkinsonism: a cluster of progressive supranuclear palsy-like tauopathy. Brain. (2002) 125:801–11. doi: 10.1093/brain/awf086
160. Steele JC. Parkinsonism-dementia complex of Guam. Mov Disord. (2005) 20:S99–S107. doi: 10.1002/mds.20547
161. Okumiya K, Wada T, Fujisawa M, Ishine M, Garcia del Saz E, Hirata Y, et al. Amyotrophic lateral sclerosis and parkinsonism in Papua, Indonesia: 2001-2012 survey results. BMJ Open. (2014) 4:e004353. doi: 10.1136/bmjopen-2013-004353
162. Kokubo Y, Ito K, Kuzuhara S. Ophthalmomyiasis-like pigmentary retinopathy in ALS/PDC in the Kii peninsula of Japan. Neurology. (2003) 60:1725–6. doi: 10.1212/01.WNL.0000061487.16841.72
163. Kokubo Y, Kuzuhara S. Neuroradiological study of patients with amyotrophic lateral sclerosis and Parkinsonism-dementia complex on the Kii Peninsula of Japan. Arch Neurol. (2003) 60:1257–61. doi: 10.1001/archneur.60.9.1257
164. Yeo T, Tan LC. ‘Hummingbird’ Sign in a patient with Guam Parkinsonism-dementia complex. J Mov Disord. (2017) 10:145–8. doi: 10.14802/jmd.17025
165. Mimuro M, Yoshida M, Kuzuhara S, Kokubo Y. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: a multiple proteinopathy? Neuropathology. (2018) 38:98–107. doi: 10.1111/neup.12434
166. Ishiura H, Takahashi Y, Mitsui J, Yoshida S, Kihira T, Kokubo Y, et al. C9ORF72 repeat expansion in amyotrophic lateral sclerosis in the Kii Peninsula of Japan. Arch Neurol. (2012) 69:1154–8. doi: 10.1001/archneurol.2012.1219
167. Dombroski BA, Galasko DR, Mata IF, Zabetian CP, Craig U-K, Garruto RM, et al. C9orf72 hexanucleotide repeat expansion and guam amyotrophic lateral sclerosis-Parkinsonism-dementia complex. JAMA Neurol. (2013) 70:742. doi: 10.1001/jamaneurol.2013.1817
168. van Bemmel K, van der Weegen K. Universal classifications, national approaches and specific situations: a comparative study on the conceptualization of nodding syndrome in Uganda and Tanzania. Anthropol Med. (2019) 26:177–96. doi: 10.1080/13648470.2017.1361652
169. Spencer PS, Mazumder R, Palmer VS, Pollanen MS. Nodding syndrome phenotypes. Rev Neurol (Paris). (2019) 175:679–85. doi: 10.1016/j.neurol.2019.09.005
170. Idro R, Opoka RO, Aanyu HT, Kakooza-Mwesige A, Piloya-Were T, Namusoke H. Nodding syndrome in Ugandan children-clinical features, brain imaging and complications: a case series. BMJ Open. (2013) 3:e002540. doi: 10.1136/bmjopen-2012-002540
171. Caparros-Lefebvre D, Golbe LI, Deramecourt V, Maurage C-A, Huin V, Buée-Scherrer V, et al. A geographical cluster of progressive supranuclear palsy in northern France. Neurology. (2015) 85:1293–300. doi: 10.1212/WNL.0000000000001997
172. Hoffman LA, Vilensky JA. Encephalitis lethargica: 100 years after the epidemic. Brain. (2017) 140:2246–51. doi: 10.1093/brain/awx177
173. Villemagne VL, Velakoulis D, Doré V, Bozinoski S, Masters CL, Rowe CC, et al. Imaging of tau deposits in adults with Niemann-Pick type C disease: a case-control study. Eur J Nucl Med Mol Imaging. (2019) 46:1132–8. doi: 10.1007/s00259-019-4273-7
174. Ortega-Aznar A, Romero-Vidal FJ, Castellví J, Ferrer JM, Codina A. Adult-onset subacute sclerosing panencephalitis: clinico-pathological findings in 2 new cases. Clin Neuropathol. (2003) 22:110–8.
