 Carolina Alquezar
Carolina Alquezar Shruti Arya
Shruti Arya Aimee W. Kao
Aimee W. Kao- Department of Neurology, Memory and Aging Center, University of California, San Francisco, San Francisco, CA, United States
Post-translational modifications (PTMs) on tau have long been recognized as affecting protein function and contributing to neurodegeneration. The explosion of information on potential and observed PTMs on tau provides an opportunity to better understand these modifications in the context of tau homeostasis, which becomes perturbed with aging and disease. Prevailing views regard tau as a protein that undergoes abnormal phosphorylation prior to its accumulation into the toxic aggregates implicated in Alzheimer's disease (AD) and other tauopathies. However, the phosphorylation of tau may, in fact, represent part of the normal but interrupted function and catabolism of the protein. In addition to phosphorylation, tau undergoes another forms of post-translational modification including (but not limited to), acetylation, ubiquitination, glycation, glycosylation, SUMOylation, methylation, oxidation, and nitration. A holistic appreciation of how these PTMs regulate tau during health and are potentially hijacked in disease remains elusive. Recent studies have reinforced the idea that PTMs play a critical role in tau localization, protein-protein interactions, maintenance of levels, and modifying aggregate structure. These studies also provide tantalizing clues into the possibility that neurons actively choose how tau is post-translationally modified, in potentially competitive and combinatorial ways, to achieve broad, cellular programs commensurate with the distinctive environmental conditions found during development, aging, stress, and disease. Here, we review tau PTMs and describe what is currently known about their functional impacts. In addition, we classify these PTMs from the perspectives of protein localization, electrostatics, and stability, which all contribute to normal tau function and homeostasis. Finally, we assess the potential impact of tau PTMs on tau solubility and aggregation. Tau occupies an undoubtedly important position in the biology of neurodegenerative diseases. This review aims to provide an integrated perspective of how post-translational modifications actively, purposefully, and dynamically remodel tau function, clearance, and aggregation. In doing so, we hope to enable a more comprehensive understanding of tau PTMs that will positively impact future studies.
Introduction
Post-translational modifications (PTMs) refer to the modifications that occur in a protein either shortly after its translation by ribosomes or after its folding and localization are complete (1). These modifications are usually catalyzed by enzymes and involve the addition of chemical groups, sugars, or proteins to specific residues of the targeted protein. Nearly all cellular processes can be regulated via PTMs. Therefore, PTMs provide a means for the natural increase in the proteome complexity and allow one gene to encode a variety of distinct protein molecules with distinct functional roles, known as proteoforms (2). PTMs alter the charge and hydrophobicity (electrostatics) of a protein, which in turn induces structural changes that influence protein function, protein-protein interactions, and protein aggregation (3, 4). In this way, PTMs also can affect the clearance of proteins by regulating the functionality of degrons, peptide sequences that target a protein for degradation (5). Essentially all proteins in eukaryotes, including the “intrinsically disordered” or “natively unfolded” proteins that tend to aggregate in neurodegenerative diseases, are susceptible to PTMs (3, 6, 7). Unlike folded proteins, natively unfolded proteins do not have a well-defined three-dimensional structure and exist as an ensemble of dynamically fluctuating conformations. The lack of tertiary structure makes natively unfolded proteins more vulnerable to PTMs (8).
Tau is a classic example of a natively unfolded protein (7, 9) that can be modified by a myriad of PTMs. Tau is a microtubule-binding protein found in neurons and glial cells, and is primarily involved in the stabilization of the cytoskeleton (10). Based on its interaction with microtubules and on the basis of its amino acid composition, the primary structure of tau can be divided into an N-terminal projection domain, a proline-rich region, a repeat region, and a C-terminal domain (11) (Figure 1). Tau has six different isoforms in human brain that differ from each other in the number of N-terminal inserts and either contain or lack one repeat (R2) of the four repeats (R1-R4) present in the microtubule-binding domain region. Because of its ability to bind to microtubules, the main function of tau is thought to be the modulation of microtubule dynamics, which consequently affects neurite outgrowth, axonal transport, and synapsis (12–14). Recent studies have shown that tau also interacts with several cell membranes, including the plasma membrane, the endoplasmic reticulum, and Golgi (15–17). Tau may also regulate intracellular signaling cascade via interactions with proteins such as 14.3.3, Pin-1, and Fyn (18), suggesting a role for tau in cell signaling (18). Although tau is primarily an axonal protein, it can be found within neuronal nuclei where it binds to either DNA or RNA and regulates gene expression (19–21). Under certain pathological circumstances, tau loses its function as a microtubule-binding protein and accumulates in the cytosol of affected cells, forming first “pre-tangles” and then insoluble cytosolic inclusions or aggregates composed of fibrillar forms of tau known as neurofibrillary tangles (NFTs).
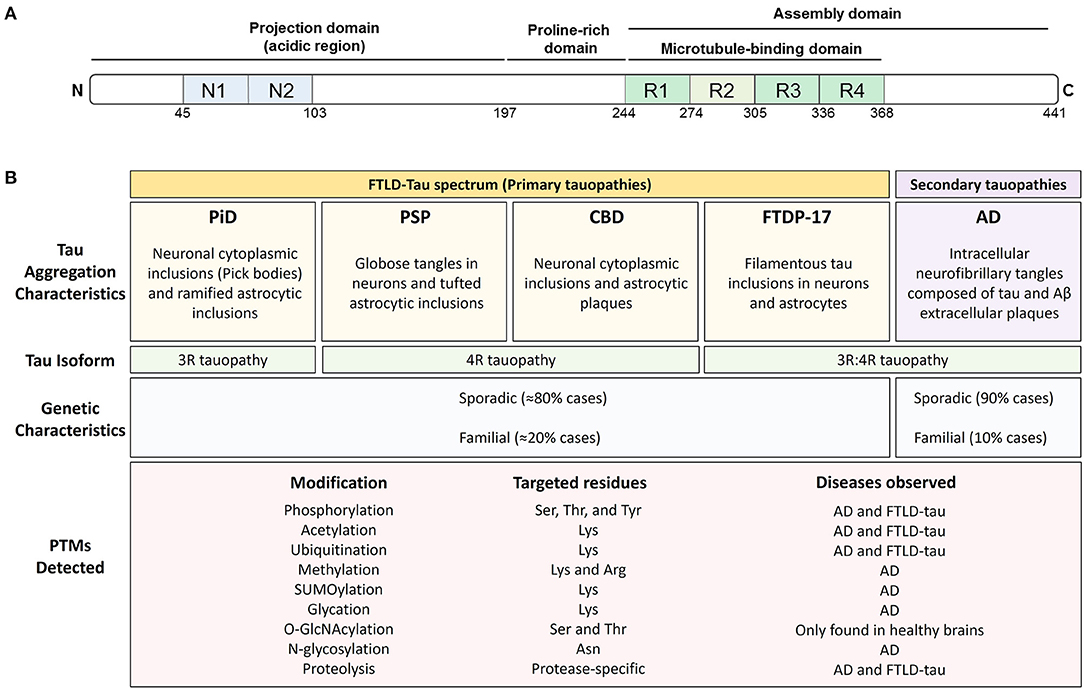
Figure 1. Post-translational modifications are found throughout tau and associate with all tauopathies. (A) Schematic representation of the structural features of 2N4R tau protein. Tau exists in 6 isoforms generated by the alternative splicing of exons 2, 3, and 10 of the MAPT gene. The isoforms include 0, 1, or 2 amino-terminal inserts (blue rectangles) and three (3R) or four (4R) microtubule-binding potential repeat domains (green rectangles). (B) Pathological and genetic classification of tauopathies as they relate to post-translational modifications (PTMs). Primary tauopathies fall under the umbrella pathological term frontotemporal lobar degeneration with tau inclusions (FTLD-Tau) and include Pick's disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and frontotemporal dementia with parkinsonism (FTDP-17). Alzheimer's disease, a secondary or mixed tauopathy, is characterized by the presence of extracellular inclusions containing Amyloid β (Aβ) protein. Tauopathies are also classified depending on tau isoform composing the intracellular inclusions. PiD is a 3R-predominat tauopathy, PSP, and CBD are 4R tauopathies and FTDP-17 and AD are 3R:4R tauopathies. The PTMs observed in tau, the targeted residues and the disease where they have been observed is also shown.
Diseases associated with the presence of tau inclusions in the brain are collectively referred to as tauopathies and include a diverse group of neurodegenerative diseases with distinct pathological characteristics (Figure 1). Tau-immunopositive intracellular aggregates are the pathognomonic hallmark of tauopathies (22). However, the forces driving tau, especially non-mutant, wild-type tau, into intracellular aggregates are incompletely understood. The cellular protein homeostasis (or proteostasis) network is fundamental to the proper functioning of tau and prevents the accumulation of tau aggregates. In principle, proteostasis encompasses the entire life cycle of a protein such as tau, including synthesis, proper folding, post-translational processing, and degradation. Thus, PTMs are a key contributor to tau proteostasis and thereby could regulate tau function, levels, and aggregation.
The same PTMs that regulate tau function also have the ability to induce alterations in its clearance, conformation, and aggregation potential (23). However, not all tau PTMs are “pathological”. Many PTMs have been identified in tau extracted from healthy brains, suggesting a normal role for PTMs in tau function (24). Among all tau PTMs, phosphorylation is the most studied, and traditionally, it was thought that increased phosphorylation was the trigger for tau intracellular aggregation (25). However, a growing body of evidence suggests that other PTMs also considerably regulate tau function and may even precede phosphorylation in the sequence of events leading to tau inclusions formation.
In this review, we first provide a synopsis of the many tau PTMs that impact its function and potentially contribute to dysfunction (section Effects of PTMs on Tau Function and Dysfunction). We then fold this information into an integrated discussion of how PTMs can regulate tau degradation (section Effect of PTMs on Tau Degradation) and aggregation (section Effect of PTMs on Tau Solubility and Aggregation). A rarely discussed but critically important subject, cross-talk, and competition between PTMs, is considered next (section Cross-Talk and Competition Between PTMs). Finally, we summarize strengths and limitations of current approaches to the study of PTMs (section Approaches and Limitations) and provide concluding remarks (section Concluding Remarks).
Effects of PTMs on Tau Function and Dysfunction
The main role of PTM addition to any protein, including tau, is to regulate and increase functional diversity usually by altering electrostatics and/or structure. Approximately 35 percent of the amino acid residues in tau are susceptible to modification peri- or post-translationally. These residues are serine (S), threonine (T), tyrosine (Y), lysine (K), arginine (R), asparagine (N), histidine (H), and cysteine (C). Like other proteins, tau undergoes PTMs that involve the addition of small chemical groups or peptides on different side chains of tau: phosphorylation on S, T, or Y; acetylation, ubiquitination, SUMOylation, and glycation on K; methylation on K or R; O-GlcNAcylation on S and T, N-glycosylation on nitrogens, nitration on Y and oxidation on carbons (Figure 2). Tau can also undergo protonation on histidine residues and proteolytic cleavage (or truncation), although these modifications are often not considered alongside traditional PTMs. The implications of all of these modifications will be discussed in this section.
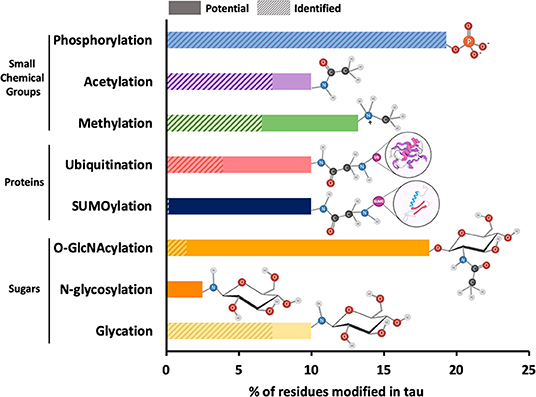
Figure 2. Relative frequency of post-translational modifications on tau. The 2N4R isoform of tau contains 441 amino acid residues, around 35 percent of which can potentially undergo PTMs. These residues include serine (forty-five), threonine (thirty-five), tyrosine (five), asparagine (three), arginine (fourteen), and lysine (forty-four). The most common PTMs in tau are shown on the y-axis of the bar plot. The x-axis of the plot shows the percentage of amino acid residues with solid bars showing the sites that have been identified within total potential sites shown in shaded bars. The functional groups associated with each PTM are also shown next to each bar. Since all potential phosphorylation sites have been identified, the shaded bar is completely masked by the solid bar. Similarly, in case of N-glycosylation, the specific N-glycosylation sites have not been reported and therefore, only potential sites represented by shaded bar have been shown.
The clustering of PTMs within short sequence motifs and functional domains suggests that tau function and subcellular localization could be regulated by a complex interplay between different PTMs (Figure 3). Since some modifications have been found only in the tau aggregates associated with disease (26, 27), it has been suggested that these modifications may be regulating tau dysfunction. Although a hypothetical sequence of events has been proposed to occur during the formation of tau inclusions (28, 29), what remains elusive is which PTMs actually drive tau self-association vs. those that are simply part of normal tau function and captured or trapped as “bystanders” in aggregates. The transformation from healthy (functional) to aggregated (or pathological) tau is most likely not a consequence of a single PTM, but rather a combination of the intrinsic structural alterations and extrinsic cellular conditions that may ultimately constitute pathogenicity. Below, we describe the PTMs that have been observed in tau and their potential impacts on tau function and dysfunction.
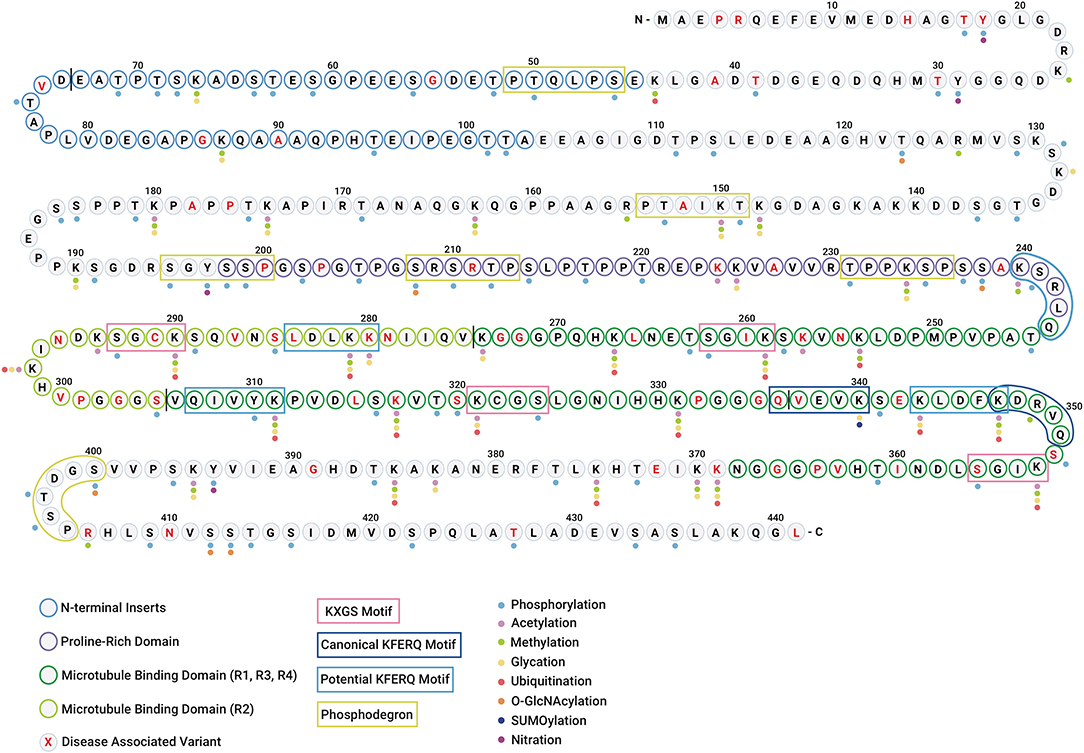
Figure 3. Tau undergoes diverse post-translational modifications that often compete for the same amino acid residues. Diagram showing the polypeptide chain of the longest tau isoform 2N4R tau along with individual amino acid residues highlighting different regions/domains. The N-terminal inserts are represented with blue residue borders, proline-rich domain is represented with purple residue borders, microtubule-binding domains are represented with green residue borders (R1, R3, R4 in dark green, and R2 in light green), and disease-associated amino acid variants are highlighted in red text. The vertical black line between residues is shown for distinction between two N-terminal inserts and four microtubule-binding repeats. The colored dots represent the PTMs identified in tau (WT and mutant) in vitro and in vivo. The color code for PTMs is as follows: phosphorylation (light blue), acetylation (pink), methylation (green), glycation (yellow), ubiquitination (red), O-GlcNAcylation (orange), SUMOylation (dark blue), and nitration (purple). N-glycosylation is omitted as no specific sites have been identified. The KXGS microtubule-binding motifs are shown in pink boxes. The canonical and potential KFERQ motifs are represented with dark blue and light blue boxes, respectively. The phosphodegron motifs are highlighted with green boxes.
