 Alejandra Malavera1
Alejandra Malavera1 Dominique A. Cadilhac2,3
Dominique A. Cadilhac2,3 Vincent Thijs3,4
Vincent Thijs3,4 Joyce Y. Lim1Brenda Grabsch3Sibilah Breen3Stephen Jan1,5Craig S. Anderson1,5,6*
Joyce Y. Lim1Brenda Grabsch3Sibilah Breen3Stephen Jan1,5Craig S. Anderson1,5,6*- 1Faculty of Medicine, The George Institute for Global Health, University of New South Wales, Sydney, NSW, Australia
- 2Stroke and Ageing Research, Department of Medicine, School of Clinical Sciences at Monash Health, Monash University, Clayton, VIC, Australia
- 3Florey Institute of Neuroscience and Mental Health, University of Melbourne, Heidelberg, VIC, Australia
- 4Department of Neurology, Austin Health Heidelberg, Heidelberg, VIC, Australia
- 5Sydney Medical School, University of Sydney, Sydney, NSW, Australia
- 6Neurology Department, Royal Prince Alfred Hospital, Sydney Health Partners, Sydney, NSW, Australia
Introduction: Fabry disease (FD) is an X-linked lysosomal storage disorder characterized by a deficiency or absence of alpha-galactosidase A (α-GAL A) enzyme, where stroke can be a serious complication. The aim of this study is to determine the feasibility of centralized screening for FD, among young stroke adults registered in the national Australian Stroke Clinical Registry (AuSCR).
Methods: The study was conducted in young (age 18 – 55 years) survivors of acute stroke of unknown etiology registered in AuSCR at hospitals in Queensland, Tasmania, New South Wales, and Victoria during 2014 – 2015; and who, at the 3-month outcome assessment, agreed to be re-contacted for future research. Descriptive analyses of case identification from responses and specific enzyme and DNA sequencing analyses were conducted for α-galactosidase A (α-GLA) from dried blood spot (DBS) testing.
Results: Of 326 AuSCR-identified patients invited to participate, 58 (18%) provided consent but six were subsequently unable to provide a blood sample and two later withdrew consent to use their data. Among the remaining 50 participants (median age 53 years [48 – 56 years]; 47% female), 67% had experienced an acute ischemic stroke. All males (n = 27) had an initial screen for α-GLA enzyme activity of whom seven with low enzyme levels had normal secondary α-GLA gene analysis. All females (n = 23) had genetic analysis, with one shown to have a pathogenic c.352C>T p.(Arg118Cys) missense mutation of the α-GLA gene for FD.
Conclusions: These findings provide logistical data for embedding a process of automated central stroke registry screening for an additional case-finding tool in FD.
Introduction
Stroke in young adults (age <50 years) has major personal, social and economic impact (1, 2). Among the various etiologies, genetic causes are important to diagnose for counseling and consideration of available therapies (3, 4). One such condition, Fabry disease, is an X-linked lysosomal storage disorder caused by mutations in the α-galactosidase A (α-GLA A) gene, which accounts for 1 – 4% of cryptogenic strokes in younger patients (5). Absent or reduced activity of α-GAL A enzyme impairs catabolism of neutral glycosphingolipids, which progressively accumulate within the lysosomes of various cell types, including the vascular endothelium (6). Affected individuals typically show manifestations of the condition in childhood, including acroparesthesias, angiokeratoma, cornea verticillate, and hypohidrosis (7), but acute stroke can occur without any pre-existing cardiac or renal complications (8). As the condition can be easily overlooked as part of investigative work-up, when only ~1 – 2% of young strokes of unknown cause have Fabry disease (9), clinical quality assurance registries may provide an alternative mechanism to support screening efforts.