175. Risacher SL, Farlow MR, Bateman DR, Epperson F, Tallman EF, Richardson R, et al. Detection of tau in Gerstmann-Sträussler-Scheinker disease (PRNP F198S) by [18F]Flortaucipir PET. Acta Neuropathol Commun. (2018) 6:114. doi: 10.1186/s40478-018-0608-z
176. Forrest SL, Kril JJ, Stevens CH, Kwok JB, Hallupp M, Kim WS, et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain. (2018) 141:521–34. doi: 10.1093/brain/awx328
177. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
178. Bourinaris T, Houlden H. C9orf72 and its relevance in Parkinsonism and movement disorders: a comprehensive review of the literature. Mov Disord Clin Pract. (2018) 5:575–85. doi: 10.1002/mdc3.12677
179. Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. (2005) 37:806–8. doi: 10.1038/ng1609
180. Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. (2010) 74:366–71. doi: 10.1212/WNL.0b013e3181ccc732
181. Goldman JS, Rademakers R, Huey ED, Boxer AL, Mayeux R, Miller BL, et al. An algorithm for genetic testing of frontotemporal lobar degeneration. Neurology. (2011) 76:475–83. doi: 10.1212/WNL.0b013e31820a0d13
182. Rohrer JD, Warren JD, Omar R, Mead S, Beck J, Revesz T, et al. Parietal lobe deficits in frontotemporal lobar degeneration caused by a mutation in the progranulin gene. Arch Neurol. (2008) 65:506. doi: 10.1001/archneur.65.4.506
183. Le Ber I, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet-Lecrux A, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. (2008) 131:732–746. doi: 10.1093/brain/awn012
184. Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, et al. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage. (2010) 53:1070–6. doi: 10.1016/j.neuroimage.2009.12.088
185. Kim EJ, Rabinovici GD, Seeley WW, Halabi C, Shu H, Weiner MW, et al. Patterns of MRI atrophy in tau positive and ubiquitin positive frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. (2007) 78:1375–8. doi: 10.1136/jnnp.2006.114231
186. Josephs KA, Whitwell JL, Parisi JE, Petersen RC, Boeve BF, Jack Jr CR, et al. Caudate atrophy on MRI is a characteristic feature of FTLD-FUS. Eur J Neurol. (2010) 17:969–75. doi: 10.1111/j.1468-1331.2010.02975.x
187. Caroppo P, Le Ber I, Camuzat A, Clot F, Naccache L, Lamari F, et al. Extensive white matter involvement in patients with frontotemporal lobar degeneration. JAMA Neurol. (2014) 71:1562. doi: 10.1001/jamaneurol.2014.1316
188. Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. (2015) 14:253–62. doi: 10.1016/S1474-4422(14)70324-2
189. Schönecker S, Neuhofer C, Otto M, Ludolph A, Kassubek J, Landwehrmeyer B, et al. Atrophy in the thalamus but not cerebellum is specific for C9orf72 FTD and ALS patients - an atlas-based volumetric MRI study. Front Aging Neurosci. (2018) 10:45. doi: 10.3389/fnagi.2018.00045
190. Lynch T, Sano M, Marder KS, Bell KL, Foster NL, Defending RF, et al. Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia- parkinsonism-amyotrophy complex. Neurology. (1994) 44:1878–8. doi: 10.1212/WNL.44.10.1878
191. McCarthy A, Lonergan R, Olszewska DA, O'Dowd S, Cummins G, Magennis B, et al. Closing the tau loop: the missing tau mutation. Brain. (2015) 138:3100–9. doi: 10.1093/brain/awv234
192. Kouri N, Carlomagno Y, Baker M, Liesinger AM, Caselli RJ, Wszolek ZK, et al. Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol. (2014) 127:271–82. doi: 10.1007/s00401-013-1193-7
193. Baba Y, Baker MC, Le Ber I, Brice A, Maeck L, Kohlhase J, et al. Clinical and genetic features of families with frontotemporal dementia and Parkinsonism linked to chromosome 17 with a P301S tau mutation. J Neural Transm. (2007) 114:947–50. doi: 10.1007/s00702-007-0632-9
194. Rossi G, Marelli C, Farina L, Laurà M, Maria Basile A, Ciano C, et al. The G389R mutation in the MAPT gene presenting as sporadic corticobasal syndrome. Mov Disord. (2008) 23:892–5. doi: 10.1002/mds.21970
195. Tacik P, DeTure M, Lin W-L, Sanchez Contreras M, Wojtas A, Hinkle KM, et al. A novel tau mutation, p.K317N, causes globular glial tauopathy. Acta Neuropathol. (2015) 130:199–. doi: 10.1007/s00401-015-1425-0
Keywords: tauopathy, movement disorders, chameleons, mimics, MAPT
Citation: Ganguly J and Jog M (2020) Tauopathy and Movement Disorders—Unveiling the Chameleons and Mimics. Front. Neurol. 11:599384. doi: 10.3389/fneur.2020.599384
Received: 27 August 2020; Accepted: 30 September 2020;
Published: 05 November 2020.
Edited by:
Sanjay Pandey, University of Delhi, IndiaReviewed by:
Prachaya Srivanitchapoom, Mahidol University, ThailandRukmini Mridula Kandadai, Nizam's Institute of Medical Sciences, India
Copyright © 2020 Ganguly and Jog. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mandar Jog, TWFuZGFyLkpvZyYjeDAwMDQwO2xoc2Mub24uY2E=