 Eva Parobkova1,2
Eva Parobkova1,2 Julie van der Zee3,4Lubina Dillen3,4
Julie van der Zee3,4Lubina Dillen3,4 Christine Van Broeckhoven3,4Robert Rusina2,5
Christine Van Broeckhoven3,4Robert Rusina2,5 Radoslav Matej1,2,6,7*
Radoslav Matej1,2,6,7*- 1Department of Pathology and Molecular Medicine, Third Faculty of Medicine, Charles University and Thomayer Hospital, Prague, Czechia
- 2National Reference Laboratory for Human Prion Diseases, Thomayer Hospital, Prague, Czechia
- 3Neurodegenerative Brain Diseases Group, VIB Center for Molecular Neurology, Vlaams Instituut voor Biotechnologie (VIB), Antwerp, Belgium
- 4Department of Biomedical Sciences, University of Antwerp, Antwerp, Belgium
- 5Department of Neurology, Third Faculty of Medicine, Charles University and Thomayer Hospital, Prague, Czechia
- 6Department of Pathology, First Faculty of Medicine, Charles University and General University Hospital, Prague, Czechia
- 7Department of Pathology, Third Faculty of Medicine, Charles University and Kralovske Vinohrady University Hospital, Prague, Czechia
Background: Sporadic Creutzfeldt–Jakob disease (sCJD) is the most common type of a group of transmissible spongiform encephalopathies (prion diseases). The etiology of the sporadic form of CJD is still unclear. sCJD can occur in combination with other neurodegenerative diseases, which further complicates the diagnosis. Alzheimer's disease (AD), e.g., is often seen in conjunction with sCJD.
Method: In this study, we performed a systematic analysis of 15 genes related to the most important neurodegenerative diseases - AD, frontotemporal dementia, amyotrophic lateral sclerosis, prion disease, and Parkinson's disease - in a cohort of sCJD and sCJD in comorbidity with AD and primary age-related proteinopathy (PART). A total of 30 neuropathologically verified cases of sCJD with and without additional proteinopathies were included in the study. In addition, we compared microtubule-associated protein tau (MAPT) haplotypes between sCJD patients and patients with sCJD and PART or sCJD and AD. Then we studied the interaction between the Apolipoprotein E gene (APOE) and PRNP in sCJD patients.
Results: We did not find any causal mutations in the neurodegenerative disease genes. We did detect a p.E318G missense variant of uncertain significance (VUS) in PSEN1 in three patients. In PRNP, we also found a previously described non-pathogenic insertion (p.P84_Q91Q).
Conclusion: Our pilot study failed to find any critical differences between pure sCJD and sCJD in conjunction with other comorbid neurodegenerative diseases. Further investigations are needed to better understand this phenomenon.
Introduction
Neurodegenerative diseases are characterized by intra-or extracellular accumulation of specific protein aggregates in the central nervous system (CNS) (1). These proteins have a predominantly β-sheet form and are found in a number of neurodegenerative diseases such as Alzheimer's disease (AD); synucleinopathies (Parkinson's disease (PD), multiple system atrophy, dementia with Lewy bodies); transmissible spongiform encephalopathies (TSE; also known as prion disease); amyotrophic lateral sclerosis and frontotemporal dementia (2). There is a significant overlap of symptoms resulting from the multiplication and tissue storage of protein aggregates in the brain, leading to progressive neuronal dysfunction and neurodegeneration (3, 4).
Creutzfeldt-Jakob disease (CJD; MIM #176640), the most common human prion disease with an estimated incidence of 2 cases per million per year, is comprised of several clinical-pathological phenotypes and occurs in four unique forms (sporadic, genetic, variant, or acquired), each with seemingly distinct etiologies (5).