Screening for Fabry disease entails measuring α-GAL A enzyme activity in peripheral blood leukocytes or plasma but, while affected males usually have low or undetectable enzyme activity, females often have normal or only mildly reduced α-GAL A enzyme activity due to random X-chromosome inactivation (10). Thus, GLA gene sequencing is critical for diagnosing Fabry disease (11). Recently, a novel screening method has been developed for measuring α-GAL A activity from dried blood spots (DBS) on filter paper (12), but data are limited on the utility of this method for screening for Fabry disease in young strokes where the cause is unknown. Herein, we report results of a study designed to test the potential of embedding a process of screening for Fabry disease in young survivors of stroke registered in the ongoing national Australian Stroke Clinical Registry (AuSCR).
Materials and Methods
Study Design and Linked Dataset
This is an investigator-initiated, prospective, study using data from AuSCR for case-finding and merging of additional project generated data using patient-level data linkage. AuSCR is an ongoing, voluntary, quality assurance, clinical registry used to capture minimal data on demographics, case mix, clinical care and 3-month outcomes for consecutive patients admitted with stroke or transient ischemic attack at Australian hospitals (13). An opt-out consent process is used whereby patients, upon admission to hospital, receive an information sheet that outlines the purpose of AuSCR, type of information collected, and requirement to assess functional status and health-related quality of life by questionnaire (or telephone interview) at 3-months post-stroke. At the time of follow-up, participants are also asked if they would be willing to be re-contacted to participate in future research. Approximately 65 – 70% of the cohort complete surveys, and all registered patients have their survival status determined annually through linkage to the National Death Index by the Australian Institute of Health and Welfare.
This study pertains to individuals with the following criteria: age 18 – 55 years at time of stroke with unknown etiology; registered in AuSCR during 2014 – 2015; living at home in Queensland, Tasmania, New South Wales and Victoria; agreed to be contacted; and last known to be alive. They were sent an invitation letter (a second letter was sent to non-responders after 4 weeks) by the AuSCR Office, based in Victoria. Those who responded returned information to The George Institute for Global Health (University of New South Wales), where researchers responsible for coordinating the screening program were based. Following provision of written consent for enzymatic and genetic testing for Fabry disease, subcontracted nurses were organized to undertake home visits to obtain DBS samples and self-reported data on socio-demographic status, major comorbid conditions, vascular risk factors, and any clinical manifestations of Fabry disease.
Screening for Fabry Disease
Eligible participants provided finger-tip blood or venous blood from an antecubital vein onto a DBS card (12) at home, facilitated by a trained nurse from an independent company that provides home-based nursing services across Australia. The DBS card was sent for enzyme and GLA gene analysis at Centogene AG laboratory, Germany, with α-GAL A enzyme activity measured in males followed by GLA gene analysis and Lyso-Gb3 levels in those with low enzyme activity. Conversely, an initial direct genetic analysis of GLA was performed in females, with Lyso-Gb3 levels only measured in those with abnormal result. Fluorometry was used to measure α-GAL A enzyme activity (normal ≥15.3 μmol/L/h) and next-generation sequencing (NGS-Illumina) was used for genetic analysis. Lyso-Gb3 levels were analyzed by liquid chromatography mass spectrometry (normal ≤1.8 ng/mL), with multiplex ligation-dependent probe amplification (MLPA). This variation of the multiplex polymerase chain reaction permits amplification of multiple targets with only a single primer pair, ideally in situations of large deletions or duplications. Results were communicated to the participants via their primary care physician (General Practitioner) or specialist, with recommendations for referral for counseling as appropriate.
Statistical Analysis
Descriptive statistics was performed for available demographic and clinical parameters. Continuous data are shown as medians, while categorical data are given as absolute counts and proportions. Analyses were conducted using STATA version 15.1 (Stata Corporation, College Station, TX).
Ethics Approval
Ethics approval for conduct of the study was granted by the Human Ethics Review Committee of Royal Prince Alfred Hospital (Protocol No X16-0120; HREC/16/RPAH/146). The independent AuSCR Research Task Group approved access to the AuSCR dataset (by AuSCR staff) to identify potential participants according to the study protocol.
