 Giona Pedrioli
Giona Pedrioli Rickie Patani3,4
Rickie Patani3,4 Paolo Paganetti
Paolo Paganetti
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Neurol. , 13 November 2020
Sec. Dementia and Neurodegenerative Diseases
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.596528
This article is part of the Research Topic Consequences of the COVID-19 Pandemic on Care for Neurological Conditions View all 77 articles
On the verge of the ongoing coronavirus pandemic, in vitro data suggested that chloroquine, and its analog hydroxychloroquine, may be useful in controlling SARS-CoV-2 infection. Efforts are ongoing in order to test this hypothesis in clinical trials. Some studies demonstrated no evidence of efficacy, whereas in some cases results were retracted after reporting. Despite the lack of scientific validation, support for the use of these compounds continues from various influencers. At the cellular level, the lysosomotropic drug chloroquine accumulates in acidic organelles where it acts as an alkalizing agent with possible downstream effects on several cellular pathways. In this perspective, we discuss a possible modulatory role of these drugs in two shared features of neurodegenerative diseases, the cellular accumulation of aberrantly folded proteins and the contribution of neuroinflammation in this pathogenic process. Certainly, the decision on the use of chloroquine must be determined by its efficacy in the specific clinical situation. However, at an unprecedented time of a potential widespread use of chloroquine, we seek to raise awareness of its potential impact in ongoing clinical trials evaluating disease-modifying therapies in neurodegeneration.
On February 4th, 2020, at the verge of a new pandemic crisis, the anti-malarial drug chloroquine (CQ), was proposed to be highly effective in controlling SARS-CoV-2 infection in vitro (1). Soon after, in March 2020, the lack of specific treatments for the rising coronavirus burden induced the U. S. Food and Drug Administration (FDA) to issue an emergency use authorization (EUA) for CQ, and its (more soluble and less toxic) analog hydroxychloroquine (HCQ), as treatments for the control of SARS-CoV-2, the severe acute respiratory syndrome caused by the new coronavirus (2). On June 2020, in light of recent scientific data and analysis, the FDA revoked the EUA for CQ/HCQ, as reported side effects “no longer outweigh the known and potential risks for the authorized use” (3). The most worrisome adverse effects, also listed in the drug labels, include heart rhythm interference related to long QT syndrome, ventricular tachycardia and fibrillation, in particular in combination with QT-prolonging drugs or pre-existing kidney or heart disorders (4–6). Likely differences in dosing regimens when using CQ/HCQ for their approved indications, which are unlikely to meet the concentrations affecting SARS-CoV-2 activity in vitro, may explain why the occurrence of these symptoms is uncommon in the medical practice. For instance, while safety profile of CQ in the treatment of rheumatic diseases have been reported up to 500 mg (once daily) (7), in a SARS-CoV-2 clinical trial assessing CQ efficacy a QT interval alteration was observed in patients treated with a higher dose (600 mg, twice daily) (4).
The EUA permission and revocation of the use of CQ/HCQ has caused a stir in the scientific community and beyond during this unstable and delicate pandemic situation. While we acknowledge the natural tendency to dismiss uncomfortable facts and the keenness to move away from CQ, reflecting on possible short and long-term neurological side-effects caused by its use are worthy of a more comprehensive scientific consideration. In particular considering that CQ was, and still is, used as a putative off-label drug to treat SARS-CoV-2, highlights that the response to this pandemic has not always been ruled by a rational and scientific approach. Nonetheless, the possible consequences of using CQ should instigate discussion and warrant a more cautious approach if a similar situation should arise in the future. Here we provide a perspective on the potential interaction of CQ and the neuronal dyshomeostasis observed in common degenerative disorders such as Alzheimer's or Parkinson's disease. We consider the pharmacodynamics and pharmacokinetic attributes of CQ, and its potential effects on the nervous and immune systems.
Historically recognized for its undeniable utility in malarial prophylaxis and treatment (8), CQ is also extensively used as a cell biology research compound based on its potent inhibitory activity on autophagic and lysosomal clearance functions. The lipophilic nature of CQ enables a rapid penetration across lipid bilayer membranes. Within the cell, CQ behaves as a lysosomotropic agent, i.e., it undergoes a protonation-based trapping when it reaches the acidic environment present in the lumen of organelles such as lysosomes. Its weak base characteristics results in its accumulation as a function of the pH gradient, the neutralization of the low pH, the inhibition of acidic hydrolases and the impairment of organelle maturation (9). This has led to defining the mode of action of CQ as an inhibitor of both enzymatic activity and organelle fusion resulting in halting autophagy flux and endo-lysosomal degradative function (10).
