 Yanqing Fei
Yanqing Fei Ruting Shi
Ruting Shi Zhi Song
Zhi Song Jinze Wu
Jinze Wu- 1Department of Neurology, The Third Xiangya Hospital, Central South University, Changsha, China
- 2Department of Rehabilitation, The Third Xiangya Hospital, Central South University, Changsha, China
Epilepsy is a common neurological disease that is not always controlled, and the ketogenic diet shows good antiepileptic effects drug-resistant epilepsy or seizures caused by specific metabolic defects via regulating the metabolism. The brain is a vital organ with high metabolic demands, and epileptic foci tend to exhibit high metabolic characteristics. Accordingly, there has been growing interest in the relationship between brain metabolism and epilepsy in recent years. To date, several new antiepileptic therapies targeting metabolic pathways have been proposed (i.e., inhibiting glycolysis, targeting lactate dehydrogenase, and dietary therapy). Promising strategies to treat epilepsy via modulating the brain's metabolism could be expected, while a lack of thorough understanding of the role of brain metabolism in the control of epilepsy remains. Herein, this review aims to provide insight into the state of the art concerning the brain's metabolic patterns and their association with epilepsy. Regulation of neuronal excitation via metabolic pathways and antiepileptic therapies targeting metabolic pathways are emphasized, which could provide a better understanding of the role of metabolism in epilepsy and could reveal potential therapeutic targets.
Introduction
Epilepsy is a brain disease with neurobiological, cognitive, psychological, and social consequences, characterized by an enduring pre-disposition to generate epileptic seizures (1). Regardless of its etiology, epilepsy is widely regarded as a disease of neuronal network excitability unbalance from altered ionic or synaptic transmission (2). Excessive synchronized discharge of neuronal networks causes epileptic seizures and is a specific manifestation of network excitability changes, such explosive electrical activity, that must be supported by enhancing metabolism (3, 4).
Most activities involve and are influenced by metabolism. Numerous individuals have various diseases caused by inborn or acquired metabolic dysfunctions. Over 500 inborn errors of metabolism were confirmed and affected ~1 in 2000 live births (5). Except for some typical metabolic endocrine diseases, many acquired diseases could involve systemic or focal metabolic changes that could be helpful in diagnosis or prognosis and as therapeutic targets (6–8). It is well-recognized that human health is strongly related to metabolism, but this has only started to attract sufficient attention in the last decade. Scientific developments have led to an in-depth understanding of metabolism and its role in pathophysiological processes. Metabolic changes both accompany diseases and can comprise therapeutic targets. Hippocrates first documented calorie restriction therapeutics to treat epilepsy (9). Based on this, the ketogenic diet emerged (10) and has played an important role in antiepileptic therapy (see section Dietary Therapy in Epilepsy).
Recently, neuroscientists have proposed several antiepileptic treatment methods that involve metabolic regulation (11–13). Technological developments have facilitated the detection of metabolic changes; then, epilepsy foci can be located (14). Today, researchers have acquired a larger understanding of the brain's metabolism and its roles in epilepsy, and recent findings have shown that there is a significant association between epilepsy and metabolism, with some researchers defining epilepsy as a metabolic disease (2, 15, 16). However, the relationship between metabolism and epilepsy has rarely been examined in detail. This review focused on the relationship between metabolism and epilepsy aiming to provide a better understanding of the role of metabolism in epilepsy and to reveal potential therapeutic targets.
Metabolic Features in the Brain
It is an established fact that the brain is the most developed part of the nervous system, controlling nearly all activity and adjusting the body to the external environment. Although the adult human brain accounts for only 2% of body weight, it consumes 20% percent of the body's oxygen. Interestingly, the mass of a child's brain (5-year-old) accounts for 6% of body weight and consumes 50% of the body's oxygen (17). Additionally, 12% of the cardiac output will flow to the brain (18). It was estimated that adenosine triphosphate (ATP) consumption in the gray matter of the brain is 30 mmol ATP/Kg tissue/min, which is close to the use of muscles in the human leg during a marathon (19). These data suggest that the brain has a stunning energy metabolism. While the metabolic properties of the brain are not limited to these, some unique metabolic characteristics are closely relevant to epilepsy treatment.
Glycolysis Is Essential for Brain/Neuronal Function
Glucose is the most important energy source in the brain, with some additional energy substrates having been reported, e.g., ketones and lactate (20, 21). These alternative energy substrates substitute glucose when glucose deficiency occurs, but they can only partially compensate for glucose. Evoked population spikes were attenuated by decreasing the glucose levels in a culture medium even without altering the intracellular concentrations of ATP or phosphocreatine (PCr); abnormal synaptic function could not fully recover by replacing glucose with pyruvate, lactate, or other energy substrates (22). This evidence showed that decreased glycolysis due to insufficient glucose concentration impairs neuronal function, and glycolysis is crucial for sustaining synaptic function. Conversely, to support increased synaptic vesicle circulation, the glycolysis levels in activated neurons were significantly increased (2-fold change from baseline), especially in the pre-synaptic terminals (23). These results may have been derived from brain slices but are suitable for the entire brain. Glucose does not consume oxygen in anaerobic glycolysis, while 1 mol of glucose consumes 6 mol of oxygen gas (O2) in aerobic oxidation: 1 glucose + 6O2 = 6CO2 + 6H2O. The ratio of O2/glucose consumed by the brain is called the oxygen–glucose index (OGI) and is maintained at a value close to six during rest, until stimulation leads to metabolic changes. Even if sufficient oxygen is delivered to the brain, the OGI decreases during brain activation by several types of stimulation, such as vigorous motor and complex cognitive tasks or by pathological conditions (seizures and depression). This marks a preferential increase in glycolysis in the activated brain, even if oxygen availability is sufficient (17). Furthermore, this may account for the antiepileptic effect of glycolysis inhibition (see section Inhibiting Glycolysis to Reduce Seizures).
Astrocyte-Neuron Lactate Shuttle (ANLS)
The cerebrum is composed of ~100 billion neurons and one trillion neuroglia cells. Astrocytic endfeet are a fundamental and important component of the blood–brain barrier (BBB); almost 99% of the surface of blood capillaries are enwrapped by astrocytes end feet (24), signifying that most of the neurons in the brain do not directly contact the capillaries. Then, how could neuronal energy uptake substrates and other nutritional materials sustain a high-intensity metabolism become a question. Although the classical theory holds that glucose is the main energy source for neurons, new viewpoints are emerging. In 1994, Pellerin and Magistretti first proposed the mechanism of ANLS (25) that has been frequently discussed in recent years and may thoroughly explain the neuroenergetics at the cellular and molecular levels.
