 Jan Lykke Scheel Thomsen
Jan Lykke Scheel Thomsen Henning Andersen
Henning Andersen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol., 23 December 2020
Sec. Neuromuscular Disorders and Peripheral Neuropathies
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.596382
This article is part of the Research TopicNews and Views in the Management of Myasthenia GravisView all 24 articles
Myasthenia gravis (MG) is a heterogeneous disorder whose clinical presentation ranges from mild ocular deficits to severe widespread weakness. This variance poses a challenge when quantifying clinical deficits. Deficits and symptoms are quantified using standardized clinical scales and questionnaires which are often used as outcome measures. The past decades have seen the development of several validated outcome measures in MG, which are used in clinical trials to obtain regulatory approval. In recent years, emphasis has moved from objective assessments to patient-reported outcomes. Despite a growing body of literature on the validity of the MG-specific outcome measures, several unresolved factors remain. As several novel therapeutics are currently in clinical development, knowledge about capabilities and limitations of outcome measures is needed. In the present paper, we describe the most widely used clinical classifications and scales in MG. We highlight the choice of outcome measures in published and ongoing trials, and we denote whether trial efficacy was reached on these outcomes. We discuss advantages and limitations of the individual scales, and discuss some of the unresolved factors relating to outcome assessments in MG.
Myasthenia gravis (MG) is an autoimmune neuromuscular disease characterized by fatigable muscle weakness due to autoantibodies targeting components of the neuromuscular junction (1). Symptoms and deficits involve ocular, bulbar, respiratory and proximal limb muscles, and they fluctuate in a diurnal and day-to-day pattern. This fluctuating nature of symptoms challenges assessments of disease severity. Deficits and symptoms are measured using validated clinical scales. The past decades have seen the development of several clinical scales reflecting objective, patient-reported and composite measures of disease severity. These validated outcome measures are frequently employed as primary and secondary efficacy parameters in randomized controlled trials (RCT). Several RCTs of currently used immunosuppressants have produced ambiguous results concerning their efficacy in MG. This lack of efficacy may be due to trial-related factors, including sample size issues (e.g., low recruitment), design (e.g., length and inclusion criteria) and insufficiently sensitive outcome measures (e.g., floor and ceiling effects) (2). Accordingly, the current use of these treatments is based on expert consensus and convincing efficacy in daily clinical use. Treatment of MG has recently entered a new era with the development of monoclonal antibodies targeting specific pathophysiologic culprits. As RCTs of these therapeutics may lead to regulatory approval of new treatments, knowledge of the capabilities and limitations of the clinical scales is imperative in understanding the efficacy of current and future treatments in MG.
MG is a heterogeneous disease with several possible classifications according to disease and patient-related factors (1). Type of autoantibody enables classification accordingly, which may directly affect treatment choice. Age at onset enables classification into early-onset and late-onset disease; the former having a female predominance and a higher frequency of thymic hyperplasia. Symptom distribution may be used to classify MG into ocular and generalized MG; and MG may be classified by presence or absence of thymoma.
Although subpopulations of MG are distinguishable, patients are often classified according to the severity of deficits using the Myasthenia Gravis Foundation of America (MGFA) Classification. In 2000, the MGFA Classification was defined as an iteration of previously used classifications (3–6). Patients are classified according to level of overall severity, spanning ocular-only (I), mild (II), moderate (III), severe (IV) and intubation (V), with additional subclassification related to axial/extremity (a) or bulbar (b) predominance. The MGFA Classification is not a recommended outcome measure owing to its poor correlation with summated rating scales (7, 8) and high dependence on physician interpretation. The MGFA Classification is a system broadly characterizing patients according to severity of disease and prognosis.
In the 1930s, the use of ephedrine (9), acetylcholine esterase inhibitors (10), pituitary extract (11) and thymectomy (12) enabled non-quantifiable individual-level descriptions of treatment-related improvements in MG. A rating scale specific to MG was not introduced until the 1980s, and the subsequent decades saw the development of several MG-specific clinical scales (Table 1). Several publications review the various measures in detail (13, 25). Currently, the QMG, the MGC, the MG-ADL, and the QOL15(r) are the most widely used scales in clinical trials. Recently, the MGII was developed and has several potential advantages, however this scale has not been used in any clinical trials yet. Accordingly, the QMG, the MGC, the MG-ADL, and the QOL15(r) will be described below. The MGII will be discussed in context of advantages and limitations of these scales.