CJD can coexist with other neurodegenerative diseases because the presence of both Aβ and tau pathology is not unusual in sporadic and genetic CJD brains (6–9). Primary age-related proteinopathy (PART) is a common pathology involving misfolded tau protein aggregates associated with human aging (10). PART can cause cognitive impairment in the absence of AD (11); additionally, the coexistence of PART and sporadic CJD (sCJD) has been reported (12). A major genetic risk factor for PART is the haplotype of the microtubule-associated protein tau (MAPT) (13). The frequency of Apolipoprotein E (APOE) ε4 is much lower in PART, being ~10% (10, 14), whereas its prevalence in AD exceeds 45% (15, 16). These studies suggest that APOE ε4 allele deficiency – in contrast to AD – is not a risk factor for PART.
Coexistence with other neurodegenerations is relatively common in sCJD patients. Since clinical symptoms of sCJD can overlap with manifestations of other comorbid disorders, establishing a clinical diagnosis in patients with rapidly progressive dementia is very difficult (17), and a definite diagnosis can only be made after a neuropathological examination of the brain.
Our goal was to identify disease-associated variants using genetic studies of sCJD patients. For this reason, we compared sCJD patients without any comorbid proteinopathies to sCJD patients with AD and sCJD patients with PART.
Materials and Methods
Study Population
Our study was designed as a retrospective study. We included patients with post-mortem confirmed sCJD, as well as information regarding clinical presentation and data from neuropsychological testing, biochemical analysis, EEG, and neuroimaging. Neuropathological diagnoses, including prion protein immunoassays, were provided according to standard protocols National CJD Research & Surveillance Unit. Protocol: Surveillance of CJD in the UK) (18) used by the National reference laboratory for human prion diseases at the Department of Pathology and Molecular Medicine, Prague, Czech Republic. Molecular genetic analyses were performed in the Neurodegenerative Brain Disease group of the VIB Center for Molecular Neurology, Antwerp, Belgium.
We divided our cohort into three subgroups: (1) isolated sCJD neuropathology, (2) sCJD and PART or early stage AD (NIA consensus criteria level “low”) (19), and (3) sCJD with more advanced AD (NIA consensus criteria level A2 and or higher).
The Molecular Diagnostics Study Group
All autopsied patients (30/30) fulfilled the WHO diagnostic criteria for definite sporadic CJD (18)1 and were genetically profiled for the most common genes (n = 15) associated with AD (APP, PSEN1, PSEN2, APOE), the FTD-ALS spectrum (MAPT, GRN, TARDBP, FUS, SOD1, VCP), prion disease (PRNP), and PD (LRRK2, PRKN, SNCA) (Supplementary Table 1).
Other available clinical data, which were designated as variables (including age at onset, age at death, gender, and symptoms occurring during the disease), were analyzed to determine how they affected the pathogenesis of sCJD and the concomitant Aβ and tau pathologies.
Genetic Screen
Mutation analyses by gene panel sequencing were performed on genomic DNA extracted from bone marrow. The targeted gene panel captured all exons of the 15 genes and flanking intronic regions to cover the splice sites. Using amplicon target amplification technology (Agilent, https://www.agilent.com), primers were designed using mPCR software (20) (Supplementary Material). Specific target regions were amplified using multiplex PCR, followed by purification of the equimolar pooled amplicons using Agencourt AMPureXP beads (Beckman Coulter, CA, USA). Individual barcodes (Illumina Nextera XT) were incorporated in a universal PCR step prior to sample pooling. Libraries were sequenced on a MiSeq platform using the v3 reagent kit with a paired-end read length of 300 bp (Illumina, San Diego, CA, USA). Non-sense, splice site, indel, and missense variants, with a minor allele frequency (MAF) ≤ 1%, were selected.
