 Michael J. Strong
Michael J. Strong Neil S. Donison
Neil S. Donison Kathryn Volkening
Kathryn Volkening
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 23 November 2020
Sec. Dementia and Neurodegenerative Diseases
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.598907
This article is part of the Research TopicTau Pathology in Neurological DisordersView all 18 articles
There is increasing acceptance that amyotrophic lateral sclerosis (ALS), classically considered a neurodegenerative disease affecting almost exclusively motor neurons, is syndromic with both clinical and biological heterogeneity. This is most evident in its association with a broad range of neuropsychological, behavioral, speech and language deficits [collectively termed ALS frontotemporal spectrum disorder (ALS-FTSD)]. Although the most consistent pathology of ALS and ALS-FTSD is a disturbance in TAR DNA binding protein 43 kDa (TDP-43) metabolism, alterations in microtubule-associated tau protein (tau) metabolism can also be observed in ALS-FTSD, most prominently as pathological phosphorylation at Thr175 (pThr175tau). pThr175 has been shown to promote exposure of the phosphatase activating domain (PAD) in the tau N-terminus with the consequent activation of GSK3β mediated phosphorylation at Thr231 (pThr231tau) leading to pathological oligomer formation. This pathological cascade of tau phosphorylation has been observed in chronic traumatic encephalopathy with ALS (CTE-ALS) and in both in vivo and in vitro experimental paradigms, suggesting that it is of critical relevance to the pathobiology of ALS-FTSD. It is also evident that the co-existence of alterations in the metabolism of TDP-43 and tau acts synergistically in a rodent model to exacerbate the pathology of either.
Our understanding of the pathobiology of amyotrophic lateral sclerosis (ALS) has evolved dramatically since its first description as a clinicopathological entity in 1874 by Charcot as a disorder of progressive muscular atrophy associated with spasticity (1). ALS was once considered a disorder restricted to the degeneration of the descending supraspinal motor neurons and those lower motor neurons subserving bulbar and spinal motor functions, with specific sparing of a number of functions (including cognition). This concept, however, has been replaced by the understanding that ALS is a multisystems disorder for which motor neuron dysfunction is one aspect of a much larger picture (2, 3).
Given this, it is not surprising that ALS is now considered to be syndromic with clinical manifestations that reflect not only a range of underlying genetic and biochemical impairments consistent with prominent disruptions in RNA metabolism and protein degradation pathways, but also with considerable non-motor involvement (4). This latter aspect has become increasingly evident as our recognition and understanding of the neuropsychological manifestations of ALS has grown. While there has also been remarkable growth in our understanding of the perturbed biochemistry underlying this aspect of the disorder, there remains a tremendous amount that is unknown. In this review, we will discuss the nature of the phenotypic heterogeneity of ALS as it applies to associated syndromes of frontotemporal dysfunction that can be associated with ALS, explore the nature of the proteinopathies that are thought to underlie the process including a critical analysis of perturbations in microtubule associated protein tau (tau) metabolism, and consider a conceptual framework in which synergism amongst co-expressed toxic proteins can drive disease phenotype.
The classic clinical and neuropathological description of ALS hinges on the evidence of motor system degeneration in which the loss of lower motor neurons drives progressive muscle atrophy while the loss of upper motor neurons is predominantly, but not exclusively, manifested as spasticity. The loss of motor function culminates in respiratory failure in the majority of individuals within 3 and 5 years of symptom onset, although there is wide variability in survivorship (5). Degenerating motor neurons in ALS classically bear the neuropathological hallmark of neuronal cytoplasmic inclusions (NCIs) composed largely of cytoskeletal proteins, predominantly of the neuronal intermediate filament family (6). Ubiquitination of these proteins is consistent with the concept of impaired proteasomal degradation in ALS as a key pathological process.
This classical conceptualization of ALS has been significantly altered through two fundamental observations: firstly, that widespread frontotemporal dysfunction can occur in a significant proportion of patients (7–11); and secondly, that a fundamental alteration in RNA metabolism, at multiple levels, is a core biological process for the majority of ALS cases (12). This latter postulate is supported by the observation of NCIs composed of a broad range of RNA binding proteins not only within degenerating motor neurons, but within cortical and subcortical neurons in ALS. It was these observations that have now provided a unifying linkage between the two major clinical aspects of ALS–progressive motor neuron degeneration and frontotemporal dysfunction. The key breakthrough that led to this transformative understanding of ALS was the discovery that TDP-43 (TAR DNA binding protein, or transactive response DNA binding protein 43 kDa) accumulates in degenerating motor neurons and within degenerating cortical neurons in frontotemporal lobar degeneration (FTLD) (13, 14). This protein is a DNA/RNA binding protein that is vital to stress and injury responses and is intricately involved in the regulation of RNA metabolism (15, 16). While pathological neuronal and glial cytoplasmic and nuclear inclusions of TDP-43 are now considered to be the neuropathological hallmark of ALS, multiple RNA binding proteins can aggregate as pathological NCIs in ALS, often within the same motor neurons, and often colocalizing to the same aggregates (17–24). This lends support to the broader concept of ALS as a disorder of RNA metabolism in which a diverse array of RNA binding proteins can be involved, likely in several different mechanisms ranging from mutations resulting in gain- or loss-of-function of RNA binding proteins [including fused in sarcoma (FUS) and TDP-43] to also including those proteins in which ALS-associated mutations confer novel RNA interacting capacity [for instance as observed with mutations in copper/zinc superoxide dismutase (mtSOD1)] (25–27). This is consistent with the conceptualization that ALS is syndromic and reflective of a significant biological heterogeneity underlying not just its pathobiology, but also its phenotypic expression.
Nowhere is this more evident than in our understanding and acceptance that individuals with otherwise classical ALS can be affected by a range of neuropsychological, behavioral, speech, and language deficits. These can range from mild to severe impairment, including frontotemporal dementia (FTD) (28, 29). These deficits are encapsulated in the most recent diagnostic criteria under the rubric of the “frontotemporal spectrum disorder” of ALS (ALS-FTSD) (7). Applying these criteria, less than half of ALS patients have a pure motor neuron disorder in which there is no evidence of degeneration outside of the motor system, and for whom the El Escorial or Awaji criteria are fully met (30, 31). Amongst the remaining patients, ~15% will also suffer from, or will have presented with, a FTD that meets the Rascovsky et al. criteria for diagnosis, including progression (32, 33). The remaining patients will exhibit one or more features of behavioral impairment, cognitive or executive dysfunction, speech, and/or language impairments, and in some cases, a mixture thereof but at levels insufficient for a diagnosis of FTD. Approximately 2% of ALS patients can also be affected by a non-FTD dementia such as Alzheimer's disease or vascular cognitive impairment. Finally, there is also a subgroup of patients with frontotemporal lobar degeneration (FTLD, the neuropathological correlate of the FTD) in whom motor neuron degeneration, typically marked by pathological TDP-43 inclusions, is only obvious postmortem.
It is this broad range of clinical deficits that has led to the current thinking that ALS and FTD are but two points on a disease continuum, with ALS-FTSD residing between these two extremes (34). Where along this continuum a patient will manifest is simply the clinical reflection of where the greatest burden of disease is impacting at that moment in time, both in terms of the extent of pathology present but also on the nature of disruption of the affected neural networks. This conceptualization is supported by the observation that the neuropathological hallmark of the vast majority of ALS cases, regardless of the presence or absence of ALS-FTSD, is the presence of pathological cytoplasmic TDP-43 inclusions reflective of disruption in the metabolism of TDP-43 and thus sharing a feature in common with a subset of FTD patients (35, 36).
To this end, the pathological characterization of FTLD is based on the primary protein associated with the neuropathology. Using this classification, approximately half of which are FTLD-TDP (37) and slightly less than half are tau predominant pathology (FTLD-tau) (36). The remaining minority of cases demonstrate neither, but rather pathology driven by alterations in the FET family of proteins [FUS; Ewing's Sarcoma (EWS) and TATA-binding protein-associated factor 15 (TAF15)] metabolism or with evidence for impaired ubiquitin-conjugated of proteins (FTLD-UPS) (37). The challenge with this categorization is that there is increasing evidence to support overlaps amongst these pathologies, and in particular between TDP-43 and tau (38). As will be discussed, there is evidence to suggest that this underlies the pathobiology of ALS-FTSD.