Beside the autophagy-lysosome pathway, experimental evidence proposes that CQ is a weak antagonist of the proteasome system, causing accumulation of ubiquitinated proteins in mammalian cells (11, 12). Mechanistically, CQ acts as an allosteric inhibitor of the enzymatic activity of the 26S proteasome degradation system (13). Together, these studies highlight a likely dual inhibitory effect of CQ in the two major metabolic systems regulating cellular proteostasis. Moreover, the presence of CQ modify the heat-shock response regulating protein chaperons expression in mammalian cells (14) with additional consequences on the mammalian proteostasis and on the drug resistance of the malaria parasite Plasmodium (15).
CQ can be administered orally as a phosphate salt and it is efficiently absorbed by the upper intestinal tract, thus permitting a high drug bioavailability. Plasma CQ concentration peaks at 8–12 h post-administration. CQ is slowly metabolized mainly in the liver by cytochrome P450 enzymes and is converted into desethylchloroquine. Further desethylation leads to the second, less frequent, metabolite bisdesethylchloroquine. CQ and its active metabolites have a remarkably slow elimination rate, which in turns may facilitate a widespread tissue exposure, indeed reflected in a large distribution volume. Although about 70% of CQ is directly cleared by the kidneys, CQ and its metabolites are detected in blood plasma for as long as 70 days, and in the urine up to 1 year post-administration. Notably, the equally active CQ enantiomers differ in their overall elimination kinetics. In animals, the concentration of CQ reaches 10-to-700 times higher levels in the liver, spleen, kidney, and lung when taking that detected in the plasma as reference (16, 17). Despite some controversy around the efficacy of CQ to penetrate the blood-brain barrier (BBB), animal studies demonstrate that this drug and its analogs can penetrate and reach a concentration that is sufficient to exert its effects within the central nervous system (CNS) (18–20). Nonetheless, reported neurological side effects of CQ and its analogs implicate a non-yet fully confirmed CNS exposure in humans (21). In particular, CQ/HCQ can have potential adverse neuropsychiatric effects, similar to symptoms occurring in neurodegenerative disorders, such as agitation, emotional instability, anxiety, irritability and, rarely, psychosis (22, 23).
Therefore, at a time were CQ is used in clinical trials or as a self-remedy, and as long it is not excluded that the CNS is a target tissue of the drug, predicting possible consequences of CQ exposure in the brain is important in order to prevent possible neurological effects, e.g., for patients affected by neurodegenerative disorders.
Although little is known regarding the direct effects of CQ on the CNS, the latter is particularly vulnerable to disruptions of the cellular degradative pathways. Indeed, terminally differentiated neurons rely on efficient quality control systems such as the autophagic-lysosomal pathway for maintaining their delicate proteostasis, which is gradually impaired as the brain ages (24). Autophagy is responsible for delivering cytoplasmic material to the lysosome for degradation. Autophagy is subdivided in three distinct processes that differ in their mechanism of recognition and delivery of substrates to lysosomes: chaperon mediated autophagy (CMA), macroautophagy and microautophagy (25). The selective clearance of aberrant proteins is primarily carried out by CMA and macroautophagy. In CMA, proteins that bear a pentapeptide degradation signal (KFERQ-like) are recognized by the chaperone heat-shock cognate 70 (HSC70) and delivered through the CMA adaptor lysosomal membrane associated protein 2A (LAMP2A) to the lysosomal lumen for degradation. In contrast, aberrantly folded proteins that are prone to self-aggregate into β-sheet-rich oligomers and higher order aggregates are sequestered by macroautophagy together with small portion of the cytoplasm. These substrates are encapsulated within an intermediate double lipid bilayer membrane organelle termed “autophagosome” and directed toward lysosomes, where upon membrane fusion, cargos are liberated in the hydrolases-enriched lysosomal lumen for enzymatic digestion (26) (Figure 1A).