Results
We analyzed data from 30 patients (n = 30) with a mean age at onset (AAO) of 58.4 ± 5 years, and a male-to-female ratio of 18:12. Ten cases had sCJD without any other comorbid proteinopathy, 10 cases had sCJD with tauopathy and/or early evolved AD, and 10 cases had sCJD with more developed AD. Family histories were available in 24 cases (82%), with only one patient (3.4%) having a positive family history for dementia. No family histories of CJD were reported. Effects of rare (MAF ≤ 1%) missense variants on protein structure and function were predicted using SIFT (http://sift.jcvi.org/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) and SNP&GO (https://snps-and-go.biocomp.unibo.it/snps-and-go/) (Supplementary Table 2).
Clinical manifestations included mild to moderate dementia with predominant executive and speech/language impairment (aphasia, dysarthria) with less impaired memory and visuospatial function. Behavioral and psychiatric manifestations (depression, apathy, irritability, anxiety, aggression, visual hallucinations, and insomnia) were described in most patients. Motor symptoms typically included Parkinsonism, spasticity, gait disturbance, and/or immobility (Supplementary Table 3).
Mutation Screening
Gene panel screening (15 genes) for variants and mutations associated with AD, FTD-ALS, and PD, revealed 4 rare, protein-modifying variants (Supplementary Table 2). Effects on protein structure and function were predicted using SIFT (http://sift.jcvi.org/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) and SNP&GO (https://snps-and-go.biocomp.unibo.it/snps-and-go/). In PSEN1, the p.E318G polymorphism in PSEN1 was found in three patients (10.3%), of which one had pure sCJD, and two were sCJD + AD. No relevant variants were observed in the other AD genes APP and PSEN2. In PRNP, the p.P84_Q91Q insertion was detected in one patient with β-amyloidopathy. This variant is considered non-pathogenic (21). Furthermore, one benign missense variant was present in GRN and one in SOD1. We did not find any potential disease-causing mutations in the PD genes SNCA, LRRK2, and PRKN; silent mutations were found (Supplementary Table 2).
Genetic Predisposing Factors - ε4 Allele of Apolipoprotein E (APOE)
APOE polymorphic variants were tested at codons 112 and 158. Of the 30 cases in our study, 10% (n = 3) carried the ε4 allele of the APOE gene (Figure 1). All three cases were AD level A2 (AAO 62, 75, 83 years). The distribution of the polymorphic codon 129 of PRNP and APO genotypes in sCJD patients are shown in Supplementary Table 4. We found no association between APOE ε4 allele status and sCJD; however, the APOE ε4 was seen in two PRNP M129M homozygotes (n = 2).
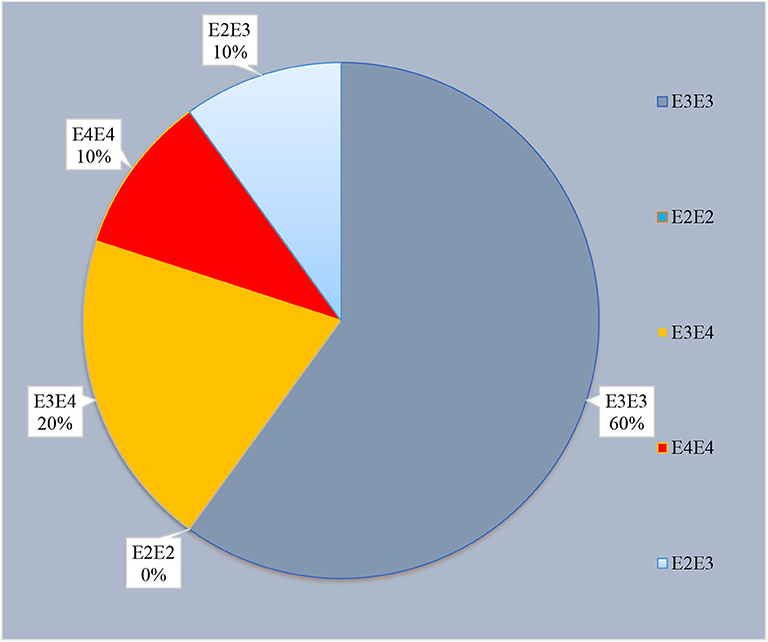
Figure 1. Distribution of APOE polymorphism carriers in our cohort (n = 30).