Phosphorylation
Protein phosphorylation is one of the most common PTMs in the proteome (30). Phosphorylation involves the reversible addition of a phosphate (PO4) group to the polar group of serine, threonine or tyrosine amino acid residues (31) (Figure 2). Protein kinases and phosphatases, the enzymes responsible for phosphorylation and dephosphorylation, respectively (32), coordinate the phosphorylation status of proteins. Phosphorylation alters protein electrostatics by introducing a negatively charged, hydrophilic group, resulting in an overall more hydrophilic protein. As a consequence of this increased hydrophilicity, phosphorylation imparts conformational changes, and regulates some important protein functions including protein-protein interactions, signaling cascades, and protein degradation (5, 33). Phosphorylation is involved in essential cellular functions such as metabolism and intracellular signaling, and therefore, is fundamental to several normal cellular processes. However, abnormal phosphorylation has also been described as one of the primary causes for the alteration of a variety of structural, functional, and regulatory proteins under disease conditions.
The longest isoform of tau, known as 2N4R tau, has 85 potential phosphorylation sites (44 S, 35 T, and five Y) (34). Therefore, almost 20 percent of tau has the potential to become phosphorylated (Figures 2, 3). Of these sites, ~20 residues have been found to undergo phosphorylation in tau extracted from healthy brains (35, 36). In contrast, around 44 residues, some of which overlap with residues found in tau from healthy brains, have been identified as “abnormally” hyperphosphorylated in individuals with a tauopathy (25). Phosphorylation of serine and threonine residues is associated with the completion of degrons (or degradation-signaling motifs) and the subsequent clearance of tau (for a more extensive discussion about phosphodegrons, see section Effect of PTMs on Tau Degradation). If tau degradation is blocked, then these normally transient phospho-sites would persist, even though they are a part of the normal metabolism of tau. Thus, the phosphorylation observed in tau extracted from diseased brains could either occur abnormally as part of disease pathogenicity or be a found as a normal event to promote the normal, but interrupted, clearance of tau. Additionally, it is also possible that many other tau phosphorylation sites exist that have not been identified in post-mortem human brain material due to rapid dephosphorylation of tau (37).
As a dynamic and highly regulated process, tau phosphorylation requires a balanced interplay of kinases and phosphatases. Several serine/threonine kinases phosphorylate tau, including glycogen synthase kinase 3 (GSK3β), cyclin dependent kinase 5 (cdk5), casein kinase 1 (CK1), cyclic AMP-dependent protein kinase (PKA), p42/p44 MAPKs (ERKs 1/2), protein kinase C (PKC), calmodulin-dependent protein kinase II (CaMKII), the brain-specific kinases 1 and 2, the tau-tubulin kinases 1 and 2, and some microtubule affinity-regulating kinases (MARKs; also known as PAR1 kinases) (38). Furthermore, tau can be phosphorylated by tyrosine kinases, such as the SRC family members (LCK, SYK, and FYN) and the ABL family members (ARG and ABL1) (39). FYN, SYK, and ABL kinases phosphorylate tau at the Y18, Y197, and Y394 residues (40), which have been associated with tau aggregation and AD pathology (41).
Tau can be dephosphorylated by protein phosphatases (PP) 1, 2A, 2B, 2C, and 5 (25). Among these phosphatases, PP2A accounts for around 70 percent of all tau dephosphorylation in the human brain (42). Interestingly, PP2A also dephosphorylates and modulates the activity of components along the ERK1/2 MAPK cascade that leads to GSK3β activation (43), suggesting potential feedback cycles. As phosphorylation is one of the most studied PTMs in tau, the kinase pathways implicated in tau phosphorylation have been extensively reviewed elsewhere (44). The regulation of the phosphorylation status of tau is essential to maintain the biological function of the protein.
The change in tau electrostatics associated with phosphorylation may have a critical role in the regulation of tau function, localization, and interaction with other molecules. The phosphorylation status of tau is developmentally regulated, implying the significance of phosphorylation in the regulation of specific function of tau in each developmental stage (45, 46). Tau phosphorylation inside the microtubule-binding domain (e.g., S262/S214) and/or the proline-rich (e.g., T231) domain reduces the affinity of tau for negatively charged microtubules (47–50). Binding of tau stabilizes microtubules, therefore tau phosphorylation regulates neuronal functions such as axonogenesis and neurite outgrowth (47, 51–53). Additionally, it has been reported that the specific phosphorylation of tau by FYN kinases induces the relocation of tau from the axon to dendrites (54). However, the effects of tau relocation in neuronal function are not yet well-understood. Additionally, tau interacts with the kinesin-associated protein JIP1 (JUN N-terminal kinase-interacting protein 1) only when phosphorylated, thereby impairing the formation of the kinesin complex that mediates axonal transport (55).
Since phosphorylation regulates the biological functions of tau, inappropriate phosphorylation may be an important contributor to the pathogenesis of tauopathies. The abnormal phosphorylation in several residues inside the KXGS motif (25) and the proline-rich domain (47, 56–58) has been identified in NFTs of tauopathy patients brains. Aberrant tau phosphorylation at these motifs may pose detrimental consequences for neurons, such as the impairment of neurite outgrowth (59) and the deregulation of axonal transport (60, 61), which may lead to synaptic dysfunction and neurodegeneration (62–64). The phosphorylation at residues outside of the microtubules-binding and proline-rich domains does not interfere with the ability of tau to bind microtubules. Some of these residues such as S396, S404, and S422, have been found abnormally “hyperphosphorylated” in diseased brains (57, 65), and it has been suggested that phosphorylation at these residues may influence tau aggregation (66). While substantial published work suggests that tau phosphorylation may contribute to development of tauopathies, the exact mechanism behind how phosphorylation regulates tau function and aggregation in vivo is still unknown. To unravel the role of abnormal tau phosphorylation in tauopathies, it will be necessary to better understand the broader impacts of phosphorylation on normal tau function.
Acetylation
The acetylation of mammalian proteins involves the N-terminal addition of an acetyl group from acetyl coenzyme A (Ac-CoA) to the lysine residue of a polypeptide chain (67) (Figure 2). Acetylation was first described on histone proteins, therefore the associated enzymes are known as histone acetyl transferases (HATs) and histone deacetylases (HDACs), although they are less discriminating in their targets than their names would suggest (67). Acetylation is considered an essential modification that plays critical roles in the function of diverse proteins, including p53 and tubulin (68). The addition of an acetyl group neutralizes the positive charge of lysine residues, thereby eliciting its downstream molecular impacts on protein structure and protein-protein recognition. Among its functional roles, acetylation is perhaps best known to regulate gene expression via modification of histone structure and chromatin accessibility (69). However, the acetylation of non-histone proteins regulates a range of cellular processes such as DNA damage repair, cell division, signal transduction, protein folding, autophagy, and metabolism (70). Incorporation of an acetyl group onto lysine residues prevents other modifications and thus, acetylation also can block the processes regulated by ubiquitination, SUMOylation, methylation, and glycation.
Acetylation has recently arisen to challenge the predominance of phosphorylation as the key PTM on tau. The longest isoform of tau protein (2N4R) contains 44 lysine residues and thus, 10 percent of the protein has the potential to be modified by acetylation (Figure 3). Among all HATs implicated in protein acetylation, p300 and its close homolog CREB-binding protein (CBP) appear competent to acetylate tau in vitro (71, 72). Moreover, the histone deacetylase 6 (HDAC6) (36, 73) and the NAD+-dependent sirtuin 1 deacetylase (SIRT1) (72) are the major tau deacetylases. Interestingly, these two HDACs have seemingly opposing effects on tau: while tau deacetylation by SIRT1 has a protective role against tau accumulation, the deacetylation of tau by HDAC6 contributes to the increase of tau phosphorylation and aggregation in vitro (74, 75). Tau has also been shown to undergo acetyl-coenzyme A–induced auto-acetylation in vitro (76). However, the role of tau as acetyltransferase remains somewhat controversial, given that other groups have not consistently identified tau acetylation in absence of HAT enzymes (26, 76, 77).
Tau acetylation was first recognized in the context of neurodegeneration in mouse models of neurodegenerative disease (72, 78), and subsequently in brain lysates from AD patients at early/moderate Braak stages (79). The dysregulation of the HATs and HDACs involved in tau acetylation was also found in diseased brains (79, 80). However, the underlying abnormalities of HAT and HDAC activity may be both context and disease specific. In this regard, while increased p300/CBP HAT activity was observed in the brains of FTLD-tau patients (80), p300/CBP levels, and activity were found to be lower in the frontal cortex and hippocampus of AD patients (81) and in a mouse model of AD (82). Although these reports failed to show the effect of p300/CBP dysregulation in tau acetylation, they suggest that the role of p300/CBP in tau acetylation may vary between different tauopathies such as FTLD-Tau and AD. Furthermore, the levels and activity of HDACs have also been measured in patients' brains. It has been observed that the expression of SIRT1 is reduced in AD patients (83, 84) while the levels of HDAC6 are increased (73), confirming that these enzymes have opposite effects on tau function and dysfunction. Altogether, this evidence demonstrates that tau acetylation may have a key role in tauopathies.
As with phosphorylation, to fully understand the role of acetylation in tau dysfunction, one must necessarily understand its role in the normal function of tau. While the neutralization of positively charged lysine residues by acetylation appears crucial for regulation of tau, the downstream functional impacts of this regulation seem to be residue specific. For example, acetylation at lysine residue K280 weakens the binding of tau to negatively charged microtubules, potentially destabilizing microtubule networks (26, 27, 85). This contrasts with acetylation at K174, which induces tau aggregation without affecting tau-microtubule binding (79). Finally, HDAC6-regulated tau acetylation at KXGS motif-associated K259, K290, K321, or K353 residues suppresses both tau phosphorylation and aggregation (36). Unlike acetylation of lysine residues that have been identified only in tauopathy brains (26, 79, 86, 87), the acetylation at these four residues occurs in normal tau but is reduced in tauopathy brains (36). Since acetylation has emerged as a key PTM in the regulation of tau function and dysfunction, mapping all the acetylation sites on tau in different biological contexts will help to delineate the ultimate roles and consequences of each modification.
Acetylation on tau is particularly interesting because many lysine residues that are targets for acetylation are also targets for other PTMs, including ubiquitination, SUMOylation, methylation, and glycation (78, 88, 89) (Figure 3). Therefore, a potential rivalry between PTMs could exist, where the addition of one chemical group to a given residue blocks the addition of another, suggesting multilayered avenues for the regulation of the biology of the protein (88). While the cross-talk between PTMs will be extensively discussed in section Cross-Talk and Competition Between PTMs, below we review the role of other lysine-associated PTMs in tau function.
Ubiquitination
Covalent modification of proteins with ubiquitin (Ub) (ubiquitination or ubiquitylation) represents one of the most common PTMs in mammalian cells (Figure 2). The small, 76 residues, protein ubiquitin is nearly ubiquitously found in eukaryotes (ergo its name) and is used to modify activity, localization, or stability of other proteins (90, 91). Ubiquitination involves a multi-step process carried out sequentially by activating (E1), conjugating (E2), and ligating (E3) enzymes and has been reviewed elsewhere (92). Similar to acetylation, ubiquitination occurs on lysine residues. In case of ubiquitination, the C-terminal carboxyl group of a lysine in the ubiquitin sequence itself is covalently attached to the ε-amino group on a lysine of the “target” protein. Substrates can be modified with a single ubiquitin (monoubiquitination) or with polymeric Ub chains of variable length (polyubiquitination). The ubiquitin protein contains seven lysine residues. Therefore, the nomenclature around ubiquitination is based upon which of these seven lysine residues serves as the initial link to the target protein (i.e., ubiquitinated chains are called K6, K11, K27, K29, K33, K48, or K63) (93). The type of ubiquitin chain determines the biological effects of these modifications. Chains linked through K6, K27, and K33 are often involved in cell proliferation, DNA damage repair, and innate immunity (91). Other linkages are directly related with protein degradation. In this regard, K48 chains generally target substrates for degradation by the 26S proteasome (94), whereas K63-polyubiquitinated proteins are directed to the degradation by the autophagy-lysosomes pathway (93–96).
As a protein rich in lysine residues, tau has a high susceptibility toward ubiquitination. Out of a total of 44 lysine residues, 17 residues of the 2N4R tau isoform have been found ubiquitinated with most of them located in the tau microtubule-binding domain (78, 97). The main role of tau ubiquitination appears to be the regulation of tau clearance by the proteasomal or lysosome-autophagy systems (98). E3 ligases confer ubiquitin residues to target proteins (99). Among ~600 E3 ligases encoded by the human genome, only three appear competent to ubiquitinate tau: the C-terminus of the Hsc70-interacting protein (CHIP), the TNF receptor-associated factor 6 (TRAF6), and axotrophin/MARCH7 (100–102). Each of these E3 ligases ubiquitinates tau through different linkages and at different residues, suggesting that each ligase modulates tau degradation by different mechanisms. CHIP ubiquitinates tau through K48 or K63 linkages and thus, regulates tau degradation via both proteasomal and autophagy systems (102–104). On the other hand, the E3 ligase TRAF6 ubiquitinates tau via K63 linkages (100), suggesting that ubiquitination mediated by this enzyme may regulate the degradation of tau in the autophagy-lysosomes pathway only (100, 105–107). Additionally, in vitro experiments have shown that tau can be monoubiquitinated by axotrophin/MARCH7 (101). However, the effect of monoubiqutination by axotrophin/MARCH7 on tau function and degradation is not yet well-understood.
While E3 ligases attach ubiquitins, deubiquitinases (dUbs) remove them. The cysteine protease Otub1 is the only dUb that has been shown to target tau. Outb1 removes the K48 polyubiquitin chains from endogenous tau, preventing tau degradation in primary neurons derived from a transgenic mouse model (108) and thus implying an important role in the regulation of tau ubiquitination.
Ubiquitin has been found in aggregated tau extracted from the brains of tauopathy patients. However, whether ubiquitin is causal, contributory, or simply a bystander in aggregation remains to be seen. Tau isolated from human AD brains has been shown to be monoubiquitinated at K254, K257, K311, and K317 (109) and polyubiquitinated at K254, K311, and K353 residues (110). Tau was also found to be ubiquitinated at K290 in a mouse model of AD (78). Interestingly, insoluble tau from AD brains is modified predominantly by K48 linkage (110), whereas soluble tau can also be ubiquitinated via K63 polyubiquitin conjugation (102), suggesting that soluble and aggregated tau are degraded by different pathways. While ubiquitin is a component of tau aggregates found in the brains AD patients (109, 111–113), tau “pre-tangles” did not exhibit positive ubiquitin immunostaining (114–116). Moreover, it has been shown that tau phosphorylation precedes ubiquitination in the NFTs of AD patients and that the formation of paired helical filaments precedes tau ubiquitination in vitro (109, 114), suggesting that ubiquitin may be linked to tau after the formation of the fibrillar inclusions. This finding potentially supports ubiquitination as a compensatory response to tau accumulation. In contrast, other groups have reported that both mono- and polyubiquitination contribute to the formation of insoluble protein inclusions present in neurodegenerative diseases (96, 117) and that the induction of tau ubiquitination in cell cultures increases aggregation (102). The somewhat contradictory nature of the current literature around ubiquitination suggests that whether this modification is causal, consequential, bystander, or context specific in disease remains to be sorted.
SUMOylation
In SUMOylation, the small ubiquitin-like modifier protein SUMO is transferred to the terminal amino group of lysine side chains of the target protein by an ATP-dependent enzymatic cascade. Analogous to ubiquitination, this SUMOylation cascade involves an E1 activating enzyme, an E2 conjugating enzyme, and an E3-type ligase (Figure 2) (118, 119). SUMO groups can be removed by specific proteases known as SUMO-specific proteases/isopeptidases (SENPs). Three main SUMO isoforms are expressed in cells: SUMO1, SUMO2, and SUMO3, of which SUMO2 and SUMO3 are more similar to each other and are different from SUMO1 (120).
SUMO is predominantly found in the nucleus and thus, SUMOylation plays a crucial role in many nuclear processes such as gene expression, genome stability (121), DNA damage response (122), protein trafficking (123), and cell cycle control (124). SUMOylation has emerged as an essential regulator of neuronal function with a growing evidence suggesting that SUMOylation of proteins inside and outside the nucleus plays an important role in neurodegenerative diseases (125). Alterations in protein SUMOylation are observed in a wide range of neurological and neurodegenerative diseases including tauopathies (126–128), and several extranuclear disease-associated proteins including tau have been shown to be directly SUMOylated (125).
In vitro studies have demonstrated that tau can be SUMOylated. Specifically, it has been found that tau is monoSUMOylated in vitro at the K340 residue, which is located within the microtubule-binding domain (128, 129). Tau becomes available for SUMOylation only after it is released from microtubules (129), suggesting that SUMOylation may be a secondary PTM that occurs after phosphorylation and/or acetylation. However, little is known about how SUMOylation interferes with the formation of tau inclusions in tauopathies. Tau SUMOylation at K340 inhibits tau ubiquitination and the subsequent proteasome-dependent degradation, suggesting that SUMOylation may block tau degradation and therefore induces tau accumulation and aggregation (128). In contrast, previous evidence indicates that the inhibition of the proteasome pathway stimulates tau ubiquitination and eliminates tau SUMOylation (129), suggesting that SUMOylated tau can be degraded by other pathways such as autophagy. In this context, a more recent study has demonstrated that SUMO1 labels lysosomes in oligodendrocytes from PSP patients containing tau inclusions (127), an indicator that SUMOylation may be regulating autophagy-lysosomes pathway in tauopathies. Additionally, the activation of autophagy-lysosomes pathway appears to reduce tau SUMOylation, tau inclusions and cortical atrophy associated with the rTg4510 mouse model of tauopathy (130). Despite the role of SUMOylation in tau function and dysfunction not being completely understood, substantive evidence support SUMOylation competing with ubiquitin for lysine residues in tau, with consequences on tau clearance in the proteasome and/or autophagy-lysosomes systems.