Most neurons in the brain do not directly contact the capillaries; astrocytes are employed as a bridge to connect the capillaries and neurons and transport energy substrates. Glucose is absorbed by astrocytes through glucose transporters from the capillaries or extracellular fluid, and then translated to lactate via glycolysis. Lactate was traditionally regarded as a waste product of glycolysis. However, new studies have indicated that lactate is an important energy substrate for normal tissues and/or tumors (26, 27); in the brain, lactate produced in astrocytes fuels the mitochondrial tricarboxylic acid (TCA) cycle of neurons in a proprietary way, i.e., the ANLS (28). According to the ANLS, astrocytes absorb glucose and convert it to lactate. Then, lactate is transferred out of the astrocytes through the type 1 and 4 of monocarboxylate transporter (MCT1, 4) and carried into the neighboring neurons through MCT2. Lactate originating from the astrocytes is again catalyzed to pyruvate by lactate dehydrogenase, and the pyruvate could be carried into the mitochondria and be utilized as an energy metabolite in the Krebs cycle (Figure 1). Although this theory remains controversial, the concentration of lactate in astrocytes was significantly higher than that in neurons, and this lactate gradient provided a pre-condition for the flow of lactate from astrocytes to neurons mediated by carriers (31). Furthermore, the ANLS theory makes perfect use of the astrocytes' support function; it reduces neuronal dependence on glycolysis, i.e., a multifarious biochemical reaction process that produces a very small amount of ATP. Hence, neurons can derive more energy with as few biochemical steps as possible (17). This pattern of energetics mainly exists in activated excitatory neurons and is very important in sustaining the energy metabolism of neurons during high synaptic activity; therefore, it may be involved in certain diseases, such as epilepsy (32). Targeting the LDH-a key enzyme in the ANLS exhibited an antiepileptic effect (13) (see section Targeting Lactate Dehydrogenase to Treat Epilepsy).
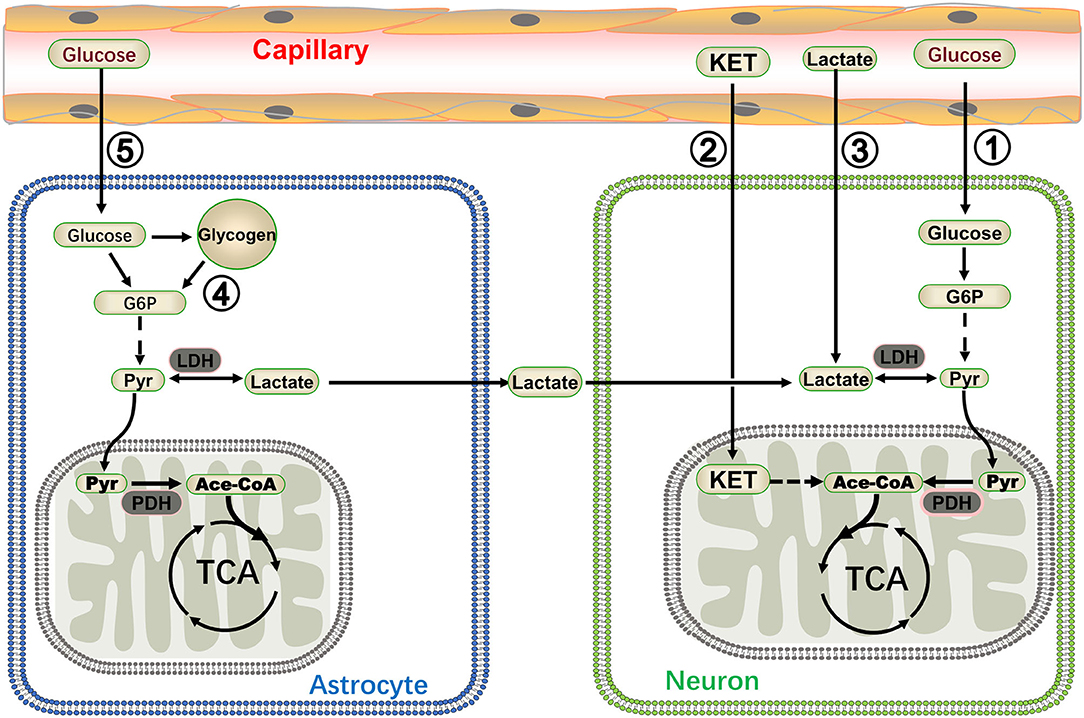
Figure 1. A summary of energy metabolism patterns in neurons. Glucose and ketone (KET) are recognized as energy substrates, which are labeled as ① and ② respectively. Lactate in the blood can also be utilized as an energy source for the brain during moderate or intense exercise (marked as ③) (20, 29). Glycogen stored in astrocytes generated lactate via glycogenolysis and lactate was then shuttled to neurons (marked as ④) (30). The astrocyte-neuron lactate shuttle is very important in sustaining the energy metabolisms of neurons during high synaptic activity (marked as ⑤) (13). G6P: glucose 6 phosphate, Pyr, pyruvate; PDH, pyruvate dehydrogenase; Ace-CoA, acetyl-coenzyme A.
Various Energetics in the Brain
Organisms can survive in various complex environments because organs can change their metabolism for adaptation purposes. Glucose in plasma or extracellular fluid remains the most important energy substrate for the brain, with numerous other substrates fueling the brain via various pathways. Glycogen stored in the liver or muscles is an important alternative energy source for the human body. It is also present in human brain tissue (33) and plays a crucial role in memory formation, learning capacity, and regulation of the sleep-wake cycle (34–36). As a matter of fact, glycogen is an energy reserve selectively localized in astrocytes (37). It is lactate that fuels the neurons through the ANLS and plays important roles in the brain. In some special circumstances, such as high-intense memory tasks, sleep deprivation, and hypoglycemia, lactate derived from astrocytic glycogen fuels the brain during exhaustive conditions (30). In general, blood glucose could contribute to brain energetics, and lactate in the blood can also be utilized as an energy source for the brain during moderate or intense exercise (20, 29). In addition, ketone bodies comprise an important alternative energy substrate for the brain and could be directly utilized to maintain the energy metabolism in a starvation condition or when following a ketogenic diet (21). Various energy supply modes enable the nervous system to adapt to various states and offer the potential to better understand and treat diseases (the energy metabolism in the brain is illustrated in Figure 1).
Changes in Metabolic Characteristics in Epilepsy
There are often metabolic changes in a diseased organ or tissue. Sometimes, these metabolic changes contribute to diagnosis or treatment, which could be applicable to epilepsy. Similar to the energy changes associated with earthquakes, epileptic foci are often accompanied by significant energy metabolism changes during seizures (4, 38). With the development of detection methods and technological progress, evidence supporting this point is accumulating.
Neuroimaging
The lesions caused by general diseases can be found by CT or MRI, while the location of epilepsy lesions is more complicated. Sometimes, epileptic lesions do not show structural abnormalities, but can be detected through differences in metabolic levels or combined with electroencephalography (EEG) to locate the epileptic foci. 18F-FDG positron-emission tomography (PET) makes it possible to intuitively observe the metabolic differences in brain regions and locate the epileptogenic foci. PET is usually used as interictal investigation, and epileptic foci show characteristic hypometabolism in the inter-ictal phase (39). PET scanning is almost impossible to perform in epileptic patients with motor symptoms during the seizure period, but it can be used for absence epilepsy. During the ictal phase of absence epilepsy, the metabolism of the thalamus is obviously enhanced, and cerebral blood flow is increased in the whole brain (40–42). Animal experiments have supported that there is enhancement of brain metabolism during epileptic seizures. In a rat seizure model induced by pilocarpine, FDG-PET imaging showed significant hypermetabolism in the area of the hippocampus and entorhinal cortex during status epilepticus (43). Single-photon emission computed tomography (SPECT) also constitutes a technique to evaluate brain metabolism and to locate epileptic foci. SPECT imaging can monitor dynamic changes in cerebral perfusion, thereby reflecting the metabolic changes before, during, and after seizures. True ictal SPECT imaging has shown that hyperperfusion emerges in the epileptogenic region, and indirectly reflects increased brain metabolism through changes in cerebral perfusion (44).