MAPT Haplotype Association With Sporadic CJD
We analyzed MAPT haplotypes in both isolated sCJD cases and in cases with sCJD and tauopathy. We identified only one case with the H2/H2 haplotype, and they were in the comorbid subgroup (Supplementary Table 5). As such, our study shows no evidence of an association between MAPT gene variations and sCJD, which could have contributed to the tau deposits in the CNS.
Discussion
In our study of 30 cases of sCJD in the Czech Republic (the annual rate of definite CJD is about 20 cases/yr.), we analyzed the most important genes related to neurodegeneration. The cognitive profile in our patients was characterized by a heterogeneous manifestation, with predominant involvement of executive and speech/language functions with a significant proportion also having behavioral manifestations (including visual hallucinations).
We did not detect any pathogenic mutations in the PRNP gene. Our study also tried to determine if there were any predisposing genetic factors that could account for the occurrence of comorbid Aβ and tau protein deposits in CJD brains. Previous studies have provided evidence that comorbid proteinopathy is not unusual in CJD brains, although the exact mechanism by which β-amyloid and tau deposits spread within brain tissue remains unclear (22). Since several studies have documented a possible spread of β-amyloid in brain tissue (23, 24), we performed a mutation analysis of APP (Aβ encoding exons) as well as the coding region of PSEN1 and PSEN2. However, we did not find any mutations in the genes that would explain the increased Aβ42 production.
There is only sparse evidence supporting the potential interaction between APOE and PRNP in sCJD. Recent studies that analyzed the influence of APOE on CJD have yielded discordant results. Three of our cases had the APOE ε4 genotype (AAO > 70 years on average), i.e., β-amyloidopathy level A2 and Methionine/Methionine homozygosity at codon 129 of the PRNP gene (M129M). Recent studies have suggested variants of PRNP129 (methionine/methionine, methionine/valine, valine/valine) as possible modifiers of AD disease (25). However, because of the small sample size of our study, this interpretation should be approached cautiously. Further studies should be carried out to assess the effects of PRNP129 in the AD phenotype. We found no influence of the APOE genotype relative to the age at onset, nor any significant differences in the distribution of the APOE ε4 and ε2 genotypes relative to those with isolated sCJD and those with sCJD and AD. Our results are consistent with other studies showing that APOE is not a risk factor for CJD (26–29).
The pathology of tau in sCJD brains is not unique, and in our cohort, this additional pathology was seen in 6 of the 30 definite CJD patients (30%) (6). Tau is encoded by the MAPT gene, and there are two common MAPT extended haplotypes, i.e., H1 and H2 (29). Only one study has investigated the role of MAPT in the etiology of sCJD (30). There is somewhat more evidence regarding the role of MAPT haplotypes (H1 and H2) in neurodegenerative diseases. H1 has been linked to FTLD and AD (31), whereas H2 is associated with a lower risk for developing late-onset AD (32). Our study shows no evidence for any association between MAPT gene haplotypes and sCJD.
The coexistence of CJD and PD is exceedingly rare. Several reported case studies show that α-synuclein amyloid deposits in CJD patients are associated with a slower disease course. The precise molecular mechanism explaining how misfolded α-synuclein accumulates and spreads in synucleinopathies is still unknown (33). Sequence or copy number variants in at least six genes (SNCA, LRRK2, PRKN, PINK1, DJ-1, and ATP13A2) have been identified to cause monogenic forms of PD (34). To date, no mutations responsible for PD have been reported in patients with CJD. Due to the low incidence of patients with proven CJD and PD, it is not clear whether there are gene interactions between CJD and PD. Our study, however, was not focused on the issue of sCJD and synucleinopathy, due to the extremely low incidence of both pathologies in comorbidity. This issue is, nevertheless, a promising direction for future research, and as such, it could help us better understand the genetic background as well as perhaps offer novel therapeutic options.