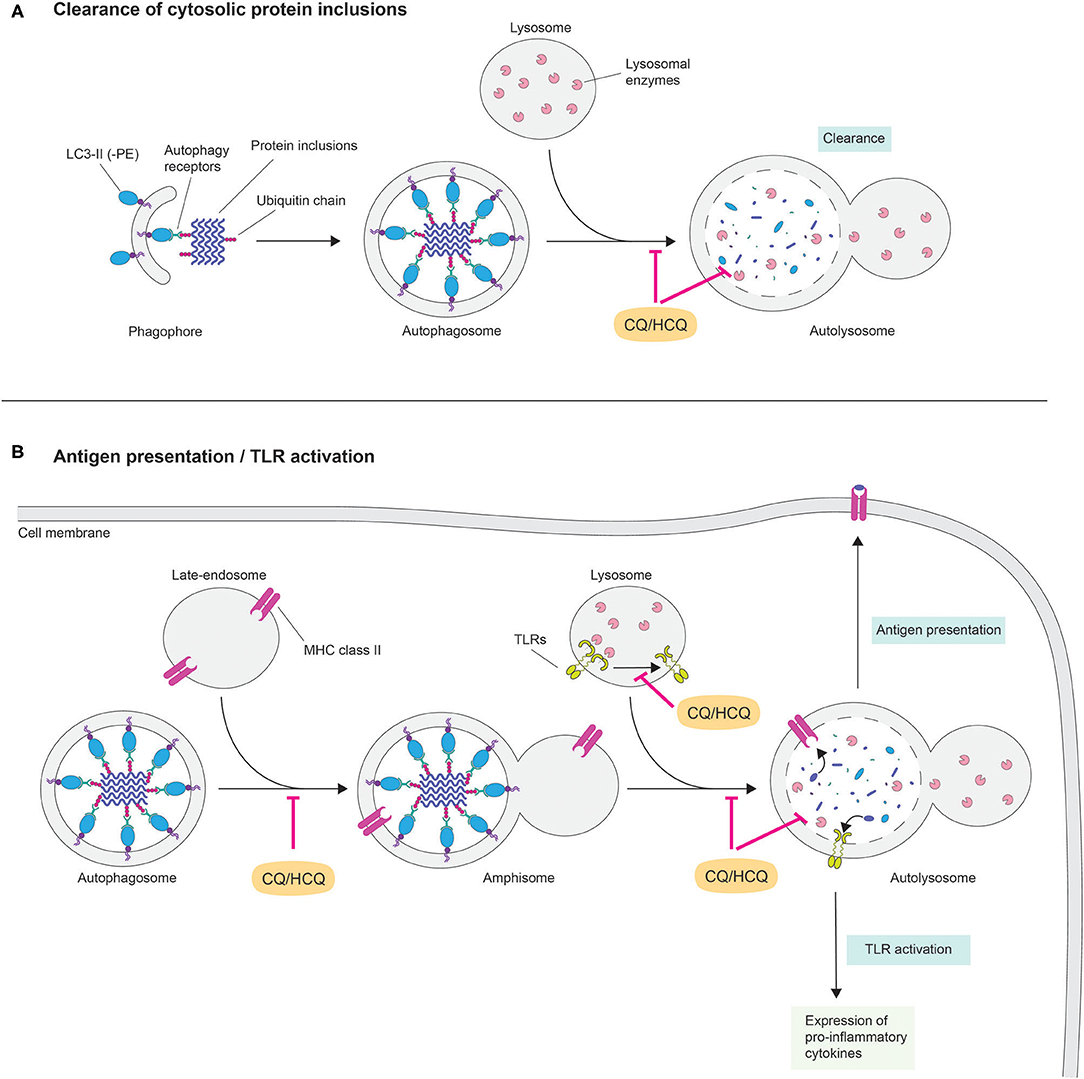
Figure 1. Potential cellular and molecular mechanisms of chloroquine in neurodegeneration. The lysosomotropic agent chloroquine (CQ) rapidly penetrates across lipid bilayer membranes and following a pH gradient accumulates within lysosomes. In these acidic organelles, CQ behaves as a weak base by increasing the pH, which in turns affects the activity of lysosomal hydrolases. Disruption of lysosomal activity prevents interaction and fusion among organelles of the autophagy-lysosome and of the endocytic pathways. This cellular condition may have dichotomic effects in the pathogenesis of neurodegenerative diseases by (A) inhibiting cytosolic clearance of aberrantly protein fibrils and (B) preventing MHC class II-mediated antigen presentation and preventing the expression of pro-inflammatory cytokines via TLR activation.
Most late onset neurodegenerative disorders share the progressive deposition of aberrantly folded, β-sheet-rich protein aggregates into ubiquitinated intraneuronal inclusions. Each disorder is characterized by the aggregation of specific proteins: examples are beta-amyloid and TAU in Alzheimer's disease (27), NACP/α-synuclein in Parkinson's disease (28), huntingtin in Huntington's disease (29), TDP-43 in amyotrophic lateral sclerosis and frontotemporal lobar degeneration (30). Nevertheless, another key pathological hallmark of these otherwise clinically and etiologically diverse disorders is the progressive impairment in the autophagy-lysosome degradation pathway. This is exemplified by the fact that mutations of genes regulating autophagy and lysosome activity are associated to the most frequent late-onset forms of neurodegeneration (31). Furthermore, experimental animal models demonstrate that autophagy deficiency accelerates protein aggregation and behavioral phenotypes of neurodegeneration. Evidence that CQ exposure on neurons may lead to a similar outcome are known since long time (32). More recently the activity of CQ on the amyloidogenic processing of the amyloid precursor protein by neurons (33, 34) as well as on huntingtin accumulation in brain (20) were reported. CQ also modulates autophagic flux (35) and mitochondrial homeostasis by an autophagic process (36). CQ is also linked to neuronal death in primary cultures (37, 38). These facts are reinforced by studies demonstrating that autophagy stimulation can clear intra-neuronal insoluble protein inclusions with amelioration of behavioral phenotypes in animal models of neurodegenerative diseases (26) (Table 1). Nevertheless, macroautophagy may also favor seeded propagation of aberrantly folded neurodegeneration-associated TAU mediated by extracellular vesicles (39).