Tau is an intrinsically disordered, highly conserved protein that is primarily enriched in axons of central neurons and whose primary function is to stabilize and promote microtubule stability (39–42). The MAPT gene located on chromosome 17q21 that encodes for human tau contains 16 exons, of which exons 2 and 3 encode for N-terminus insertions of 29 amino acids in length while exon 10 encodes for one of four microtubule binding repeats (Figure 1). Through alternative splicing of the tau mRNA, tau can therefore exist as 6 isoforms ranging from 352 to 441 amino acid residues with apparent molecular weights of 48–67 kDa (predicted 36–45 kDa without modifications) and characterized by the presence or absence of one or two N-terminus inserts (yielding 0N, 1N, or 2N isoforms) accompanied by the expression of either 3 or 4 microtubule binding repeats (3R or 4R) (48). Tau is a largely unstructured soluble protein that in vitro takes on a “paperclip” conformation such that the N-terminus, C-terminus, and microtubule binding region (MTBR) are in close approximation (Figure 1C) (43–46, 49, 50). The relative expression of 3R to 4R isoforms is highly regulated and when altered is associated with a range of neurodegenerative disease states characterized by FTLD with pathological tau deposition (36). Tau undergoes extensive post-translational modification, with phosphorylation arguably being the most crucial to its interaction with microtubules (48, 51). Site specific tau phosphorylation modulates the interaction between tau and microtubules, with pathological phosphorylation dissociating the two and giving rise to elevated soluble tau isoforms which are then free to dimerize into stable tau oligomers, which then continue to polymerize into protomers and lead to fibril formation.
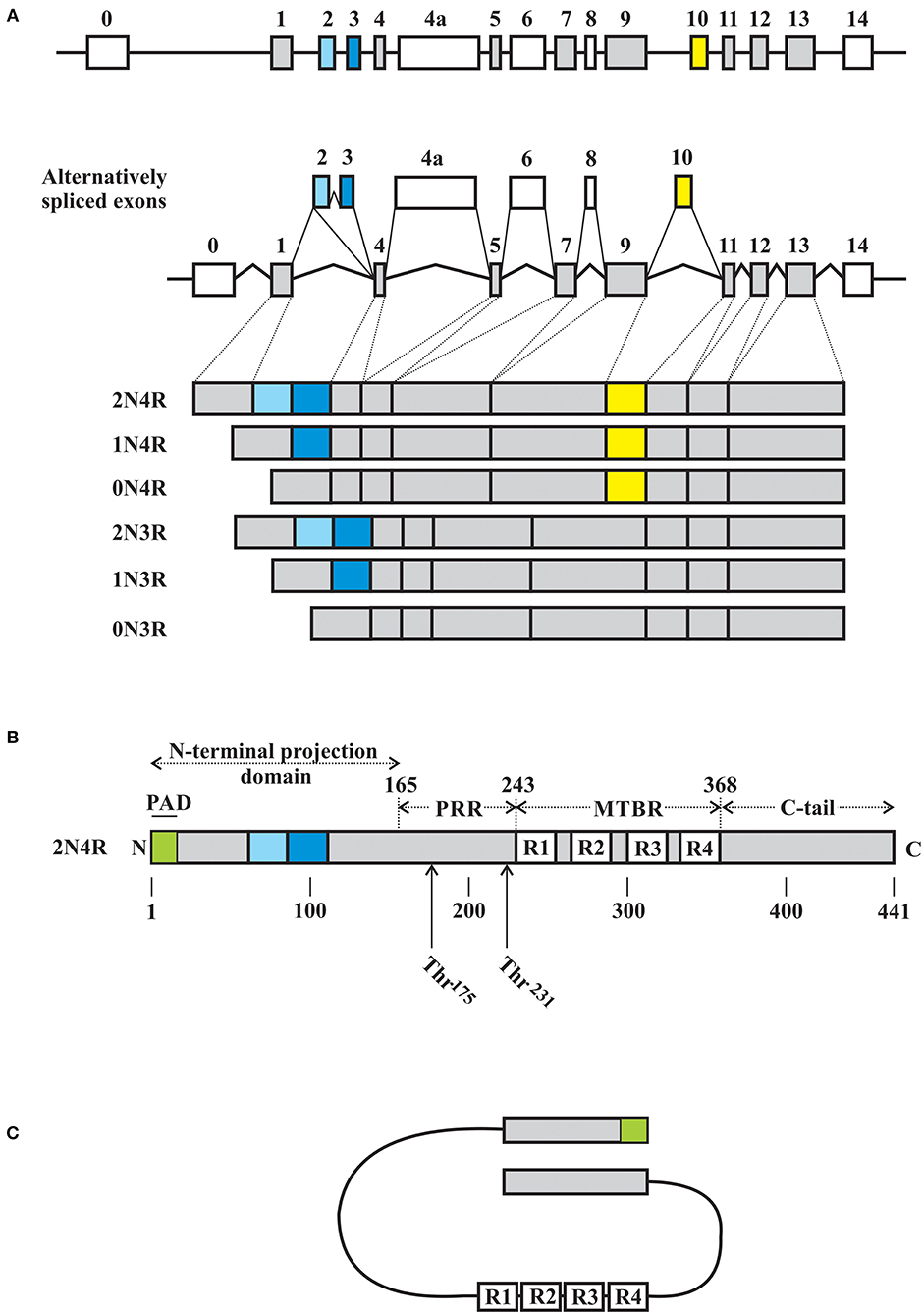
Figure 1. Schematic representations of MAPT gene and tau. (A) Tau is encoded by a single gene on chromosome 17q.31, with 16 exons, including the alternatively spliced exons 2, 3, 4a, 6, 8, and 10. Start and stop codons are located in exons 1 and 13, respectively. Exons 2, 3, and 10 are alternatively expressed in the adult human brain, giving rise to 6 tau isoforms based on the inclusion of 0, 1, or 2 N-terminus inserts encoded by exons 2 or 3 (exon 2 can be alternatively spliced independent of exon 3, while exon 3 is always expressed with an exon 2 encoded insert) with 3 or 4 microtubule binding repeats (MTBR) depending on the expression of an exon 10 encoded repeat. As such, the 6 isoforms are described as 0N, 1N, or 2N with either 3R or 4R MTBRs. Exon 4A and 6 are expressed in the peripheral nervous system giving rise to additional tau isoforms. To date, exon 8 does not appear to be expressed. (B) Using numbering based on the 2N4R isoform of tau of 441 amino acid residues, tau has four major domains, including the N-terminus domain (residues 1–165) with the phosphatase activating domain (PAD) localized to the extreme N-terminus region, a proline rich domain (PRR, residues 166–242), a microtubule assembly domain (residues 243–367) consisting of either 3 or 4 microtubule binding repeats (R1 thru R4, each of ~31 amino acid residues), and a C-terminus domain (residues 368–441). (C) Schematic representation of the proposed paperclip conformation of soluble tau in which the N-terminus domain, C-terminus domain, and MTBR are in close approximation. Schematic illustrations based on the following references: (43–47).
While the primary function of tau is to promote microtubule stability, tau also appears to be involved in several other non-cytoskeletal related activities within cells. Phosphorylation of tau plays a role in determining the localization of tau throughout neurons, with soma localized tau exhibiting high levels of phosphorylation, and a gradual loss of phosphorylation occurring as tau is located closer to the growth cone of an axon (52). It was originally thought that tau phosphorylation was altered as tau was transported through the cell; however, evidence also supports a model in which new tau is synthesized and immediately phosphorylated, with this pattern of phosphorylation dictating where in the cell that tau will ultimately be localized (49, 53, 54).
A tau isoform that lacks phosphorylation at S195-S202 is localized to the nucleus, where it is involved in DNA binding (either through association with AT-rich DNA, or with the minor groove of AT-rich DNA sequences) (55, 56), heterochromatin stability, transcription repression of ribosomal DNA, long non-coding RNA production, or possibly in repressing gene expression (55, 57, 58). The isoform of tau expressed is also important in that compartmentalization appears to prefer one isoform type to another, with isoforms bearing 0N or 2N preferentially located outside of the nucleus, while 1N isoforms are found preferentially in the nucleus (59).
In response to cell stress, nuclear tau translocates to the nucleolus, where it is postulated to increase RNA binding protein interactions with TIA1 (T-cell intracellular antigen) (60). Cell stress also elicits the formation of stress granules (SG) within neurons. TIA1 translocates to SG in the cytosol, where these TIA1 positive SG have been detected to localize with tau that is highly phosphorylated, or “hyper” phosphorylated. However, not all SG are TIA1 positive, and other populations of SG do not appear to colocalize with hyperphosphorylated tau, for example the G3BP1-positive SG (61). SG formation has been noted to be upregulated in ALS, with both TDP-43 and FUS proteins playing important roles in the formation and maintenance of SG [reviewed in (62)]. Proteins with low complexity/highly disordered domains appear to be integral to SG formation, and dysregulation of the expression of these proteins, the presence of mutations, or alterations in the localization of these proteins appear to have a profound effects on SG dynamics in neurodegenerative disease including ALS and FTLD [reviewed in (63, 64)]. The role(s) of tau in SG formation is unclear; however evidence shows that if tau is overexpressed there is an increase in SG formation (65–67). There is also evidence suggesting that the deposition of tau can induce the condensation of RNA binding proteins through liquid-liquid phase separation (LLPS). LLPS has been shown to involve highly disordered protein domains [including the DNA/RNA binding protein TDP-43 (68)] that tend to fall into an insoluble state spontaneously when in high enough concentrations. Tau can experience LLPS and as such may induce the formation of toxic tau conformations (69–72). It would not be unreasonable to postulate that tau may help to initiate LLPS of RNA binding proteins that are contained within the same SG. Consistent with this concept, there is evidence that the RNA binding proteins Musashi 1 and 2 (MSI 1 and MSI 2, respectively), both highly expressed in mature neurons, form intranuclear complexes with oligomeric tau proteins in a concentration dependant manner (73). In doing so, these complexes significantly impair nucleocytoplasmic transport of NLS-containing cargo, a process that has been postulated to fundamental to the pathobiology of ALS and FTD (74). It is of direct relevance therefore that both intracellular and extracellular MSI 1 and MSI 2 complexes are present in both ALS and FTD cortical tissue, and that this occurs in the presence of increased oligomeric tau (73). It is often inferred that the metabolism of tau protein is altered in any given disease process based largely on the presence of tau immunoreactive NCIs; the finding that this immunoreactivity is also for specific tau phospho epitopes normally observed in the presence of a disease state supports this. This is further augmented by the biochemical characterization of the tau protein following separation into detergent soluble or insoluble fractions, with characterization then based on the relative preponderance of either or both the 3R or 4R isoforms. However, the deposition of tau protein can be either a primary process as in neurodegeneration or a secondary process as in a stress response (60, 61, 75).