Biochemical Changes in Epilepsy
Epilepsy manifests as a change in consciousness and/or behavior, with abnormal EEG activities being at the root of seizure causality. The electrical activity must be accompanied by changes in energy; in fact, the conversion between substances will produce energy changes. Therefore, epilepsy must be accompanied by changes in energy-related substances; some abnormal neurochemical changes occur in the epileptic brain. However, elevated peripheral blood lactate levels are considered to be correlated with the extent of disease or injury (45). Comparing with the non-epileptogenic hippocampus, the concentration of lactate in the epileptogenic hippocampus increased from 4.6 ± 0.4 to 6.8 ± 0.7 mM (P < 0. 001) (46–48). An increase in anaerobic glycolysis was also shown in the epileptogenic brain. In addition, status epilepticus causes a significant increase in cerebrospinal fluid (CSF) lactate, and the magnitude of lactate elevation could play an important role in predicting the morbidity and mortality of status epilepticus (49).
Metabolism Dysfunction Leads to Epilepsy
We so far discussed the metabolic characteristics of the brain and energy metabolism changes in epilepsy. Additionally, metabolic disorders can lead to epilepsy. Next, we introduce several types of common epileptic diseases, including the glucose transporter (GLUT1) deficiency syndrome, hypoglycemia, creatine deficits, and mitochondrial encephalomyopathies.
GLUT1 Deficiency Syndrome
As discussed above, the brain is an important organ with high energy demand and glucose uptake via GLUT1 on the endothelial cells of the BBB. Although there are several subtypes of GLUTs in the brain, mainly including GLUT1 on the endothelial cells of the BBB, GLUT2 on astrocytes, and GLUT3 on neurons (50, 51), GLUT1 is the one most importantly associated with epilepsy (52). Defects of GLUT1 will impair glucose transport into the brain and result in Glut-1 deficiency syndrome (53). Glut1 deficiency syndrome is an autosomal dominant hereditary neurologic disorder characterized by low glucose (<40 mg/dL) and low lactate levels in the CSF. Seizures, which often initiate in the first 4 months of life, are the most common presenting symptom in this disorder. Patients also often have stunting, acquired microcephaly, ataxia, and muscle spasms. Sudden onset of confusion, lethargy, sleep disturbances, and headaches may also occur. The extent of cognitive impairment could vary with the condition. Epilepsy caused by Glut1 deficiency syndrome shows different types: complex-partial seizures (53), absence epilepsy (54), generalized tonic-clonic seizures (55), and others. In addition, mutations in GLUT1 (also known as SLC2A1) can also cause a syndrome called focal epilepsy (FE) and paroxysmal exercise-induced dyskinesia (PED) (56), whose attacks may be associated with increased glucose consumption caused by exercise. Ketogenic diet therapy is the gold standard treatment for patients with GLUT1 deficiency syndrome (57). The age at the correct diagnosis is the most important factor determining prognosis, and early diagnosis is very important as well as initiating the ketogenic diet as soon as possible (58).
Hypoglycemia
Hypoglycemia is a common, acute life-threatening illness in diabetic patients that may cause seizures. Not all patients with hypoglycemia have epileptic seizures; neonates and children appear to be more susceptible to epileptic seizures induced by hypoglycemia, which may be associated with brain immaturity (59). Furthermore, hypoglycemia-induced seizures may be linked to genetic susceptibility. In the background of DBA mice prone to epileptic seizures, insulin injection that reduced blood sugar levels to 60%, increased spike-and-wave discharge (SWD) by >300%, whereas the same dose of insulin could not induce SWD events in C57Bl6 mice with epileptic seizure resistance (60). This difference decided by genetic susceptibility has not been confirmed in the population, but patients with systemic epilepsy are more sensitive to hypoglycemia; Increased cortical excitation was much more obvious in epileptic patients than in healthy individuals under starvation conditions, which indicated that the neuronal networks in the epileptic brain were more susceptible to hypoglycemia (61). The mechanism of this sensitivity difference is not clear at present. In general, most epileptic seizures caused by hypoglycemia can be quickly relieved after blood glucose level recovery. However, attention is required in that children with diabetes with hypoglycemic convulsions could eventually develop perpetual abnormal EEG. Good control of blood glucose did not improve the abnormal EEG caused by hypoglycemia. Severe hypoglycemia in infantile or early-onset diabetes mellitus appears to be an important risk factor for persistent EEG abnormalities (59). Hence, the prevention of hypoglycemia is very important, especially in infants.
Creatine Deficits
Creatine deficiency syndrome comprises a group of disorders caused by creatine (Cr) synthesis or transport defects. The main symptoms of creatine deficiency syndrome include intellectual impairment, severe language delay, behavioral abnormalities, and seizures (62). Creatine is the most important material for the synthesis of the high energy compound, creatine phosphate. L-arginine glycine amidine transferase (AGAT) and guanidine acetate methyltransferase (GAMT) are key enzymes in creatine synthesis. Creatine synthesized in the liver is transported by creatine transporter 1 (CT1) to muscle and brain tissues with high energy metabolism (63). Hence, deficits in the synthesis or transport of creatine can lead to creatine deficiency. The biosynthesis dysfunction of creatine comprises two autosomal recessive disorders: AGAT deficiency and GAMT deficiency.
AGAT deficiency is a rare disease with only individual cases having been reported and involves non-specific symptoms including intellectual disability and epilepsy (63). Various GAMT mutations have been reported (64, 65). GAMT deficiency can lead to severe early epileptic encephalopathy and a range of developmental, behavioral, and motor disorders. Approximately 50% of patients develop seizures, which are the second most common symptom in GAMT deficiency. The most common types of epilepsy reported include febrile convulsions, generalized tonic-clonic seizures, and myoclonic seizures (63). CT1 defect is a relatively common X-linked disease due to SLC6A8 mutation. Intellectual disability and epilepsy are common in CT1 defect, and the types of seizures include febrile, myoclonic, generalized tonic-clonic seizures; convulsive status epilepticus, and partial seizures with secondary generalization (63, 65). Oral creatine supplementation was shown to effectively improve the clinical symptoms of disorders of creatine biosynthesis (66). Treatment for GAMT deficiency that corrects creatine depletion and reduces the accumulation of the enzyme product guanyl acetic acid (GAA) can be successful (64, 67). Most evidence suggests that creatine supplementation is not effective in patients with CT1 defect, and conventional antiepileptic drugs could control well epilepsy in CT1 defect (68).