Methylation
Methylation is a biochemical process that involves the transfer a methyl group (CH3) to DNA or protein targets (Figure 2). DNA methylation was discovered contemporaneously with the discovery of DNA as genetic material (131, 132). On DNA, methylation is regarded as an epigenetic means of regulating gene expression through blocking transcription factor binding or recruiting transcriptional repressors (133). Protein methylation was first described in histones. This was followed by decades of limited interest until the physiological role for protein methylation was documented in the late 1990s and non-histone proteins were also found to be methylated (134). Currently, methylation is fourth on the list of PTMs in terms of overall abundance in the proteome, with more than 18,000 methylation sites described in ~7,400 proteins (135). The N-methylation of histone and non-histone proteins occurs primarily on lysine and arginine residues. In the process of DNA or protein methylation, methyl groups from an S-adenosyl methionine (SAM) donor are conferred by methyltransferases and removed by demethylases (136). Lysine residues can be methylated up to three times, resulting in a mono-, di-, or trimethyl-lysine, which each give rise to distinctive biological consequences. Indeed, although the methyl group is one of the smallest post-translational modifications, each methylation event removes a proton from the ε-amino group and thereby decreases the hydrogen-bonding potential of lysine. Therefore, methylation also increases the hydrophobicity and bulkiness of lysine side-chains. Based on the electrostatic properties of methylation, this PTM has been shown to regulate different aspects of proteins function such as protein–protein and protein–nucleic acid interactions, protein stability, subcellular localization, and enzyme activity, which in turn affect essential cellular processes including transcription, protein synthesis, signal transduction, and metabolism (137, 138).
Tau methylation is a relatively recent discovery (89). Results from mass spectrometric analyses show that several lysine residues are methylated on tau extracted from either healthy individuals or AD patient brains (89, 139–141). Moreover, in mouse models tau is also methylated at arginine residues R126, R155, and R349 (78). The number of methyl residues in tau protein is relatively low compared to other modifications such as phosphorylation and acetylation (140). Tau from both healthy and AD patient brains can be mono and dimethylated (141, 142). Interestingly the methylation status of tau changes qualitatively with aging and disease progression (89, 140, 143). Inherent in this finding is the possibility that aging alone, rather than disease per se, dictates tau methylation, analogous to the “Aging Clock” described for DNA methylation (144).
Until now, the specific enzymes involved in tau methylation have not been identified, and the exact role of methylation in tau function and pathogenesis is unclear. Methylation changes the electrostatics of tau protein. Given that tau is methylated within the KXGS motifs, regions that are essential for interactions with microtubules, lysine methylation may suppress tau's microtubule-binding function (139). Since tau aggregates from AD patients are methylated (89), it has also been suggested that methyl residues can bind to tau after aggregation and perhaps impair ubiquitination and UPS-mediated degradation (89). Future investigation of site-dependent tau methylation in vivo, and determination of its relationship to the DNA methylation “Aging Clock”, would advance our understanding of the role of this modification in tau function and pathology.
Glycosylation and Glycation
Glycosylation involves the addition of carbohydrate chains to proteins and lipids, of which we will focus on the former. Protein glycosylation is classified into two subtypes depending on the type of reaction involved: enzymatic glycosylation and non-enzymatic glycosylation (aka glycation). Enzymatic glycosylation typically occurs on secreted proteins or those that remain in membrane-bound organelles, hence this process occurs in the endoplasmatic reticulum (ER)/Golgi system. In addition, a specialized enzymatic glycosylation called O-GlcNAcylation occurs almost exclusively on cytoplasmic and nuclear (non-secreted) proteins (145). Given that the addition of sugar groups has steric effects on proteins, both enzymatic and non-enzymatic glycation have been described to play a major role in determining the structure and stability of proteins (146). However, while enzymatic glycosylation is an important PTM that exerts functional effects on glycoproteins, glycation typically results in dysfunctional or defective biomolecules (146, 147). Both enzymatic and non-enzymatic glycosylation of tau function are discussed below.
Enzymatic Glycosylation
Enzymatic glycosylation is a process in which glycosyltransferase enzymes attach activated sugar donor groups (monosaccharides) to proteins via covalent, glycosidic linkages (148–150). Sugar groups are linked to either asparagine (N-glycosylation) or serine/threonine (O-glycosylation) residues (Figure 2) (146). N-glycosylation begins as a co-translational event in the ER. There, the oligosaccharyltransferase enzyme adds an immature polysaccharide to a nascent polypeptide chain. After the further maturation of this polysaccharide, the resulting glycoprotein is transferred to the Golgi apparatus, where the sugar groups acquire an even more complex structure during a process called “terminal glycosylation” (149). Conversely, O-glycosylation is achieved post-translationally in the cis-Golgi compartment (151). N- and O-glycosylation also differ in the linker-residue of the targeted protein. N-glycosylation is characterized by the enzymatic addition of N-glycans to asparagine residues in the sequence N-X-S/T (where X is any amino acid except P or D), being X any amino acid except proline (149). O-glycosylation occurs on S/T residues with a β-turn in the proximity of a proline residue, however the O-glycosylation domains are not exactly defined (152). Both N- and O-glycosylation impact the structure, stability, folding, oligomerization, and solubility of glycoproteins (143, 153, 154).
Some proteins can additionally undergo a specialized and highly dynamic form of O-glycosylation called O-GlcNAcylation or O-GlcNAc. O-GlcNAcylation differs from other types of glycosylation in that the glycan is not further processed after the addition of a single GlcNAc to S/T residues. It also occurs almost exclusively on intracellular (nuclear and cytoplasmic) rather than secreted proteins (151). O-GlcNAcylation is highly regulated by two enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) that add and remove sugar groups, respectively (145). Because O-GlcNAcylation affects S/T residues, many known O-GlcNAcylation sites are nearby or overlapping with phosphorylation sites and thus it has been suggested that these two post-translational modifications can regulate each other (145, 155–159).
Enzymatic glycosylation may contribute to tauopathies by modifying proteins other than tau, such as APP and the β-site APP-cleaving enzyme 1 (BACE1). APP and BACE1 have been found to be O-glycosylated (160–162) and N-glycosylated (163, 164) in tauopathy patients' brains. Tau also has the potential to be modified by N-glycosylation and by the specialized form of O-glycosylation, O-GlcNAcylation (165, 166). Interestingly, to date tau has been found to be N-glycosylated only in AD subjects (167–169), while O-GlcNAcylation levels are relatively elevated in brains from normal controls (155, 166, 170). To date, at least five O-GlcNAc sites have been mapped in tau (T123, S208, S238, S400, and one of S409/412/413) (171–173) and all of them are also susceptible to phosphorylation. Therefore, it has been speculated that O-glycosylation may compete with or protect from phosphorylation (155, 174). Since tau O-GlcNAcylation is decreased in diseased brain (155, 170), it has been suggested that this PTMs has a protective role. This hypothesis is potentially supported by the fact that that O-GlcNAcylation increases the interaction of tau with microtubules (175), increases tau degradation (166), and suppresses tau aggregation (171, 175, 176).
Despite tau being N-glycosylated, the exact residues have not been identified. Furthermore, the relevance of N-glycosylation in tau function is a matter of debate since tau is a cytosolic protein, but the N-glycosylation process occurs in the ER-Golgi system (177). Therefore, tau N-glycosylation is thought to be associated with aberrant subcellular localization of tau. However, the mechanisms and consequences behind tau relocation are still elusive. It has been proposed that N-glycosylation precedes and accelerates tau phosphorylation (169), either by suppressing tau dephosphorylation (178) and/or inducing tau phosphorylation by protein kinase A (PKA) (169, 178). While a direct impact of tau N-glycosylation on microtubule polymerization and stability has not been observed (167), the removal of N-glycans and phosphate groups of tau seems to restore its microtubule polymerization activity (164). Although little is known about the role of N-glycosylation in tau biology, this evidence suggests that aberrant N-glycosylation of tau may be involved in neurodegenerative disease pathogenesis.
Glycation or Non-enzymatic Glycosylation
Glycation (or non-enzymatic glycosylation) is a PTM in which sugars or sugar-derived metabolites are covalently attached to the side chain of lysine residues (Figure 2). The glycation process involves a set of chemically heterogeneous modifications that lead to the formation of advanced glycation end-products (AGEs) (179). AGEs are produced by the irreversible cross-link between glycated and non-glycated proteins and are considered glycotoxins with significance in aging and age-related diseases (180–182). Both early stage glycation and AGEs have been shown to impair protein activity. Moreover, AGEs deposition has been shown to correlate with age and disease progression in AD (183). Although the study of AGEs is an active area of investigation in the field of aging research (184), it is still unknown how proteins are selected to be glycated and the biological role of glycation in neurodegeneration is not well-understood (185).
Thirty-two lysine residues in 2N4R tau have been identified in vitro as potential glycation sites (186, 187) (Figure 3). Although most of these sites were detected in both 3R and 4R isoforms, K280 and K281 are absent in the 3R tau, and this difference seems sufficient to slow the glycation of 3R relative to 4R isoforms (186, 187). In AD, glycation has been detected in aggregated tau purified from human AD brains but not in soluble tau (188). The molecular functions for glycation, the sites of these reactions and whether or how they contribute to tau aggregation remain subjects for debate. Since the glycation of tau in microtubule binding domains reduces tau affinity for microtubules in vitro (189), it has been suggested that glycation sterically blocks tau-microtubule interactions. Glycation may also induce the accumulation of tau by blocking the ubiquitination of tau at lysine residues and consequently its degradation (189, 190). Furthermore, the late glycosylation product known as AGEs have also been found colocalized with pathological tau aggregates in the brain of sporadic AD cases (191). Similarly, AGEs colocalize with NFTs from PSP, PiD, and ALS patients (180, 191). Although, the role of glycosylation in tau function is not well-understood, evidence presented here implies that this PTM can play an important role in tauopathies.
Proteolysis
Proteolysis refers to the breakdown of proteins into peptides and amino acids through the hydrolysis of peptide bonds via the action of proteases. Most proteases are highly specific and cleave their protein substrates from the N-terminus (aminopeptidases), C-terminus (carboxypeptidases), or more centralized regions (endopeptidases) of a protein. Furthermore, based on their catalytic mechanisms for substrate hydrolysis and the residues associated with their active sites, proteases are categorized into six types: aspartyl, cysteine, glutamic, metallo, serine, and threonine proteases. Proteolysis is a critical modification that can lead to alterations in protein function with important outcomes in many biological processes including signaling pathways and apoptosis. It can regulate the concentration of a protein, transform a protein into an active form, or process a protein to provide the amino acids required to synthesize a different protein. Proteolysis also plays an important function in removing damaged or unnecessary proteins from cells.
Several cytosolic proteases responsible for tau proteolysis have been identified, including caspases, calpains, and thrombin (192). Caspases are a family of evolutionarily conserved cysteine-dependent proteases that cleave proteins after specific aspartic acid residues (193). These proteases play a crucial role in important biological process such as apoptosis and inflammation (194). Thirteen different caspases have been described in humans, and their functional classification is reviewed elsewhere (193).
The role of proteolysis in tau function is incompletely understood. Tau proteolysis could be protective and may promote the removal of abnormal tau or could favor the abnormal accumulation of cleaved tau in the cell (195). It is also possible that tau cleavage is neither protective nor damaging, but is merely a bystander effect of tau accumulation. The proteolytic cleavage of tau has been observed both in vitro and in vivo (192, 195). Prior research on tau proteolysis has also shown that the proteolytic cleavage of tau precedes its aggregation in vitro (196–198). Furthermore, the N-terminal truncation of tau has been shown to alter the cellular localization of the protein from the cytosol to the nucleus (199), indicating a possible link between proteolysis and tau localization. Given that it has been shown that tau fragments induce death in cultured neurons (200), it has been suggested that tau cleavage may induce neurodegeneration. However, the toxic nature of tau fragments has not been demonstrated in vivo and therefore, it remains unclear if these fragments are indeed toxic.
The in vitro cleavage of tau by caspase-3,−7,−8, and less efficiently by caspases-1, and−6 resulting in the formation of an N-terminal fragment, tau1-421 (Tau-C3) (198, 201, 202) has been demonstrated. Tau-C3 fragment has been observed in AD brains (198) and interestingly, the active forms of both caspase-3 and caspase-6 are elevated in AD brains compared to control brains (203–205). While the exact function of Tau-C3 fragment is unknown, it has been shown that Tau-C3 fragment induce toxicity in neuronal cultures (201, 206, 207), and that the caspase-mediated tau cleavage precedes tangle formation in tau transgenic mice (Tg4510) (208). However, whether caspase-mediated cleavage of tau is toxic or protective for the neurons still remains a matter of debate.
While the proteolytic cleavage of tau by caspases has been extensively studied, the cleavage of tau by calpains and thrombin remains elusive. Calpains are calcium-activated cytosolic cysteine proteases implicated in a variety of calcium associated cellular functions like cell proliferation, migration, invasion, apoptosis, and signal transduction (209). Dysregulation of calcium homeostasis has been proposed to induce abnormal activation of calpains in a number of neurodegenerative diseases (209). The cleavage of tau by calpains, has been demonstrated in vitro and in neuronal cultures (210, 211). According to a recent study, calpain-mediated cleavage events are considered a part of normal tau protein processing (212). However, there are studies that have suggested that calpain-mediated proteolysis of tau may induce toxicity, as seen in cell cultures and a Drosophila tauopathy model (210, 213). Thrombin is a cytosolic serine protease that is expressed within neurons and astrocytes (195). The cleavage of tau by thrombin has been shown in vitro and in cell models of tauopathy (214, 215). Based on cell culture-based studies, thrombin cleavage products of tau are believed to be potentially pathogenic (216, 217). However, due to contradictory reports and lack of in vivo evidence for tau proteolysis mediated by these enzymes, the relevance of thrombin, and calpains in tau function remains unclear. Naturally, tau cleavage also occurs in the proteasome and lysosomes, which will be discussed further in section Effect of PTMs on Tau Degradation.
Other PTMs
In addition to the PTMs discussed above, several other post-translational modifications occur on tau, which are less well-recognized. Below, we discuss three different tau PTMs that fall in this category: protonation, oxidation, and nitration.
Protonation
Protonation involves enzyme-independent, rapid, and reversible addition or removal of protons. Protonation adds a positive charge to otherwise uncharged amino acid side chains and therefore can lead to dynamic changes in protein conformation and function (218). The optimum protonation state of a protein is critical to its functioning. Dysregulation in local pH can lead to disruption in the protonation state of a protein and has recently been shown to have implications with neurodegenerative diseases (219, 220). Among several protein sites that can participate in the protonation/deprotonation process, only a few can significantly undergo this modification within the normal cytosolic pH range. Histidine side chains, with a nominal pKa of ~6.5, are among the most attractive residues for protonation within cytosolic pH range.
Tau contains highly-conserved histidine residues near the C-terminus of each microtubule-binding tau repeat (Figure 3) (221). These residues have been proposed to act as pH sensors and influence tau–microtubule interaction within the physiological intracellular pH range (221). The histidines are positively charged at pH <g7.5 and show increased tau-microtubule binding (221). However, at pH >7.5, histidine residues in tau are deprotonated and bind microtubules with lower affinity (221). Furthermore, it has been proposed that a highly conserved histidine residue, H299 (Figure 3), near the R2 C-terminus seems to contribute to tau–microtubule binding. The labile nature and minimal molecular weight change of protonation renders it difficult to detect by traditional approaches, such as mass spectroscopy or gel electrophoresis, used to detect PTMs. Nonetheless, given its effects on protein structure, solubility and function, protonation should be considered as a post-translational protein modification like the others in this review.
Oxidation
Like protonation, protein conformation is also sensitive to oxidation-reduction (redox) changes. Redox homeostasis is achieved by the regulation of appropriate levels of reactive oxygen (ROS) and nitrogen (RNS) species, that are considered important physiological regulators of intracellular signaling pathways (222). The dysregulation of ROS is a major contributor to oxidative damage and it has been proposed that age-related accumulation of oxidized proteins may contribute to the aging process (223). However, the importance of protein oxidation in the progression of aging still remains poorly understood. Cysteine residues are the prime amino acid that can exist in oxidized or reduced states. The unique properties of cysteine side chain allows it to undergo various oxidative PTMs, which can potentially have diverse regulatory effects. The oxidation of cysteine residues leading to disulfide bond formation is one of the well-established mechanisms underlying the redox regulation of protein conformation and hence its function.
On tau, there exists a pair of cysteine residues that can undergo oxidation (Figure 3). While the effect of oxidation on tau function is unknown, its role in tau aggregation has been studied in vitro (224) and is discussed in section Effect of PTMs on Tau Solubility and Aggregation of this review. Tau oxidation has only been described in vitro, and therefore the status of oxidized tau in vivo remains unclear.
Nitration
Nitration involves the addition of nitrogen dioxide (NO2) onto tyrosine residues. The mechanism underlying protein nitration is not clear but it most likely involves the presence of ROS/RNS-like peroxynitrite and therefore nitration can also occur during oxidative damage (225). Several lines of evidence suggest a role of nitration in physiological processes like signal transduction (226, 227). Additionally, increased protein nitration has been identified in a large variety of diseases and have also been implicated in the process of aging (227).