Results
Of 326 patients who met the initial eligibility criteria in the AuSCR database and were invited to participate, 58 (18%) provided consent but six were unable to provide a blood sample and another two later withdrew their consent to provide blood samples. Thus, DBS tests for enzyme/genetic analysis were obtained from 50 participants (Figure 1). The Table 1 outlines their demographic characteristics (median age 53 years [range 19–53]; 47% female) and clinical features, with over two thirds with a history of ischemic stroke. There was a wide range of vascular risk factors and potential early manifestations of Fabry disease, and three (6%) participants reported a genetic disorder (Lynch syndrome, mitochondrial disease and Leiden thrombophilia).
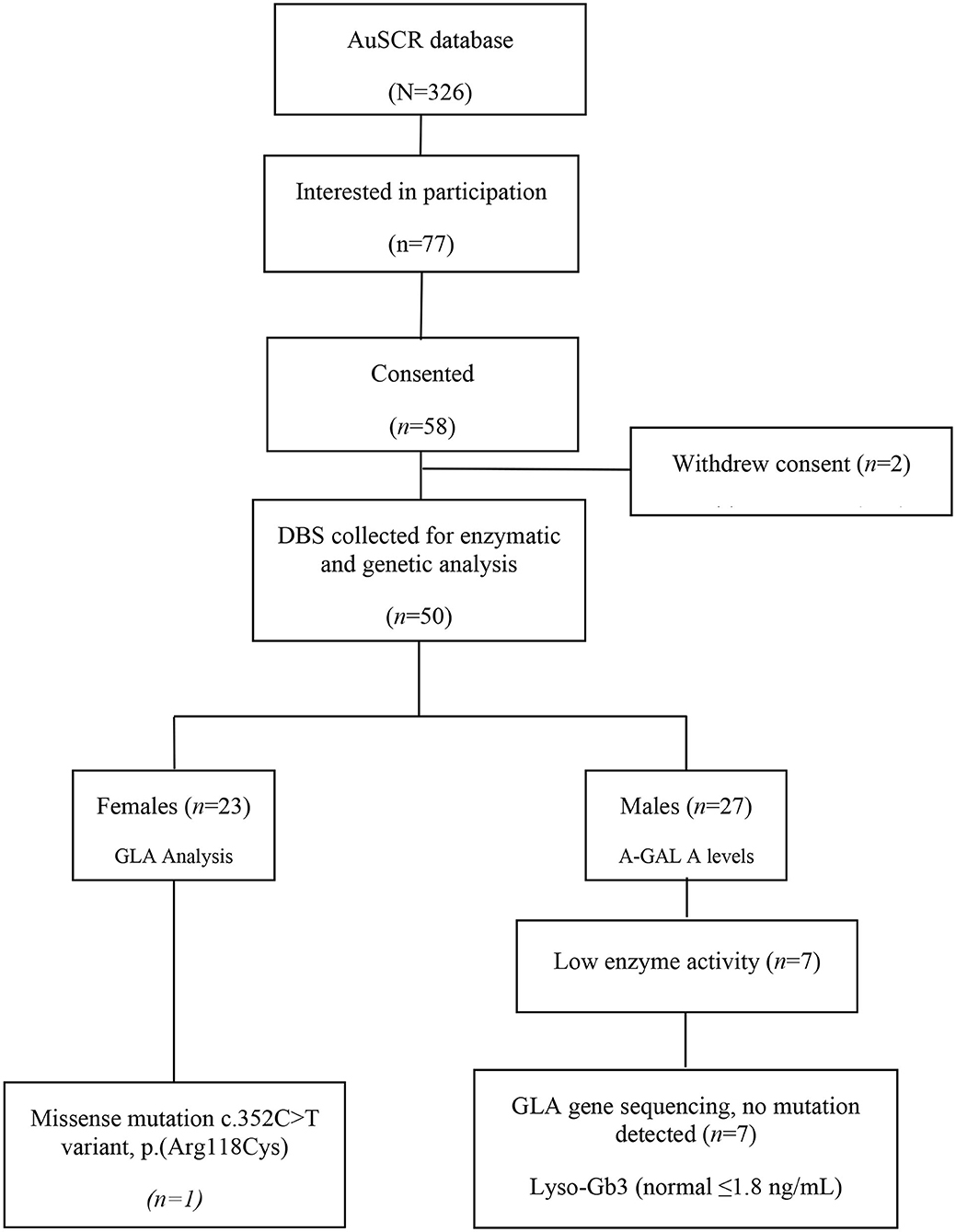
Figure 1. Case identification for genetic screening for Fabry disease in young stroke.