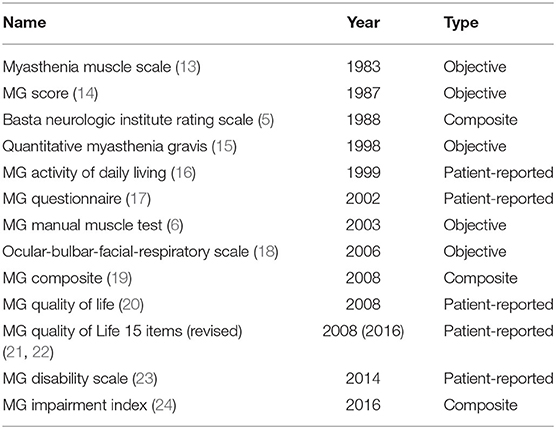
Table 1. MG-specific outcome measures.
The Quantitative Myasthenia Gravis (QMG) scale was introduced in 1998, serving as an objective measure of disease severity (16). The QMG encompassed eight items in the first version (26). It was later expanded to include 13 items (15). In a subsequent revision, the patient-reported items were replaced by physician examinations resulting in its current version (16). The QMG assesses muscle strength and fatigability using objective measures of double vision, ptosis, facial muscles, dysphagia, dysarthria, proximal limb, hand muscles, neck muscles and respiratory function. These assessments are somewhat time consuming and require equipment. Accordingly, in daily clinical practice use of the QMG is challenging. Each item is given a score of 0–3, resulting in an unweighted total score of 0–39. A higher score corresponds to more severe disease. Based on data from the cyclosporine trials (15, 16, 27), a 3-point change is considered clinically meaningful, with a modification in milder cases where a 2-point change is considered sufficient (13). Reliability is high and interobserver variability is low (16, 28, 29).
The MG Activity of Daily Living (MG-ADL) scale is a patient-reported outcome developed in 1999 (17) as a quickly administered set of questions examining frequency and severity of key MG symptoms. The MG-ADL was constructed as an expanded version of the patient-reported sub-items from another scale (15). Using a recall period of a few weeks, eight questions assess ocular function, speech, chewing, swallowing, respiratory function, and strength of proximal upper and lower extremities. Each item is scored from 0 to 3, which results in an unweighted total score of 0–24 points. A higher score indicates more severe symptoms. Based on a longitudinal study on the MG-ADL, the QOL15 and the physician impression of change (30), a 2-point change is considered clinically meaningful. Reliability is high (30).
The MG Composite (MGC) scale was developed in 2008 (20). It was constructed using the top performing items of the QMG, the MG-ADL and the Manual Muscle Test during a trial of mycophenolate. Six physician-assessed examinations evaluate ocular, neck and proximal limb muscles. Furthermore, four patient-reported items assess speech, chewing, swallowing and respiratory function. All patient-reported items are from the MG-ADL. A group of MG experts decided on item-score weighting based on symptom severity. Total score spans from 0 to 50; a higher score indicating more severe disease. A 3-point change is considered clinically meaningful based on physician's impression of change (31). The MGC has been reported to have a high reliability (31).
The MG Quality of Life 15-items (QOL15) was developed in 2008 as a patient-reported outcome (22). It was based on a large 60-item MG questionnaire (21). The current 15 questions were based on feedback from patients and on responsiveness of the individual items during a trial of mycophenolate. Using a recall period of a few weeks [originally 4 weeks (22)]), these 15 questions assess ocular symptoms, swallowing, speech, proximal limb function, mobility, personal grooming, work, social life, activities, fluctuations and psychological items. Scoring is qualitative. Each question is scored from 0 to 4, resulting in a total score in the range of 0–60; a higher score indicates poorer quality of life. The QOL15 score was slightly revised to its present version during subsequent international validation (23). The QOL15r retains the original 15 questions using a slight re-phrasing of some items and reducing the item score to a range of 0–2. Reliability is high (23, 32). The responsiveness has not been studied or published. The questionnaire has been validated in various languages and cultures.
Choice of primary and secondary endpoint(s) vary among the published and ongoing RCTs. In Table 2, trials with more than 30 participants are summarized, and their results are denoted according to the prespecified analysis.

Table 2. RCTs in MG with ≥30 participants and available results.
The prespecified endpoints have not been reached in several trials (Table 2). This may be due to a lack of efficacy; however, lack of efficacy may also result from sample-size issues, trial design and choice of statistical analysis.