In the following, we will examine the neuropathological, biochemical, and experimental evidence in support of pathological tau metabolism amongst a subset of ALS patients.
Tau dysmetabolism has been most clearly documented in the Western Pacific variant of ALS in which affected individuals developed either a Parkinson-Dementia-ALS complex or ALS alone, at rates several thousand fold greater than observed amongst the rest of the world (76). The characteristic neuropathological feature was the widespread deposition of tau protein throughout the neuroaxis, including spinal motor neurons (77, 78). This latter feature is not traditionally observed in other ALS variants with the exception of those individuals with chronic traumatic encephalopathy (CTE) who also develop clinical and electrophysiological features consistent with a diagnosis of ALS (CTE-ALS) (79–81).
Beyond this, the evidence for alterations in tau metabolism in ALS has been scant and largely limited to individual case reports or unique inherited disorders in which ALS has been described amongst a broader range of neurological and neuropathological deficits or in association with rare MAPT mutations (82–87). However, two instructive exceptions drawn from the spectrum of genetic mutations associated with ALS, ALS-FTD and FTD are the pathological hexanucleotide expansions of C9orf 72 and the ALS-associated mutations of FUS (88–90). Both provide further support for the concept of a biological continuum underlying the pathogenesis of ALS and FTD in addition to insights into mechanisms of altered tau metabolism.
Pathological hexanucleotide repeat (GGGGCC)n expansion of C9orf 72 is the most common hereditary molecular cause of ALS, ALS-FTD and FTD. The neuropathological hallmark of the associated FTLD includes foci of dinucleotide-peptide repeats (DPRs) in specific neuronal subpopulations in association with more widespread pathological TDP-43 deposition [reviewed in (36, 91)] in the absence of an associated tauopathy or when present, as a non-specific finding (92). Although the exact mechanism by which these pathological expansions give rise to a toxic variant of C9orf 72, there is evidence to support a role in fundamentally altering RNA metabolism through a variety of avenues [reviewed in (91)]. However, there is also emerging evidence that pathological C9orf 72 expansion may act in a synergistic manner to increase the propensity for tau pathology by increasing cdk5-mediated tau phosphorylation, including pathological phosphorylation (93–95).
In contrast, mutations in FUS are an uncommon molecular cause of familial ALS, rarely observed in sporadic ALS, and found in only a single case report in consanguineous twins with FTD (17, 18, 90, 96–98). While FUS has an array of both cytoplasmic and nuclear functions, it plays a critical role in regulating alternative splicing through its association with the spliceosome, including in regulating the physiological splicing of MAPT exon 10 (99). The loss of FUS activity results in the preferential inclusion of exon 10 and an increased expression of 4R tau (100). FUS forms a complex with splicing factor, proline- and glutamine-rich (SFPQ) through an RNA-dependant interaction such that the loss of either FUS or SFPQ results in the preferential expression of 4R tau and an accompanying increase in ptau expression, including pathological tau isoforms (101–103). The importance of this is highlighted by the finding of reduced interactions between FUS and SFPQ across a broad range of FTLDs, including ALS-FTD (102). The recent report of a single atypical case of sporadic ALS manifesting with predominantly upper motor neuron dysfunction with extrapyramidal features who at autopsy demonstrated pyramidal neuron FUS basophilic inclusions that colocalized with AT8 tau immunoreactivity in motor neurons, in addition to a 4R predominant tauopathy, provides support for such a proposed role of FUS in contributing to alterations in tau metabolism in ALS (104).
More recently, a 17 kDa neurotoxic tau fragment (tau45−230) has been observed in extracts of both the brain and ventral spinal cord of sporadic ALS patients, but not in controls (105, 106). This fragment is generated by calpain cleavage of full length tau and has been observed both in vitro and in vivo across a broad range of tauopathies (107, 108).
Both tau immunoreactive inclusions and pathological tau phosphorylation disproportionate for age have been observed in ALS patients affected with cognitive or executive dysfunction (ALSci), typically in the form of dystrophic neurites, neurofibrillary tangle-like structures and pre-tangles, neuritic granules, and tau-immunoreactive tufted astrocytes (109–111). Tau isolated from mesial frontal cortex and subcortical at postmortem demonstrates a significantly greater propensity for fibril formation in a thioflavin S assay, partitions into the sarkosyl-insoluble fraction as all 6 isoforms (distinguishing it from Alzheimer's disease tau, 3R or 4R tau disorders), and is pathologically phosphorylated at Thr175 (pThr175tau) (112). Using a polyclonal antibody recognizing pThr175tau, widespread tau deposition, sparing the motor neurons, was observed to be in association with a diffuse increase in TDP-43 immunoreactivity (110). In the same study, it was also observed that of those patients without evidence of neuropsychological impairment, approximately half demonstrated pThr175tau immunoreactive pathology although significantly less pronounced than observed in those with evidence of ALS-FTSD. Of note, the active isoform of GSK3β (pGSK3β) was also upregulated in those regions most extensively affected with tau pathology (113). The observation of pThr175tau pathology was subsequently confirmed in a separate cohort of motor neuron disease (MND) patients with concomitant FTD, including approximately 10% with non-descript neuronal aggregates and as with the earlier study, associated with pathological TDP-43 expression (114). Although not examined for the pThr175tau phospho epitope, ptau (pSer396, pSer214, and pSer404) has been described in ALS cervical spinal cord and motor cortex in the cytosol and nuclei of motor neurons as diffuse immunoreactivity in the absence of fibril formation (115). The presence of nuclear tau in this latter series may be reflective of a neuronal stress response although no comment was made with respect to the presence or absence of nucleolar tau staining. To date, no other tau phospho-epitopes have been examined or characterized in either ALS or ALS-FTSD to this extent.
In addition to this evidence supporting alterations in tau metabolism in sporadic ALS, a subset of CTE patients (in whom a tauopathy is the neuropathological hallmark) will have evidence of ALS either antemortem with clinical or electrophysiological evidence of diffuse motor neuron degeneration, or at autopsy (80, 81, 116). Amongst these patients, pThr175tau, pThr231tau, and oligomeric tau (T22) immunoreactive inclusions are readily observed in both cortical and spinal motor neurons (79).
Given the broad array of pathological site specific tau phospho epitopes associated with a range of neurodegenerative disorders, the observation of pThr175tau immunoreactive inclusions suggests but does not prove that phosphorylation at this epitope is also pathologically important amongst a number of tauopathies (117). However, pThr175tau is not observed in fetal human tissue although tau exhibits significantly higher molar phosphorylation in the fetus, nor is it observed in the healthy aged (117, 118). Hence, its presence suggests a role in the genesis of pathological tau fibrils.
Elevations in cerebrospinal fluid tau and/or TDP-43 have been widely documented and suggested as potential biomarkers for ALS although for the former these have in general reflected total tau (119, 120). However, others have failed to find similar increases in either CSF total tau or ptau levels in ALS patients (121–124). Thus, it remains unclear if CSF tau can be used as a surrogate marker for a concomitant tauopathy in ALS.
Supporting the hypothesis of pathological significance of pThr175tau, the introduction of a pseudophosphorylated 2N4R human tau construct mimicking pThr175tau (Thr175Asp-tau) induces pathological tau fibril formation in both HEK293T and Neuro2a cells, significantly more so than when either wild-type human tau or tau in which phosphorylation at Thr175 was inhibited (Thr175Ala-tau) were expressed (125). The induction of pathological tau fibril formation in vitro was dependent on up-regulation of pGSK3β activity, which in turn induced phosphorylation at Thr231tau (pThr231tau)—one of several key phosphorylation sites in the cascade of driving tau oligomer formation (Figure 2) (126). The activation of GSK3β is dependent on the dephosphorylation of pSer9 which removes inhibition, allowing for GSK3β activity on its substrate (127, 128). We have shown that the phosphorylation of Thr175 of tau is associated with an enhanced exposure of the N-terminus of tau, increasing access to the tau phosphatase activating domain (PAD) located at amino acids 2–18 (129, 130). PAD exposure activates protein phosphatase-1 (PP1) which dephosphorylates pSer9 of GSK3β enhancing GSK3β activity through a relative increase in exposure of pTyr216 while phosphorylation of tau residue 18 (Y18) by the non-receptor tyrosine kinase fyn inhibits the PP1-GSK3β cascade (129, 131). While it remains unknown how tau phosphorylation at Thr175 leads to an increased PAD exposure, Thr175 resides within the proline-rich domain of tau in which the phosphorylation of other amino acids induces an opening of the tau paperclip conformation, leading to increased PAD exposure (43, 46, 47, 132). It is reasonable to anticipate that pThr175 would act similarly.