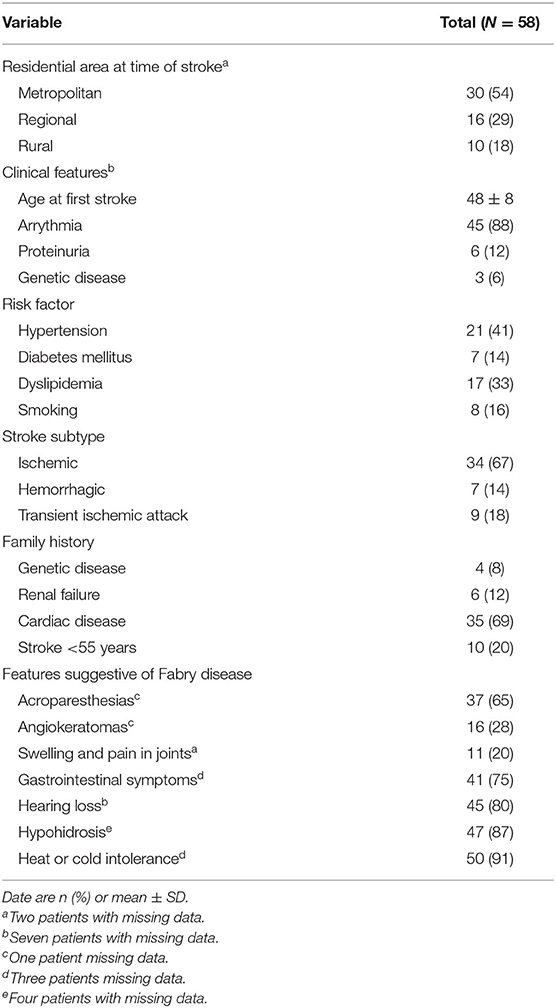
Table 1. Demographic and clinical characteristics of young survivors of stroke.
All males (27/50) had their α-GAL A enzyme level measured on DBS and, although this was low in seven, sequencing of GLA gene did not identify a clinically relevant mutation for Fabry disease. Moreover, all levels of the biomarker Lyso-Gb3 were normal (≤1.8 ng/mL) and MLPA analysis in two patients did not show any deletion or duplication.
For the 23 females, only one had a pathogenic GLA mutation c.352C>T variant, p.(Arg118Cys), despite having a normal level of Lyso-Gb3. She had a history of spontaneous intracerebral hemorrhage at the age of 49 years and reported fatigue, a history of proteinuria and cystic kidney, and a paternal history of premature vascular disease (father with chronic renal failure and brother who had a stroke at a young age) but no other risk factors. Her management included an antiplatelet agent and a statin for secondary stroke prevention.
Discussion
In this study we identified only one potential case of Fabry disease among 326 young stroke survivors of unknown etiology who were screened in AuSCR, a Figure 1 which is consistent with other studies reporting Fabry disease in 0.5% to 3.9% of cases of cryptogenic stroke (7, 14) Similarly, large screening studies involving mixed patients with ischemic stroke, transient ischemic attack, and intracranial hemorrhage, report Fabry disease in 0.4–1% of young strokes (3, 15). Among patients who consent, the DBS test is simple, reliable and relatively rapid in obtaining a result for Fabry disease (16) in an “at-risk” patient group identified through a national prospective clinical registry.