In conclusion, we failed to find any association between the investigated genes and the accumulation of specific protein aggregates in the examined brain tissue. These findings suggest that comorbid neurodegenerative disorders in sCJD behave as if they were independent processes taking place within the same brain; additionally, the underlying pathophysiology of comorbid protein deposits in CJD appears to have a complex multifactorial origin.
It would, however, be promising in the future to examine other risk genes for AD, FTD, and PD, and their potential association with CJD (Supplementary Table 6) (35). The search for genetic evidence of clinical, pathological, and possible molecular overlap between neurodegenerative diseases certainly needs to continue and would be best done with a larger multicenter cohort.
Data Availability Statement
The data analyzed in this study is subject to the following licenses/restrictions: This manuscript utilizes proprietary data. Requests to access these datasets should be directed to Julie van der Zee, anVsaWUudmFuZGVyemVlQHVhbnR3ZXJwZW4udmliLmJl, Neurodegenerative Brain Diseases Group, VIB Center for Molecular Neurology, VIB, Antwerp, Belgium. Primers for the targeted assay were designed using proprietary mPCR software from Agilent (previously Multiplicom) (20).
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author Contributions
JZ created the concept and design of the project. CVB provided laboratory facilities for the practical implementation of the entire project, provided complete instrumentation of the VIB Center for Molecular Neurology. JZ and CVB critically revised the manuscript. LD provided recommendations and specific approaches to the samples analysis (analysis tools) as an expert technician in the laboratory. RR diagnosed in detail the neurological cases mentioned in the article. RM verified the neuropathological diagnosis of neurodegenerations and approved the final version to be published. All authors participated in the manuscript.
Funding
This study was supported by the Ministry of Health, Czech Republic (Conceptual development of research organization VFN64165, General University Hospital in Prague and Thomayer Hospital in Prague, TN64190), by the Grants Agency of the Ministry of Health (NV19-04-00090 and NV18-04-00179), by Charles University (Project Progress Q27/LF1, Q35/LF3 and Erasmus + Third Faculty of Medicine) and by the AVASTipendium (Alzheimer Foundation Czech Republic). Molecular genetic analyses were performed in the Neurodegenerative Brain Diseases Group of the VIB Center for Molecular Neurology, Antwerp, Belgium, and funded by a Flanders Methusalem Excellence Program of the Flemish Government (CVB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Thomas Secrest for the revision of the English version of this article.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.596108/full#supplementary-material
Footnotes
1. ^www.cjdsupport.org/, https://www.cjdsupport.org.au/site/wp-content/uploads/2017/04/PRNP-Guidelines-160417.pdf
References
1. Ross CA, Poirier MA. Protein aggregation sand neurodegenerative disease. Nat Med. (2004) 10:S10–7. doi: 10.1038/nm1066
2. Santiago JA, Bottero V, Potashkin JA. Dissecting the molecular mechanisms of neurodegenerative diseases through network biology. Front Aging Neurosci. (2017) 9:166. doi: 10.3389/fnagi.2017.00166
3. Braak H. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/BF00308809