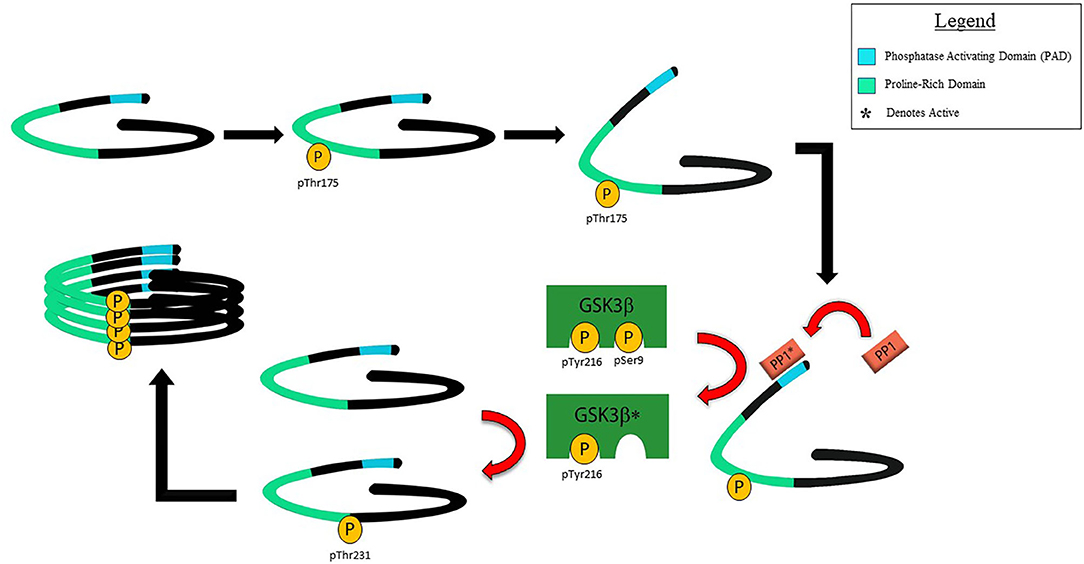
Figure 2. Schematic of proposed mechanism for the toxicity of pThr175tau. Under normal physiological conditions, tau is thought to exist in a closed paperclip conformation. Following phosphorylation of Thr175 by a kinase or kinases yet to be defined, the closed paperclip structure opens sufficiently to allow for exposure of the PAD which in turn activates protein phosphatase 1 (PP1). PP1 in turn dephosphorylates Ser9 of GSK3β, allowing for increased pGSK3β by unmasking pTyr216. pGSK3β in turn phosphorylates Thr231tau in a process that does not require priming by phosphorylation of Ser235tau. It is not known whether pThr231 exists in either the cis or trans isoform, but our evidence suggests that the presence of pThr231 is associated with increased tau oligomer formation and its subsequent polymerization into tau fibrils.
PAD exposure has been observed to be a common feature of the pathological inclusions across a broad range of tauopathies (133) and has been associated with neurotoxicity through the inhibition of fast axonal transport as an early contribution to the process of neurodegeneration (130, 133–135). In this mechanism, PAD exposure and activation of GSK3β results in phosphorylation of the kinesin light chain which promotes the release of cargo from kinesin 1 (130).
In addition, pThr231tau can exist in either trans or cis conformation (trans-pThr231 and cis-pThr231, respectively), in which cis-pThr231 is specific to the induction of tau pathology and cannot be readily degraded, in contrast to trans-pThr231 (136). Under normal physiological conditions, the isomerase peptidyl-propyl cis-trans isomerase NIMA-interacting 1 (PIN1) catalyzes the isomerization of cis-pThr231 to trans-pThr231, allowing for the dephosphorylation of this site by PP2A (protein phosphatase 2) and the subsequent ability for tau to exert its physiological activity in microtubule polymerization (137, 138). The pathological relevance of cis-pThr231 is highlighted by its observation in association with pathological tau deposition in AD and CTE (both experimental and human) (139). In accordance to this, it has also been demonstrated that there are reduced levels of PIN1 in human AD brains, resulting in decreased activity and thus the inability to isomerize cis-tau (138, 140). Additionally, genetic polymorphisms resulting in a decrease in PIN1 levels are associated with an increased risk of late-onset AD, whereas another polymorphism which leads to increased PIN1 expression has a neuroprotective effect (141, 142). The relative expression of cis vs. trans-tau and PIN1 activity in ALS or ALS-FTSD has not been examined to date.
It had been previously observed that the use of somatic gene transfer of tau harboring the P301L mutation that is causal for frontotemporal dementia with parkinsonism (FTDP-17) using a recombinant adeno-associated virus (rAAV9) injected into the hippocampus of rats induced both tau pathology and impairments in spatial working memory (143). Given this, we examined whether Thr175Asp-tau, when inoculated using somatic gene transfer with rAAV9 could similarly induce both pathology and behavioral deficits in rats. We observed that when Thr175-Asp tau was inoculated into the hippocampus of young adult female Sprague-Dawley rats, tau pathology was evident as early as 1 month post inoculation and consistently so by 1 year post-inoculation (144). While this was not associated with a behavioral or motor phenotype, it was observed that the burden of neuropathology was localized to the CA2 region, a region for which the testing paradigm employed was insensitive. It was of note however that the induction of tau pathology was accompanied by an upregulation of TDP-43 expression in this model.
Given the observation of pThr175tau, pGSK3β, pThr231tau, and oligomeric tau in affected neurons of individuals with CTE-ALS, we also examined whether a single subconcussive injury administered to Sprague-Dawley rats would also induce pathological tau fibril formation. Indeed this was the case, suggesting that this specific pathway of pathological tau fibril formation was a primary event (79).
As has been alluded to, concomitant pathologies of both TDP-43 and tau are increasingly recognized, including AD and the aforementioned ALS, CTE-ALS and in vivo models of pThr175 tau pathology (145–147). To begin to understand how these two pathologies inter-relate, we repeated the somatic gene transfer AAV9 hippocampal inoculums in rats also expressing the ALS-associated mutant human TDP-43 (TDP-43M337V). In these rats, constitutive cholinergic neuronal expression was driven by a ChAT promoter with a tTA-dependant tetracycline response element driver (148, 149). In the presence of doxycycline, no mtTDP-43 expression occurred whereas ChAT-tTA/TRE-TDP-43M337V rats, in the absence of doxycycline, developed a fulminant motor phenotype with prominent motor neuron NCI formation. Six months following stereotactic hippocampal inoculums of rAAV9 GFP-tagged tauT175D2N4R, a 50% withdrawal of doxycycline was undertaken. Rats were sacrificed 30 days later. Within 3 weeks, all ChAT-tTA/TRE-TDP-43M337V rats had developed motor deficits. At 30 days, a 2-fold increase in the extent of tau pathology was observed in the hippocampus of those rats inoculated with the pseudophosphorylated tau. This was associated with immunoreactivity to a monoclonal antibody recognizing tau phosphorylated at Ser202 and Thr205 (AT8) (150), suggesting that the tau construct had been further modified to a pathological tau phospho-isoform in the presence of the mutant TDP-43. There was also a trend toward an increase in the extent of spinal motor neuron pathology, a finding that may serve to explain the shortened survival in ALS-FTSD patients when contrasted to patients with ALS alone (151).
This inter-relationship between tau and TDP-43 has been further characterized in a murine model of CTE using a repetitive mild traumatic brain injury (rmTBI) in C57Bl6 mice using a repeated weight drop. In this model, a prominent tauopathy developed, including cis-ptau accumulations at 6 months post injury in association with tau oligomers, tangles, and TDP-43 immunoreactive NCIs (139). As described earlier, the cis conformation of pThr231tau prevents pThr231 dephosphorylation and the ability of tau to stabilize microtubules. The intraperitoneal administration of a monoclonal neutralizing antibody to cis-ptau eliminated the pathological tau accumulation and subsequent TDP-43 pathology, suggesting a linkage between the two pathologies. However, the induction of TDP-43 pathology may in this case represent a physiological stress response marked by an upregulation in its expression as previously observed following axonal injury, and thus only indirectly linked to the induction of the tau pathology (15).