The 2N4R tau isoform has five tyrosine residues, and the nitration at four of them has been shown in vitro: Y-18, Y-29, Y-197, and Y-394 (Figure 3). These studies have shown that residues Y-18 and T-29 are more susceptible toward nitration than residues Y-197 and Y-394. The nitration of tau at Y-197 has been observed in the healthy brain and therefore has been proposed to have important physiological functions (228). However, the nitration of Y-18, Y-29, and Y-394 has only been observed in AD or other tauopathies (229). It is believed that nitration of these tyrosine residues alters tau conformation and reduces its ability to bind to microtubules (230). While there are some interesting insights on tau nitration, most of these studies are based on in vitro experiments. Therefore, studies focusing on this PTM will provide additional useful insights into its role in tau pathobiology.
Effect of PTMs on Tau Degradation
Protein degradation is important for the maintenance of proteostasis and for preventing the accumulation of misfolded or aggregated protein species (231). There are two major protein degradation systems in the cell: the ubiquitin-proteasome system (UPS) and the autophagy-lysosomes pathway (231). Both protein degradation pathways are key effectors of the proteostasis network and are strongly influenced by PTMs. Protein clearance is highly regulated and utilizes ubiquitin as a tag for direct protein degradation in both proteasomal and autophagy systems. However, other PTMs also affect the process of protein degradation indirectly. This is the case for phosphorylation and acetylation, both of which can modify amino acid sequences known as degrons, which are important for protein degradation.
A subset of degrons are regulated by phosphorylation and thus are called phosphodegrons (232, 233). Phosphodegrons are short linear amino acid sequences, D/E-S/T-GXX-S/T-P or LL-S/T-PXX-S/T-P, where the phosphorylation of S/T residues promotes protein clearance through stimulation of subsequent ubiquitination and proteasomal degradation (232, 233). Proteins can also contain other motifs that promote degradation by the autophagy-lysosomes system. These pentapeptide motifs are known as KFERQ motifs as they are composed of one or two positively charged residues (K, R), one or two hydrophobic residues (I, L, V, F), one negatively charged residue (D, E), and one bookending glutamine (Q) (234). “Canonical” KFERQ motifs are complete without modification while “potential” KFERQ motifs require completion via a PTM such as phosphorylation (235) or acetylation (234, 236) (Figure 3).
Tau homeostasis is typically maintained via degradation by both UPS and autophagy-lysosome systems (96, 110). Yet at some point in the pathobiology of tauopathies, the local concentration of tau must increase to favor the formation of fibrils, oligomers, and aggregates. Does self-association of potentially abnormally modified tau lead to damage and subsequent impairment of protein degradation systems? In this model, tau accumulation is causal for neurodegenerative disease. Or, alternatively, does tau accumulation result from decreased efficiency or failure of the protein degradation systems? In this case, tau accumulation is a consequence of other pathobiology in neurodegeneration. This type of binary, chicken, or egg approach to the genesis of tauopathies may be over-simplifications of a complex, biological milieu that combines aspects of both tau accumulation as cause and consequence. Nonetheless, the answers to these questions are key, as they dictate certain nodes to target for disease therapy. In this section, we will delineate the pathways for tau clearance and describe how PTMs could play active roles in these processes.
Tau Clearance by the Ubiquitin-Proteasome Pathway
The UPS pathway is responsible for the clearance of soluble, intracellular proteins (237, 238). The proteasome is a highly organized and complex molecular machinery responsible for the selective and efficient degradation of client proteins, whose basic science and function has been reviewed elsewhere (239–242). The 26S proteasome is responsible for the majority of protein clearance in mammalian cells and degrades proteins that are tagged with polyubiquitin chains (195). Therefore, the fact that tau is a target of ubiquitination (as discussed above) (109, 110) suggests that it can be degraded by the proteasome.
While ubiquitination is the proximal signal for protein degradation in the UPS, other PTMs such as phosphorylation are often required for tau ubiquitination to occur (243). For example, tau phosphorylated at proline-directed serine/threonine sites is selectively ubiquitinated by CHIP (103, 243), and thus phosphorylation in these sites increases tau ubiquitination and UPS-mediated degradation. Given that tau has six phosphodegron sequences (Figure 3), phosphorylation also regulates the ubiquitination and degradation of tau through this means (233). Phosphorylation of tau degrons is highly regulated by different kinases such as cdk5 and GSK3β (244), which work together in order to phosphorylate tau at specific residues and prepare the molecule for ubiquitination and degradation (245). However, phosphorylation of tau at alternative sites prevents ubiquitination and clearance. As a case in point, phosphorylation of tau at the KXGS motifs in the C-terminal microtubule binding repeat domains seems to prevent the ubiquitination and degradation of tau (243).
The dysfunction of the UPS and its related PTMs are associated with the development of tauopathies. Insoluble tau aggregates isolated from human AD brains contain proteasome subunits (246). Ubiquitin ligases such as CHIP and TRAF6 also colocalize with tau in AD brains (102, 103). Ubiquitinated tau is a component of NFTs (246). Further, a recent study from our group showed that the PSP-related A152T tau variant increases tau phosphorylation at threonine 153, potentially interfering with recognition of the local phosphodegron and thereby UPS degradation (233). Impairment of UPS activity has also been observed in postmortem human AD brains (246–250). While together these data implicate the UPS in tauopathies, they nonetheless represent correlative, rather than causal evidence. To our knowledge, no studies have established UPS failure as necessary and sufficient in promoting tau accumulation, aggregation, neuronal loss, and neurodegeneration.
Tau Clearance by Autophagy
Autophagy is the process by which unnecessary or damaged cellular components are proteolytically degraded in the endosomal-lysosomal pathway. Based on the mechanisms by which the substrate is delivered for degradation, there are three major types of autophagy: chaperone-mediated autophagy (CMA), endosomal microautophagy (e-MI), and macroautophagy. Long-lived proteins or protein aggregates that are too large to be processed through the proteasome are typically degraded via autophagy (242). Tau, being a long-lived protein, is anticipated to be degraded in lysosomes (242) and indeed, it has been shown that both soluble and insoluble tau can be degraded by the three forms of autophagy (251, 252). Furthermore, it has been observed that the use of autophagy inhibitors, such as ammonium chloride (NH4Cl), chloroquine, and 3-methyladenine (3-MA), as well as the inhibition of lysosomal proteases, delays tau clearance, and enhances tau accumulation (253–255), confirming the importance of autophagy in tau degradation.
Not only is tau cleared by autophagy, but PTMs play distinct roles in modulating these processes. Within the amino acid sequence of the tau protein are found two KFERQ motifs (336QVEVK340 and 347KDRVQ351) (Figure 3). These consensus sequences direct tau toward either chaperone-mediated autophagy or endosomal-microautophagy (252, 256). In addition to the two canonical KFERQ motifs, tau contains four potential KFERQ motifs that can become canonical in the presence of phosphorylation, acetylation, or a combination of both (Figure 3) (KFERQ finder) (234), emphasizing that PTMs play an important role in the degradation of tau by these two systems. CMA-mediated protein degradation is a selective form of protein clearance in which the KFERQ motifs are recognized by the Hsc70 chaperone. Upon recognition, the proteins are transported to the lysosomal lumen after binding to the lysosome-associated membrane protein-2 (LAMP-2) (257). Tau protein can be efficiently degraded by CMA as long as the KFERQ motifs are not blocked by PTMs (252).
In addition to initiating CMA, the binding of Hsc70 to KFERQ motifs also targets cytosolic proteins for selective degradation by e-MI. This form of protein degradation occurs in late endosomes/multivesicular bodies instead of lysosomes and involves sequestering substrate proteins into intraluminal vesicles budding in toward the lumen of multivesicular endosomes (234, 256). Unlike CMA, in mammals, the presence of KFERQ motifs is necessary but not sufficient for e-MI (256). In addition to protein cargo delivery by Hsc70, the e-MI pathway relies on the ESCRT (endosomal sorting complexes required for transport) I and III complexes that are required for the formation of vesicles which internalizes the cytosolic cargo (256). A recent study has analyzed the influence of two different PTMs, oxidation and phosphorylation, on the e-MI-dependent degradation of tau (251). A substantial decrease in the association and internalization of tau in late endosomes via e-MI was observed upon oxidation of mutant tau, which suggests that the oxidation of tau at C291/C322 is a prerequisite for completing the internalization of tau through e-MI (251). Moreover, in the same study, the authors found that phosphorylating tau at S262, S293, S324, and S356 inside microtubule-binding domains diminishes its degradation by e-MI (251). In contrast, another study showed e-MI mediated degradation of total tau and of specific phosphorylated species (258). Thus, PTMs regulate the clearance of tau degradation by both CMA and e-MI.
Non-soluble, larger or aggregated species of tau are cleared via macroautophagy, a form of autophagy that involves the sequestration of cytoplasmic cargo into double-membrane vesicles known as autophagosomes, which fuse with the lysosome to degrade its contents (259). Several reports have confirmed that tau aggregates can be degraded by macroautophagy. For example, the aggregates generated by the overexpression of the tau repeat domain with an FTD-17 mutation (TauRDΔK280) (252) as well as the proteolytically cleaved tau isoform D421 (Tau-C3) (260) are degraded by macroautophagy in cell models of tauopathy. Interestingly, tau phosphorylated at KXGS motifs escapes degradation by the UPS but is efficiently degraded by macroautophagy (252), suggesting the that wild type tau can also be degraded by macroautophagy. As PTMs contribute to tau aggregation (see section Effect of PTMs on Tau Solubility and Aggregation), they likely regulate tau clearance via macroautophagy.
Failure of tau degradation by the autophagy-lysosomes pathway has been linked to tauopathies. For example, there are several studies suggesting a defect in macroautophagy in tauopathy patients (261–263). Furthermore, it has been shown that tau in NFTs colocalizes with lysosomes and lysosomal markers in human AD, CBD, and PSP brains (263–267), suggesting that tau is directed toward macroautophagy but fails to be degraded. Moreover, evidence from mouse and Drosophila models of FTLD-Tau indicates that the microtubule destabilization associated with the abnormal phosphorylation of tau leads to the disruption of the axonal vesicle transport by impairing the dynein-dynactin complex, vesicle trafficking, and autophagic flux (268, 269). Alterations in CMA may also contribute to tauopathies, since it has been described that tau carrying the FTD-related mutation P301L reduces tau's susceptibility for degradation by CMA (251). Overall, the regulation of the autophagy-lysosomes pathway seems to be essential for maintaining tau homeostasis in health and disease, and PTMs in tau influence the autophagic degradation of tau in multiple ways.
Effect of PTMs on Tau Solubility and Aggregation
The degradation pathways discussed above represent a major component of the proteostasis network and ensure that, under normal conditions, tau is maintained in an optimally functional state (231). However, in tauopathies, there is a breakdown in tau homeostasis that in turn leads to aberrant accumulation, misfolding, and aggregation of tau (45, 242, 270). Recent studies suggest that PTMs contribute in sometimes surprising ways to altering tau solubility and thereby promoting its aggregation.
Various biophysical techniques have demonstrated that under physiological conditions, tau exists in a “natively unfolded” or “intrinsically disordered” state (45, 271, 272). Despite being intrinsically disordered, tau shows a preference for global interactions between its negatively charged N-terminus and positively charged repeat domains, leading to an energetically favored “paperclip” like conformation (273). In contrast to soluble tau, tau aggregates are composed of β-sheet rich structures known as amyloid fibrils (274–277). Amyloid fibrils are highly heterogeneous in nature and are not easily amenable to study via traditional methods used in structural biology. Recent breakthroughs in cryo-electron microscopy (cryo-EM) technology, however, have made it possible to determine high resolution three-dimensional structure of tau amyloid fibrils (278, 279). Determination of the atomic structures of tau aggregates may provide a better understanding of the mechanisms underlying their formation, spreading, and clearance.
To date, cryo-EM has elucidated the structure of tau filaments isolated from brains of patients with several different forms of tauopathies (279–284). According to these studies, each tauopathy is associated with a unique tau filament fold (279). The origin of such structural specificity is not yet known, but could emanate from PTMs (279). For example, tau is phosphorylated at S262 in tau filaments extracted from AD and CBD brains, but not in the filaments of PiD patients (11). Consistent with this phospho-modified residue contributing to aggregate structure, S262 is present in the tau filament core in PiD but not AD (279). Therefore, distinctive patterns of post-translational modification in different tauopathies may underlie not only the structural specificity of amyloids observed in cryo-EM structures, but could even contribute to selective vulnerability to these diseases.
To understand the fundamental role of PTMs in tau aggregation, it is important to account for the influence of PTMs over the charge composition and distribution within a polypeptide chain. The net charge on any protein is dependent on the content of ionizable groups. Net charge is 0 when the pH of the surrounding environment equals that protein's isoelectric point (pI). Similarly, net charge is positive at a pH below pI, and is negative at pH above its pI. Tau contains around 29 percent charged residues, has low hydrophobicity and has a low net charge of +2 at physiological pH. While tau is overall slightly positively charged, the amino-terminal residues (including the two N-terminal inserts) are more negatively charged, the microtubule-binding repeat region is predominantly positive, and the distal carboxy-terminal residues are mostly neutral (285). Such asymmetric distribution of charges has been proposed to play a crucial role in tau interactions with microtubules and other binding partners (45). It may also explain the inability of tau to undergo aggregation in vitro without polyanionic aggregation inducers (286–288). The inducers of tau aggregation are not typically present in neurons but their effect may be achieved through the masking of charged residues in tau via PTMs.
PTMs that make tau more negatively charged are phosphorylation, acetylation, and nitration. These PTMs, when present in the microtubule-binding domains, have been proposed to initiate tau aggregation by first weakening its interaction with negatively charged microtubules (26, 45, 289, 290) The reduced affinity of tau toward microtubules upon these modifications would increase the “free” pool of soluble modified tau molecules that might have a higher tendency to self-assemble into aggregates, as has been observed in case of phosphorylated tau (189, 291). The aggregation might be further promoted by electrostatic interactions between negatively charged, modified tau and positively charged, unmodified tau. The effects of phosphorylation on tau aggregation, however, seem to be site-dependent. For example, phosphorylation sites close to the N-terminus tend to prevent tau aggregation but phosphorylation sites in the vicinity of the C-terminus and microtubule-binding region seem to accelerate filament formation in vitro (285). A similar site-dependence has been observed in tau nitration, where depending on the residues that are modified, nitration can either promote or inhibit tau aggregation (230, 289). Interestingly, the core of tau filaments isolated from tauopathy patients contains multiple lysine residues that are susceptible to acetylation (280, 281, 283). Since the microtubule binding region constitutes a major part of the core of tau filaments, acetylation of lysine residues in this region likely plays an important role in the assembly of tau filaments.
Unlike phosphorylation, acetylation, and nitration that alter the charge of tau, methylation preserves the positive charge on lysine residues for a neutral effect on net charge of the protein. The role of methylation in tau aggregation is controversial and it is unclear whether tau methylation induces tau aggregation or has a protective role. While some studies have remarked that methylation at lysine residues facilitates abnormal protein aggregation (292, 293), further experimentation using recombinant tau has shown opposite results, demonstrating that methylation reduces the tendency of tau toward aggregation and promotes tubulin assembly (141). Interestingly, the methylation status of tau changes from dimethyl- to monomethyl-lysine with aging and disease (140). Therefore, it is possible that high stoichiometry (number of methyl groups/protein molecule) of methylation reduces the propensity of tau toward aggregation in vitro and promote tubulin assembly (140).
Another important modification in tau that does not affect charge but can affect tau aggregation is oxidation. The oxidation at one of the two cysteine residues C-322 (present in R3 repeat) (Figure 3) seems to promote tau aggregation into paired helical filaments (224). However, aggregation is inhibited under reduced conditions, upon mutation of cysteine to alanine and upon the formation of an intramolecular disulfide bond between C-322 and C-291 (224, 294).
While the PTMs described above involve the addition of small chemical groups to tau, other PTMs involve the addition of large sugar groups (glycosylation and glycation) and protein macromolecules (ubiquitination and SUMOylation) (Figure 2). These large molecules can have profound steric effects on the native conformation of tau and therefore can strongly influence misfolding and aggregation. The mechanisms by which glycation and glycosylation regulate tau solubility and aggregation are poorly understood. Glycation appears to promote the polymerization and stabilization of aggregated tau (295). Additionally, the N-glycosylation of tau is thought to be associated with maintaining the structure of NFTs (167). On the other hand, O-GlcNAcylation has been proposed to protect tau against aggregation (175). In fact, a study on tau peptides has shown that O-GlcNAcylation in proline-rich sequences favors a more disordered or extended conformation of tau (296).
Like sugars, the effects of ubiquitin and SUMO addition to tau on aggregation propensity have not been extensively studied. However, both mono- and polyubiquitination can contribute to the formation of insoluble tau inclusions (96, 117). In addition, a recent cryo-electron microscopy (cryo-EM) and mass spectrometry-based study performed on tau filaments isolated from the brains of tauopathy patients, has proposed that incorporation of ubiquitin into tau filaments mediates specific inter-protofilament packing by providing additional contacts between tau molecules in each protofilament via ubiquitin chains (282). SUMOylation is analogous to ubiquitination and has been shown to decrease the solubility of tau (128). However, the mechanisms by which SUMOylation reduces tau solubility and most likely promotes its aggregation, remain unknown.