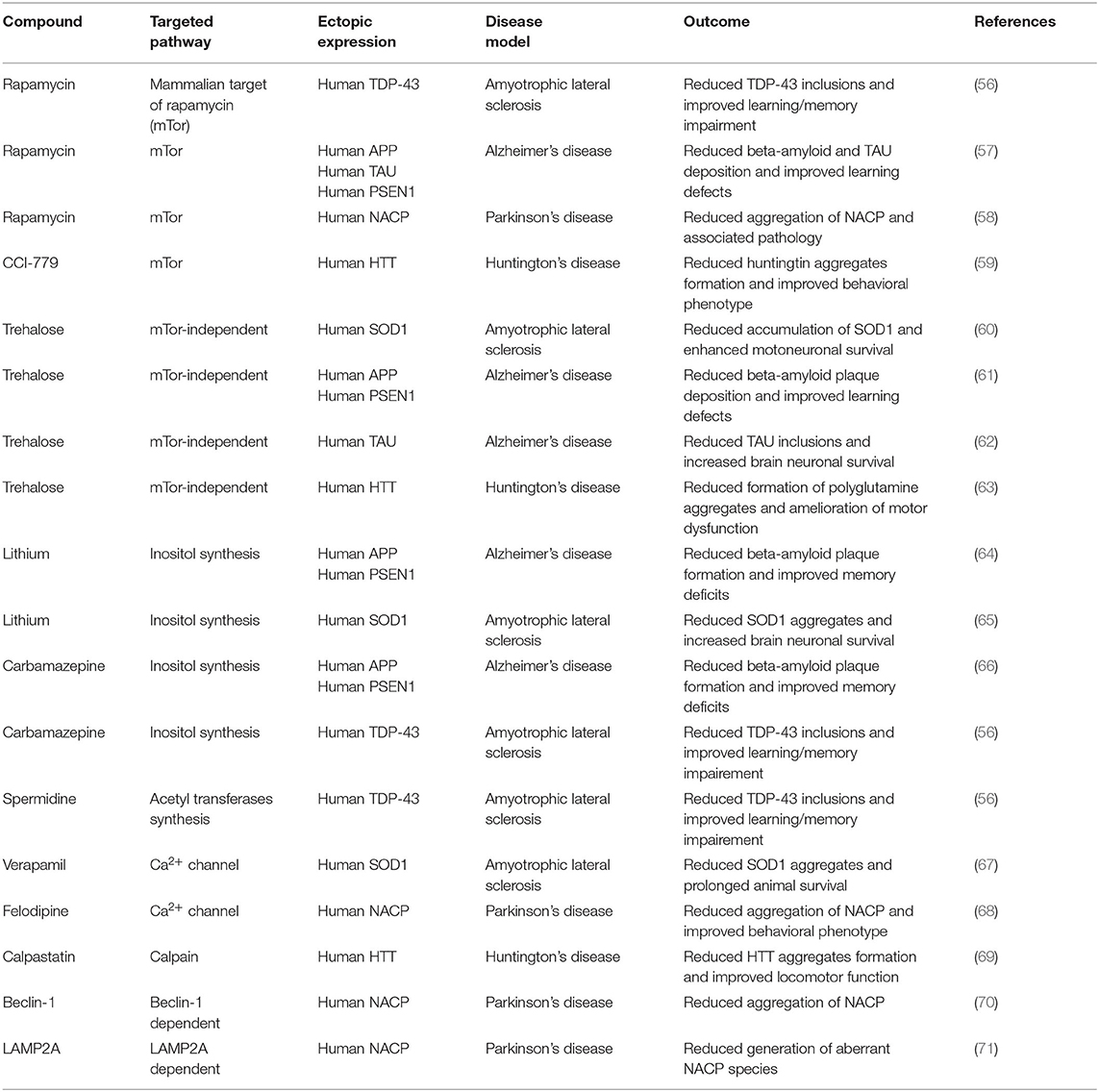
Table 1. Examples of evidence for beneficial effect of autophagy stimulation in murine brain.