Of note, both tau tubulin kinase 1 (TTBK1) and tau tubulin kinase 2 (TTBK2) have been shown to extensively phosphorylate both TDP-43 (152) and tau (153, 154). Both kinases have been observed to co-localize with TDP-43 and tau aggregates in FTLD-tau and FTLD-TDP cases (152, 155). These observations provide further support for the concept of a pathological commonality underlying these two proteinopathies and a mechanism to drive both pathological tau and TDP-43 phosphorylation concomitantly (156).
The conventional concept of ALS as a pure motor neuron disorder and a discrete disease has long been replaced by one of a multisystem disorder that is syndromic in nature and for which progressive neuropsychological deficits, including a frontotemporal spectrum disorder, are common. As this conceptualization of ALS has evolved, so too has our understanding of its underlying pathobiology as reflected in the broad range of not only causative gene mutations associated with both familial and sporadic variants of the disorder, but the broad range of protein aggregates which typify its pathology (4). Embedded within this has been the recognition that FTD and ALS are increasingly recognized to share not only a clinical phenotypic spectrum, but also an underlying pathobiology as reflected not only in alterations in the metabolism of TDP-43, but increasingly in the metabolism of tau.
While the classical role of tau is the stabilization of microtubules through interactions mediated by its MBTR, there is increasing evidence to suggest a much broader repertoire of function of tau under both normal physiological states as well as in response to stress. These functions are critically dependant on a complex array of determinants including the somatopically-specific subcellular distribution of individual tau isoforms that includes an emerging role in gene expression and inter-relationships with RNA binding proteins, the impact of cellular stress upon tau expression, the conformational structure of tau and the extent of its post-translational modification (44, 49, 157). Although a broad array of post-translational modifications and their relationship to specific disease states have been well-characterized [reviewed in (44)], we have focused on the specific role of pathological phosphorylation at Thr175 and the mechanisms by which this culminates in the generation of oligomeric tau and a subsequent tauopathy, including a synergistic role with pathological TDP-43 deposition.
However, several pivotal questions remain in order to further clarify the role of alterations in tau metabolism in ALS, including:
• In the context of understanding that ALS-FTSD represents a broad array of neuropsychological, behavioral, speech, and language deficits, and that our initial studies characterizing pThr175-tau expression were undertaken in ALS patients with cognitive impairment (ALSci) while those of Behrouzi et al. were undertaken of individuals with FTD and ALS (114), clarifying the spectrum of clinicopathological correlates to ALS-FTSD and whether alterations in tau metabolism are specific to one or more variants within the spectrum;
• Linked to this, further defining the nature of TDP-43 deposition in ALS-FTSD in tau NCI-bearing neurons with a view to understanding whether the TDP-43 accumulation bears hallmarks of pathological tau deposition or that observed in a stress response;
• Identifying the kinase or kinases responsible for the induction of phosphorylation at Thr175, the conditions under which the pathway becomes activated, and whether these differ amongst the range of FTLDs in which pThr175tau has been observed;
• Determining whether the pThr231tau exists in either cis or trans conformation and the associated activity of PIN1 in ALS-FTSD and its in vivo model as demonstrated using somatic gene transfer with rAAV9, including in the presence or absence of pathological ALS-associated TDP-43 mutations;
• Further understanding the inter-relationships between alterations in the expression of specific tau isoforms and alterations in TDP-43 metabolism, in particular in relationship to the role of TDP-43 in mediating gene expression through its nuclear functions as an RNA binding protein; and
• Determining whether therapeutic strategies targeted toward restoring physiological tau/microtubule interactions would be of therapeutic benefit in ALS-FTSD.
MS: conceptualization and original draft preparation. MS, ND, and KV: writing, review, and editing. All authors have read and agreed to the published version of the manuscript.
Portions of this research were funded by the Ontario Neurodegeneration Research Initiative (ONDRI) funding and the National Hockey League Players Association (NHLPA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Charcot JM, Joffroy A. Deux cas d'atrophie musculaire progressive avec lésions de la substance grise et des faisceaux antérolatéraux de la moelle épinière. Arch Physiol Norm Pathol. (1869) 2:354–744.
2. Strong MJ. The evidence for ALS as a multisystems disorder of limited phenotypic expression. Can J Neurol Sci. (2001) 28:283–98. doi: 10.1017/S0317167100001505
3. Strong MJ. Revisiting the concept of amyotrophic lateral sclerosis as a multisystems disorder of limited phenotypic expression. Curr Opin Neurol. (2017) 30:599–607. doi: 10.1097/WCO.0000000000000488
4. Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. (2020) 27:1918–29. doi: 10.1111/ene.14393
5. Strong MJ. The basic aspects of therapeutics in amyotrophic lateral sclerosis. Pharmacol Ther. (2003) 98:379–414. doi: 10.1016/S0163-7258(03)00040-8
6. Strong MJ, Kesavapany S, Pant HC. The pathobiology of amyotrophic lateral sclerosis: a proteinopathy? J Neuropathol Exp Neurol. (2005) 64:649–64. doi: 10.1097/01.jnen.0000173889.71434.ea
7. Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotr Lateral Scler Frontotemporal Degener. (2017) 18:153–74. doi: 10.1080/21678421.2016.1267768
8. Elamin M, Phukan J, Bede P, Jordan N, Byrne S, Pender N, et al. Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology. (2011) 76:1263–9. doi: 10.1212/WNL.0b013e318214359f
9. Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. (2012) 83:102–8. doi: 10.1136/jnnp-2011-300188
10. Montuschi A, Iazzolino B, Calvo A, Moglia C, Lopiano L, Restagno G, et al. Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry. (2015) 86:168–73. doi: 10.1136/jnnp-2013-307223
11. Oh SI, Park A, Kim HJ, Oh KW, Choi H, Kwon MJ, et al. Spectrum of cognitive impairment in Korean ALS patients without known genetic mutations. PLoS ONE. (2014) 9:e87163. doi: 10.1371/journal.pone.0087163
12. Strong MJ. The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS). J Neurol Sci. (2010) 288:1–12. doi: 10.1016/j.jns.2009.09.029
13. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. (2006) 351:602–11. doi: 10.1016/j.bbrc.2006.10.093
14. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. (2006) 314:130–3. doi: 10.1126/science.1134108
15. Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, Strong MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy. Brain Res. (2009) 1249:202–11. doi: 10.1016/j.brainres.2008.10.021
16. Wang IF, Wu LS, Chang HY, Shen CKJ. TDP-43, the signature protein of FTLD-U, is a neuronal activity responsive factor. J Neurochem. (2008) 105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x
17. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. (2009) 323:1205–8. doi: 10.1126/science.1166066
18. Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. (2009) 323:1208–11. doi: 10.1126/science.1165942
19. Couthouis J, Hart MP, Shorter J, Dejesus-Hernandez M, Erion R, Oristano R, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci USA. (2011) 108:20881–90. doi: 10.1073/pnas.1109434108
20. Collins M, Riascos D, Kovalik T, An J, Krupa K, Krupa K, et al. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. (2012) 124:717–32. doi: 10.1007/s00401-012-1045-x
21. Droppelmann CA, Keller BA, Campos-Melo D, Volkening K, Strong MJ. Rho guanine nucleotide exchange factor is an NFL mRNA destabilizing factor that forms cytoplasmic inclusions in amyotrophic lateral sclerosis. Neurobiol Aging. (2013) 34:242–62. doi: 10.1016/j.neurobiolaging.2012.06.021
22. Keller BA, Volkening K, Droppelmann CA, Ang LC, Rademakers R, Strong MJ. Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol. (2012) 124:733–47. doi: 10.1007/s00401-012-1035-z
23. Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. (2013) 77:639–46. doi: 10.1016/j.neuron.2013.02.004
24. Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. (2013) 126:881–93. doi: 10.1007/s00401-013-1189-3
25. Ge WW, Wen W, Strong WL, Leystra-Lantz C, Strong MJ. Mutant copper/zinc superoxide dismutase binds to and destabilizes human low molecular weight neurofilament mRNA. J Biol Chem. (2005) 280:118–24. doi: 10.1074/jbc.M405065200
26. Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. (2010) 19:R46–64. doi: 10.1093/hmg/ddq137
27. Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. (2009) 136:1001–4. doi: 10.1016/j.cell.2009.03.006
28. Strong MJ. The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2008) 9:323–38. doi: 10.1080/17482960802372371
29. Bak TH, Hodges JR. Motor neurone disease, dementia and aphasia; coincidence, co-occurrence or continuum? J Neurol. (2001) 248:260–70. doi: 10.1007/s004150170199
30. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
31. de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, et al. The Awaji criteria for diagnosis of ALS. Muscle Nerve. (2011) 44:456–7. doi: 10.1002/mus.22175
32. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. (2011) 134:2456–77. doi: 10.1093/brain/awr179
33. Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. (2002) 59:1077–9. doi: 10.1212/WNL.59.7.1077
34. Lillo P, Savage S, Mioshi E, Kiernan MC, Hodges JR. Amyotrophic lateral sclerosis and frontotemporal dementia: a behavioural and cognitive continuum. Amyotroph Lateral Scler. (2012) 13:102–9. doi: 10.3109/17482968.2011.639376
35. Cykowski MD, Powell SZ, Peterson LE, Appel JW, Rivera AL, Takei H, et al. Clinical significance of TDP-43 neuropathology in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. (2017) 76:402–13. doi: 10.1093/jnen/nlx025
36. Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. (2015) 129:469–91. doi: 10.1007/s00401-014-1380-1
37. Mackenzie IR, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. (2016) 138(Suppl. 1):54–70. doi: 10.1111/jnc.13588
38. Chornenkyy Y, Fardo DW, Nelson PT. Tau and TDP-43 proteinopathies: kindred pathologic cascades and genetic pleiotropy. Lab Invest. (2019) 99:993–1007. doi: 10.1038/s41374-019-0196-y
39. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. (1975) 72:1858–62. doi: 10.1073/pnas.72.5.1858
40. Baas PW, Pienkowski TP, Cimbalnik KA, Toyama K, Bakalis S, Ahmad FJ, et al. Tau confers drug stability but not cold stability to microtubules in living cells. J Cell Sci. (1994) 107:135–43.
41. Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. (1990) 343:461–3. doi: 10.1038/343461a0
42. Knops J, Kosik KS, Lee G, Pardee JD, Cohen-Gould L, McConlogue L. Overexpression of tau in a nonneuronal cell induces long cellular processes. J Cell Biol. (1991) 114:725–33. doi: 10.1083/jcb.114.4.725
43. Jeganathan S, von BM, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. (2006) 45:2283–93. doi: 10.1021/bi0521543
44. Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull. (2016) 126:238–92. doi: 10.1016/j.brainresbull.2016.08.018
45. Li D, Scarano S, Lisi S, Palladino P, Minunni M. Real-time tau protein detection by sandwich-based piezoelectric biosensing: exploring tubulin as a mass enhancer. Sensors. (2018) 18:946. doi: 10.3390/s18040946
46. Zabik NL, Imhof MM, Martic-Milne S. Structural evaluations of tau protein conformation: methodologies and approaches. Biochem Cell Biol. (2017) 95:338–49. doi: 10.1139/bcb-2016-0227
47. Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. (2008) 283:32066–76. doi: 10.1074/jbc.M805300200
48. Fichou Y, Al-Hilaly YK, Devred F, Smet-Nocca C, Tsvetkov PO, Verelst J, et al. The elusive tau molecular structures: can we translate the recent breakthroughs into new targets for intervention? Acta Neuropathol Commun. (2019) 7:31. doi: 10.1186/s40478-019-0682-x
49. Zempel H, Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. (2014) 37:721–32. doi: 10.1016/j.tins.2014.08.004
50. Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, et al. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. (2009) 7:e34. doi: 10.1371/journal.pbio.1000034
51. Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer's disease. Neurochem Int. (2011) 58:458–71. doi: 10.1016/j.neuint.2010.12.023
52. Mandell JW, Banker GA. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. (1996) 16:5727–40. doi: 10.1523/JNEUROSCI.16-18-05727.1996
53. Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. (2010) 68:1067–81. doi: 10.1016/j.neuron.2010.11.030
54. Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. (2002) 156:1051–63. doi: 10.1083/jcb.200108057
55. Sultan A, Nesslany F, Violet M, Begard S, Loyens A, Talahari S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. (2011) 286:4566–75. doi: 10.1074/jbc.M110.199976
56. Wei Y, Qu MH, Wang XS, Chen L, Wang DL, Liu Y, et al. Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS ONE. (2008) 3:e2600. doi: 10.1371/journal.pone.0002600
57. Benhelli-Mokrani H, Mansuroglu Z, Chauderlier A, Albaud B, Gentien D, Sommer S, et al. Genome-wide identification of genic and intergenic neuronal DNA regions bound by Tau protein under physiological and stress conditions. Nucleic Acids Res. (2018) 46:11405–22. doi: 10.1093/nar/gky929
58. Bukar Maina M, Al-Hilaly YK, Serpell LC. Nuclear Tau and its potential role in Alzheimer's disease. Biomolecules. (2016) 6:9. doi: 10.3390/biom6010009
59. Liu C, Gotz J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE. (2013) 8:e84849. doi: 10.1371/journal.pone.0084849
60. Maina MB, Bailey LJ, Wagih S, Biasetti L, Pollack SJ, Quinn JP, et al. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol Commun. (2018) 6:70. doi: 10.1186/s40478-018-0565-6
61. Silva JM, Rodrigues S, Sampaio-Marques B, Gomes P, Neves-Carvalho A, Dioli C, et al. Dysregulation of autophagy and stress granule-related proteins in stress-driven tau pathology. Cell Death Differ. (2019) 26:1411–27. doi: 10.1038/s41418-018-0217-1
62. Fernandes N, Eshleman N, Buchan JR. Stress granules and ALS: a case of causation or correlation? Adv Neurobiol. (2018) 20:173–212. doi: 10.1007/978-3-319-89689-2_7
63. Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. (2013) 201:361–72. doi: 10.1083/jcb.201302044
64. Fan AC, Leung AK. RNA granules and diseases: a case study of stress granules in ALS and FTLD. Adv Exp Med Biol. (2016) 907:263–96. doi: 10.1007/978-3-319-29073-7_11
65. Meier S, Bell M, Lyons DN, Ingram A, Chen J, Gensel JC, et al. Identification of novel tau interactions with endoplasmic reticulum proteins in Alzheimer's disease brain. J Alzheimers Dis. (2015) 48:687–702. doi: 10.3233/JAD-150298
66. Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, et al. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. (2012) 32:8270–83. doi: 10.1523/JNEUROSCI.1592-12.2012
67. Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, et al. Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. (2016) 15:1455–66. doi: 10.1016/j.celrep.2016.04.045
68. Babinchak WM, Haider R, Dumm BK, Sarkar P, Surewicz K, Choi JK, et al. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J Biol Chem. (2019) 294:6306–17. doi: 10.1074/jbc.RA118.007222
69. Hernandez-Vega A, Braun M, Scharrel L, Jahnel M, Wegmann S, Hyman BT, et al. Local nucleation of microtubule bundles through tubulin concentration into a condensed tau phase. Cell Rep. (2017) 20:2304–12. doi: 10.1016/j.celrep.2017.08.042
70. Wegmann S, Eftekharzadeh B, Tepper K, Zoltowska KM, Bennett RE, Dujardin S, et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. (2018) 37:e98049. doi: 10.15252/embj.201798049
71. Ambadipudi S, Biernat J, Riedel D, Mandelkow E, Zweckstetter M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat Commun. (2017) 8:275. doi: 10.1038/s41467-017-00480-0
72. Kanaan NM, Hamel C, Grabinski T, Combs B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat Commun. (2020) 11:2809. doi: 10.1038/s41467-020-16580-3
73. Montalbano M, McAllen S, Puangmalai N, Sengupta U, Bhatt N, Johnson OD, et al. RNA-binding proteins musashi and tau soluble aggregates initiate nuclear dysfunction. Nat Commun. (2020) 11:4305. doi: 10.1038/s41467-020-18022-6
74. Kim HJ, Taylor JP. Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron. (2017) 96:285–97. doi: 10.1016/j.neuron.2017.07.029
75. van der Harg JM, Nolle A, Zwart R, Boerema AS, van Haastert ES, Strijkstra AM, et al. The unfolded protein response mediates reversible tau phosphorylation induced by metabolic stress. Cell Death Dis. (2014) 5:e1393. doi: 10.1038/cddis.2014.354
76. Garruto RM. Amyotrophic lateral sclerosis and parkinsonism-dementia of guam: clinical, epidemiological and genetic patterns. Am J Human Biol. (1989) 1:367–82. doi: 10.1002/ajhb.1310010317
77. Hirano A. Neuropathology of amyotrophic lateral sclerosis and parkinsonism-dementia complex on Guam. In: Luthy L, Bischoff A, editors. Proceedings of the Fifth International Congress of Neuropathology. Amsterdam: Excerpta Medica (1966). p. 190–4.