Mitochondrial Encephalomyopathies
Traditionally, mitochondrial diseases refer to a group of hereditary disorders in which mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) deficiencies cause oxidative phosphorylation dysfunction of the mitochondrial respiratory chain (69). Mitochondrial encephalomyopathies are the most serious mitochondrial disease involving the brain and muscles. Myopathy, encephalopathy, lactate acidosis, and stroke-like episodes (MELAS), myoclonus epilepsy and ragged red fibers (MERRF), and polymerase gamma (POLG)-related disease are the three most common mitochondrial encephalomyopathies and are closely associated with epilepsy (70). In MELAS, epilepsy is one of the most frequent and early onset symptoms that mainly occurs in the group of patients with stroke-like lesions, and seizures are often accompanied by migraine-like headaches. Patients with MELAS are prone to status epilepticus, which may be the first symptom (70). Just as its name implies, patients with MERRF are usually characterized by progressive myoclonic seizures, and epilepsy in most patients tends to develop to generalized tonic-clonic seizures (71). As for POLG-related disease, it is caused by POLG defect and related to Alpers-Hüttenlocher syndrome or to adult-onset encephalopathy, spinocerebellar ataxia, and epilepsy (72). Myoclonic seizures are a common and obligatory feature in both multisystemic disorders, and epilepsy in POLG-related disease is often resistant to drug therapy (73).
Possible Mechanism of Seizures Caused by Metabolism Dysfunction
A common characteristic of these relatively widespread metabolic disorders leading to epileptic seizures is a decrease in energy substances, with the ultimate result being a decrease in ATP levels. Continued severe energy crisis could lead to epileptic seizures. The possible mechanisms are as follows. First, the resting potential of neurons determines neuronal excitation. The sodium-potassium pump (Na+-K+ ATPase) plays an important role in retaining the resting potential through ATP consumption. The function of the sodium-potassium pump will become impaired when ATP produced in neurons decreases, which will lead to a decrease in the absolute value of the resting potential. The relative depolarization of neurons increases excitability and leads to epileptic discharges (74). Second, neural networks in the hippocampus can be inhibited by the activation of intermediate neurons by purine receptors. ATP reduction may reduce the inhibition effect, facilitating the spreading of excitation in the neural network (75, 76). Furthermore, Na+-K+ ATPase dysfunction reduces the GABAergic potentials, thereby enhancing the excitatory post-synaptic potentials and spike firing leading to reduced inhibition and increased excitation; this is also an important mechanism underlying hyperexcitability and is associated with increased epilepsy sensitivity (77). Therefore, energy metabolism dysfunction will lead to epileptic seizures, especially for energy deficiency in normal cells.
Metabolites Affect Neuronal Excitability
Epilepsy can cause systemic or focal metabolic changes, and some metabolic disorders can lead to seizures. It is the metabolites that affect the excitability of neurons. Next, we will introduce the effects of two important metabolites on neuronal excitation.
Lactate
Traditionally, lactate was considered a metabolic waste produced by anaerobic glycolysis and a sign of the severity of some diseases (49). However, a recent study suggested that lactate is the energy substrate of some cancer or normal tissue cells, and its priority may be higher than glucose (26). In the past two decades, lactate was in the spotlight, as researchers focused on its metabolic role in the brain. Besides this still debated metabolic role, lactate could act as a signal molecule in brain cells (78). In 2008, lactate was found to be a natural ligand for the hydrocarboxylic acid-1 receptor [HCAR1, also named G protein-coupled receptor (GPR) 81 receptor], a GPR primarily regulating the cAMP signaling pathway (79). Thus, the possibility that lactate plays a physiological and pathological role through G protein-coupled receptors was greatly expanded. Moreover, increasing numbers of studies have shown that lactate is a by-product of metabolism and an important signal molecule in regulating neuronal excitation. However, its effects on neuronal excitation in different brain regions differ, even being completely opposite. Although exogenous lactate was excitatory to locus coeruleus neurons, further research showed it may act through an unknown receptor or pathway; however, it is highly unlikely that GPR81, which was described in the adipose tissue previously, could be responsible for the effects on LC neurons induced by lactate (Figure 2A) (80). Lactate over physiological levels acted as an inhibitor for cortical neurons as it inhibited the excitability of cortical neurons through a metabolic or a GPR81-mediated pathway (Figure 2B) (81–83). Exogenous lactate may also act as an inhibitor in hippocampal neurons. According to a recent research, exogenous lactate induced outward currents mediated by G protein–gated inwardly rectifying potassium(GIRK) through activating GPR81, resulting in hyperpolarization and epileptiform firing decreasing in the subicular neurons of hippocampal slices (Figure 2C) (84). Furthermore, lactate could regulate some ion channels by decreasing pH. Lactate increases TREK1 channel activity by reducing intracellular pH (86), and reduced the damage to neurons caused by hyperexcitation. However, acid-sensing ion channel-1a (ASIC1a) is sensitive to extracellular pH and regulates neuronal excitation (Figure 2D). It was revealed that ASIC1a on inhibitory interneurons is activated by reducing extracellular pH and could terminate seizures (85). Lactate as a substrate for energy metabolism affects neuronal excitation. The latter will be explained in section Control of Epilepsy by Regulating Metabolic Pathways.
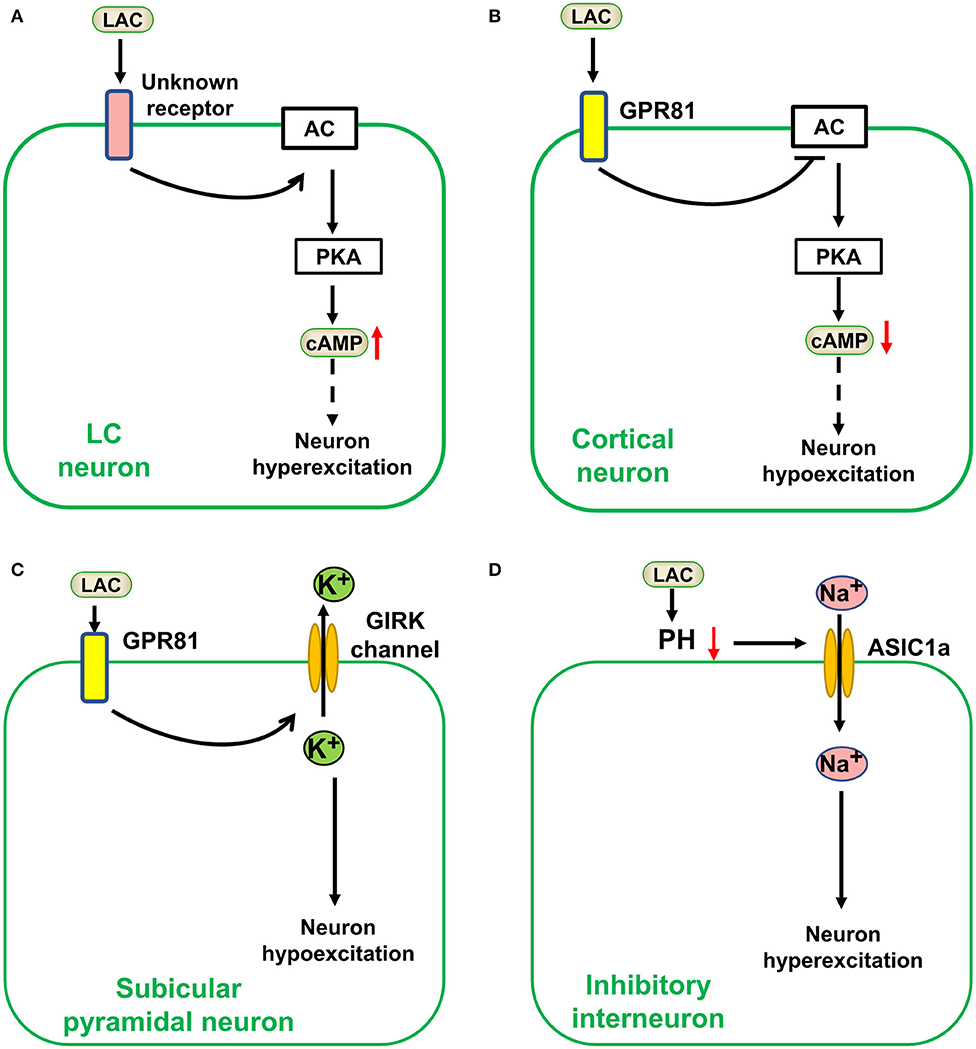
Figure 2. Lactate affects neuronal excitation as a signal molecule in the brain. Lactate (LAC) affects neuronal excitation as a signal molecule in the brain. (A) Exogenous lactate was excitatory to locus coeruleus (LC) neurons via an unknown receptor-mediated pathway (80). (B) Lactate over physiological levels acted as an inhibitor for cortical neurons a GPR81-mediated pathway (81–83). (C) Exogenous lactate (over physiological levels) induced an outward current mediated by G protein–gated inwardly rectifying potassium (GIRK) through activating GPR81, resulting in hyperpolarization and epileptiform firing decrease in the subicular neurons (84). (D) Lactate could regulate acid-sensing ion channel−1a (ASIC1a) by decreasing pH to Stimulate inhibitory neuronal excitation (85). AC, adenylate cyclase; PKA, protein kinase A; cAMP, cyclic adenosine monophosphate.