Another increasingly documented feature of neurodegenerative disorders is the chronic inflammation of the CNS (neuroinflammation). Although a causal relationship has not yet been demonstrated, there are studies reporting a correlation between prolonged treatment with non-steroidal anti-inflammatory drugs and decreased risk for Alzheimer's and Parkinson's disease (40, 41). Activation of CNS-resident macrophages (microglia cells) around senile plaques has been documented in transgenic mouse model of Alzheimer's disease (42). These phagocytic cells actively uptake beta-amyloid and acquire an activated phenotype characterized by morphological changes and by an increased production of pro-inflammatory modulators such as the major histocompatibility complex (MHC) class II, several interleukins and tumor necrosis factor alpha. Persistent microglial activation is associated with cellular senescence, neurotoxicity and subsequent disease progression (43). Recent studies suggest that this may also involve deleterious reactive transformation in astrocytes (44). Notably, elimination of senescent glial cells, which are known to release proinflammatory modulators, is beneficial (45–47).
Against this background, HCQ's clinical efficacy in treating autoimmune inflammatory diseases, such as rheumatoid arthritis and systemic lupus erythematosus, is well-documented (48). Current hypotheses in the field are linked to an indirect effect of HCQ in modulating the inflammatory response (Figure 1B). Specifically, interference of lysosomal activity might affect several immunomodulatory pathways. One intuitive mechanism is the inhibition of antigen presentation via the autophagy-lysosome pathway. As lysosomes are the main organelles for hydrolytic processing, they reside at the intersection between different pathways delivering intracellular and extracellular cargos on route to degradation (49). This context provides a unique cellular environment for the binding of antigens to MHC class II. For instance, a recent report suggests that extracellular proteins are hydrolysed in endocytic compartments and delivered to MHC class II-containing lysosomes as antigenic peptides before being presented to CD4+ T cells (50). Nevertheless, functional lysosomes are required for antigenic peptide-binding to MHC class II molecules and the alkalizing properties that HCQ exerts in these organelles might impair this process. Another possibility is that HCQ interferes in Toll-like receptor (TLR) signaling. In mammals, TLRs are a group of transmembrane pattern-recognition receptors that initiate innate immune response to infection by sensing pathogen macromolecules. However, TLRs can also be activated in the absence of pathogen infection (51). Indeed, activated microglia surrounding beta-amyloid plaques in Alzheimer's disease brains display up-regulated levels of TLRs (52, 53). A recent report indicates that, in order to be functional, TLR7 requires proteolytic cleavage in lysosomes (48). Thus, interfering with lysosomal pH via lysosomotropic agents may prevent activation of TLRs. Moreover, evidence exists for a mode of action of CQ/HCQ independently of its effect on lysosomal function, as shown for its ability to interference with interleukin-2 production (54). Although the precise mechanism(s), by which CQ/HCQ inhibits inflammatory response, requires further investigation, its potential role in disrupting the integrity of the CNS immune system in neurodegenerative disorders is an intriguing and noteworthy hypothesis. Evidence for a possible role of CQ in modulating inflammation and autophagic death of neurons in the brain exists (55).
Given the demographic, in particular associated to aging, of people affected by neurodegenerative disorders and patients more vulnerable to develop a serious SARS-CoV-2 disease course, the possibility that CQ, or one of its analogs, will be prescribed/self-consumed by patients enrolled in clinical trials (or outside this context and off license) is worth considering. However, the use of CQ and its analogs must be determined by clinical need, so that prescribing CQ may be opportune and take priority depending on specific clinical context. However, at a time of a potential widespread use of CQ, in order to mitigate the risk of potential misinterpretation in ongoing clinical trials evaluating disease-modifying therapies in neurodegeneration, we seek to raise awareness and caution that the use of CQ and its analogs needs to be clearly documented and carefully considered in interpreting trial outcomes in this arena and beyond.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
PP was funded by the Synapsis Foundation, the Gelu Foundation, and the Mecri Foundation. RP holds an MRC Senior Clinical Fellowship [MR/S006591/1].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the whole laboratory for support and advice.
CQ, Chloroquine; FDA, U. S. Food and Drug Administration; EUA, Emergency use authorization; CNS, Central nervous system; CMA, Chaperon mediated autophagy; HSC70, Chaperone heat-shock cognate 70; LAMP2A, Lysosomal membrane associated protein 2A; MHC, Major histocompatibility complex; TLR, Toll-like receptor.
1. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. (2020) 30:269–71. doi: 10.1038/s41422-020-0282-0
2. Administration U,.S.F.a.D. Coronavirus (COVID-19) Update: Daily Roundup March 30, 2020. (2020). Available online at: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-daily-roundup-march-30-2020 (accessed April 3, 2020).
3. Administration U,.S.F.a.D. Coronavirus (COVID-19) Update: FDA Revokes Emergency Use Authorization for Chloroquine Hydroxychloroquine. (2020). Available online at: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-chloroquine-and (accessed June 16, 2020).