78. Hirano A, Malamud N, Elizan TS, Kurland LT. Amyotrophic lateral sclerosis and parkinsonism-dementia complex on Guam. Further pathologic studies. Arch Neurol. (1966) 15:35–51. doi: 10.1001/archneur.1966.00470130039004
79. Moszczynski AJ, Strong W, Xu K, McKee A, Brown A, Strong MJ. Pathological Thr175 tau phosphorylationin CTE and CTE with ALS. Neurology. (2018) 90:e380–e7. doi: 10.1212/WNL.0000000000004899
80. McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. (2009) 68:709–35. doi: 10.1097/NEN.0b013e3181a9d503
81. Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA. (2017) 318:360–70. doi: 10.1001/jama.2017.8334
82. Dobson-Stone C, Luty AA, Thompson EM, Blumbergs P, Brooks WS, Short CL, et al. Frontotemporal dementia-amyotrophic lateral sclerosis syndrome locus on chromosome 16p12.1-q12.2: genetic, clinical and neuropathological analysis. Acta Neuropathol. (2013) 124:523–33. doi: 10.1007/s00401-013-1078-9
83. Nakamura S, Wate R, Kaneko S, Ito H, Oki M, Tsuge A, et al. An autopsy case of sporadic amyotrophic lateral sclerosis associated with the I113T SOD1 mutation. Neuropathology. (2014) 34:58–63. doi: 10.1111/neup.12049
84. Orrell RW, King AW, Hilton DA, Campbell MJ, Lane RJ, de Belleroche JS. Familial amyotrophic lateral sclerosis with a point mutation of SOD-1: intrafamilial heterogeneity of disease duration associated with neurofibrillary tangles. J Neurol Neurosurg Psychiatry. (1995) 59:266–70. doi: 10.1136/jnnp.59.3.266
85. Soma K, Fu YJ, Wakabayashi K, Onodera O, Kakita A, Takahashi H. Co-occurrence of argyrophilic grain disease in sporadic amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. (2012) 38:54–60. doi: 10.1111/j.1365-2990.2011.01175.x
86. Takeuchi R, Toyoshima Y, Tada M, Tanaka H, Shimizu H, Shiga A, et al. Globular glial mixed four repeat tau and TDP-43 proteinopathy with motor neuron disease and frontotemporal dementia. Brain Pathol. (2016) 26:82–94. doi: 10.1111/bpa.12262
87. Origone P, Geroldi A, Lamp M, Sanguineri F, Caponnetto C, Cabona C, et al. Role of MAPT in pure motor neuron disease: report of a recurrent mutation in Italian patients. Neurodegener Dis. (2018) 18:310–4. doi: 10.1159/000497820
88. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
89. Dejesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
90. Ferrer I, Legati A, Garcia-Monco JC, Gomez-Beldarrain M, Carmona M, Blanco R, et al. Familial behavioral variant frontotemporal dementia associated with astrocyte-predominant tauopathy. J Neuropathol Exp Neurol. (2015) 74:370–9. doi: 10.1097/NEN.0000000000000180
91. Vatsavayai SC, Nana AL, Yokoyama JS, Seeley WW. C9orf72-FTD/ALS pathogenesis: evidence from human neuropathological studies. Acta Neuropathol. (2019) 137:1–26. doi: 10.1007/s00401-018-1921-0
92. Tacik P, Sanchez-Contreras M, Rademakers R, Dickson DW, Wszolek ZK. Genetic disorders with tau pathology: a review of the literature and report of two patients with tauopathy and positive family histories. Neurodegener Dis. (2016) 16:12–21. doi: 10.1159/000440840
93. Bieniek KF, Murray ME, Rutherford NJ, Castanedes-Casey M, Dejesus-Hernandez M, Liesinger AM, et al. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol. (2013) 125:289–302. doi: 10.1007/s00401-012-1048-7
94. King A, Al-Sarraj S, Troakes C, Smith BN, Maekawa S, Iovino M, et al. Mixed tau, TDP-43 and p62 pathology in FTLD associated with a C9ORF72 repeat expansion and p.Ala239Thr MAPT (tau) variant. Acta Neuropathol. (2013) 125:303–10. doi: 10.1007/s00401-012-1050-0
95. He H, Huang W, Wang R, Lin Y, Guo Y, Deng J, et al. Amyotrophic lateral sclerosis-associated GGGGCC repeat expansion promotes tau phosphorylation and toxicity. Neurobiol Dis. (2019) 130:104493. doi: 10.1016/j.nbd.2019.104493
96. Sproviero W, La Bella V, Mazzei R, Valentino P, Rodolico C, Simone IL, et al. FUS mutations in sporadic amyotrophic lateral sclerosis: clinical and genetic analysis. Neurobiol Aging. (2012) 33:837.e1–5. doi: 10.1016/j.neurobiolaging.2011.10.005
97. Lai SL, Abramzon Y, Schymick JC, Stephan DA, Dunckley T, Dillman A, et al. FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. (2011) 32:550.e1–4. doi: 10.1016/j.neurobiolaging.2009.12.020
98. Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. (2013) 34:812–26. doi: 10.1002/humu.22319
99. Orozco D, Edbauer D. FUS-mediated alternative splicing in the nervous system: consequences for ALS and FTLD. J Mol Med. (2013) 91:1343–54. doi: 10.1007/s00109-013-1077-2
100. Orozco D, Tahirovic S, Rentzsch K, Schwenk BM, Haass C, Edbauer D. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep. (2012) 13:759–64. doi: 10.1038/embor.2012.90
101. Ishigaki S, Sobue G. Importance of functional loss of FUS in FTLD/ALS. Front Mol Biosci. (2018) 5:44. doi: 10.3389/fmolb.2018.00044
102. Ishigaki S, Riku Y, Fujioka Y, Endo K, Iwade N, Kawai K, et al. Aberrant interaction between FUS and SFPQ in neurons in a wide range of FTLD spectrum diseases. Brain. (2020) 143:2398–405. doi: 10.1093/brain/awaa196
103. Ishigaki S, Fujioka Y, Okada Y, Riku Y, Udagawa T, Honda D, et al. Altered tau isoform ratio caused by loss of FUS and SFPQ function leads to FTLD-like phenotypes. Cell Rep. (2017) 18:1118–31. doi: 10.1016/j.celrep.2017.01.013
104. Wharton SB, Verber NS, Wagner BE, Highley JR, Fillingham DJ, Waller R, et al. Combined fused in sarcoma-positive (FUS+) basophilic inclusion body disease and atypical tauopathy presenting with an amyotrophic lateral sclerosis/motor neurone disease (ALS/MND)-plus phenotype. Neuropathol Appl Neurobiol. (2019) 45:586–96. doi: 10.1111/nan.12542
105. Vintilescu CR, Afreen S, Rubino AE, Ferreira A. The neurotoxic TAU45-230 fragment accumulates in upper and lower motor neurons in amyotrophic lateral sclerosis subjects. Mol Med. (2016) 22:477–86. doi: 10.2119/molmed.2016.00095
106. Lang AE, Riherd Methner DN, Ferreira A. Neuronal degeneration, synaptic defects, and behavioral abnormalities in tau45−230 transgenic mice. Neuroscience. (2014) 275:322–39. doi: 10.1016/j.neuroscience.2014.06.017
107. Ferreira A, Bigio EH. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med. (2011) 17:676–85. doi: 10.2119/molmed.2010.00220
108. Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci. (2005) 25:5365–75. doi: 10.1523/JNEUROSCI.1125-05.2005
109. Yang W, Sopper MM, Leystra-Lantz C, Strong MJ. Microtubule-associated tau protein positive neuronal and glial inclusions in amyotrophic lateral sclerosis. Neurology. (2003) 61:1766–73. doi: 10.1212/01.WNL.0000099372.75786.F8
110. Yang W, Strong MJ. Widespread neuronal and glial hyperphosphorylated tau deposition in ALS with cognitive impairment. Amyotroph Lateral Scler. (2012) 13:178–93. doi: 10.3109/17482968.2011.622405
111. Yang W, Ang LC, Strong MJ. Tau protein aggregation in the frontal and entorhinal cortices as a function of aging. Dev Brain Res. (2005) 156:127–38. doi: 10.1016/j.devbrainres.2005.02.004
112. Strong MJ, Yang W, Strong WL, Leystra-Lantz C, Jaffe H, Pant HC. Tau protein hyperphosphorylation in sporadic ALS with cognitive impairment. Neurology. (2006) 66:1770–1. doi: 10.1212/01.wnl.0000218161.15834.db
113. Yang W, Leystra-Lantz C, Strong MJ. Upregulation of GSK3beta expression in frontal and temporal cortex of ALS with cognitive impairment (ALSci). Brain Res. (2008) 1196:131–9. doi: 10.1016/j.brainres.2007.12.031
114. Behrouzi R, Liu X, Wu D, Robinson AC, Tanaguchi-Watanabe S, Rollinson S, et al. Pathological tau deposition in motor neurone disease and frontotemporal lobar degeneration associated with TDP-43 proteinopathy. Acta Neuropathol Commun. (2016) 4:33. doi: 10.1186/s40478-016-0301-z
115. Stevens CH, Guthrie NJ, van Roijen M, Halliday GM, Ooi L. Increased tau phosphorylation in motor neurons from clinically pure sporadic amyotrophic lateral sclerosis patients. J Neuropathol Exp Neurol. (2019) 78:605–14. doi: 10.1093/jnen/nlz041
116. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. (2016) 131:75–86. doi: 10.1007/s00401-015-1515-z
117. Moszczynski AJ, Yang W, Hammond R, Ang LC, Strong MJ. Threonine175, a novel pathological phosphorylation site on tau protein linked to multiple tauopathies. Acta Neuropathol Commun. (2017) 5:6. doi: 10.1186/s40478-016-0406-4