4. Arneson D, Zhang Y, Yang X, Narayanan M. Shared mechanisms among neurodegenerative diseases: from genetic factors to gene networks. J Genet. (2018) 97:795–806. doi: 10.1007/s12041-018-0963-3
5. Sikorska B, Knight R, Ironside JW, Liberski PP. Creutzfeldt-Jakob disease. Adv Exp Med Biol. (2012) 724:76–90. doi: 10.1007/978-1-4614-0653-2_6
6. Kovacs GG, Rahimi J, Ströbel T, Lutz MI, Regelsberger G, Streichenberger N, et al. Tau pathology in Creutzfeldt-Jakob disease revisited. Brain Pathol. (2017) 27:332–44. doi: 10.1111/bpa.12411
7. Reiniger L, Lukic A, Linehan J, Rudge P, Collinge J, Mead S, et al. Tau, prions and Aβ: the triad of neurodegeneration. Acta Neuropathol. (2011) 121:5–20. doi: 10.1007/s00401-010-0691-0
8. Kovacs GG, Seguin J, Quadrio I, Höftberge R, Kapás I, Streichenberger N, et al. Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol. (2011) 121:39–57. doi: 10.1007/s00401-010-0713-y
9. Tousseyn T, Bajsarowicz K, Sanchez H, Gheyara A, Oehler A, Geschwind M, et al. Prion disease induces alzheimer disease-like neuropathologic changes. J Neuropathol Exp Neurol. (2015) 74:873–88. doi: 10.1097/NEN.0000000000000228
10. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. (2014) 128:755–66. doi: 10.1007/s00401-014-1349-0
11. Bell WR, An Y, Kageyama Y, English C, Rudow GL, Pletnikova O, et al. Neuropathologic, genetic, and longitudinal cognitive profiles in primary age-related tauopathy (PART) and Alzheimer's disease. Alzheimers Dement. (2019) 15:8–16. doi: 10.1016/j.jalz.2018.07.215
12. Rossi M, Kai H, Baiardi S, Bartoletti AS, Carla B, Zenesini C, et al. The characterization of AD/PART co-pathology in CJD suggests independent pathogenic mechanisms and no cross-seeding between misfolded Aβ and prion proteins. Acta Neuropathol Commun. (2019) 7:53. doi: 10.1186/s40478-019-0706-6
13. Kim D, Kim HS, Choi SM, Kim BC, Lee MC, Lee KH, et al. Primary age-related tauopathy: an elderly brain pathology frequently encountered during autopsy. J Pathol Transl Med. (2019) 53:159–63. doi: 10.4132/jptm.2019.03.14
14. Josephs KA, Murray ME, Tosakulwong N, Whitwel JL, Knopman DS, Machulda MM, et al. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol. (2017) 133:705–15. doi: 10.1007/s00401-017-1681-2
15. Jicha GA, Parisi JE, Dickson DW, Cha RH, Johnson KA, Smith GE, et al. Age and apoE associations with complex pathologic features in Alzheimer's disease. J Neurol Sci. (2008) 273:34–9. doi: 10.1016/j.jns.2008.06.008
16. Alzheimer's Association 2016. Alzheimer's disease facts and figures. Alzheimer Dement. (2016) 12:459–509. doi: 10.1016/j.jalz.2016.03.001
17. Mackenzie G, Will R. Creutzfeldt-Jakob disease: recent developments. F1000Res. (2017) 6:2053. doi: 10.12688/f1000research.12681.1
18. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. (2012) 132:2659–68. doi: 10.1093/brain/awp191
19. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on aging-Alzheimer's association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. (2012) 8:1–13. doi: 10.1016/j.jalz.2011.10.007
20. Goossens D, Moens LN, Nelis E, Lenaerts AS, Glassee W, Kalbe W, et al. Simultaneous mutation and copy number variation (CNV) detection by multiplex PCR-based GS-FLX sequencing. Hum Mutat. (2009) 30:472–6. doi: 10.1002/humu.20873
21. Bagyinszky E, Giau VV, Youn YC, An SSA, Kim S. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr Dis Treat. (2018) 14:2067–85. doi: 10.2147/NDT.S165445