Prior to 2017, the primary endpoint was mainly objective assessments (15, 27, 33, 35–37, 39, 42), antibody titers (15, 27) and the steroid-sparing effect (27, 34, 38, 40–42). The REGAIN trial evaluating eculizumab (43) was published in 2017 and was the first trial to introduce the MG-ADL as a primary endpoint. Currently, most ongoing phase 3 trials rely on the MG-ADL as a primary endpoint (Table 2). A trial of rituximab applies a composite measure of QMG and steroid-sparing effect, and a trial of oral Salbutamol is using the QOL15 as primary endpoint. Recently, the QMG is mostly used as a secondary endpoint in phase 3 trials (Table 2) and as a primary endpoint in pilot studies and phase 2 trials, including trials of mycophenolate (2003) (47), terbutaline (2009) (48), eculizumab (2013) (49), belimumab (2018) (50), rozanolixizumab (2019) (46), iscalimab (2019), and zilucoplan (2020) (51).
In recent years, the regulatory authorities have emphasized the use of patient-reported outcomes as primary efficacy parameter in clinical trials. Accordingly, several ongoing trials in MG use patient-reported outcomes as primary endpoint (Table 2). Symptoms fluctuate in MG; hence, objective assessments may not necessarily reflect patients' experienced symptom burden. Consequently, patient-reported outcomes are preferred as primary outcomes in MG trials.
Few patient-reported scales have been developed in MG (Table 1). The MG-ADL is validated, it has been tested in several trials, it is quick and easy to administer, and it assesses disease severity using questions specifically addressing MG symptoms. However, several symptoms of MG are not assessed, and the negative consequences of treatment (e.g., side-effects) are not addressed. Despite improvements in symptoms during treatment, the overall quality of life may be more severely affected due to, e.g., intolerable side-effects. Therefore, health-related quality of life measures may be considered more relevant outcome parameters. Using the QOL15 score introduces new challenges as factors unrelated to MG symptoms may affect quality-of-life scores (22, 52–55). Hence, relying on the QOL15 as primary endpoint may result in inadequate power to detect improvements in core MG-related symptoms. This may, in turn, result in issues relating to adequate trial recruitment. Improvements in the QOL15(r) score should therefore be considered as supplementary information when using the MG-ADL as primary endpoint. The use of a single patient-reported question assessing perceived degree of normal (Single Simple Question, SSQ) (56) has shown a high degree of correlation with the QOL15 and other MG measures, however this has not been tested prospectively. The Myasthenia Gravis Impairment Index (MGII) (57) is a newly developed composite outcome measure consisting of patient-reported items and physical examinations. The patient-reported subitems have excellent reliability as a stand-alone scale (57), however responsiveness and clinical meaningful change has only been published on the composite measure (58).
Some MG symptoms are poorly reflected by the MG-ADL. Neck weakness is not addressed although it is a debilitating symptom in some patients. Assessment of limb muscle fatigability is restricted to few shoulder and hip activities, although fatigability is one of the most relevant symptoms in patients with MG (59) potentially affecting several ADL functions. The QMG scale specifically addresses both complaints. The QMG is a well-established test providing evidence of responsiveness during various treatments; however, the QMG may be more sensitive to changes in ocular, limb and axial muscles than to changes in bulbar and respiratory functions (60). Thus, the QMG provides valuable objective information complementing the patient-reported outcomes, however objective assessments of respiratory and bulbar functions are still lacking.
MG symptoms contribute differently to the degree of clinical disability. Obviously, respiratory failure is more medically severe than persistent ocular symptoms. Hence, weighted scores as used in the MG-Composite may capture more clinically relevant information concerning disease severity. Thus, the MG-Composite may serve as an alternative to linear disease measures, complementing both the patient-reported outcomes and the QMG.
Degree of clinical disability is heterogeneous; hence, clinical scores should cover the entire spectrum ranging from mild to severely affected cases. However, there is considerable floor-effect in the MG-ADL (61) limiting its use in milder cases. The MGII shows less floor-effect than both the MG-ADL and the MGC (57), and it was recently shown to provide clinically relevant supplementary information to the MG-ADL (61). Interestingly, the MGII correlates only moderately with the QMG and the QOL15 during follow-up (58). Until now, the MGII has not been used as an outcome measure in trials, but it has the potential as an attractive alternative to other secondary outcomes. Due to the emphasis by regulatory authorities on patient-reported outcomes, the MGII is currently best suited as a secondary endpoint. MGII may enable superior assessment of efficacy covering a larger spectrum of disease severity if used as a primary endpoint, however this remains to be studied in RCTs. Further, the responsiveness and clinical meaningful change of the MGII patient-reported items as a stand-alone scale is unsettled.