In sum, lactate can affect neuronal excitation as a signal molecule. However, its physiological concentration is not sufficient to activate the known GPR81 receptor (78) and its direct effect on epilepsy as a signal molecule is uncertain.
ATP
ATP is the universal energy currency of life. However, it is an important signaling molecule involved in apoptosis (87) and autophagy (88). ATP in the nervous system is involved in ischemia, epilepsy, Parkinson's disease, infection, amyotrophic lateral sclerosis, Alzheimer's disease, etc (89). As a signal molecule, ATP plays an important role in affecting neuronal excitation through intracellular or extracellular pathways. The intracellular pathway regulates the excitability of neurons mainly by regulating the opening of ATP-sensitive potassium (KATP) channels. A KATP channel opens when ATP levels are decreased, potassium ion outflows, and the cell membrane becomes hyperpolarized, leading to neuronal excitation decrease (90). Studies have shown that epilepsy treatment by modulating metabolism mainly acted through this intracellular ATP/KATP signaling pathway (13, 91). Regarding the extracellular pathway, ATP derived exogenously or from glia is a widely distributed cell-to-cell signaling molecule in the brain. It is the endogenous ligand of purine receptors (P2Xs, P2Ys). The P2XRs receptors may mediate chronic neuromodulation of the entire nervous system by acting on glia, especially on astrocytes (92). Thus, the scope of its impact is larger, and its specific role is unclear. While the role of ATP in activating P2Ys receptors is clear, extracellular ATP could activate the intermediate neurons mediated by P2Y1 receptors in the hippocampus (76). Therefore, extracellular ATP acts as an inhibitor in the hippocampal neuronal network (75). Theoretically speaking, activation of interneurons contributes to inhibition of the excitability of the neural network and, thus, reduces seizures, but the anti-epileptic effect of targeting purine receptors is uncertain. Simultaneously, extracellular ATP is unstable and easy to be decomposed to adenosine (93). In terms of extracellular pathways, ATPergic signaling is complex and its role in epilepsy remains unclear.
Control of Epilepsy by Regulating Metabolic Pathways
Epilepsy treatment by inhibiting metabolism may originate from the antiepileptic effect of the ketogenic diet, which mimics a state of starvation or calorie restriction and regulates neuronal excitation via energy metabolism modulation (94, 95). Therefore, avenues of treating epilepsy by targeting metabolism are opened.
Inhibiting Glycolysis to Reduce Seizures
In section Glycolysis Is Essential for Brain/Neuronal Function, we discussed the importance of glycolysis in maintaining neuronal function. Glycolysis may preferentially increase when the brain is in a certain pathological state, such as seizures. Inhibition of glycolysis reduces abnormal neuronal activity. 2-Deoxy-d-glucose (2-DG), a typical glycolysis inhibitor that can significantly reduce glucose uptake and glycolysis (96), has exhibited antiepileptic effects in several animal models (11, 91). The mechanisms of glycolysis inhibitor 2-DG in reducing epileptic seizures vary. First, 2-DG could activate the KATP channel (91), an inward rectifier potassium channel, which is sensitive to and regulated by intracellular ATP concentration. High and low ATP concentration in the cytoplasm inhibits and opens KATP channels, respectively, resulting in hyperpolarization of the neuronal cell membrane by potassium ion outflow and decreasing neuronal excitation. Thus, it plays a role in inhibiting epilepsy (91, 97). Second, in a rat kindling model of temporal lobe epilepsy, 2-DG reduced seizure progression by reducing the expression of brain-derived neurotrophic factor (BDNF) and its receptor, TrkB; this reduced expression is mediated by the decreasing of transcription factor NRSF caused by 2-DG administration (11). Previous studies have shown that conditional TrkB knockout (98) or BDNF signaling pathway reduction through transgenic technology (99) could reduce seizures in kindled animal models. These mechanisms can recur neuronal circuits that promote hyperexcitability (100), thus reducing the BDNF pathway, may have antiepileptic effects. In addition, 2-DG promotes the production of nicotinamide adenine dinucleotide phosphate (NADPH) by enhancing pentose phosphate pathway (PPP) metabolism in cells. The higher concentration of NADPH will potentiate the biosynthesis of neurosteroids via enhancing the activity of 5α-reductase (5α-R), a crucial enzyme catalyzing the steroid precursors into neurosteroids (101), resulting in potentiating the GABAergic tonic inhibition (91, 101) (Figure 3).