Our study provides some support for embedding a routine genetic screening process within a national stroke registry. However, future implementation of this approach requires greater economic scrutiny over more opportunistic screening at the time of initial hospital management. AuSCR allows direct contact to be made to individuals who had expressed interest in future research, but the 18% response was lower than we had expected (17). This likely reflects our retrospective interrogation of the database for timely identification of a cohort of adequate sample size which consequently delayed the time from the index stroke event, raising the potential for compromised interest and changes to mailing addresses. Although community nurses were contracted to undertake home visits to collect the DBS samples from participants, this was for the purpose of a research project with time and funding constraints. Response rates could potentially have been higher with alternative prospective “real time” case finding, such as early coordination/communication between the AuSCR office and service providers in combination with increased awareness of Fabry disease among stroke clinicians, for detecting cases soon after the occurrence of stroke.
Although Fabry disease is relatively rare, it is important not to miss the diagnosis as enzyme replacement therapy or, more recently, oral pharmacological chaperone therapy (Migalastat) for specific mutant forms of GLA (18), are available in Australia through the Life Saving Drugs Program, a fully subsidized program for life threatening and rare diseases (19). Screening for Fabry disease also provides an opportunity for family members to be identified and managed before complications arise (7), and to be provided with appropriate expert multidisciplinary care. However, as we have not undertaken a formal economic analysis, we recognize the overall costs of screening, incorporating counseling and expensive therapy, need to be considered in the context of broader economic pressures on health care system (20).
Our study found one female with a missense GLA mutation c.352C>T, p.(Arg118Cys), which has been reported in newborns (21), patients with renal failure (22) and young stroke (14). Early studies have shown mixed results on the pathogenicity of this variant, with variable residual α-GAL A enzyme activity and Gb3 deposits in tissues (23). While further research is necessary to clarify the relevance of this variant to late-onset Fabry disease and elevated vascular risk, it is noteworthy that our female case had proteinuria, history of intracerebral hemorrhage, and family history of young stroke, complications that have been described elsewhere (5). We found an absence of genetic mutation and normal levels of Lyso-Gb3 (≤1.8 ng/mL) in seven males with low enzyme level activity. Despite the probably false-positive also reported in other studies (15), measurement of enzyme levels activity using DBS is a reliable method for screening for FD in males (7), but normal results have been reported in about one third of heterozygous females due to the random X-chromosome inactivation (24). The approach of genetic testing those with low enzyme activity is recommended to confirm the diagnosis of Fabry disease (25).
Given that as many as one-third of females with Fabry disease are missed with α-GAL A enzyme activity screening due to random X-inactivation, identification of any pathogenic GLA mutation is essential to define diagnosis and carrier status in females (10). Although Fabry disease is considered rare in heterozygous female carriers, studies have shown that they can still develop complications (8). Moreover, as we have shown with the female case in our study, this approach is more useful for screening and monitoring response to therapy as levels of Lyso-Gb3 are often within the normal range in females compared to males (26). Additionally, Lyso-Gb3 has recently been shown to be an independent risk factor for the cerebral white matter lesions in males with Fabry disease, whilst plasma lyso-Gb3 concentration correlates better with overall disease severity in females (27).
We acknowledge that our study is limited by selection bias and small sample size that has prohibited a reliable assessment of the frequency of Fabry disease in the wider population. Furthermore, we were unable to verify, or obtain further details, of the reported medical history and self-reported symptoms and signs in participants.
In conclusion, we found very few young strokes identified with probable Fabry disease through retrospective screening in a national stroke registry. Our findings draw attention to the complexities of an alternative, automated approach to screening, using a national stroke registry. Further adaption for prospective linkage to clinicians based in participating hospitals might increase case identification early after an acute event.
Data Availability Statement
The primary data supporting the conclusions of this article will be made available by the authors, without undue reservation. The secondary use of data from the Australian Stroke Clinical Registry that partially support the findings of this study are available from the Florey Institute but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available.