Response to treatment is variable, and the overall treatment-effect consists of patients with both minor and larger improvements. Accordingly, the point-change required for a clinically meaningful improvement has been established on the MG-ADL, the QMG, and the MGC. This enables responder-analysis and assessments of clinical meaningful effects while negating minor placebo-effects and natural fluctuations. The pooled QMG response of several RCTs (62) detected significant effects over placebo on both continuous and categorical analysis. The MGFA Post Intervention System (MGFA-PIS) apply this required point-change on the MGC (recommended) or the QMG in order to address whether patients improve or deteriorate (2). Only few studies have applied the MGFA-PIS as an outcome measure (Table 2), however assessments or minimal manifestation and clinical remission are also included in the MGFA-PIS. Recently, to obtain patient acceptable symptom states (Patient Acceptable Symptoms Score, PASS) the cut-off values required on several clinical scales (the QMG, the MGC, the MG-ADL, the QOL15 and the MGII) were analyzed (63). It is currently unsettled whether dichotomized assessments of minimal manifestation or PASS is feasible in clinical trials.
No prospective study has analyzed the relations between the four most frequently used scales (the QMG, the MGC, the MG-ADL and the QOL15). Correlations between some of the scales have previously been published (30–32, 57, 58, 64), and the relations between objective (QMG and MGC) and patient-reported measures (MG-ADL and QOL15) seem attenuated during treatment and follow-up. One study (58) has applied the QMG, the MGC, the MG-ADL and the QOL15 to the same population; however, between-scale correlations were not published. It is unknown whether improvements on objective scores are accompanied by equal improvements on patient-reported outcomes (e.g., MG-ADL and QOL15/QOL15r).
There is a lack of information concerning how outcome measures are affected by basic patient characteristics and how the scales perform in various subpopulations. Such information is crucial in design of clinical studies, and it is critical when determining relevant change in burden of symptoms and deficits during routine care. Sex differences characterize early and late-onset subpopulations of MG; hence, females often have longer disease duration than males. Further, studies report sex differences in rates of refractory MG (65–67). Most recent and ongoing trials focus on severe or refractory patients; hence, trial populations may consist mainly of females, and participants may have long-standing disease. It is unsettled whether sex and disease duration affect potential for improvement on current outcome measures, and it is unknown whether current outcome measures are equally applicable in the various MG subpopulations.
When applying the current outcome measures, a major challenge is inability to capture all clinically relevant factors in MG. Fatigue is a relevant feature of MG in addition to muscular fatigability (68). Being a subjective feeling of exhaustion, fatigue is preferably quantified using patient-reported outcomes. Several generic fatigue scales have been used in MG, including the Neuro-QOL Fatigue Scale (68) and the Chalder Fatigue Scale (53). Only the REGAIN trial included fatigue as a secondary outcome (69). Although the QOL15 was not designed to specifically incorporate fatigue, a high degree of correlation has been established between fatigue and QOL15 (53, 69). This suggests some responsiveness to improvements in fatigue in addition to MG specific symptoms. Further, the patient-reported subitems of the MGII incorporate fatigue (57). Change-correlations between the MGII and the Neuro-QOL Fatigue Scale are moderate and equally directed (58). Whether fatigue scores complement improvement captured by the QOL15 or MGII scores remains to be studied.
Use of treatment as well as presence and severity of side effects are not systematically assessed in any of the outcome measures despite their clinical relevance. Steroids are frequently used during MG exacerbations and as effective bridging therapies when tapering immunosuppressive agents. Some patients require chronic steroid therapy due to inadequate symptomatic control. Several trials have used the steroid-sparing effect as an outcome measure (Table 2). Due to the side-effect profile of chronic steroid exposure, a reduced steroid dose is equated to improvement on MG scales. Reduction in other therapies (e.g., pyridostigmine or immunosuppressive agents) or a change in therapy (e.g., intravenous to subcutaneous immunoglobulin) may result in better quality of life despite stability in MG symptoms; however, this is only indirectly assessed by sub-items of the QOL15(r) and not addressed by any of the symptom-orientated scales. Risk of side effects may result in significant psychological stress, especially when considering cancer risk in young patients requiring long-term treatment or potentially teratogenic effects in fertile woman. Since MG is a chronic disease usually requiring treatment for decades, treatment satisfaction may be considered as important as symptomatic control. Treatment satisfaction is not systematically assessed using any of the current outcome measures.
In coming years, the use of tele-medicine will likely increase, especially due to the current global pandemic when monitoring immunocompromised patients. Further, virtual care may increase patient willingness to participate in RCTs owing to fewer physical attendances. Accordingly, validated measures assessing MG functioning through virtual care are needed. It is unsettled how the current MG scales function in a virtual setting. Some objective assessments are feasible, especially of ocular and bulbar involvement, however pure patient-reported measures will likely result in the most robust assessments. This area currently merits further research.