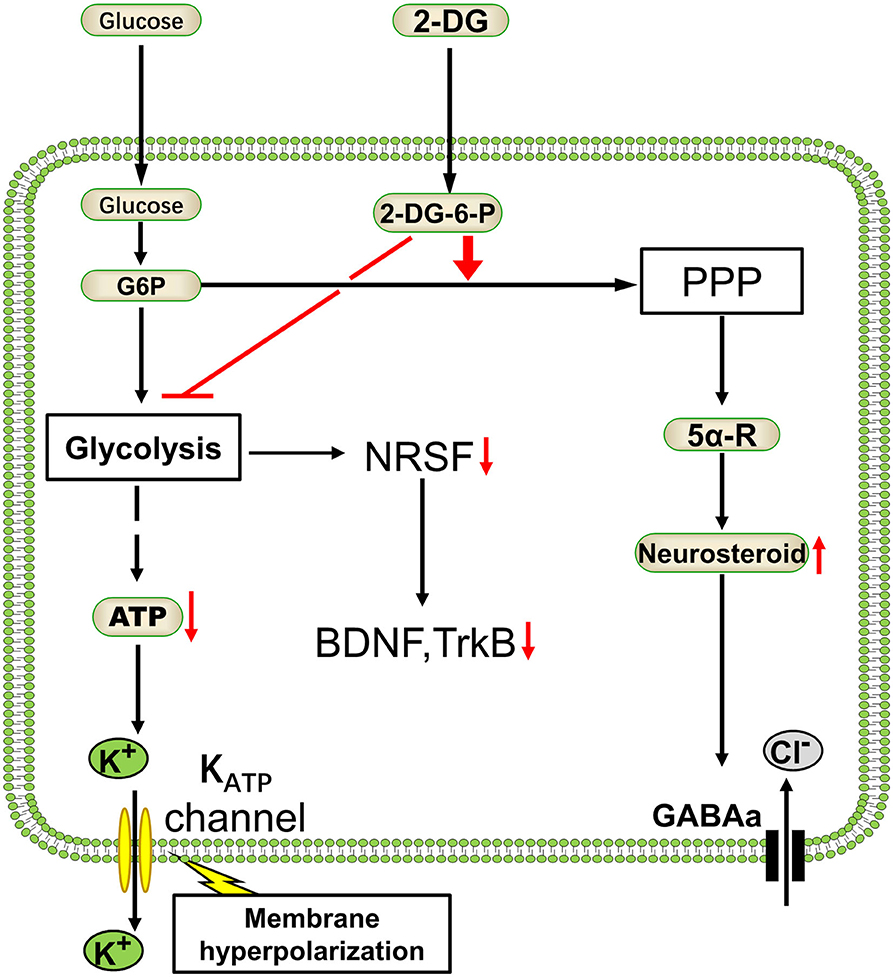
Figure 3. Antiepileptic mechanism of glycolysis inhibitor 2-deoxyglucose. As a classical glycolysis inhibitor, 2-deoxyglucose (2-DG) would inhibit the energetics of glucose and decrease the ATP. Glycolysis reduction will enhance the pentose phosphate pathway (PPP) metabolism and resulting in GABAergic strength (91). On the other hand, 2-DG reduces epilepsy progression by NRSF–dependent metabolic regulation of BDNF and TrkB (11). Glc, glucose; G-6-P, glucose-6-phosphate; 2-DG-6-P, 2-deoxyglucose-6-phosphate; NRSF, neural restrictive silencing factor; BDNF, brain-derived neurotrophic factor; TrkB, tyrosine kinase receptor B; 5α-R, 5α-reductase.
The antiepileptic effect of 2-DG by inhibiting glycolysis has also been confirmed by other studies. The Bcl-2-associated death promoter (BAD) is a member of the BCL-2 family with dual functions in proapoptosis and glucose metabolism. Gene knockout or alteration of BAD will impair glucose metabolism in brain cells and activate the ATP-sensitive KATP channels on the neuronal membrane, leading to an induced resistance to seizures (12, 102). A high dose of Fructose-1,6-Bisphosphate (F1, 6BP) was also demonstrated to suppress seizures in animals [(103–106)]. F1, 6BP becomes an inhibitor of PFK1, a rate-limiting enzyme in glycolysis. Therefore, excessive exogenous F1,6BP may play an inhibitory role in neurons (103). In conclusion, glycolysis inhibition, such as by 2-DG, is a promising treatment for epilepsy [(107, 108)].
Targeting Lactate Dehydrogenase to Treat Epilepsy
Lactate dehydrogenase (LDH), one of the most abundant proteins in the cell cytoplasm, is a type of enzyme widely existing in tissues. LDH is known as a biomarker of disease and tissue damage (109, 110). Increased LDH activity was found in some epileptic kindling models (111). It catalyzes the mutual conversion between lactate and pyruvate. Pyruvate is the final product of aerobic glycolysis and generates ATP in the mitochondria. Presently, lactate is no longer regarded as metabolic waste, as it fuels various tissues even under fully aerobic conditions (26, 27). Lactate in the brain acts as an energy substrate through the ANLS (discussed in section Metabolic Features in the Brain). Glucose derived from glycogenolysis or peripheral circulation is metabolized by glycolysis to produce pyruvate or lactate. Pyruvate, which is the end product of glycolysis, can be utilized by the mitochondria of astrocytes to generate ATP. Conversely, it can be transformed into lactate by LDH. Then, the lactate produced in astrocytes is shuttled from astrocytes to the adjacent neurons by monocarboxylic acid transporters (MCTs). After shuttling into the neurons, lactate is again converted to pyruvate by LDH, and pyruvate could be used by the mitochondria to feed the tricarboxylic acid cycle. ATP is produced through oxidative phosphorylation within the mitochondrial respiratory chain (MRC), and then released into the cytoplasm (45). LDH plays a crucial role in this pathway, and its inhibition will interfere with the ANLS to decrease ATP production in neurons. A high concentration of ATP in the cytoplasm inhibits KATP channels, while KATP channels open when intracellular ATP concentration decreases, resulting in hyperpolarization of neuronal-cell membrane by potassium ion outflow and decreasing neuronal excitation. Thus, LDH inhibits neuronal discharges and plays a role in inhibiting epilepsy (97) (Figure 4). Inhibition of LDH by oxamate had been demonstrated in mice. Another study found that stiripentol was also an LDH inhibitor, and LDH inhibition may be one of its important antiepileptic mechanisms (13). Interestingly, the mTOR pathway, i.e., an important signaling pathway involved in epilepsy, was also inhibited by LDH inhibitor oxamate (112, 113). In addition, activation of the mTOR pathway increased the expression of LDH (114, 115). Furthermore, the rapamycin, an classical mTOR inhibitor, could decrease the activity and expression of LDH, resulting in reduced lactate concentration (116, 117). Drugs targeting mTOR, such as rapamycin, can inhibit epileptic seizures in type II focal cortical dysplasia (FCD II) (118). Therefore, LDH may be a promising potential target for the treatment of epilepsy, and these discoveries have contributed to broaden our understanding of epilepsy and to develop new therapies.