Overall, PTMs on tau critically influence the assembly of natively unfolded tau into highly ordered β-sheet rich aggregates. It is likely that PTMs drive this process by altering the distinctive charge distribution on tau. It is worth emphasizing that while PTMs undoubtedly affect tau solubility and intermolecular interactions, the effect of PTMs on processes such as protein localization and degradation would also contribute, in a discrete and upstream manner, to when, how and why tau aggregation events occur.
Cross-Talk and Competition Between PTMs
The PTM code hypothesis proposes that the combination of PTMs in proteins generates a dynamic “code” that can be translated into complex biological consequences (297). This hypothesis may apply to tau since it undergoes many modifications, often targeting the same amino acid residue (Figure 3). Such heterogeneity and competition between PTMs suggest that tau function and aggregation could be a consequence of the combination of several PTMs and/or the substitution of one PTM by another at the same position. In this regard, and based on the evidence described above, it is likely that while some PTM ensembles are essential for the regulation of tau function and degradation, others are more deterministic for formation of tau aggregates (Figure 4).
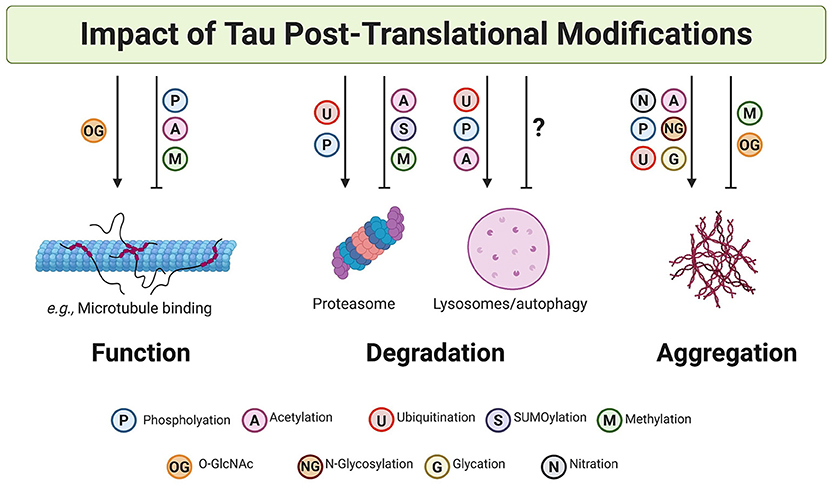
Figure 4. Post-translational modifications on tau impact tau function, degradation, and aggregation. PTMs can both promote and inhibit tau function, clearance, and aggregation. A non-exhaustive list of examples are illustrated here. In many cases, it is likely that cross-talk and competition between PTMs results in the specific and dynamic regulation of tau.
One level at which competing biological outcomes can be coded is via competition of different PTMs for the same amino acid. For example, S and T residues are modified by phosphorylation and O-GlcNAcylation. O-GlcNAcylation in some S/T residues protects tau from phosphorylation and thus blocks the effects of phosphorylation, which impacts microtubule-binding affinity and aggregation propensity, on these residues (155, 165, 298).
Perhaps even more interesting are lysine residues, which are susceptible to five different PTMs—acetylation, ubiquitination, methylation, SUMOylation, and glycation. Each one of these modifications have potentially different consequences for tau structure, function and aggregation. Since ubiquitination occurs on lysine residues, other lysine-based PTMs may prevent ubiquitination and therefore impair tau degradation (128, 139). In fact, many lysine residues that competent for ubiquitination are also targets for other PTMs (Figure 3). For example, K254 residue in tau can undergo ubiquitination and methylation. In fibrillar tau, K254 was found to be primarily methylated and ubiquitinated to a lesser extent, suggesting that methylation may block UPS-mediated tau degradation and lead to an increase in tau levels (89). Like methylation, acetylation could potentially prevent lysine ubiquitination, resulting in the insufficient turnover endogenous tau (72). The veritable log jam of PTMs that can be found on lysine residues marks this amino acid as a particularly interesting and likely important site at which cells orchestrate the biological outcomes they wish to achieve with tau and other proteins.
In addition to competition, crosstalk between PTMs can also be achieved through cooperativity. Some studies suggest that certain PTMs make tau more amenable to other modifications. In vitro tau SUMOylation seemingly enhances phosphorylation at residues such as T231 and S262 (128). Other studies have shown that tau methylation at K267 may result in subsequent and possibly abnormal phosphorylation of tau at S262 (89). It has also been proposed that N-glycosylation precedes tau phosphorylation (299). In each of these cases, though, a reasonable alternative interpretation is that SUMOylation, methylation, or N-glycosylation may impair tau clearance, enhancing the half-life of tau in the cytosol, and thus, providing more opportunities for phosphorylation.
On the other hand, phosphorylation can also affect the propensity of tau for other PTMs. For example, in specific experimental conditions the in vitro phosphorylation of tau in the microtubule binding domain activates tau autoacetyltransferase activity and therefore promote its acetylation (76). Furthermore, a protective role of the switch between tau phosphorylation and acetylation has been described. The acetylation of tau at KXGS motifs inhibits phosphorylation and thus prevents the detachment of tau from microtubules and its aggregation (34, 247). Additionally, the phosphorylation of tau at S422 seems to precede and block the cleavage of tau at D421 by caspase-3 (300), suggesting that phosphorylation at S422 could be a protective mechanism for the inhibition of the caspase-mediated cleavage of tau.
Taken together, the current literature suggests that crosstalk, combinations, and competition between PTMs play a critical role in tau function, degradation, and aggregation. An old truism states that “We are the company we keep.” If so, then future studies dedicated to documenting and decoding ensembles of tau PTMs will rise in importance in regards to understanding and developing interventions for tauopathies.
Approaches and Limitations
The complete understanding of the role of PTMs in tau function and dysfunction is obstructed by several limitations in the methodology and models used. Historically, PTMs were identified using immunochemistry-based approaches. Using this method, several phosphorylation sites were identified and thus, a large number of anti-phospho tau antibodies are available (301). While immunochemistry methods are routinely used and are highly effective for the characterization tauopathies, they do not provide a comprehensive and quantitative description of PTMs, nor do they distinguish between single molecules with many PTMs vs. many molecules with single PTMs (302). Some of these problems can be solved by the use of quantitative mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy, which provide a more detailed description of PTMs and allow the simultaneous analyses of a variety of PTMs (89, 110, 303–308). However, most of these methods are biased toward the peptides of highest intensity, and prevent the accurate determination of site-specific stoichiometry, associated site-specific dynamics and identification of labile PTMs like protonation (302). The high percentage of PTM sites in tau adds to the complexity, making it incredibly challenging to design and carry out such studies. To overcome some of these challenges, analytical methods including one known as FLEXITau (Full-Length Expressed stable Isotope-labeled tau) have been recently developed (309). This method allows for unbiased analyses of tau PTMs in a more quantitative manner. In this regard, a study using FLEXITau has reported 95 PTMs on tau isolated from postmortem human tissue from AD and has demonstrated that these modifications occur in an ordered manner and lead to tau aggregation (310).
The choice of models to use for studying PTMs also influences the understanding of the effect of PTMs in tau biology. The vast majority of PTMs have been identified in vitro, using biochemical assays where purified tau protein was incubated with specific enzymes (71) or using tau/enzymes overexpression systems in cell models (128). Some PTMs have also been confirmed or identified in vivo using tauopathy mouse models. However, most of these models are based on the expression of mutant forms human tau, and whether those findings and insights into PTMs extends to wild-type tau and sporadic tauopathies remains to be seen (79).
Tau PTMs have also been studied in post-mortem human brains using immunochemistry techniques and cryo-EM (282). It is important to keep in consideration that some PTMs can be lost or degraded during the process of brain extraction, fixation, storage, and analysis. Therefore, it is possible that in studies performed on human brain extracts, some tau PTMs may not be identified because of the caveats associated with brain preparation processes (37).
The cross-talk between PTMs is very important for the regulation of function, structure, and degradation of the proteins. Many studies on tau PTMs have focused only on one type of modification. Extrapolation of results obtained from single PTM studies lack context in regard to cross-talk and competition. As a result, a major caveat of many studies is the inability to differentiate between causes, consequences and bystander effects of these PTMs. The understanding of and solution to these limitations may enable the discovery of a “PTM code” in tau that will help to define sensitive biomarkers and lead to insights into disease pathobiology. In addition, unravelling the specific effects of PTMs in tau biology will pave the road toward new therapeutic approaches for tau-related neurodegenerative diseases.
Concluding Remarks
PTMs are essential to the normal function of tau and therefore alterations in the pattern of PTMs has the potential to lead to tau dysfunction, accumulation, and abnormal aggregation. PTMs exert their function either through altering protein electrostatics or conferring steric alterations. Among all tau PTMs, phosphorylation has been studied most extensively and historically, was thought to hold primacy in regulating tau function and pathogenesis of tauopathies. However, increasing evidence suggests that many other PTMs contribute to dynamic regulation of tau. Cross-talk and competition between PTMs, in particular at lysine residues, introduce an intriguing new layer of complexity to orchestration of tau function and dysfunction. At the same time, combinatorial ensembles of PTMs may emerge as a means by which cells achieve differential biological outcomes involving tau. As a robust and fast-moving area of investigation, tau post-translational modification will likely impact the basic, pre-clinical, and clinical-translational aspects of tau biology in interesting ways for years to come.
Author Contributions
CA and SA performed the literature review. CA, SA, and AK wrote the manuscript. All authors agreed to the final version of the manuscript.
Funding
Tau consortium, P30 P0531421, R01 NS095257, R01 AG059052, Chan Zuckerberg Initiative grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge Jacqueline Jia for artistic contributions to figure preparation. We thank Dr. Alma Burlingame and Dr. William Seeley for critical review of the manuscript.
References
1. Walsh CT. Posttranslational Modification of Proteins: Expanding Nature's Inventory - Christopher Walsh - Google Libros. Greenwood Village, CO: Roberts and Company, Publishers (2006) Available online at: https://books.google.com/books?hl=es&lr=&id=JGBfQXIzdwgC&oi=fnd&pg=PP21&ots=v8ZAw__wOI&sig=owJxdEEdGvj8IHuiAf6pMUkowWM#v=onepage&q&f=false (accessed August 14, 2020).
2. Smith LM, Kelleher NL. Proteoform: a single term describing protein complexity. Nat Methods. (2013) 10:186–7. doi: 10.1038/nmeth.2369
3. Darling AL, Uversky VN. Intrinsic disorder and posttranslational modifications: the darker side of the biological dark matter. Front Genet. (2018) 9:158. doi: 10.3389/fgene.2018.00158
4. Barber KW, Rinehart J. The ABCs of PTMs. Nat Chem Biol. (2018) 14:188–92. doi: 10.1038/nchembio.2572
5. Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. (2008) 9:679–89. doi: 10.1038/nrm2468
6. Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. (2002) 11:739–56. doi: 10.1110/ps.4210102
7. Uversky VN. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front Aging Neurosci. (2015) 7:18. doi: 10.3389/fnagi.2015.00018
8. Dyson HJ. Expanding the proteome: disordered and alternatively folded proteins. Q Rev Biophys. (2011) 44:467–518. doi: 10.1017/S0033583511000060
9. Jeganathan S, Von Bergen M, Mandelkow EM, Mandelkow E. The natively unfolded character of tau and its aggregation to Alzheimer-like paired helical filaments. Biochemistry. (2008) 47:10526–39. doi: 10.1021/bi800783d
10. Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. (2000) 33:95–130. doi: 10.1016/S0165-0173(00)00019-9
11. Goedert M, Eisenberg DS, Crowther RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci. (2017) 40:189–210. doi: 10.1186/s13041-017-0298-7
12. Dixit R, Ross JL, Goldman YE, Holzbaur ELF. Differential regulation of dynein and kinesin motor proteins by tau. Science. (2008) 319:1086–9. doi: 10.1126/science.1152993
13. Zuo YC, Li HL, Xiong NX, Shen JY, Huang YZ, Fu P, et al. Overexpression of Tau rescues nogo-66-induced neurite outgrowth inhibition in vitro. Neurosci Bull. (2016) 32:577–84. doi: 10.1007/s12264-016-0068-z
14. Merino-Serrais P, Benavides-Piccione R, Blazquez-Llorca L, Kastanauskaite A, Rá A, Avila JS, et al. The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer's disease. Brain. (2013) 136:1913–28. doi: 10.1093/brain/awt088
15. Brandt R, Léger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol. (1995) 131:1327–40. doi: 10.1083/jcb.131.5.1327
16. Farah CA, Perreault S, Liazoghli D, Desjardins M, Anton A, Lauzon M, et al. Tau interacts with golgi membranes and mediates their association with microtubules. Cell Motil Cytoskeleton. (2006) 63:710–24. doi: 10.1002/cm.20157
17. Meier S, Bell M, Lyons DN, Ingram A, Chen J, Gensel JC, et al. Identification of novel tau interactions with endoplasmic reticulum proteins in Alzheimer's disease brain. J Alzheimer's Dis. (2015) 48:687–702. doi: 10.3233/JAD-150298
18. Noble W, Hanger DP, Miller CCJ, Lovestone S. The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol. (2013) 4:83. doi: 10.3389/fneur.2013.00083
19. Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. (1996) 399:344–9. doi: 10.1016/S0014-5793(96)01386-5
20. Krylova SM, Musheev M, Nutiu R, Li Y, Lee G, Krylov SN. Tau protein binds single-stranded DNA sequence specifically - the proof obtained in vitro with non-equilibrium capillary electrophoresis of equilibrium mixtures. FEBS Lett. (2005) 579:1371–5. doi: 10.1016/j.febslet.2005.01.032
21. Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. (2011) 286:4566–75. doi: 10.1074/jbc.M110.199976
22. Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull. (2016) 126:238–92. doi: 10.1016/j.brainresbull.2016.08.018
23. Park S, Lee JH, Jeon JH, Lee MJ. Degradation or aggregation: the ramifications of post-translational modifications on tau. BMB Rep. (2018) 51:265–23. doi: 10.5483/BMBRep.2018.51.6.077
24. Haj-Yahya M, Lashuel HA. Protein semisynthesis provides access to Tau disease-associated post-translational modifications (PTMs) and paves the way to deciphering the tau PTM code in health and diseased states. J Am Chem Soc. (2018) 140:6611–21. doi: 10.1021/jacs.8b02668
25. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. (2009) 15:112–9. doi: 10.1016/j.molmed.2009.01.003
26. Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. (2011) 2:252. doi: 10.1038/ncomms1255
27. Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VMY, et al. Acetylated tau, a novel pathological signature in Alzheimer's disease and other tauopathies. Brain. (2012) 135:807–18. doi: 10.1093/brain/aws013
28. Guillozet-Bongaarts AL, Glajch KE, Libson EG, Cahill ME, Bigio E, Berry RW, et al. Phosphorylation and cleavage of tau in non-AD tauopathies. Acta Neuropathol. (2007) 113:513–20. doi: 10.1007/s00401-007-0209-6
29. Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, et al. Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol Aging. (2005) 26:1015–22. doi: 10.1016/j.neurobiolaging.2004.09.019
30. Sacco F, Perfetto L, Castagnoli L, Cesareni G. The human phosphatase interactome: an intricate family portrait. FEBS Lett. (2012) 586:2732–9. doi: 10.1016/j.febslet.2012.05.008
31. Humphrey SJ, James DE, Mann M. Protein phosphorylation: a major switch mechanism for metabolic regulation. Trends Endocrinol Metab. (2015) 26:676–87. doi: 10.1016/j.tem.2015.09.013
32. Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. (1995) 80:225–36. doi: 10.1016/0092-8674(95)90405-0
33. Ardito F, Giuliani M, Perrone D, Troiano G, Muzio LL. The crucial role of protein phosphorylation in cell signalingand its use as targeted therapy. Int J Mol Med. (2017) 40:271–80. doi: 10.3892/ijmm.2017.3036
34. Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase- 3β. J Neurochem. (2000) 74:1587–95. doi: 10.1046/j.1471-4159.2000.0741587.x
35. Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, et al. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. (1995) 270:823–9. doi: 10.1074/jbc.270.2.823
36. Cook C, Stankowski JN, Carlomagno Y, Stetler C, Petrucelli L. Acetylation: a new key to unlock tau's role in neurodegeneration. Alzheimer's Res Ther. (2014). 6:29. doi: 10.1186/alzrt259
37. Matsuo ES, Shin RW, Billingsley ML, van deVoorde A, O'Connor M, Trojanowski JQ, et al. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron. (1994) 13:989–1002. doi: 10.1016/0896-6273(94)90264-X
38. Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, et al. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimers disease and tauopathies. Curr Alzheimer Res. (2005) 2:3–18. doi: 10.2174/1567205052772713
39. Ji C, Tang M, Zeidler C, Höhfeld J, Johnson GVW. BAG3 and SYNPO (synaptopodin) facilitate phospho-MAPT/Tau degradation via autophagy in neuronal processes. Autophagy. (2019) 15:1199–213. doi: 10.1080/15548627.2019.1580096
40. Derkinderen P, Scales TME, Hanger DP, Leung KY, Byers HL, Ward MA, et al. Tyrosine 394 is phosphorylated in Alzheimer's paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci. (2005) 25:6584–93. doi: 10.1523/JNEUROSCI.1487-05.2005
41. Vega IE, Cui L, Propst JA, Hutton ML, Lee G, Yen SH. Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Mol Brain Res. (2005) 138:135–44. doi: 10.1016/j.molbrainres.2005.04.015
42. Gong C -X, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. (1993) 61:921–7. doi: 10.1111/j.1471-4159.1993.tb03603.x
43. Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC. Protein phosphatases and Alzheimer's disease. Prog Mol Biol Transl Sci. 106:343–79. doi: 10.1016/B978-0-12-396456-4.00012-2
44. Wadhwa P, Jain P, Jadhav HR. Glycogen synthase kinase 3 (GSK3): its role and inhibitors. Curr Top Med Chem. (2020) 20:1522–34. doi: 10.2174/1568026620666200516153136
45. Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Biol. (2012) 4:ea006247. doi: 10.1101/cshperspect.a006247
46. Yu Y, Run X, Liang Z, Li Y, Liu F, Liu Y, et al. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem. (2009) 108:1480–94. doi: 10.1111/j.1471-4159.2009.05882.x
47. Biernat J, Mandelkow E-M. The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol Biol Cell. (1999) 10:727–40. doi: 10.1091/mbc.10.3.727
48. Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt-Ulms G, Meyer HE, et al. Microtubule-associated protein/microtubule affinity-regulating kinase (p110(mark)). a novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer- specific site serine 262. J Biol Chem. (1995) 270:7679–88. doi: 10.1074/jbc.270.13.7679
49. Biernat J, Gustke N, Drewes G, Mandelkow E, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. (1993) 11:153–63. doi: 10.1016/0896-6273(93)90279-Z
50. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. (2016) 17:5–21. doi: 10.1038/nrn.2015.1
51. Mandel JE, Banker GA. Microtubule-associated proteins, phosphorylation gradients, and the establishment of neuronal polarity. Perspect Dev Neurobiol. (1996) 4:125–35.