Patient-reported outcomes are often used as primary endpoints in establishing efficacy of novel treatments. Several of the recent trials focus on medically severe and refractory patients. However, a large proportion of patients are mild to moderately affected. New therapeutic options are warranted addressing unmet medical needs in this large group of patients. None of the current patient-reported outcomes enables detection of improvement on the entire severity continuum. In addition, no single patient-reported scale captures both the quantitative and qualitative aspect of improvement in MG symptoms during treatment.
Several MG-specific outcome measures have been developed, reflecting objective disease burden, patient-reported symptom severity and health-related quality of life. Each scale has distinct advantages relating to MG assessments and complements information obtained from other outcome measures. Detailed assessments of treatment efficacy should currently incorporate patient-reported assessments (e.g., MG-ADL), quality-of-life measurements [e.g., QOL15(r)], objective assessments (e.g., QMG) and composite measures (e.g., MGC or MGII). Fatigue measures (e.g., NeuroQOL) may provide additional and relevant information. However, several clinically relevant issues are not addressed by any of the current scales, and the relation of several basic patient characteristics to current outcome measures remain unsettled. This restricts thorough assessment of treatment efficacy and may limit conclusions concerning validity across subpopulations in MG.
JT wrote and revised the manuscript. HA co-authored and revised the manuscript for intellectual content. Both authors contributed to the article and approved the submitted version.
JT has received speaker honorarium from Alexion. HA has received research support from Sanofi Genzyme and CSL Behring, and received travel support and speaker honoraria from Novo, Alexion, Sanofi Genzyme, Octapharma, and CSL Behring and served as consultant on advisory board for NMD Pharma.
1. Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM. Myasthenia gravis. Nat Rev Dis Primers. (2019) 5:30. doi: 10.1038/s41572-019-0079-y
2. Benatar M, Sanders DB, Burns TM, Cutter GR, Guptill JT, Baggi F, et al. Recommendations for myasthenia gravis clinical trials. Muscle Nerve. (2012) 45:909–917. doi: 10.1002/mus.23330
3. Osserman KE, Kornfeld P, Cohen E, Genkins G, Mendelow H, Goldberg H, et al. Studies in myasthenia gravis: review of two hundred eighty-two cases at The Mount Sinai Hospital, New York City. AMA Arch Int Med. (1958) 102:72–81. doi: 10.1001/archinte.1958.00260190074008
4. Oosterhuis HJGH. Studies in myasthenia gravis. Part 1. A clinical study of 180 patients. J Neurol Sci. (1964) 1:512–46. doi: 10.1016/0022-510X(64)90171-6
5. Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteriod treatment of myasthenia gravis: report of 116 patients. Ann Neurol. (1984) 15:291–8. doi: 10.1002/ana.410150316
6. Mantegazza R, Antozzi C, Peluchetti D, Sghirlanzoni A, Cornelio F. Azathioprine as a single drug or in combination with steroids in the treatment of myasthenia gravis. J Neurol. (1988) 235:449–53. doi: 10.1007/BF00314245
7. Sanders DB, Tucker-Lipscomb B, Massey JM. A simple manual muscle test for myasthenia gravis. Ann N Y Acad Sci. (2003) 998:440–4. doi: 10.1196/annals.1254.057
8. Jaretzki A, Barohn RJ, Ernstoff RM, Kaminski HJ, Keesey JC, Penn AS, et al. Myasthenia gravis: recommendations for clinical research standards. Neurology. (2000) 55:16–23. doi: 10.1212/WNL.55.1.16
9. Edgeworth H. A report of progress on the use of ephedrine in a case of myasthenia gravis. J Am Med Assoc. (1930) 94:1136. doi: 10.1001/jama.1930.27120410003009c
10. Walker MB. Treatment of myasthenia gravis with physostigmine. Lancet. (1934) 223:1200–1. doi: 10.1016/S0140-6736(00)94294-6
11. Simon HE. Myasthenia gravis: effect of treatment with anterior pituitary extract: preliminary report. J Am Med Assoc. (1935) 104:2065–6. doi: 10.1001/jama.1935.02760230013004
12. Blalock A, Mason MF, Morgan HJ, Riven SS. Myasthenia gravis and tumors of the thymic region. Ann Surg. (1939) 110:544–61. doi: 10.1097/00000658-193910000-00005
13. Barnett C, Herbelin L, Dimachkie MM, Barohn RJ. Measuring clinical treatment response in myasthenia gravis. Neurol Clin. (2018) 36:339–53. doi: 10.1016/j.ncl.2018.01.006
14. Gajdos P, Simon N, de Rohan-Chabot P, Raphael JC, Goulon M. [Long-term effects of plasma exchange in myasthenia. Results of a randomized study]. Presse Med. (1983) 12:939–42.