Ethics Statement
The studies involving human participants were reviewed and approved by Human Ethics Review Committee of Royal Prince Alfred Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CSA, DAC, and VT: conducted the study, and responsible for the concept and design. BG and SB: contributed to the identification of participants in the AuSCR database registry according to the selection criteria. JL and AM: were responsible for the data acquisition and follow-up of test results. AM: analyzed the data and drafted the main manuscript for intellectual content. CSA, DAC, VT, JL, BG, SB, and SJ: critically reviewed the manuscript for intellectual content and elaboration of the discussion. All authors listed read and approved the final manuscript.
Funding
This study was funded by an unrestricted grant from Shire, Australia, who had no role in study design, data collection, analysis or decision over submission for publication. The following authors received support from the National Health and Medical Research Council: DAC (1063761 co-funded Heart Foundation, and 1154273) and CSA (1081356).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
The Handling Editor declared a past co-authorship with one of the authors, VT. The reviewer MAB declared a past co-authorship with one of the authors, VT to the handling Editor.
Acknowledgments
We thank all participants, the ASCOTT nurses; Centogene AG for the diagnostic laboratory services; and AuSCR office staff.
References
1. Maaijwee NA, Rutten-Jacobs LC, Arntz RM, Schaapsmeerders P, Schoonderwaldt HC, van Dijk EJ, et al. Long-term increased risk of unemployment after young stroke: a long-term follow-up study. Neurology. (2014) 83:1132–8. doi: 10.1212/WNL.0000000000000817
2. Feigin VL, Roth GA, Naghavi M, Parmar P, Krishnamurthi R, Chugh S, et al. Global burden of stroke and risk factors in 188 countries, during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol. (2016) 15:913–24. doi: 10.1016/S1474-4422(16)30073-4
3. Rolfs A, Fazekas F, Grittner U, Dichgans M, Martus P, Holzhausen M, et al. Acute cerebrovascular disease in the young: the Stroke in Young Fabry Patients study. Stroke. (2013) 44:340–9. doi: 10.1161/STROKEAHA.112.663708
4. Smajlovic D. Strokes in young adults: epidemiology and prevention. Vasc Health Risk Manag. (2015) 11:157–64. doi: 10.2147/VHRM.S53203
5. Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. (2009) 40:788–94. doi: 10.1161/STROKEAHA.108.526293
6. Wanner C. Fabry disease model: a rational approach to the management of Fabry disease. Clin Ther. (2007) 29(Suppl. A):S2–5. doi: 10.1016/S0149-2918(07)80115-9
7. Zarate YA, Patterson L, Yin H, Hopkin RJ. A case of minimal change disease in a Fabry patient. Pediatr Nephrol. (2010) 25:553–6. doi: 10.1007/s00467-009-1353-0
8. Rolfs A, Bottcher T, Zschiesche M, Morris P, Winchester B, Bauer P, et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet. (2005) 366:1794–6. doi: 10.1016/S0140-6736(05)67635-0
9. Mitsias P, Levine SR. Cerebrovascular complications of Fabry's disease. Ann Neurol. (1996) 40:8–17. doi: 10.1002/ana.410400105
11. Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. (2008) 93:112–28. doi: 10.1016/j.ymgme.2007.09.013
12. Chamoles NA, Blanco M, Gaggioli D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta. (2001) 308:195–6. doi: 10.1016/S0009-8981(01)00478-8
13. Cadilhac DA, Kilkenny MF, Lannin NA, Dewey HM, Levi CR, Hill K, et al. Outcomes for Patients with in-hospital stroke: a multicenter study from the Australian Stroke Clinical Registry (AuSCR). J Stroke Cerebrovasc Dis. (2019) 28:1302–10. doi: 10.1016/j.jstrokecerebrovasdis.2019.01.026
14. Baptista MV, Ferreira S, Pinho EMT, Carvalho M, Cruz VT, Carmona C, et al. Mutations of the GLA gene in young patients with stroke: the PORTYSTROKE study–screening genetic conditions in Portuguese young stroke patients. Stroke. (2010) 41:431–6. doi: 10.1161/STROKEAHA.109.570499
15. Brouns R, Thijs V, Eyskens F, Van den Broeck M, Belachew S, Van Broeckhoven C, et al. Belgian Fabry study: prevalence of Fabry disease in a cohort of 1000 young patients with cerebrovascular disease. Stroke. (2010) 41:863–8. doi: 10.1161/STROKEAHA.110.579409
16. Lukacs Z, Hartung R, Beck M, Keil A, Mengel E. Direct comparison of enzyme measurements from dried blood and leukocytes from male and female Fabry disease patients. J Inherit Metab Dis. (2007) 30:614. doi: 10.1007/s10545-007-0679-7
17. Denham AMJ, Halpin S, Twyman L, Guillaumier A, Bonevski B. Prevent 2(nd) Stroke: a pilot study of an online secondary prevention program for stroke survivors. Aust N Z J Public Health. (2018) 42:484–90. doi: 10.1111/1753-6405.12794
18. Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, et al. Treatment of Fabry's disease with the pharmacologic chaperone migalastat. N Engl J Med. (2016) 375:545–55. doi: 10.1056/NEJMoa1510198
19. Life Saving Drugs Program (LSDP) Guidelines for Initial Application and Annual Reapplication for Subsidised Treatment for Fabry Disease. Canberra, ACT: Department of Health; Australian Government (2018).