4. Borba MGS, Val FFA, Sampaio VS, Alexandre MAA, Melo GC, Brito M, et al. Effect of high vs low doses of chloroquine diphosphate as adjunctive therapy for patients hospitalized with Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) infection: a randomized clinical trial. JAMA Netw Open. (2020) 3:e208857. doi: 10.1001/jamanetworkopen.2020.8857
5. Chorin E, Dai M, Shulman E, Wadhwani L, Bar-Cohen R, Barbhaiya C, et al. The QT interval in patients with COVID-19 treated with hydroxychloroquine and azithromycin. Nat Med. (2020) 26:808–9. doi: 10.1038/s41591-020-0888-2
6. Mercuro NJ, Yen CF, Shim DJ, Maher TR, McCoy CM, Zimetbaum PJ, et al. Risk of QT interval prolongation associated with use of hydroxychloroquine with or without concomitant azithromycin among hospitalized patients testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:1036–41. doi: 10.1001/jamacardio.2020.1834
7. Klinger G, Morad Y, Westall CA, Laskin C, Spitzer KA, Koren G, et al. Ocular toxicity and antenatal exposure to chloroquine or hydroxychloroquine for rheumatic diseases. Lancet. (2001) 358:813–4. doi: 10.1016/S0140-6736(01)06004-4
8. Pullman TN, Craige B, Alving AS, Whorton CM, Jones R, Eichelberger L. Comparison of chloroquine, quinacrine (atabrine), and quinine in the treatment of acute attacks of sporozoite-induced vivax malaria (chesson strain). J Clin Invest. (1948) 27:46–50. doi: 10.1172/JCI101970
9. Al-Bari MA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother. (2015) 70:1608–21. doi: 10.1093/jac/dkv018
10. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K-J, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. (2018) 14:1435–55. doi: 10.1080/15548627.2018.1474314
11. Qiao L, Zhang J. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett. (2009) 456:15–9. doi: 10.1016/j.neulet.2009.03.085
12. Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. (2011) 286:22426–40. doi: 10.1074/jbc.M110.149252
13. Sprangers R, Li X, Mao X, Rubinstein JL, Schimmer AD, Kay LE. TROSY-based NMR evidence for a novel class of 20S proteasome inhibitors. Biochemistry. (2008) 47:6727–34. doi: 10.1021/bi8005913
14. Neznanov N, Gorbachev AV, Neznanova L, Komarov AP, Gurova KV, Gasparian AV, et al. Anti-malaria drug blocks proteotoxic stress response: anti-cancer implications. Cell Cycle. (2009) 8:3960–70. doi: 10.4161/cc.8.23.10179
15. Daniyan MO, Przyborski JM, Shonhai A. Partners in mischief: functional networks of heat shock proteins of Plasmodium falciparum and their influence on parasite virulence. Biomolecules. (2019) 9:925. doi: 10.3390/biom9070295
16. Popert AJ. Chloroquine: a review. Rheumatol Rehabil. (1976) 15:235–8. doi: 10.1093/rheumatology/15.3.235
17. Adelusi SA, Salako LA. Tissue and blood concentrations of chloroquine following chronic administration in the rat. J Pharm Pharmacol. (1982) 34:733–5. doi: 10.1111/j.2042-7158.1982.tb06211.x
18. Osifo NG. The regional uptake of chloroquine in the rat brain. Toxicol Appl Pharmacol. (1979) 50:109–14. doi: 10.1016/0041-008X(79)90498-8
19. Baudry S, Pham YT, Baune B, Vidrequin S, Crevoisier C, Gimenez F, et al. Stereoselective passage of mefloquine through the blood-brain barrier in the rat. J Pharm Pharmacol. (1997) 49:1086–90. doi: 10.1111/j.2042-7158.1997.tb06047.x
20. Vodicka P, Lim J, Williams DT, Kegel KB, Chase K, Park H, et al. Assessment of chloroquine treatment for modulating autophagy flux in brain of WT and HD mice. J Huntigtons Dis. (2014) 3:159–74. doi: 10.3233/JHD-130081
21. Quinn JC. Complex membrane channel blockade: a unifying hypothesis for the prodromal and acute neuropsychiatric sequelae resulting from exposure to the antimalarial drug mefloquine. J Parasitol Res. (2015) 2015:368064. doi: 10.1155/2015/368064