15. Tindall RSA, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. New Engl J Med. (1987) 316:719–24. doi: 10.1056/NEJM198703193161205
16. Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan WW. Reliability testing of the quantitative myasthenia gravis scorea. Ann N Y Acad Sci. (1998) 841:769–72. doi: 10.1111/j.1749-6632.1998.tb11015.x
17. Wolfe GI, Herbelin L, Nations SP, Foster B, Bryan WW, Barohn RJ. Myasthenia gravis activities of daily living profile. Neurology. (1999) 52:1487–9. doi: 10.1212/WNL.52.7.1487
18. Padua L, Evoli A, Aprile I, Caliandro P, Batocchi AP, Punzi C, et al. Myasthenia gravis outcome measure: development and validation of a disease-specific self-administered questionnaire. Neurol Sci. (2002) 23:59–68. doi: 10.1007/s100720200027
19. Farrugia ME, Robson MD, Clover L, Anslow P, Newsom-Davis J, Kennett R, et al. MRI and clinical studies of facial and bulbar muscle involvement in MuSK antibody-associated myasthenia gravis. Brain. (2006) 129:1481–92. doi: 10.1093/brain/awl095
20. Burns TM, Conaway MR, Cutter GR, Sanders DB. Construction of an efficient evaluative instrument for myasthenia gravis: the MG composite. Muscle Nerve. (2008) 38:1553–62. doi: 10.1002/mus.21185
21. Mullins LL, Carpentier MY, Paul RH, Sanders DB, McDermott M, Thornton C, et al. Disease-specific measure of quality of life for myasthenia gravis. Muscle Nerve. (2008) 38:947–56. doi: 10.1002/mus.21016
22. Burns TM, Conaway MR, Cutter GR, Sanders DB. Less is more, or almost as much: a 15-item quality-of-life instrument for myasthenia gravis. Muscle Nerve. (2008) 38:957–63. doi: 10.1002/mus.21053
23. Burns TM, Sadjadi R, Utsugisawa K, Gwathmey KG, Joshi A, Jones S, et al. International clinimetric evaluation of the MG-QOL15, resulting in slight revision and subsequent validation of the MG-QOL15r. Muscle Nerve. (2016) 54:1015–22. doi: 10.1002/mus.25198
24. Raggi A, Schiavolin S, Leonardi M, Antozzi C, Baggi F, Maggi L, et al. Development of the MG-DIS: an ICF-based disability assessment instrument for myasthenia gravis. Disabil Rehabil. (2014) 36:546–55. doi: 10.3109/09638288.2013.804591
25. Burns TM. History of outcome measures for myasthenia gravis. Muscle Nerve. (2010) 42:5–13. doi: 10.1002/mus.21713
26. Besinger UA, Toyka KV, Hömberg M, Heininger K, Hohlfeld R, Fateh-Moghadam A. Myasthenia gravis: long-term correlation of binding and bungarotoxin blocking antibodies against acetylcholine receptors with changes in disease severity. Neurology. (1983) 33:1316–21. doi: 10.1212/WNL.33.10.1316
27. Tindall RSA, Phillips JT, Rollins JA, Wells L, Hall K. A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci. (1993) 681:539–51. doi: 10.1111/j.1749-6632.1993.tb22937.x
28. Barnett C, Merkies ISJ, Katzberg H, Bril V. Psychometric properties of the quantitative myasthenia gravis score and the myasthenia gravis composite scale. J Neuromusc Dis. (2015) 2:301–11. doi: 10.3233/JND-150082
29. Sharshar T, Chevret S, Mazighi M, Chillet P, Huberfeld G, Berreotta C, et al. Validity and reliability of two muscle strength scores commonly used as endpoints in assessing treatment of myasthenia gravis. J Neurol. (2000) 247:286–90. doi: 10.1007/s004150050585
30. Muppidi S, Wolfe GI, Conaway M, Burns TM, MG Composite and MG-QOL15 Study Group. MG-ADL: still a relevant outcome measure. Muscle Nerve. (2011) 44:727–31. doi: 10.1002/mus.22140
31. Burns TM, Conaway M, Sanders DB, MG Composite and MG-QOL15 Study Group. The MG Composite: a valid and reliable outcome measure for myasthenia gravis. Neurology. (2010) 74:1434–40. doi: 10.1212/WNL.0b013e3181dc1b1e
32. Burns TM, Grouse CK, Wolfe GI, Conaway MR, Sanders DB. The MG-QOL15 for following the health-related quality of life of patients with myasthenia gravis. Muscle Nerve. (2011) 43:14–8. doi: 10.1002/mus.21883
33. Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C. Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Ann Neurol. (1997) 41:789–96. doi: 10.1002/ana.410410615
34. Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Neurology. (1998) 50:1778–83. doi: 10.1212/WNL.50.6.1778
35. Gajdos P, Tranchant C, Clair B, Bolgert F, Eymard B, Stojkovic T, et al. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: a randomized double-blind clinical trial. Arch Neurol. (2005) 62:1689–93. doi: 10.1001/archneur.62.11.1689
36. Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology. (2007) 68:837–41. doi: 10.1212/01.wnl.0000256698.69121.45
37. Sanders DB. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology. (2008) 71:394–9. doi: 10.1212/01.wnl.0000312373.67493.7f
38. Sanders DB, Hart IK, Mantegazza R, Shukla SS, Siddiqi ZA, de Baets MHV, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology. (2008) 71:400–6. doi: 10.1212/01.wnl.0000312374.95186.cc
39. Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology. (2011) 76:2017–23. doi: 10.1212/WNL.0b013e31821e5505
40. Yoshikawa H, Kiuchi T, Saida T, Takamori M. Randomised, double-blind, placebo-controlled study of tacrolimus in myasthenia gravis. J Neurol Neurosurg Psychiatry. (2011) 82:970–7. doi: 10.1136/jnnp-2011-300148
41. Pasnoor M, He J, Herbelin L, Burns TM, Nations S, Bril V, et al. A randomized controlled trial of methotrexate for patients with generalized myasthenia gravis. Neurology. (2016) 87:57–64. doi: 10.1212/WNL.0000000000002795
42. Wolfe GI, Kaminski HJ, Aban IB, Minisman G, Kuo H-C, Marx A, et al. Randomized trial of thymectomy in myasthenia gravis. New Engl J Med. (2016) 375:511–22. doi: 10.1056/NEJMoa1602489
43. Howard JF, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. (2017) 16:976–86. doi: 10.1016/S1474-4422(17)30369-1
44. Zhou L, Liu W, Li W, Li H, Zhang X, Shang H, et al. Tacrolimus in the treatment of myasthenia gravis in patients with an inadequate response to glucocorticoid therapy: randomized, double-blind, placebo-controlled study conducted in China. Ther Adv Neurol Disord. (2017) 10:315–25. doi: 10.1177/1756285617721092
45. Howard JF, Bril V, Burns TM, Mantegazza R, Bilinska M, Szczudlik A, et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology. (2019) 92:e2661–73. doi: 10.1212/WNL.0000000000007600
46. Bril V, Benatar M, Brock M, Greve B, Kiessling P, Woltering F, et al. Proof-of-concept and safety of the anti-FcRn antibody rozanolixizumab in patients with moderate-to-severe generalized myasthenia gravis (GMG): a phase 2a study (S43.001). Neurology. (2019) 92.
47. Meriggioli MN, Rowin J, Richman JG, Leurgans S. Mycophenolate mofetil for myasthenia gravis: a double-blind, placebo-controlled pilot study. Ann N Y Acad Sci. (2003) 998:494–9. doi: 10.1196/annals.1254.064
48. Soliven B, Rezania K, Gundogdu B, Harding-Clay B, Oger J, Arnason BGW. Terbutaline in myasthenia gravis: a pilot study. J Neurol Sci. (2009) 277:150–4. doi: 10.1016/j.jns.2008.09.033
49. Howard JF, Barohn RJ, Cutter GR, Freimer M, Juel VC, Mozaffar T, et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. (2013) 48:76–84. doi: 10.1002/mus.23839
50. Hewett K, Sanders DB, Grove RA, Broderick CL, Rudo TJ, Bassiri A, et al. Randomized study of adjunctive belimumab in participants with generalized myasthenia gravis. Neurology. (2018) 90:E1425–34. doi: 10.1212/WNL.0000000000005323
51. Duda PW, Macdougall JE, Farzaneh-far R, Kaminski HJ. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis. JAMA Neurol. (2020) 7026:582–92. doi: 10.1001/jamaneurol.2019.5125
52. Jones SM, Gwathmey KG, Burns TM. Quality of life measures for myasthenia gravis and evaluation of non-motor symptoms. Clin Exp Neuroimmunol. (2015) 6:32–9. doi: 10.1111/cen3.12174