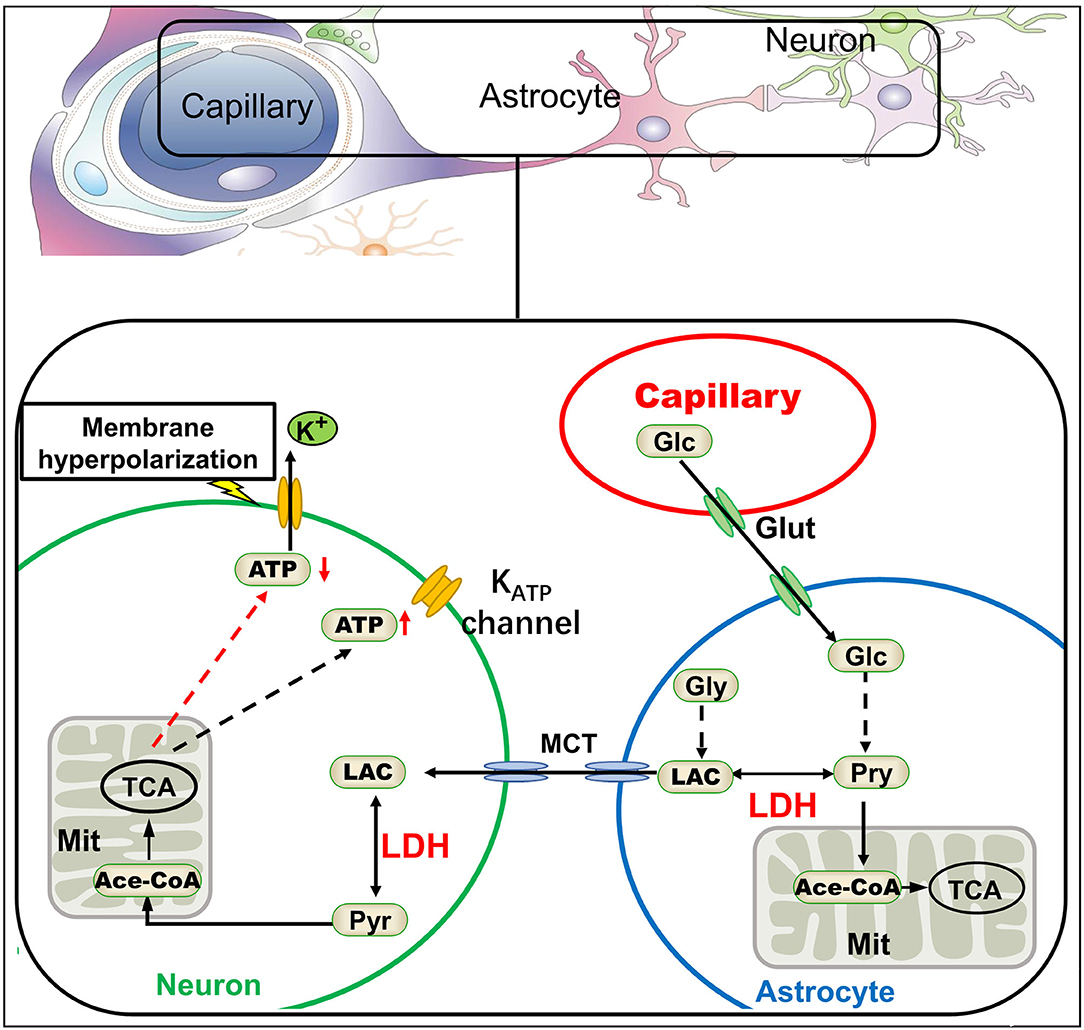
Figure 4. The astrocyte-neuron lactate shuttle and antiepileptic mechanism of LDH inhibition. According to the astrocyte-neuron lactate shuttle, lactate dehydrogenase (LDH) plays a key role in the energy supply of neurons. Inhibition of LDH would lead to the decrease of ATP in neurons, thus activating KATP channels on the neural membrane and potassium efflux, finally making the neural membrane hyperpolarization (13). Glc, glucose; Glut, glucose transporter; Pyr, pyruvate; Gly, glycogen; Lac, lactate; TCA, tricarboxylic acid cycle; MCT, monocarboxylic acid transporter.
Dietary Therapy in Epilepsy
The history of dietary therapy for epilepsy is quite long. The ketogenic diet is a high-fat and low-carbohydrate diet structure and is considered the oldest dietary therapy initially used to treat epilepsy (119). Its effectiveness in the treatment of epilepsy, especially for some types of intractable epilepsy in children has been demonstrated (120); compared to children randomized to usual care, children randomized to KDs were three times more likely to attain seizure freedom and nearly six times more likely to attain a 50% or greater reduction in seizure frequency (121). Subsequently, various dietary options for epilepsy were developed, including the medium-chain triglyceride ketogenic diet (MCT-KD), modified Atkin's diet (MAD), and low glycemic index treatment. These modified ketogenic diets have an efficacy close to that of the classical ketogenic diet (122). In addition, diet therapy was also evaluated for adjuvant treatment of obesity, type 2 diabetes, and some cancers (123–125). Moreover, diet therapy was attempted in a wide variety of neurological diseases, including Alzheimer's disease, multiple sclerosis, Parkinson's disease, and brain tumors (126), showing that it is widely potential. However, the most widely used and effective field of ketogenic diet is in the treatment of epilepsy.
As the name implies, ketogenic diet can produce ketone bodies in vivo: acetoacetic acid, beta-hydroxybutyric acid, and acetone. Studies have also shown that ketone bodies may directly exert pharmacological effects (127). The ketone body acetoacetate inhibited the transport of glutamate into synaptic vesicles by vesicular glutamate transporter 2, thus decreasing glutamate release into the synaptic cleft (128). Glutamate is a major excitatory amino acid in the brain; its signaling is inhibited by ketone bodies potentially reducing neuronal excitation and epilepsy. Conversely, ketone bodies can reduce the expression of adenosine kinase and enhance adenosine A1 receptors (A1R) and mediate the signaling pathway activated by adenosine (129), A1R activation showed anticonvulsant effects in mice and rats (130). Other studies have reported that the antiepileptic effect of the KD was due to metabolic changes caused by the conversion of energy substrates. When converting from a normal diet to a ketogenic diet, blood glucose levels decreased and ketone body concentrations significantly increased (131). At this time, the main energy substrates of the brain were converted from blood glucose to ketone bodies. The ATP production of neurons was derived from glycolysis decreases, which may lead to KATP channel activation, neuronal cell-membrane hyperpolarization, and excitability decrease (132) (Figure 5). Even in the presence of sufficient glucose, ketone bodies could inhibit neuronal firing through opening the KATP channels (133).
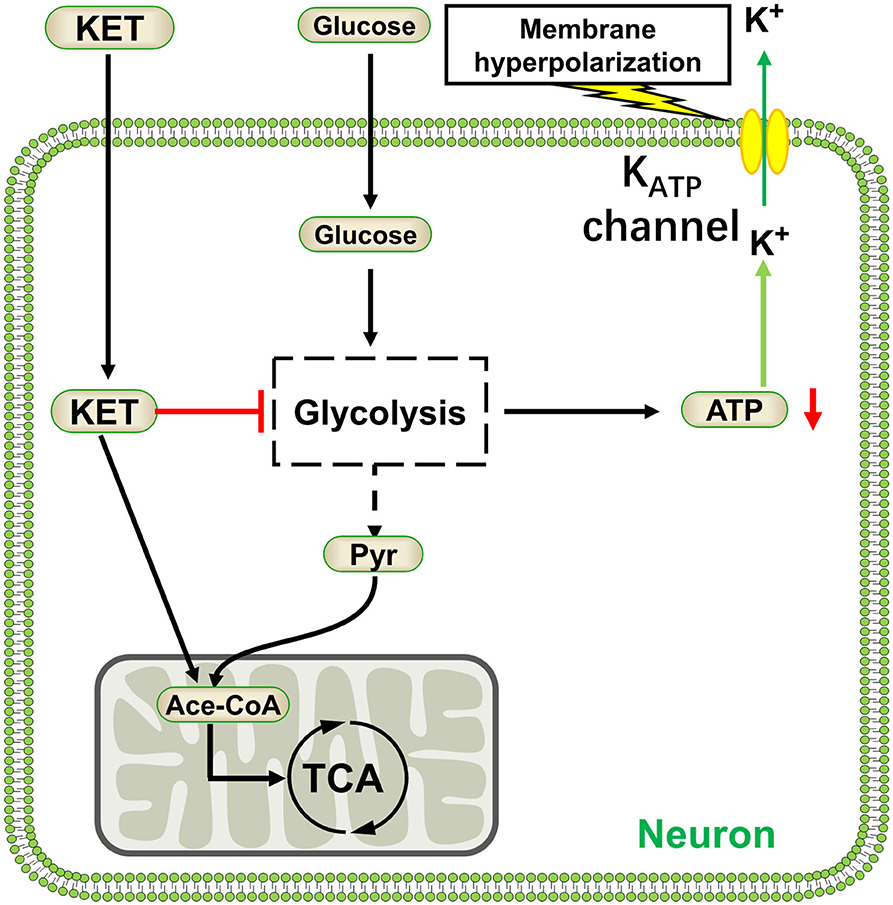
Figure 5. Ketone bodies activate KATP channels by inhibiting glycolysis. KET, ketone body; Pyr, pyruvate; Ace-CoA, acetyl coenzyme A.