20. Lambe J, Noone I, Lonergan R, Tubridy N. Auditing the frequency and the clinical and economic impact of testing for Fabry disease in patients under the age of 70 with a stroke admitted to Saint Vincent's University Hospital over a 6-month period. Ir J Med Sci. (2018) 187:189–92. doi: 10.1007/s11845-017-1625-9
21. Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. (2006) 79:31–40. doi: 10.1086/504601
22. Gaspar P, Herrera J, Rodrigues D, Cerezo S, Delgado R, Andrade CF, et al. Frequency of Fabry disease in male and female haemodialysis patients in Spain. BMC Med Genet. (2010) 11:19. doi: 10.1186/1471-2350-11-19
23. Ferreira S, Ortiz A, Germain DP, Viana-Baptista M, Caldeira-Gomes A, Camprecios M, et al. The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: data from individual patients and family studies. Mol Genet Metab. (2015) 114:248–58. doi: 10.1016/j.ymgme.2014.11.004
24. Linthorst GE, Poorthuis BJ, Hollak CE. Enzyme activity for determination of presence of Fabry disease in women results in 40% false-negative results. J Am Coll Cardiol. (2008) 51:2082. doi: 10.1016/j.jacc.2008.02.050
25. Stiles AR, Zhang H, Dai J, McCaw P, Beasley J, Rehder C, et al. A comprehensive testing algorithm for the diagnosis of Fabry disease in males and females. Mol Genet Metab. (2020) 130:209–14. doi: 10.1016/j.ymgme.2020.04.006
26. van Breemen MJ, Rombach SM, Dekker N, Poorthuis BJ, Linthorst GE, Zwinderman AH, et al. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim Biophys Acta. (2011) 1812:70–6. doi: 10.1016/j.bbadis.2010.09.007
Keywords: fabry disease, α-galactosidase A, screening, blood spot test, GLA gene, young stroke
Citation: Malavera A, Cadilhac DA, Thijs V, Lim JY, Grabsch B, Breen S, Jan S and Anderson CS (2020) Screening for Fabry Disease in Young Strokes in the Australian Stroke Clinical Registry (AuSCR). Front. Neurol. 11:596420. doi: 10.3389/fneur.2020.596420
Received: 19 August 2020; Accepted: 02 November 2020;
Published: 24 November 2020.
Edited by:
Antonio Arauz, Manuel Velasco Suárez Instituto Nacional de Neurología y Neurocirugía, MexicoReviewed by:
Miguel A. Barboza, University of Costa Rica, Costa RicaFernando Gongora-Rivera, Dr. José Eleuterio Gonzalez University Hospital, Mexico
Copyright © 2020 Malavera, Cadilhac, Thijs, Lim, Grabsch, Breen, Jan and Anderson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig S. Anderson, Y2FuZGVyc29uQGdlb3JnZWluc3RpdHV0ZS5vcmcuYXU=