53. Hoffmann S, Ramm J, Grittner U, Kohler S, Siedler J, Meisel A. Fatigue in myasthenia gravis: risk factors and impact on quality of life. Brain Behav. (2016) 6:e00538. doi: 10.1002/brb3.538
54. masuda M, Utsugisawa K, Suzuki S, Nagane Y, Kabasawa C, Suzuki Y, Shimizu Y, et al. The MG-QOL15 Japanese version: validation and associations with clinical factors. Muscle Nerve. (2012) 46:166–73. doi: 10.1002/mus.23398
55. Twork S, Wiesmeth S, Klewer J, Pöhlau D, Kugler J. Quality of life and life circumstances in German myasthenia gravis patients. Health Qual Life Outcomes. (2010) 8:129. doi: 10.1186/1477-7525-8-129
56. Abraham A, Breiner A, Barnett C, Katzberg HD, Bril V. The utility of a single simple question in the evaluation of patients with myasthenia gravis. Muscle Nerve. (2018) 57:240–4. doi: 10.1002/mus.25720
57. Barnett C, Bril V, Kapral M, Kulkarni A, Davis AM. Development and validation of the myasthenia gravis impairment index. Neurology. (2016) 87:879–86. doi: 10.1212/WNL.0000000000002971
58. Barnett C, Bril V, Kapral M, Kulkarni AV, Davis AM. Myasthenia gravis impairment index: responsiveness, meaningful change, and relative efficiency. Neurology. (2017) 89:2357–64. doi: 10.1212/WNL.0000000000004676
59. Barnett C, Bril V, Kapral M, Kulkarni A, Davis AM. A conceptual framework for evaluating impairments in myasthenia gravis. PLoS ONE. (2014) 9:e98089. doi: 10.1371/journal.pone.0098089
60. Barnett TC, Bril V, Davis AM. Performance of individual items of the quantitative myasthenia gravis score. Neuromusc Disord. (2013) 23:413–7. doi: 10.1016/j.nmd.2013.02.008
61. de Meel RHP, Barnett C, Bril V, Tannemaat MR, Verschuuren JJGM. Myasthenia gravis impairment index: sensitivity for change in generalized muscle weakness. J Neuromusc Dis. (2020) 7:297–300. doi: 10.3233/JND-200484
62. Frisaldi E, Shaibani A, Vollert J, Ferrero B, Carrino R, Ibraheem HD, et al. The placebo response in myasthenia gravis assessed by quantitative myasthenia gravis score: a meta-analysis. Muscle Nerve. (2019) 59:671–8. doi: 10.1002/mus.26469
63. Mendoza M, Tran C, Bril V, Katzberg HD, Barnett C. Patient-acceptable symptom states in myasthenia gravis. Neurology. (2020) 95: e1617–28. doi: 10.1212/WNL.0000000000010574
64. Wolfe GI, Barohn RJ, Sanders DB, McDermott MP. Comparison of outcome measures from a trial of Mycophenolate mofetil in myasthenia gravis. Muscle Nerve. (2008) 38:1429–33. doi: 10.1002/mus.21142
65. Suh J, Goldstein JM, Nowak RJ. Clinical characteristics of refractory myasthenia gravis patients. Yale J Biol Med. (2013) 86:255–60.
66. Boscoe AN, Xin H, L'Italien GJ, Harris LA, Cutter GR. Impact of refractory myasthenia gravis on health-related quality of life. J Clin Neuromusc Dis. (2019) 20:173–81. doi: 10.1097/CND.0000000000000257
67. Engel-Nitz NM, Boscoe A, Wolbeck R, Johnson J, Silvestri NJ. Burden of illness in patients with treatment refractory myasthenia gravis. Muscle Nerve. (2018) 58:99–105. doi: 10.1002/mus.26114
68. Tran C, Bril V, Katzberg HD, Barnett C. Fatigue is a relevant outcome in patients with myasthenia gravis. Muscle Nerve. (2018) 58:197–203. doi: 10.1002/mus.26069
Keywords: myasthenia gravis, classification, clinical trials, review, outcome measure, rating scale
Citation: Thomsen JLS and Andersen H (2020) Outcome Measures in Clinical Trials of Patients With Myasthenia Gravis. Front. Neurol. 11:596382. doi: 10.3389/fneur.2020.596382
Received: 19 August 2020; Accepted: 06 November 2020;
Published: 23 December 2020.
Edited by:
Nils Erik Gilhus, University of Bergen, NorwayReviewed by:
Hans Katzberg, University of Toronto, CanadaCopyright © 2020 Thomsen and Andersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Lykke Scheel Thomsen, amF0aG1zQHJtLmRr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.