22. Manzo C, Gareri P, Castagna A. Psychomotor agitation following treatment with hydroxychloroquine. Drug Saf Case Rep. (2017) 4:6. doi: 10.1007/s40800-017-0048-x
23. Ali SS, Jones H. 23. An adverse neuropsychiatric reaction following treatment with hydroxychloroquine: a case report. Rheumatol Adv Pract. (2018) 2:rky033.014. doi: 10.1093/rap/rky033.014
24. Son JH, Shim JH, Kim K-H, Ha J-Y, Han JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med. (2012) 44:89. doi: 10.3858/emm.2012.44.2.031
25. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. doi: 10.1038/nature06639
26. Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. (2015) 47:e147. doi: 10.1038/emm.2014.117
27. Selkoe DJ. Alzheimer's disease. Cold Spring Harb Perspect Biol. (2011) 3:a004457. doi: 10.1101/cshperspect.a004457
28. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the -synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
29. Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. (2011) 10:83–98. doi: 10.1016/S1474-4422(10)70245-3
30. Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. (2011) 7:603–15. doi: 10.1038/nrneurol.2011.150
31. Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. (2017) 93:1015–34. doi: 10.1016/j.neuron.2017.01.022
32. Tischner K. Effects of chloroquine on neurons of long-term cultures of peripheral and central nervous system. Acta Neuropathol. (1974) 28:233–42. doi: 10.1007/BF00719028
33. Caporaso GL, Gandy SE, Buxbaum JD, Greengard P. Chloroquine inhibits intracellular degradation but not secretion of Alzheimer beta/A4 amyloid precursor protein. Proc Natl Acad Sci USA. (1992) 89:2252–6. doi: 10.1073/pnas.89.6.2252
34. Bahr BA, Abai B, Gall CM, Vanderklish PW, Hoffman KB, Lynch G. Induction of β-amyloid-containing polypeptides in hippocampus: evidence for a concomitant loss of synaptic proteins and interactions with an excitotoxin. Exp Neurol. (1994) 129:81–94. doi: 10.1006/exnr.1994.1149
35. Redmann M, Wani WY, Volpicelli-Daley L, Darley-Usmar V, Zhang J. Trehalose does not improve neuronal survival on exposure to alpha-synuclein pre-formed fibrils. Redox Biol. (2017) 11:429–37. doi: 10.1016/j.redox.2016.12.032
36. Redmann M, Benavides GA, Berryhill TF, Wani WY, Ouyang X, Johnson MS, et al. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. (2017) 11:73–81. doi: 10.1016/j.redox.2016.11.004
37. Zaidi AU, McDonough JS, Klocke BJ, Latham CB, Korsmeyer SJ, Flavell RA, et al. Chloroquine-induced neuronal cell death is p53 and bcl-2 family-dependent but caspase-independent. J Neuropathol Exp Neurol. (2001) 60:937–45. doi: 10.1093/jnen/60.10.937
38. Cui D, Sun D, Wang X, Yi L, Kulikowicz E, Reyes M, et al. Impaired autophagosome clearance contributes to neuronal death in a piglet model of neonatal hypoxic-ischemic encephalopathy. Cell Death Dis. (2017) 8:e2919. doi: 10.1038/cddis.2017.318
39. Pedrioli G, Barberis M, Molinari M, Morone D, Papin S, Paganetti P. Extracellular vesicles hijack the autophagic pathway to induce tau accumulation in endolysosomes. bioRxiv [Preprint]. (2020). doi: 10.1101/2020.05.27.118323
40. Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. (1997) 48:626–32. doi: 10.1212/WNL.48.3.626
41. Chen H, Zhang SM, Hernán MA, Schwarzschild MA, Willett WC, Colditz GA, et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol. (2003) 60:1059. doi: 10.1001/archneur.60.8.1059
42. Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, et al. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. (1998) 152:307–17.
43. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. (2018) 21:1359–69. doi: 10.1038/s41593-018-0242-x
44. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
45. Bussian TJ, Aziz A, Meyer CF, Swenson BL, Van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. (2018) 562:578–82. doi: 10.1038/s41586-018-0543-y
46. Angelova DM, Brown DR. Microglia and the aging brain: are senescent microglia the key to neurodegeneration? J Neurochem. (2019) 151:676–88. doi: 10.1111/jnc.14860
47. Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat Neurosci. (2019) 22:719–28. doi: 10.1038/s41593-019-0372-9
48. Schrezenmeier E, Dörner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol. (2020) 16:155–66. doi: 10.1038/s41584-020-0372-x
49. Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. (2007) 8:622–32. doi: 10.1038/nrm2217
50. Chen C, Li HQ, Liu YJ, Guo ZF, Wu HJ, Li X, et al. A novel size-based sorting mechanism of pinocytic luminal cargoes in microglia. J Neurosci. (2015) 35:2674–88. doi: 10.1523/JNEUROSCI.4389-14.2015
51. Zhang Z, Schluesener HJ. Mammalian toll-like receptors: from endogenous ligands to tissue regeneration. Cell. Mol. Life Sci. (2006) 63:2901–7. doi: 10.1007/s00018-006-6189-1
52. Letiembre M, Hao W, Liu Y, Walter S, Mihaljevic I, Rivest S, et al. Innate immune receptor expression in normal brain aging. Neuroscience. (2007) 146:248–54. doi: 10.1016/j.neuroscience.2007.01.004
53. Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell. Physiol. Biochem. (2007) 20:947–56. doi: 10.1159/000110455
54. Landewe RBM, Miltenburg AMM, Verdonk MJA, Verweij CL, Breedveld FC, Daha MR, et al. Chloroquine inhibits T cell proliferation by interfering with IL-2 production and responsiveness. Clin Exp Immunol. (1995) 102:144–51. doi: 10.1111/j.1365-2249.1995.tb06648.x
55. Cui CM, Gao JL, Cui Y, Sun LQ, Wang YC, Wang KJ, et al. Chloroquine exerts neuroprotection following traumatic brain injury via suppression of inflammation and neuronal autophagic death. Mol Med Rep. (2015) 12:2323–8. doi: 10.3892/mmr.2015.3611
56. Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CK. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. (2012) 109:15024–9. doi: 10.1073/pnas.1206362109
57. Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. (2010) 285:13107–20. doi: 10.1074/jbc.M110.100420
58. Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. (2010) 5:e9313. doi: 10.1371/journal.pone.0009313
59. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. (2004) 36:585–95. doi: 10.1038/ng1362
60. Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, Court FA, van Zundert B, Hetz C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. (2013) 9:1308–20. doi: 10.4161/auto.25188
61. Du J, Liang Y, Xu F, Sun B, Wang Z. Trehalose rescues Alzheimer's disease phenotypes in APP/PS1 transgenic mice. J Pharm Pharmacol. (2013) 65:1753–6. doi: 10.1111/jphp.12108
62. Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain. (2012) 135:2169–77. doi: 10.1093/brain/aws143
63. Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. (2004) 10:148–54. doi: 10.1038/nm985
64. Zhang X, Heng X, Li T, Li L, Yang D, Zhang X, Du Y, Doody RS, Le W. Long-term treatment with lithium alleviates memory deficits and reduces amyloid-β production in an aged Alzheimer's disease transgenic mouse model. J Alzheimers Dis. (2011) 24:739–49. doi: 10.3233/JAD-2011-101875
65. Fornai F, Longone P, Cafaro L, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. (2008) 105:2052–7. doi: 10.1073/pnas.0708022105
66. Li L, Zhang S, Zhang X, Li T, Tang Y, Liu H, Yang W, Le W. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-β pathology in a mouse model of Alzheimer's disease. Curr Alzheimer Res. (2013) 10:433–41. doi: 10.2174/1567205011310040008
67. Zhang X, Chen S, Lu K, et al. Verapamil Ameliorates Motor Neuron Degeneration and Improves Lifespan in the SOD1G93A Mouse Model of ALS by Enhancing Autophagic Flux. Aging Dis. (2019) 10:1159–73. doi: 10.14336/AD.2019.0228
68. Siddiqi FH, Menzies FM, Lopez A, Stamatakou E, Karabiyik C, Ureshino R, Ricketts T, Jimenez-Sanchez M, Esteban MA, Lai L, Tortorella MD, Luo Z, Liu H, Metzakopian E, Fernandes HJR, Bassett A, Karran E, Miller BL, Fleming A, Rubinsztein DC. Felodipine induces autophagy in mouse brains with pharmacokinetics amenable to repurposing. Nat Commun. (2019) 10:1817. doi: 10.1038/s41467-019-09494-2
69. Menzies FM, Garcia-Arencibia M, Imarisio S, O'Sullivan NC, Ricketts T, Kent BA, Rao MV, Lam W, Green-Thompson ZW, Nixon RA, Saksida LM, Bussey TJ, O'Kane CJ, Rubinsztein DC. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. (2015) 22:433–44. doi: 10.1038/cdd.2014.151
70. Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci. (2009) 29:13578–88. doi: 10.1523/JNEUROSCI.4390-09.2009
Keywords: chloroquine, COVID-19, coronavirus, neurodegeneration, clinical trials
Citation: Pedrioli G, Patani R and Paganetti P (2020) Chloroquine, the Coronavirus Crisis, and Neurodegeneration: A Perspective. Front. Neurol. 11:596528. doi: 10.3389/fneur.2020.596528
Received: 19 August 2020; Accepted: 02 October 2020;
Published: 13 November 2020.
Edited by:
Jordi A. Matias-Guiu, Hospital Clínico San Carlos, SpainReviewed by:
Ulises Gomez-Pinedo, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos, SpainCopyright © 2020 Pedrioli, Patani and Paganetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Paganetti, cGFvbG8ucGFnYW5ldHRpQGVvYy5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.