52. Mandell JW, Banker GA. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. (1996) 16:5727–40. doi: 10.1523/JNEUROSCI.16-18-05727.1996
53. Biernat J, Wu YZ, Timm T, Zheng-Fischhöfer Q, Mandelkow E, Meijer L, et al. Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol Biol Cell. (2002) 13:4013–28. doi: 10.1091/mbc.02-03-0046
54. Li C, Götz J. Somatodendritic accumulation of tau in Alzheimer's disease is promoted by Fyn-mediated local protein translation. EMBO J. (2017) 36:3120–38. doi: 10.15252/embj.201797724
55. Ittner LM, Ke YD, Götz J. Phosphorylated tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J Biol Chem. (2009) 284:20909–16. doi: 10.1074/jbc.M109.014472
56. Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. (1999) 399:784–8. doi: 10.1038/21650
57. Cho JH, Johnson GVW. Glycogen synthase kinase 3β phosphorylates tau at both primed and unprimed sites: differential impact on microtubule binding. J Biol Chem. (2003) 278:187–93. doi: 10.1074/jbc.M206236200
58. Cho JH, Johnson GVW. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3β (GSK3β) plays a critical role in regulating tau's ability to bind and stabilize microtubules. J Neurochem. (2004) 88:349–58. doi: 10.1111/j.1471-4159.2004.02155.x
59. Dawson HN, Ferreira A, Ghoshal N, Eyster MV, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from τ deficient mice. J Cell Sci. (2001) 114:1179–87.
60. Spittaels K, van Den Haute C, van Dorpe J, Geerts H, Mercken M, Bruynseels K, et al. Glycogen synthase kinase-3β phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. (2000) 275:41340–9. doi: 10.1074/jbc.M006219200
61. Tatebayashi Y, Haque N, Tung YC, Iqbal K, Grundke-Iqbal I. Role of tau phosphorylation by glycogen synthase kinase-3β in the regulation of organelle transport. J Cell Sci. (2004) 117:1653–63. doi: 10.1242/jcs.01018
62. Jaworski T, Kügler S, van Leuven F. Modeling of tau-mediated synaptic and neuronal degeneration in Alzheimer's disease. Int J Alzheimer's Dis. (2010) 2010:573138. doi: 10.4061/2010/573138
63. Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. (2010) 68:1067–81. doi: 10.1016/j.neuron.2010.11.030
64. Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. (2007) 27:2896–907. doi: 10.1523/JNEUROSCI.4674-06.2007
65. Hasegawa M, Jakes R, Crowther RA, Lee VMY, Ihara Y, Goedert M. Characterization of mAb AP422, a novel phosphorylation-dependent monoclonal antibody against tau protein. FEBS Lett. (1996) 384:25–30. doi: 10.1016/0014-5793(96)00271-2
66. Haase C, Stieler JT, Arendt T, Holzer M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J Neurochem. (2004) 88:1509–20. doi: 10.1046/j.1471-4159.2003.02287.x
67. Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta - Proteins Proteomics. (2016) 1864:1372–401. doi: 10.1016/j.bbapap.2016.06.007
68. Ali I, Conrad RJ, Verdin E, Ott M. Lysine acetylation goes global: from epigenetics to metabolism and therapeutics. Chem Rev. (2018) 118:1216–52. doi: 10.1021/acs.chemrev.7b00181
69. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. (2000) 403:41–5. doi: 10.1038/47412
70. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. (2019) 20:156–74. doi: 10.1038/s41580-018-0081-3
71. Kamah A, Huvent I, Cantrelle FX, Qi H, Lippens G, Landrieu I, et al. Nuclear magnetic resonance analysis of the acetylation pattern of the neuronal tau protein. Biochemistry. (2014) 53:3020–32. doi: 10.1021/bi500006v
72. Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. (2010) 67:953–66. doi: 10.1016/j.neuron.2010.08.044
73. Ding H, Dolan PJ, Johnson GVW. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J Neurochem. (2008) 106:2119–30. doi: 10.1111/j.1471-4159.2008.05564.x
74. Min SW, Sohn PD, Li Y, Devidze N, Johnson JR, Krogan NJ, et al. SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of tauopathy. J Neurosci. (2018) 38:3680–8. doi: 10.1523/JNEUROSCI.2369-17.2018
75. Carlomagno Y, Chung D eun C, Yue M, Castanedes-Casey M, Madden BJ, Dunmore J, et al. An acetylation–phosphorylation switch that regulates tau aggregation propensity and function. J Biol Chem. (2017) 292:15277–86. doi: 10.1074/jbc.M117.794602
76. Cohen TJ, Friedmann D, Hwang AW, Marmorstein R, Lee VMY. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat Struct Mol Biol. (2013) 20:756–62. doi: 10.1038/nsmb.2555
77. Paik WK, Pearson D, Lee HW, Kim S. Nonenzymatic acetylation of histones with acetyl-CoA. BBA Sect Nucleic Acids Protein Synth. (1970) 213:513–22. doi: 10.1016/0005-2787(70)90058-4
78. Morris M, Knudsen GM, Maeda S, Trinidad JC, Ioanoviciu A, Burlingame AL, et al. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci. (2015) 18:1183–9. doi: 10.1038/nn.4067
79. Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. (2015) 21:1154–62. doi: 10.1038/nm.3951
80. Chen X, Li Y, Wang C, Tang Y, Mok SA, Tsai RM, et al. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol Neurodegener. (2020) 15:2. doi: 10.1186/s13024-019-0354-0
81. Schueller E, Paiva I, Blanc F, Wang XL, Cassel JC, Boutillier AL, et al. Dysregulation of histone acetylation pathways in hippocampus and frontal cortex of Alzheimer's disease patients. Eur Neuropsychopharmacol. (2020) 33:101–16. doi: 10.1016/j.euroneuro.2020.01.015
82. Chatterjee S, Cassel R, Schneider-Anthony A, Merienne K, Cosquer B, Tzeplaeff L, et al. Reinstating plasticity and memory in a tauopathy mouse model with an acetyltransferase activator. EMBO Mol Med. (2018) 10:e8587. doi: 10.15252/emmm.201708587
83. Julien C, Tremblay C, Émond V, Lebbadi M, Salem N, Bennett DA, Calon F. Sirtuin 1 reduction parallels the accumulation of tau in alzheimer disease. J Neuropathol Exp Neurol. (2009) 68:48–58. doi: 10.1097/NEN.0b013e3181922348
84. Rizzi L, Roriz-Cruz M. Sirtuin 1 and Alzheimer's disease: an up-to-date review. Neuropeptides. (2018) 71:54–60. doi: 10.1016/j.npep.2018.07.001
85. Luo Y, Ma B, Nussinov R, Wei G. Structural insight into tau protein's paradox of intrinsically disordered behavior, self-acetylation activity, and aggregation. J Phys Chem Lett. (2014) 5:3026–31. doi: 10.1021/jz501457f
86. Irwin DJ, Cohen TJ, Grossman M, Arnold SE, McCarty-Wood E, van Deerlin VM, et al. Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am J Pathol. (2013) 183:344–351. doi: 10.1016/j.ajpath.2013.04.025
87. Grinberg LT, Wang X, Wang C, Sohn PD, Theofilas P, Sidhu M, et al. Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta Neuropathol. (2013) 125:581–93. doi: 10.1007/s00401-013-1080-2
88. Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. (2008) 31:449–61. doi: 10.1016/j.molcel.2008.07.002
89. Thomas SN, Funk KE, Wan Y, Liao Z, Davies P, Kuret J, et al. Dual modification of Alzheimer's disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol. (2012) 123:105–17. doi: 10.1007/s00401-011-0893-0
90. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. (1998) 67:425–79. doi: 10.1146/annurev.biochem.67.1.425
91. Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol. (2011) 18:520–8. doi: 10.1038/nsmb.2066
92. Cappadocia L, Lima CD. Ubiquitin-like protein conjugation: structures, chemistry, and mechanism. Chem Rev. (2018) 118:889–918. doi: 10.1021/acs.chemrev.6b00737
93. Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. “Protein modifications: beyond the usual suspects” Review series. EMBO Rep. (2008) 9:536–42. doi: 10.1038/embor.2008.93
94. Nathan JA, Tae Kim H, Ting L, Gygi SP, Goldberg AL. Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J. (2013) 32:552–65. doi: 10.1038/emboj.2012.354
95. Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. (2007) 315:201–5. doi: 10.1126/science.1127085
96. Tan JMM, Wong ESP, Kirkpatrick DS, Pletnikova O, Ko HS, Tay S-P, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. (2008) 17:431–9. doi: 10.1093/hmg/ddm320
97. Munari F, Barracchia CG, Parolini F, Tira R, Bubacco L, Assfalg M, et al. Semisynthetic modification of tau protein with di-ubiquitin chains for aggregation studies. Int J Mol Sci. (2020) 21:4400. doi: 10.3390/ijms21124400
98. Harris LD, Jasem S, Licchesi JDF. The ubiquitin system in Alzheimer's disease. Adv Exp Med Biol. (2020) 1233:195–221. doi: 10.1007/978-3-030-38266-7_8
99. Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. (2017) 86:129–57. doi: 10.1146/annurev-biochem-060815-014922
100. Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. (2005) 94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x
101. Flach K, Ramminger E, Hilbrich I, Arsalan-Werner A, Albrecht F, Herrmann L, et al. Axotrophin/MARCH7 acts as an E3 ubiquitin ligase and ubiquitinates tau protein in vitro impairing microtubule binding. Biochim Biophys Acta - Mol Basis Dis. (2014) 1842:1527–38. doi: 10.1016/j.bbadis.2014.05.029
102. Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. (2004) 13:703–14. doi: 10.1093/hmg/ddh083
103. Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. (2004) 279:4869–76. doi: 10.1074/jbc.M305838200
104. Zhang YJ, Xu YF, Liu XH, Li D, Yin J, Liu YH, et al. Carboxyl terminus of heat-shock cognate 70-interacting protein degrades tau regardless its phosphorylation status without affecting the spatial memory of the rats. J Neural Transm. (2008) 115:483–91. doi: 10.1007/s00702-007-0857-7
105. Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. (2007) 32:95–100. doi: 10.1016/j.tibs.2006.12.002
106. Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. (2004) 24:8055–68. doi: 10.1128/MCB.24.18.8055-8068.2004
107. Seibenhener ML, Geetha T, Wooten MW. Sequestosome 1/p62 - more than just a scaffold. FEBS Lett. (2007) 581:175–9. doi: 10.1016/j.febslet.2006.12.027
108. Wang P, Joberty G, Buist A, Vanoosthuyse A, Stancu IC, Vasconcelos B, et al. Tau interactome mapping based identification of Otub1 as tau deubiquitinase involved in accumulation of pathological tau forms in vitro and in vivo. Acta Neuropathol. (2017) 133:731–49. doi: 10.1007/s00401-016-1663-9
109. Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. (1993) 10:1151–60. doi: 10.1016/0896-6273(93)90063-W
110. Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. (2006) 281:10825–38. doi: 10.1074/jbc.M512786200
111. Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. (1987) 235:1641–4. doi: 10.1126/science.3029875
112. Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci USA. (1987) 84:3033–6. doi: 10.1073/pnas.84.9.3033
113. Riederer IM, Schiffrin M, Kövari E, Bouras C, Riederer BM. Ubiquitination and cysteine nitrosylation during aging and Alzheimer's disease. Brain Res Bull. (2009) 80:233–41. doi: 10.1016/j.brainresbull.2009.04.018
114. Bancher C, Grundke-Iqbal I, Iqbal K, Fried VA, Smith HT, Wisniewski HM. Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res. (1991) 539:11–8. doi: 10.1016/0006-8993(91)90681-K
115. García-Sierra F, Jarero-Basulto JJ, Kristofikova Z, Majer E, Binder LI, Ripova D. Ubiquitin is associated with early truncation of tau protein at aspartic acid 421 during the maturation of neurofibrillary tangles in Alzheimer's disease. Brain Pathol. (2012) 22:240–50. doi: 10.1111/j.1750-3639.2011.00525.x
116. Iwatsubo T, Hasegawa M, Esaki Y, Ihara Y. Lack of ubiquitin immunoreactivities at both ends of neuropil threads possible bidirectional growth of neuropil threads. Am J Pathol. (1992) 140:277–82.
117. Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, et al. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci. (2006) 26:6985–96. doi: 10.1523/JNEUROSCI.0746-06.2006
118. Desterro JMP, Thomson J, Hay RT. Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. (1997) 417:297–300. doi: 10.1016/S0014-5793(97)01305-7
119. Gong L, Li B, Millas S, Yeh ETH. Molecular cloning and characterization of human AOS1 and UBA2, components of the sentrin-activating enzyme complex. FEBS Lett. (1999) 448:185–9. doi: 10.1016/S0014-5793(99)00367-1
120. Sarge KD, Park-Sarge OK. Sumoylation and human disease pathogenesis. Trends Biochem Sci. (2009) 34:200–5. doi: 10.1016/j.tibs.2009.01.004
121. Müller S, Ledl A, Schmidt D. SUMO: a regulator of gene expression and genome integrity. Oncogene. (2004) 23:1998–2008. doi: 10.1038/sj.onc.1207415
122. Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. (2013) 49:795–807. doi: 10.1016/j.molcel.2013.01.017
123. Melchior F, Schergaut M, Pichler A. SUMO: ligases, isopeptidases and nuclear pores. Trends Biochem Sci. (2003) 28:612–8. doi: 10.1016/j.tibs.2003.09.002
124. Hickey CM, Wilson NR, Hochstrasser M. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol. (2012) 13:755–66. doi: 10.1038/nrm3478
125. Henley JM, Carmichael RE, Wilkinson KA. Extranuclear SUMOylation in neurons. Trends Neurosci. (2018) 41:198–210. doi: 10.1016/j.tins.2018.02.004
126. Pountney DL, Huang Y, Burns RJ, Haan E, Thompson PD, Blumbergs PC, et al. SUMO-1 marks the nuclear inclusions in familial neuronal intranuclear inclusion disease. Exp Neurol. (2003) 184:436–46. doi: 10.1016/j.expneurol.2003.07.004
127. Wong MB, Goodwin J, Norazit A, Meedeniya ACB, Richter-Landsberg C, Gai WP, et al. SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotox Res. (2013) 23:1–21. doi: 10.1007/s12640-012-9358-z
128. Luo H Bin, Xia YY, Shu XJ, Liu ZC, Feng Y, Liu XH, et al. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc Natl Acad Sci USA. (2014) 111:16586–91. doi: 10.1073/pnas.1417548111
129. Dorval V, Fraser PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and α-synuclein. J Biol Chem. (2006) 281:9919–24. doi: 10.1074/jbc.M510127200
130. Hernandez I, Luna G, Rauch JN, Reis SA, Giroux M, Karch CM, et al. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci Transl Med. (2019) 11:eaat3005. doi: 10.1126/scitranslmed.aat3005
131. Avery OT, Macleod CM, McCarty M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type iii. J Exp Med. (1944) 79:137–58.
132. MacCarty M, AVery OT. Studies on the chemical nature of the substance inducing transformation of pneumococcoal types: effect of desoxyribonuclease on the biological activit of the transforming substance. J Exp Med. (1946) 83:89–96.
133. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. (2013) 38:23–38. doi: 10.1038/npp.2012.112
134. Murn J, Shi Y. The winding path of protein methylation research: milestones and new frontiers. Nat Rev Mol Cell Biol. (2017) 18:517–27. doi: 10.1038/nrm.2017.35
135. Serre NBC, Alban C, Bourguignon J, Ravanel S. An outlook on lysine methylation of non-histone proteins in plants. J Exp Bot. (2018) 69:4569–81. doi: 10.1093/jxb/ery231
136. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. (2004) 119:941–53. doi: 10.1016/j.cell.2004.12.012
137. Lanouette S, Mongeon V, Figeys D, Couture JF. The functional diversity of protein lysine methylation. Mol Syst Biol. (2014) 10:724. doi: 10.1002/msb.134974
138. Falnes P, Jakobsson ME, Davydova E, Ho A, Malecki J. Protein lysine methylation by seven-β-strand methyltransferases. Biochem J. (2016) 473:1995–2009. doi: 10.1042/BCJ20160117
139. Kontaxi C, Piccardo P, Gill AC. Lysine-directed post-translational modifications of tau protein in Alzheimer's disease and related tauopathies. Front Mol Biosci. (2017) 4:56. doi: 10.3389/fmolb.2017.00056
140. Huseby CJ, Hoffman CN, Cooper GL, Cocuron JC, Alonso AP, Thomas SN, et al. Quantification of tau protein lysine methylation in aging and Alzheimer's disease. J Alzheimer's Dis. (2019) 71:979–91. doi: 10.3233/JAD-190604
141. Funk KE, Thomas SN, Schafer KN, Cooper GL, Liao Z, Clark DJ, et al. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem J. (2014) 462:77–88. doi: 10.1042/BJ20140372
142. Sposito T, Preza E, Mahoney CJ, Setó-Salvia N, Ryan NS, Morris HR, et al. Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum Mol Genet. (2015) 24:5260–9. doi: 10.1093/hmg/ddv246
143. Shental-Bechor D, Levy Y. Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc Natl Acad Sci USA. (2008) 105:8256–61. doi: 10.1073/pnas.0801340105
144. Bell CG, Lowe R, Adams PD, Baccarelli AA, Beck S, Bell JT, et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. (2019) 20:1–24. doi: 10.1186/s13059-019-1824-y
145. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. (2017) 18:452–65. doi: 10.1038/nrm.2017.22
146. Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol. (2019) 15:346–66. doi: 10.1038/s41581-019-0129-4
147. Kim C-S, Park S, Kim J. The role of glycation in the pathogenesis of aging and its prevention through herbal products and physical exercise. J Exerc Nutr Biochem. (2017) 21:55–61. doi: 10.20463/jenb.2017.0027
148. Liu L, Xu YX, Hirschberg CB. The role of nucleotide sugar transporters in development of eukaryotes. Semin Cell Dev Biol. (2010) 21:600–8. doi: 10.1016/j.semcdb.2010.02.002
149. Aebi M. N-linked protein glycosylation in the ER. Biochim Biophys Acta Mol Cell Res. (2013) 1833:2430–7. doi: 10.1016/j.bbamcr.2013.04.001
150. Sharma A, Vijayan M. Influence of glycosidic linkage on the nature of carbohydrate binding in β-prism I fold lectins: an X-ray and molecular dynamics investigation on banana lectin-carbohydrate complexes. Glycobiology. (2010) 21:23–33. doi: 10.1093/glycob/cwq128
151. Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. (2002) 12:43R−56R. doi: 10.1093/glycob/12.4.43R
152. Haltiwanger RS, Kelly WG, Roquemore EP, Blomberg MA, Dong L-YD, Kreppel L, et al. Glycosylation of nuclear and cytoplasmic proteins is ubiquitous and dynamic. Biochem Soc Trans. (1992) 20:264–9. doi: 10.1042/bst0200264
153. Mitra N, Sinha S, Ramya TNC, Surolia A. N-linked oligosaccharides as outfitters for glycoprotein folding, form and function. Trends Biochem Sci. (2006) 31:156–63. doi: 10.1016/j.tibs.2006.01.003
154. O'Connor SE, Imperiali B. Modulation of protein structure and function by asparagine-linked glycosylation. Chem Biol. (1996) 3:803–12. doi: 10.1016/S1074-5521(96)90064-2
155. Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci USA. (2004) 101:10804–9. doi: 10.1073/pnas.0400348101
156. Cheng X, Cole RN, Zaia J, Hart GW. Alternative O-glycosylation/O-phosphorylation of the murine estrogen receptor β. Biochemistry. (2000) 39:11609–20. doi: 10.1021/bi000755i
157. Comer FI, Hart GW. Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry. (2001) 40:7845–52. doi: 10.1021/bi0027480
158. Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol. (2006) 8:1074–83. doi: 10.1038/ncb1470
159. Tarrant MK, Rho HS, Xie Z, Jiang YL, Gross C, Culhane JC, et al. Regulation of CK2 by phosphorylation and O-GlcNAcylation revealed by semisynthesis. Nat Chem Biol. (2012) 8:262–9. doi: 10.1038/nchembio.771
160. Cole SL, Vassar R. The Alzheimer's disease β-secretase enzyme, BACE1. Mol Neurodegener. (2007) 2:22. doi: 10.1186/1750-1326-2-22
161. Kizuka Y, Kitazume S, Fujinawa R, Saito T, Iwata N, Saido TC, et al. An aberrant sugar modification of BACE 1 blocks its lysosomal targeting in A lzheimer's disease. EMBO Mol Med. (2015) 7:175–89. doi: 10.15252/emmm.201404438
162. Chun YS, Kwon OH, Chung S. O-GlcNAcylation of amyloid-β precursor protein at threonine 576 residue regulates trafficking and processing. Biochem Biophys Res Commun. (2017) 490:486–91. doi: 10.1016/j.bbrc.2017.06.067
163. Palmigiano A, Barone R, Sturiale L, Sanfilippo C, Bua RO, Romeo DA, et al. CSF N-glycoproteomics for early diagnosis in Alzheimer's disease. J Proteomics. (2016) 131:29–37. doi: 10.1016/j.jprot.2015.10.006
164. Kizuka Y, Kitazume S, Taniguchi N. N-glycan and Alzheimer's disease. Biochim Biophys Acta Gen Subj. (2017) 1861:2447–54. doi: 10.1016/j.bbagen.2017.04.012
165. Gong C-X, Liu F, Iqbal K. O-GlcNAcylation: a regulator of tau pathology and neurodegeneration. Alzheimer's Dement. (2016) 12:1078–89. doi: 10.1016/j.jalz.2016.02.011
166. Shane Arnold C, Johnson GVW, Cole RN, Dong DLY, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O- linked N-acetylglucosamine. J Biol Chem. (1996) 271:28741–4. doi: 10.1074/jbc.271.46.28741
167. Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer's disease. Nat Med. (1996) 2:871–5. doi: 10.1038/nm0896-871
168. Sato Y, Naito Y, Grundke-Iqbal I, Iqbal K, Endo T. Analysis of N-glycans of pathological tau: Possible occurrence of aberrant processing of tau in Alzheimer's disease. FEBS Lett. (2001) 496:152–60. doi: 10.1016/S0014-5793(01)02421-8
169. Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Merkle RK, Gong CX. Role of glycosylation in hyperphosphorylation of tau in Alzheimer's disease. FEBS Lett. (2002) 512:101–6. doi: 10.1016/S0014-5793(02)02228-7
170. Robertson LA, Moya KL, Breen KC. The potential role of tau protein O-glycosylation in Alzheimer's disease. J Alzheimer's Dis. (2004) 6:489–95. doi: 10.3233/JAD-2004-6505
171. Yuzwa SA, Shan X, MacAuley MS, Clark T, Skorobogatko Y, Vosseller K, et al. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. (2012) 8:393–9. doi: 10.1038/nchembio.797
172. Yuzwa SA, Yadav AK, Skorobogatko Y, Clark T, Vosseller K, Vocadlo DJ. Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody. Amino Acids. (2011) 40:857–68. doi: 10.1007/s00726-010-0705-1
173. Smet-Nocca C, Broncel M, Wieruszeski JM, Tokarski C, Hanoulle X, Leroy A, et al. Identification of O-GlcNAc sites within peptides of the tau protein and their impact on phosphorylation. Mol Biosyst. (2011) 7:1420–9. doi: 10.1039/c0mb00337a
174. Lefebvre T, Ferreira S, Dupont-Wallois L, Bussière T, Dupire MJ, Delacourte A, et al. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of tau proteins - a role in nuclear localization. Biochim Biophys Acta Gen Subj. (2003) 1619:167–76. doi: 10.1016/S0304-4165(02)00477-4
175. Yuzwa SA, Cheung AH, Okon M, McIntosh LP, Vocadlo DJ. O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J Mol Biol. (2014) 426:1736–52. doi: 10.1016/j.jmb.2014.01.004
176. Yu CH, Si T, Wu WH, Hu J, Du JT, Zhao YF, et al. O-GlcNAcylation modulates the self-aggregation ability of the fourth microtubule-binding repeat of tau. Biochem Biophys Res Commun. (2008) 375:59–62. doi: 10.1016/j.bbrc.2008.07.101
177. Schedin-Weiss S, Winblad B, Tjernberg LO. The role of protein glycosylation in Alzheimer disease. FEBS J. (2014) 281:46–62. doi: 10.1111/febs.12590
178. Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Gong CX. Aberrant glycosylation modulates phosphorylation of tau by protein kinase A and dephosphorylation of tau by protein phosphatase 2A and 5. Neuroscience. (2002) 115:829–37. doi: 10.1016/S0306-4522(02)00510-9
179. Cho S-J, Roman G, Yeboah F, Konishi Y. The road to advanced glycation end products: a mechanistic perspective. Curr Med Chem. (2007) 14:1653–71. doi: 10.2174/092986707780830989
180. Sasaki N, Fukatsu R, Tsuzuki K, Hayashi Y, Yoshida T, Fujii N, et al. Advanced glycation end products in Alzheimer's disease and other neurodegenerative diseases. Am J Pathol. (1998) 153:1149–55. doi: 10.1016/S0002-9440(10)65659-3
181. Yamagishi SI, Nakamura N, Matsui T. Glycation and cardiovascular disease in diabetes: a perspective on the concept of metabolic memory. J Diabetes. (2017) 9:141–8. doi: 10.1111/1753-0407.12475
182. Simm A, Müller B, Nass N, Hofmann B, Bushnaq H, Silber RE, et al. Protein glycation - between tissue aging and protection. Exp Gerontol. (2015) 68:71–5. doi: 10.1016/j.exger.2014.12.013
183. Luth H-JL, Ogunlade V, Kuhla BB, Kientsch-Engel R, Stahl P, Webster J, et al. Age-and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer's disease brains. Cereb Cortex. (2005) 15:211–20. doi: 10.1093/cercor/bhh123
184. Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D, Bose N, et al. The role of advanced glycation end products in aging and metabolic diseases: bridging association and causality. Cell Metab. (2018) 28:337–52. doi: 10.1016/j.cmet.2018.08.014
185. Ramasamy R, Vannucci SJ, Yan SS Du, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. (2005) 15:16R−28R. doi: 10.1093/glycob/cwi053
186. Nacharaju P, Ko L, Yen S-HC. Characterization of in vitro glycation sites of tau. J Neurochem. (1997) 69:1709–19. doi: 10.1046/j.1471-4159.1997.69041709.x
187. Liu K, Liu Y, Li L, Qin P, Iqbal J, Deng Y, et al. Glycation alter the process of tau phosphorylation to change tau isoforms aggregation property. Biochim Biophys Acta Mol Basis Dis. (2016) 1862:192–201. doi: 10.1016/j.bbadis.2015.12.002
188. Ko LW, Ko EC, Nacharaju P, Liu WK, Chang E, Kenessey A, et al. An immunochemical study on tau glycation in paired helical filaments. Brain Res. (1999) 830:301–13. doi: 10.1016/S0006-8993(99)01415-8
189. Ledesma MD, Medina M, Avila J. The in vitro formation of recombinant τ polymers - effect of phosphorylation and glycation. Mol Chem Neuropathol. (1996) 27:249–58. doi: 10.1007/BF02815107
190. Smith MA, Siedlak SL, Richey PL, Nagaraj RH, Elhammer A, Perry G. Quantitative solubilization and analysis of insoluble paired helical filaments from Alzheimer disease. Brain Res. (1996) 717:99–108. doi: 10.1016/0006-8993(95)01473-X
191. Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS, et al. Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci USA. (1994) 91:7787–91. doi: 10.1073/pnas.91.16.7787
192. Quinn JP, Corbett NJ, Kellett KAB, Hooper NM. Tau proteolysis in the pathogenesis of tauopathies: neurotoxic fragments and novel biomarkers. J Alzheimer's Dis. (2018) 63:13–33. doi: 10.3233/JAD-170959
193. van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. (2019) 50:1352–64. doi: 10.1016/j.immuni.2019.05.020
194. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. (2013) 5:ea008656. doi: 10.1101/cshperspect.a008656
195. Chesser AS, Pritchard SM, Johnson GVW. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front Neurol. (2013) 4:122. doi: 10.3389/fneur.2013.00122
196. Fasulo L, Visintin M, Novak M, Cattaneo A. Tau truncation in Alzheimer's disease: expression of a fragment encompassing PHF core. Alzheimer's Rep. (1998) 1:25–32.
197. Novák M. Truncated tau protein as a new marker for Alzheimer's disease. Acta Virol. (1994) 38:173–89.
198. Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA. (2003) 100:10032–7. doi: 10.1073/pnas.1630428100
199. Paholikova K, Salingova B, Opattova A, Skrabana R, Majerova P, Zilka N, et al. N-terminal truncation of microtubule associated protein tau dysregulates its cellular localization. J Alzheimer's Dis. (2015) 43:915–26. doi: 10.3233/JAD-140996
200. Corsetti V, Florenzano F, Atlante A, Bobba A, Ciotti MT, Natale F, et al. NH 2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer's disease. Hum Mol Genet. (2015) 24:3058–81. doi: 10.1093/hmg/ddv059
201. Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, et al. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis. (2001) 8:162–72. doi: 10.1006/nbdi.2000.0335
202. Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. (2004) 114:121–30. doi: 10.1172/JCI200420640
203. Rohn TT, Rissman RA, Head E, Cotman CW. Caspase activation in the Alzheimer's disease brain: tortuous and torturous. Drug News Perspect. (2002) 15:549–57. doi: 10.1358/dnp.2002.15.9.740233
204. Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW. Activated caspase-3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res. (2001) 898:350–7. doi: 10.1016/S0006-8993(01)02018-2
205. Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer's disease. Am J Pathol. (2004) 165:523–31. doi: 10.1016/S0002-9440(10)63317-2
206. Matthews-Roberson TA, Quintanilla RA, Ding H, Johnson GVW. Immortalized cortical neurons expressing caspase-cleaved tau are sensitized to endoplasmic reticulum stress induced cell death. Brain Res. (2008) 1234:206–12. doi: 10.1016/j.brainres.2008.07.111
207. Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnsion GVW. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of alzheimer disease. J Biol Chem. (2009) 284:18754–66. doi: 10.1074/jbc.M808908200
208. De Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature. (2010) 464:1201–4. doi: 10.1038/nature08890
209. Suzuki K, Hata S, Kawabata Y, Sorimachi H. Structure, activation, and biology of calpain. Diabetes. (2004). 53(Suppl.):S12–8. doi: 10.2337/diabetes.53.2007.S12
210. Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates β-amyloid-induced neurodegeneration. J Neurosci. (2005) 25:5365–75. doi: 10.1523/JNEUROSCI.1125-05.2005
211. Liu MC, Kobeissy F, Zheng W, Zhang Z, Hayes RL, Wang KKW. Dual vulnerability of tau to calpains and caspase-3 proteolysis under neurotoxic and neurodegenerative conditions. ASN Neuro. (2011) 3:25–36. doi: 10.1042/AN20100012
212. Chen HH, Liu P, Auger P, Lee SH, Adolfsson O, Rey-Bellet L, et al. Calpain-mediated tau fragmentation is altered in Alzheimer's disease progression. Sci Rep. (2018) 8:16725. doi: 10.1038/s41598-018-35130-y
213. Reinecke JB, deVos SL, McGrath JP, Shepard AM, Goncharoff DK, Tait DN, et al. Implicating calpain in tau-mediated toxicity in vivo. PLoS ONE. (2011) 6:e23865. doi: 10.1371/journal.pone.0023865
214. Olesen OF. Proteolytic degradation of microtubule-associated protein τ by thrombin. Biochem Biophys Res Commun. (1994) 201:716–21. doi: 10.1006/bbrc.1994.1759
215. Khlistunova I, Biernat J, Wang Y, Pickhardt M, Von Bergen M, Gazova Z, et al. Inducible expression of tau repeat domain in cell models of tauopathy: Aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. (2006) 281:1205–14. doi: 10.1074/jbc.M507753200
216. Arai T, Guo JP, McGeer PL. Proteolysis of non-phosphorylated and phosphorylated tau by thrombin. J Biol Chem. (2005) 280:5145–53. doi: 10.1074/jbc.M409234200
217. Suo Z, Wu M, Citron BA, Palazzo RE, Festoff BW. Rapid tau aggregation and delayed hippocampal neuronal death induced by persistent thrombin signaling. J Biol Chem. (2003) 278:37681–9. doi: 10.1074/jbc.M301406200
218. Schönichen A, Webb BA, Jacobson MP, Barber DL. Considering protonation as a posttranslational modification regulating protein structure and function. Annu Rev Biophys. (2013) 42:289–314. doi: 10.1146/annurev-biophys-050511-102349
219. Harguindey S, Reshkin S, Orive G, Luis Arranz J, Anitua E. Growth and trophic factors, pH and the Na+/H+ exchanger in Alzheimers disease, other neurodegenerative diseases and cancer: new therapeutic possibilities and potential dangers. Curr Alzheimer Res. (2007) 4:53–65. doi: 10.2174/156720507779939841
220. Syntichaki P, Samara C, Tavernarakis N. The vacuolar H+-ATPase mediates intracellular acidification required for neurodegeneration in C. elegans. Curr Biol. (2005) 15:1249–54. doi: 10.1016/j.cub.2005.05.057
221. Charafeddine RA, Cortopassi WA, Lak P, Tan R, McKenney RJ, Jacobson MP, et al. Tau repeat regions contain conserved histidine residues that modulate microtubule-binding in response to changes in pH downloaded from. J Biol Chem. (2019) 294:8779–90. doi: 10.1074/jbc.RA118.007004
222. Chung HS, Wang SB, Venkatraman V, Murray CI, van Eyk JE. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res. (2013) 112:382–92. doi: 10.1161/CIRCRESAHA.112.268680
223. Reeg S, Grune T. Protein oxidation in aging: does it play a role in aging progression? Antioxidants Redox Signal. (2015) 23:239–55. doi: 10.1089/ars.2014.6062
224. Schweers O, Mandelkow EM, Biernat J, Mandelkow E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein τ controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci USA. (1995) 92:8463–7. doi: 10.1073/pnas.92.18.8463
225. Reynolds MR, Berry RW, Binder LI. Nitration in neurodegeneration: deciphering the “hows” “nYs.” Biochemistry. (2007) 46:7325–36. doi: 10.1021/bi700430y
226. Tedeschi G, Cappelletti G, Nonnis S, Taverna F, Negri A, Ronchi C, et al. Tyrosine nitration is a novel post-translational modification occurring on the neural intermediate filament protein peripherin. Neurochem Res. (2007) 32:433–41. doi: 10.1007/s11064-006-9244-2
227. Radi R. Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc Chem Res. (2013) 46:550–9. doi: 10.1021/ar300234c
228. Reyes JF, Fu Y, Vana L, Kanaan NM, Binder LI. Tyrosine nitration within the proline-rich region of tau in Alzheimer's disease. Am J Pathol. (2011) 178:2275–85. doi: 10.1016/j.ajpath.2011.01.030
229. Reyes JF, Geula C, Vana L, Binder LI. Selective tau tyrosine nitration in non-AD tauopathies. Acta Neuropathol. (2012) 123:119–32. doi: 10.1007/s00401-011-0898-8
230. Reyes JF, Reynolds MR, Horowitz PM, Fu Y, Guillozet-Bongaarts AL, Berry R, et al. A possible link between astrocyte activation and tau nitration in Alzheimer's disease. Neurobiol Dis. (2008) 31:198–208. doi: 10.1016/j.nbd.2008.04.005
231. Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. (2019) 20:421–35. doi: 10.1038/s41580-019-0101-y
232. Aoki K, Yoshida K. “Biological Consequences of Priming Phosphorylation in Cancer Development,” in Protein Phosphorylation (InTech).