118. Kenessey A, Yen SH. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. (1993) 629:40–6. doi: 10.1016/0006-8993(93)90478-6
119. Süssmuth S, Tumani H, Ecker D, Ludolph AC. Amyotrophic lateral sclerosis: disease stage related changes of tau protein asn S100 beta in cerebrospinal fluid and creatine kinase in serum. Neurosci Lett. (2003) 353:57–60. doi: 10.1016/j.neulet.2003.09.018
120. Bourbouli M, Rentzos M, Bougea A, Zouvelou V, Constantinides VC, Zaganas I, et al. Cerebrospinal fluid TAR DNA-binding protein 43 combined with tau proteins as a candidate biomarker for amyotrophic lateral sclerosis and frontotemporal dementia spectrum disorders. Dement Geriatr Cogn Disord. (2017) 44:144–52. doi: 10.1159/000478979
121. Paladino P, Valentino F, Piccoli T, Piccoli F, La Bella V. Cerebrospinal fluid tau protein is not a biological marker in amyotrophic lateral sclerosis. Eur J Neurol. (2009) 16:257–61. doi: 10.1111/j.1468-1331.2008.02405.x
122. Sjogren M, Davidsson P, Wallin A, Granerus AK, Grundstrom E, Askmark H, et al. Decreased CSF-beta-amyloid 42 in Alzheimer's disease and amyotrophic lateral sclerosis may reflect mismetabolism of beta-amyloid induced by disparate mechanisms. Dement Geriatr Cogn Disord. (2002) 13:112–8. doi: 10.1159/000048642
123. Jiménez-Jiménez FJ, Hernánz A, Medina-Acebrón S, de Bustos F, Zurdo JM, Alonso H, et al. Tau protein concentrations in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neurol Scand. (2005) 111:114–7. doi: 10.1111/j.1600-0404.2005.00370.x
124. Brettschneider J, Petzold A, Sussmuth SD, Ludolph AC, Tumani H. Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology. (2006) 66:852–6. doi: 10.1212/01.wnl.0000203120.85850.54
125. Gohar M, Yang W, Strong WL, Volkening K, Leystra-Lantz C, Strong MJ. Tau phosphorylation at 175Thr leads to fibril formation. Implications for the tauopathy of amyotrophic lateral sclerosis. J Neurochem. (2009) 108:634–43. doi: 10.1111/j.1471-4159.2008.05791.x
126. Moszczynski AJ, Gohar M, Volkening K, Leystra-Lantz C, Strong W, Strong MJ. Thr175-phosphorylated tau induces pathologic fibril formation via GSK3beta-mediated phosphorylation of Thr231 in vitro. Neurobiol Aging. (2015) 36:1590–9. doi: 10.1016/j.neurobiolaging.2014.12.001
127. Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. (2001) 359:1–16. doi: 10.1042/bj3590001
128. Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO. (1993) 12:803–8. doi: 10.1002/j.1460-2075.1993.tb05715.x
129. Hintermayer MA, Volkening K, Moszczynski AJ, Donison N, Strong MJ. Tau protein phosphorylation at Thr(175) initiates fibril formation via accessibility of the N-terminal phosphatase-activating domain. J Neurochem. (2019) 2019:e14942. doi: 10.1111/jnc.14942
130. Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y, et al. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci. (2011) 31:9858–68. doi: 10.1523/JNEUROSCI.0560-11.2011
131. Kanaan NM, Morfini G, Pigino G, LaPointe NE, Andreadis A, Song Y, et al. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol Aging. (2012) 33:826–30. doi: 10.1016/j.neurobiolaging.2011.06.006
132. Avila J, Jimenez JS, Sayas CL, Bolos M, Zabala JC, Rivas G, et al. Tau structures. Front Aging Neurosci. (2016) 8:262. doi: 10.3389/fnagi.2016.00262
133. Combs B, Kanaan NM. Exposure of the amino terminus of tau is a pathological event in multiple tauopathies. Am J Pathol. (2017) 187:1222–9. doi: 10.1016/j.ajpath.2017.01.019
134. Ward SM, Himmelstein DS, Lancia JK, Binder LI. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans. (2012) 40:667–71. doi: 10.1042/BST20120134
135. Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC. Characterization of early pathological tau conformations and phosphorylation in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. (2016) 75:19–34. doi: 10.1093/jnen/nlv001
136. Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, et al. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell. (2012) 149:232–44. doi: 10.1016/j.cell.2012.02.016
137. Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. (1996) 380:544–7. doi: 10.1038/380544a0
138. Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. (1999) 399:784–8. doi: 10.1038/21650
139. Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun. (2017) 8:1000. doi: 10.1038/s41467-017-01068-4
140. Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. (2003) 424:556–61. doi: 10.1038/nature01832
141. Ma SL, Tang NL, Tam CW, Lui VW, Lam LC, Chiu HF, et al. A PIN1 polymorphism that prevents its suppression by AP4 associates with delayed onset of Alzheimer's disease. Neurobiol Aging. (2012) 33:804–13. doi: 10.1016/j.neurobiolaging.2010.05.018
142. Segat L, Pontillo A, Annoni G, Trabattoni D, Vergani C, Clerici M, et al. PIN1 promoter polymorphisms are associated with Alzheimer's disease. Neurobiol Aging. (2007) 28:69–74. doi: 10.1016/j.neurobiolaging.2005.11.009
143. Mustroph ML, King MA, Klein RL, Ramirez JJ. Adult-onset focal expression of mutated human tau in the hippocampus impairs spatial working memory of rats. Behav Brain Res. (2012) 233:141–8. doi: 10.1016/j.bbr.2012.04.034
144. Moszczynski AJ, Gopaul J, McCunn P, Volkening K, Harvey M, Bartha R, et al. Somatic gene transfer using a recombinant adenoviral vector (rAAV9) encoding pseudophosphorylated human Thr175 tau in adult rat hippocampus induces tau pathology. J Neuropathol Exp Neurol. (2018) 77:685–95. doi: 10.1093/jnen/nly044
145. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. (2010) 69:918–29. doi: 10.1097/NEN.0b013e3181ee7d85
146. Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, et al. Staging TDP-43 pathology in Alzheimer's disease. Acta Neuropathol. (2014) 127:441–50. doi: 10.1007/s00401-013-1211-9
147. Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. (2014) 127:811–24. doi: 10.1007/s00401-014-1269-z
148. Zhou H, Huang C, Chen H, Wang D, Landel CP, Xia PY, et al. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. (2010) 6:e1000887. doi: 10.1371/journal.pgen.1000887
149. Moszczynski AJ, Harvey M, Fulcher N, de Oliveira C, McCunn P, Donison N, et al. Synergistic toxicity in an in vivo model of neurodegeneration through the co-expression of human TDP-43(M337V) and tau(T175D) protein. Acta Neuropathol Commun. (2019) 7:170. doi: 10.1186/s40478-019-0816-1
150. Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. (1995) 189:167–9. doi: 10.1016/0304-3940(95)11484-E
151. Olney RK, Murphy J, Forshew D, Garwood E, Miller BL, Langmore S, et al. The effects of executive and behavioral dysfunction on the course of ALS. Neurology. (2005) 65:1774–7. doi: 10.1212/01.wnl.0000188759.87240.8b
152. Liachko NF, McMillan PJ, Strovas TJ, Loomis E, Greenup L, Murrell JR, et al. The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genet. (2014) 10:e1004803. doi: 10.1371/journal.pgen.1004803
153. Sato S, Cerny RL, Buescher JL, Ikezu T. Tau-tubulin kinase 1 (TTBK1), a neuron-specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J Neurochem. (2006) 98:1573–84. doi: 10.1111/j.1471-4159.2006.04059.x
154. Tomizawa K, Omori A, Ohtake A, Sato K, Takahashi M. Tau-tubulin kinase phosphorylates tau at Ser-208 and Ser-210, sites found in paired helical filament-tau. FEBS Lett. (2001) 492:221–7. doi: 10.1016/S0014-5793(01)02256-6
155. Taylor LM, McMillan PJ, Liachko NF, Strovas TJ, Ghetti B, Bird TD, et al. Pathological phosphorylation of tau and TDP-43 by TTBK1 and TTBK2 drives neurodegeneration. Mol Neurodegener. (2018) 13:7. doi: 10.1186/s13024-018-0237-9
156. Taylor LM, McMillan PJ, Kraemer BC, Liachko NF. Tau tubulin kinases in proteinopathy. FEBS J. (2019) 286:2434–46. doi: 10.1111/febs.14866
Keywords: TDP-43, phosphorylation, traumatic encephalopathy, motor neuron, frontotemporal
Citation: Strong MJ, Donison NS and Volkening K (2020) Alterations in Tau Metabolism in ALS and ALS-FTSD. Front. Neurol. 11:598907. doi: 10.3389/fneur.2020.598907
Received: 25 August 2020; Accepted: 27 October 2020;
Published: 23 November 2020.
Edited by:
Sonia Do Carmo, McGill University, CanadaReviewed by:
Antonio Giuliano Zippo, Institute of Neuroscience, ItalyCopyright © 2020 Strong, Donison and Volkening. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Strong, bXN0cm9uZ0B1d28uY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.