22. Morales R, Callegari K, Soto C. Prion-like features of misfolded Aβ and tau aggregates. Virus Res. (2015) 207:106–12. doi: 10.1016/j.virusres.2014.12.031
23. Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. (2013) 501:45–51. doi: 10.1038/nature12481
24. Eisele YS, Duyckaerts C. Propagation of Aß pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. (2016) 131:5–25. doi: 10.1007/s00401-015-1516-y
25. Calero O, Bullido MJ, Clarimón J, Frank-García A, Martínez-Martín P, Lleó A, et al. Genetic cross-interaction between APOE and PRNP in sporadic Alzheimer's and Creutzfeldt-Jakob diseases. PLoS ONE. (2011) 6:e22090. doi: 10.1371/journal.pone.0022090
26. Salvatore M, Seeber AC, Nacmias B, Petraroli P, D'Alessandro M, Sorbi S, et al. Apolipoprotein E in sporadic and familial creutzfeldt-Jakob disease. Neurosci Lett. (1995) 199:95–98. doi: 10.1016/0304-3940(95)12030-8
27. Amouyel P, Vidal O, Launay JM, Laplanche JL. The apolipoprotein E alleles as major susceptibility factors for Creutzfeldt-Jakob disease. The French research group on epidemiology of human spongiform encephalopathies. Lancet. (1994) 344:1315–18. doi: 10.1016/S0140-6736(94)90691-2
28. Wei Y, Tang Y, He W, Qu Z, Zeng J, Qin Ch. APOE gene polymorphisms and susceptibility to Creutzfeldt-Jakob disease. J Clin Neurosci. (2004) 21:390t4. doi: 10.1016/j.jocn.2013.07.019
29. Verpillat P, Camuzat A, Hannequin D, Thomas AC, Puel M, Belliard S, et al. Association between the extended tau haplotype and frontotemporal dementia. Arch Neurol. (2002) 59:935–9. doi: 10.1001/archneur.59.6.935
30. Sánchez-Valle R, Pastor P, Yagüe J, Ribalta T, Graus F, Tolosa E, et al. Analysis of the exon 1 polymorphism in the Tau gene in transmissible spongiform encephalopathies. J Neurol. (2002) 249:938–9. doi: 10.1007/s00415-002-0717-1
31. Myers AJ, Kaleem M, Marlowe L, Pittman AM, Lees AJ, Fung HC, et al. The H1c haplotype at the MAPT locus is associated with Alzheimer's disease. Hum Mol Genet. (2005) 14:2399–2404. doi: 10.1093/hmg/ddi241
32. Allen M, Kachadoorian M, Quicksall Z, Zou F, Seng Chai H, Younkin C, et al. Association of MAPT haplotypes with Alzheimer's disease risk and MAPT brain gene expression levels. Alzheimers Res Ther. (2014) 6:39. doi: 10.1186/alzrt268
33. Aulić S, Masperone L, Narkiewicz J, Isopi E, Bistaffa E, Ambrosetti E, et al. α-synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci Rep. (2017) 7:10050. doi: 10.1038/s41598-017-10236-x
34. Crosiers D, Theuns J, Cras P, Van Broeckhoven C. Parkinson disease: insights in clinical, genetic and pathological features of monogenic disease subtypes. J Chem Neuroanat. (2011) 42:131–41. doi: 10.1016/j.jchemneu.2011.07.003
Keywords: Creutzfeldt-Jakob disease, Alzheimer's disease, β amyloid, tau protein, neurodegenerative disease
Citation: Parobkova E, van der Zee J, Dillen L, Van Broeckhoven C, Rusina R and Matej R (2020) Sporadic Creutzfeldt-Jakob Disease and Other Proteinopathies in Comorbidity. Front. Neurol. 11:596108. doi: 10.3389/fneur.2020.596108
Received: 18 August 2020; Accepted: 23 October 2020;
Published: 30 November 2020.
Edited by:
Claudia Kimie Suemoto, University of São Paulo, BrazilReviewed by:
Jerusa Smid, University of São Paulo, BrazilEva Bagyinszky, Gachon University, South Korea
Copyright © 2020 Parobkova, van der Zee, Dillen, Van Broeckhoven, Rusina and Matej. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Radoslav Matej, cmFkb3NsYXYubWF0ZWpAZnRuLmN6