233. Butler VJ, Salazar DA, Soriano-Castell D, Alves-Ferreira M, Dennissen FJA, Vohra M, et al. Tau/MAPT disease-associated variant A152T alters tau function and toxicity via impaired retrograde axonal transport. Hum Mol Genet. (2019) 28:1498–514. doi: 10.1093/hmg/ddy442
234. Kirchner P, Bourdenx M, Madrigal-Matute J, Tiano S, Diaz A, Bartholdy BA, et al. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. (2019) 17:e3000301. doi: 10.1371/journal.pbio.3000301
235. Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. (2009) 187:1083–1099. doi: 10.1083/jcb.200909067
236. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. (2011) 42:719–30. doi: 10.1016/j.molcel.2011.04.025
237. Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. (2009) 30:191–296. doi: 10.1016/j.mam.2009.04.001
238. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. (2002) 82:373–428. doi: 10.1152/physrev.00027.2001
239. Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Biol. (2009) 10:104–15. doi: 10.1038/nrm2630
240. Coux O, Tanaka K, Goldberg AL. Structure and Functions of the 20S and 26S proteasomes. Annu Rev Biochem. (1996) 65:801–47. doi: 10.1146/annurev.bi.65.070196.004101
241. Jariel-Encontre I, Bossis G, Piechaczyk M. Ubiquitin-independent degradation of proteins by the proteasome. BBA Rev Cancer. (2008) 1786:153–77. doi: 10.1016/j.bbcan.2008.05.004
242. Wang Y, Mandelkow E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans. (2012) 40:644–52. doi: 10.1042/BST20120071
243. Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. (2007) 117:648–58. doi: 10.1172/JCI29715
244. Holt LJ. Regulatory modules: Coupling protein stability to phopshoregulation during cell division. FEBS Lett. (2012) 586:2773–7. doi: 10.1016/j.febslet.2012.05.045
245. Engmann O, Giese KP. Crosstalk between Cdk5 and GSK3β: implications for Alzheimer's disease. Front Mol Neurosci. (2009) 2:9. doi: 10.3389/neuro.02.002.2009
246. Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem. (2003) 85:115–22. doi: 10.1046/j.1471-4159.2003.01642.x
247. Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. (2000) 75:436–9. doi: 10.1046/j.1471-4159.2000.0750436.x
248. López Salon M, Morelli L, Castaño EM, Soto EF, Pasquini JM. Defective ubiquitination of cerebral proteins in Alzheimer's disease. J Neurosci Res. (2000) 62:302–10. doi: 10.1002/1097-4547(20001015)62:2<302::AID-JNR15>3.0.CO;2-L
249. Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. (2001) 292:1552–5. doi: 10.1126/science.292.5521.1552
250. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. (2003) 426:895–9. doi: 10.1038/nature02263
251. Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, et al. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell. (2018) 17:e12692. doi: 10.1111/acel.12692
252. Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. (2009) 18:4153–70. doi: 10.1093/hmg/ddp367
253. Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C, et al. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. (2008) 27:1119–30. doi: 10.1111/j.1460-9568.2008.06084.x
254. Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, et al. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy. (2010) 6:182–3. doi: 10.4161/auto.6.1.10815
255. Kim SI, Lee WK, Kang SS, Lee SY, Jeong MJ, Lee HJ, et al. Suppression of autophagy and activation of glycogen synthase kinase 3beta facilitate the aggregate formation of tau. Korean J Physiol Pharmacol. (2011) 15:107–14. doi: 10.4196/kjpp.2011.15.2.107
256. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. (2011) 20:131–9. doi: 10.1016/j.devcel.2010.12.003
257. Gorantla NV, Chinnathambi S. Autophagic pathways to clear the tau aggregates in Alzheimer's disease. Cell Mol Neurobiol. (2020). doi: 10.1007/s10571-020-00897-0. [Epub ahead of print].
258. Vaz-Silva J, Gomes P, Jin Q, Zhu M, Zhuravleva V, Quintremil S, et al. Endolysosomal degradation of tau and its role in glucocorticoid-driven hippocampal malfunction. EMBO J. (2018) 37:e99084. doi: 10.15252/embj.201899084
259. Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. (2005) 12:1542–52. doi: 10.1038/sj.cdd.4401765
260. Dolan PJ, Johnson GVW. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem. (2010) 285:21978–87. doi: 10.1074/jbc.M110.110940
261. Rohn TT, Wirawan E, Brown RJ, Harris JR, Masliah E, Vandenabeele P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer's disease brain. Neurobiol Dis. (2011) 43:68–78. doi: 10.1016/j.nbd.2010.11.003
262. Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. (2005) 64:113–22. doi: 10.1093/jnen/64.2.113
263. Piras A, Collin L, Grüninger F, Graff C, Rönnbäck A. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun. (2016) 4:22. doi: 10.1186/s40478-016-0292-9
264. Caccamo A, Magrì A, Medina DX, Wisely E V, López-Aranda MF, Silva AJ, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer's disease and other tauopathies. Aging Cell. (2013) 12:370–80. doi: 10.1111/acel.12057
265. Ikeda K, Akiyama H, Arai T, Kondo H, Haga C, Tsuchiya K, et al. Neurons containing Alz-50-immunoreactive granules around the cerebral infarction: evidence for the lysosomal degradation of altered tau in human brain? Neurosci Lett. (2000) 284:187–9. doi: 10.1016/S0304-3940(00)01009-0
266. Ikeda K, Akiyama H, Arai T, Kondo H, Haga C, Iritani S, et al. Alz-50/Gallyas-positive lysosome-like intraneuronal granules in Alzheimer's disease and control brains. Neurosci Lett. (1998) 258:113–6. doi: 10.1016/S0304-3940(98)00867-2
267. D'Agostino C, Nogalska A, Cacciottolo M, King Engel W, Askanas V. Abnormalities of NBR1, a novel autophagy-associated protein, in muscle fibers of sporadic inclusion-body myositis. Acta Neuropathol. (2011) 122:627–36. doi: 10.1007/s00401-011-0874-3
268. Majid T, Ali YO, Venkitaramani DV, Jang MK, Lu HC, Pautler RG. in vivo axonal transport deficits in a mouse model of fronto-temporal dementia. NeuroImage Clin. (2014) 4:711–7. doi: 10.1016/j.nicl.2014.02.005
269. Butzlaff M, Hannan SB, Karsten P, Lenz S, Ng J, Voßfeldt H, et al. Impaired retrograde transport by the dynein/dynactin complex contributes to tau-induced toxicity. Hum Mol Genet. (2015) 24:3623–37. doi: 10.1093/hmg/ddv107
270. Young ZT, Mok SA, Gestwicki JE. Therapeutic strategies for restoring tau homeostasis. Cold Spring Harb Perspect Med. (2018) 8:ea024612. doi: 10.1101/cshperspect.a024612
271. Schweers O, Schonbrunn-Hanebeck E, Man A, Mandelkows E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. (1994) 269:24290–7.
272. Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, et al. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. (2009) 7:e1000034. doi: 10.1371/journal.pbio.1000034
273. Jeganathan S, Von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. (2006) 45:2283–93. doi: 10.1021/bi0521543
274. Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. (1999) 24:329–32. doi: 10.1016/S0968-0004(99)01445-0
276. Knowles TPJ, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. (2014) 15:384–96. doi: 10.1038/nrm3810
277. Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem. (2017) 86:27–68. doi: 10.1146/annurev-biochem-061516-045115
278. Fitzpatrick AW, Saibil HR. Cryo-EM of amyloid fibrils and cellular aggregates. Curr Opin Struct Biol. (2019) 58:34–42. doi: 10.1016/j.sbi.2019.05.003
279. Scheres SH, Zhang W, Falcon B, Goedert M. Cryo-EM structures of tau filaments. Curr Opin Struct Biol. (2020) 64:17–25. doi: 10.1016/j.sbi.2020.05.011
280. Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, et al. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature. (2018) 561:137–40. doi: 10.1038/s41586-018-0454-y
281. Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature. (2017) 547:185–90. doi: 10.1038/nature23002
282. Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, et al. Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell. (2020) 180:633–44.e12. doi: 10.1016/j.cell.2020.01.027
283. Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. (2019) 568:420–3. doi: 10.1038/s41586-019-1026-5
284. Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, et al. Novel tau filament fold in corticobasal degeneration. Nature. (2020) 580:283–7. doi: 10.1038/s41586-020-2043-0
285. Fontaine SN, Sabbagh JJ, Baker J, Martinez-Licha CR, Darling A, Dickey CA. Cellular factors modulating the mechanism of tau protein aggregation. Cell Mol Life Sci. (2015) 72:1863–79. doi: 10.1007/s00018-015-1839-9
286. Wille H, Drewes G, Biernat J, Mandelkow EM, Mandelkow E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J Cell Biol. (1992) 118:573–84. doi: 10.1083/jcb.118.3.573
287. Crowther RA, Olesen OF, Smith MJ, Jakes R, Goedert M. Assembly of Alzheimer-like filaments from full-length tau protein. FEMS Lett. (1994) 337:135–8. doi: 10.1016/0014-5793(94)80260-2
288. Wilson DM, Binder LI. Polymerization of microtubule-associated protein tau under near-physiological conditions. J Biol Chem. (1995) 270:24306–14. doi: 10.1074/jbc.270.41.24306
289. Reynolds MR, Reyes JF, Fu Y, Bigio EH, Guillozet-Bongaarts AL, Berry RW, et al. Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer's disease and other tauopathies. J Neurosci. (2006) 26:10636–45. doi: 10.1523/JNEUROSCI.2143-06.2006
290. Trzeciakiewicz H, Tseng JH, Wander CM, Madden V, Tripathy A, Yuan CX, et al. A dual pathogenic mechanism links tau acetylation to sporadic tauopathy. Sci Rep. (2017) 7:44102. doi: 10.1038/srep44102
291. Avila J. Tau aggregation into fibrillar polymers: taupathies. FEBS Lett. (2000) 476:89–92. doi: 10.1016/S0014-5793(00)01676-8
292. Sinha S, Lopes DHJ, Du Z, Pang ES, Shanmugam A, Lomakin A, et al. Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J Am Chem Soc. (2011) 133:16958–69. doi: 10.1021/ja206279b
293. West LE, Gozani O. Regulation of p53 function by lysine methylation. Epigenomics. (2011) 3:361–9. doi: 10.2217/epi.11.21
294. Bhattacharya K, Rank KB, Evans DB, Sharma SK. Role of cysteine-291 and cysteine-322 in the polymerization of human tau into Alzheimer-like filaments. Biochem Biophys Res Commun. (2001) 285:20–6. doi: 10.1006/bbrc.2001.5116
295. Necula M, Kuret J. Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. J Biol Chem. (2004) 279:49694–703. doi: 10.1074/jbc.M405527200
296. Brister MA, Pandey AK, Bielska AA, Zondlo NJ. OGlcNAcylation and phosphorylation have opposing structural effects in tau: phosphothreonine induces particular conformational order. J Am Chem Soc. (2014) 136:3803–16. doi: 10.1021/ja407156m
297. Lothrop AP, Torres MP, Fuchs SM. Deciphering post-translational modification codes. FEBS Lett. (2013) 587:1247–57. doi: 10.1016/j.febslet.2013.01.047
298. Li X, Lu F, Wang J-Z, Gong C-X. Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur J Neurosci. (2006) 23:2078–86. doi: 10.1111/j.1460-9568.2006.04735.x
299. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. (2017) 133:665–704. doi: 10.1007/s00401-017-1707-9
300. Guillozet-Bongaarts AL, Cahill ME, Cryns VL, Reynolds MR, Berry RW, Binder LI. Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo. J Neurochem. (2006) 97:1005–14. doi: 10.1111/j.1471-4159.2006.03784.x
301. Kimura T, Sharma G, Ishiguro K, Hisanaga S. Phospho-tau bar code: analysis of phosphoisotypes of tau and its application to tauopathy. Front Neurosci. (2018) 12:44. doi: 10.3389/fnins.2018.00044
302. Mair W, Muntel J, Tepper K, Tang S, Biernat J, Seeley WW, et al. FLEXITau: quantifying post-translational modifications of tau protein in vitro and in human disease. Anal Chem. (2016) 88:3704–14. doi: 10.1021/acs.analchem.5b04509
303. Hanger DP, Byers HL, Wray S, Leung KY, Saxton MJ, Seereeram A, et al. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem. (2007) 282:23645–54. doi: 10.1074/jbc.M703269200
304. Dammer EB, Lee AK, Duong DM, Gearing M, Lah JJ, Levey AI, et al. Quantitative phosphoproteomics of Alzheimer's disease reveals cross-talk between kinases and small heat shock proteins. Proteomics. (2015) 15:508–19. doi: 10.1002/pmic.201400189
305. Kang MJ, Kim C, Jeong H, Cho BK, Ryou AL, Hwang D, et al. Synapsin-1 and tau reciprocal O-GlcNAcylation and phosphorylation sites in mouse brain synaptosomes. Exp Mol Med. (2013) 45:e29. doi: 10.1038/emm.2013.56
306. Hanger DP, Betts JC, Loviny TLF, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (Paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J Neurochem. (2002) 71:2465–76. doi: 10.1046/j.1471-4159.1998.71062465.x
307. Doll S, Burlingame AL. Mass spectrometry-based detection and assignment of protein posttranslational modifications. ACS Chem Biol. (2015) 10:63–71. doi: 10.1021/cb500904b
308. Theillet FX, Smet-Nocca C, Liokatis S, Thongwichian R, Kosten J, Yoon MK, et al. Cell signaling, post-translational protein modifications and NMR spectroscopy. J Biomol NMR. (2012) 54:217–36. doi: 10.1007/s10858-012-9674-x
309. Singh S, Springer M, Steen J, Kirschner MW, Steen H. FLEXIQuant: a novel tool for the absolute quantification of proteins, and the simultaneous identification and quantification of potentially modified peptides. J Proteome Res. (2009) 8:2201–10. doi: 10.1021/pr800654s
Keywords: phosphorylation, acetylation, ubiquitination, methylation, sumoylation, glycosylation, glycation, proteolysis
Citation: Alquezar C, Arya S and Kao AW (2021) Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 11:595532. doi: 10.3389/fneur.2020.595532
Received: 16 August 2020; Accepted: 07 December 2020;
Published: 07 January 2021.
Edited by:
Sonia Do Carmo, McGill University, CanadaReviewed by:
Chaur-Jong Hu, Taipei Medical University, TaiwanIsabelle Landrieu, INSERM U1167 Facteurs de risque et déterminants moléculaires des maladies liées au vieillissement, France
Copyright © 2021 Alquezar, Arya and Kao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aimee W. Kao, YWltZWUua2FvQHVjc2YuZWR1
†These authors have contributed equally to this work