Fats in the traditional ketogenic diet are mainly long-chain triglycerides. To improve the traditional ketogenic diet, an MCT KD derived from the traditional ketogenic diet was first proposed in 1971 (134). The antiepileptic effect is similar to that of the traditional ketogenic diet (135, 136). The novelty is that the therapeutic effect of MCT KD may be due to the decanoic acid produced by the decomposition of medium-chain triglycerides rather than by ketone bodies. Decanoic acid has a strong direct inhibitory effect on glutamate receptor α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) (137). The AMPA receptor is an excitatory ionotropic glutamate receptor mediating the bulk of the generation of excitatory post-synaptic potentials (EPSPs). EPSPs are responsible for synchronous discharges in epileptic foci, and thus AMPA receptors are critically important in the spread of epileptic discharges across brain regions (138). A highly selective AMPA receptor antagonist, perampanel (Pyramidopane, trade name Fycompa), has been promoted in the United States and the European Union as an antiepileptic drug (139). Decanoic acid produced by MCT KD directly antagonizes AMPA receptors and produces antiepileptic effects in animals, broadening our understanding of the possible antiepileptic mechanism of the ketogenic diet.
Diet therapy is an essential metabolic regulation for patients with epilepsy (95). Although dietary therapy has not fundamentally changed the status quo of epilepsy treatment, it still provides an alternative and beneficial treatment option. More importantly, it may provide inspiration for novel treatments for epilepsy.
Status and Future of Antiepileptic Drugs
Drug therapy plays a dominant role in epilepsy treatment. Surgical treatment can be used for epilepsy caused by definite etiology and lesions, and diet therapies are usually used for children with intractable epilepsy. Some new therapies are emerging, such as neuromodulation therapy (140). However, the overall effect is not satisfactory; epilepsy cannot be effectively controlled in approximately one-third of patients. Although the new drugs are more advantageous in terms of safety and side effects, this does not appear to improve the seizure control rate (141). Antiepileptic drugs have been developed to the third generation, and more than 20 types of drugs have been used in the clinic. However, the targets or mechanisms of existing drugs are mainly focused on inhibiting excitatory NMDA receptors, voltage-dependent ion-channels, or enhancing GABAergic activity (142) (Table 1). Levetiracetam and brivaracetam bring us to a whole new antiepileptic target-synaptic vesicle protein 2A (SV2A). They display a high and selective affinity for SV2A in the brain and thus inhibit synaptic transmission, which may have antiepileptic effects (143, 144). However, the precise mechanism by which levetiracetam and brivaracetam exert their antiepileptic activity is unknown. Another marketed antiepileptic drug, stiripentol, which came on the market in 2002, was considered a GABA potentiation and sodium channel blocker (145). Targets for epilepsy treatment appear to lack diversity and innovation. However, stiripentol was proven to be a lactate dehydrogenase inhibitor in 2015. Its antiepileptic effect might be partly due to its metabolic-related enzyme inhibitory effect (13). This provided strong evidence for epilepsy controlled by metabolic modulation. In addition, 2-deoxyglucose, as a glycolysis inhibitor, has shown promising anti-epileptic effects in many pre-clinical studies (146), highlighting the prospect of controlling epileptic seizures by regulating metabolism. Research on the treatment of epilepsy has been confined to certain types of targets, such as ion channels and excitatory or inhibitory receptors of certain neuronal membranes for a long time. Epilepsy therapy calls for new targets, and metabolic pathway-related targets deserve more attention.
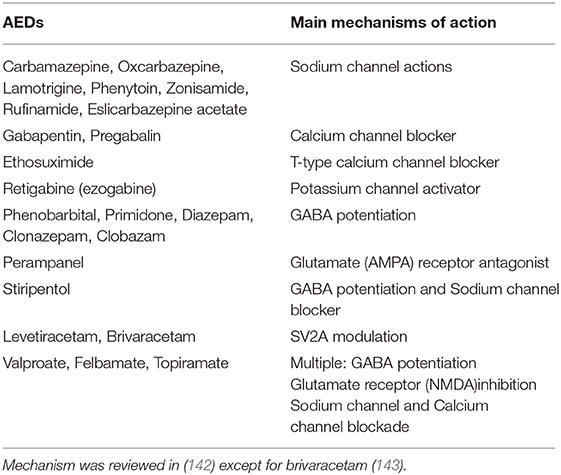
Table 1. Mechanisms of commonly used antiepileptic drugs.
Conclusion
Metabolism is the basis of brain function. For a long time, epilepsy has been regarded as a disease characterized by overexcitation of neural networks. The high metabolism of neurons provides energy for the hyperexcitation of neural networks. Neuroimaging and neurobiochemical findings have also confirmed that epileptic foci have high metabolic characteristics, and some metabolic disorders will lead to epileptic seizures. These fully illustrate the close relationship between epilepsy and metabolism. Epilepsy has even been defined as a metabolic disease. Recognizing that the ketogenic diet, an ancient dietary therapy for epilepsy, may be essentially a metabolic regulation method, suggesting that epilepsy may be controlled by targeted metabolism pathways. Drugs that reduce metabolisms, such as 2-deoxyglucose and stiripentol, have antiepileptic effects, showing the prospects of metabolic control of epilepsy. Therefore, the relationship between metabolism and epilepsy may be bi-directional. Hypometabolism could initiate convulsions or epilepsy, such as hypoglycemia, GLUT1 deficiency, and mitochondrial dysfunction. Supplementing neuronal energy is an effective treatment in such conditions. However, there is a relatively high metabolic and energy-consuming state in the epileptic seizure period caused by various primary pathological changes; thus, reducing the metabolic level during epileptic seizures is helpful for epilepsy control. Epilepsy therapy calls for more effective new targets, and the metabolic pathway may be a promising alternative. Epilepsy control by metabolism regulation may provide the means to explore new targets of epilepsy treatment. Thus, the treatment of epilepsy by metabolic modulation is a rising panacea for epilepsy. In the future, we need to further elucidate the metabolic processes taking place in the highly complex brain as well as metabolic changes under pathological conditions in order to locate key targets.
Author Contributions
YF put forward the conception of the review and wrote the manuscript. RS participated in the proposal of the concept and revised the manuscript. JW and ZS proposed suggestions for revision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant number: 81671296), Natural Science Foundation of Hunan (grant number: 2018JJ3803), and the New Xiangya Talent Project of The Third Xiangya Hospital of Central South University (grant number: JY201621).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Elsevier Author Services for providing language help.
References
1. Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. (2014) 55:475–82. doi: 10.1111/epi.12550
2. Patel M. A metabolic paradigm for epilepsy. Epilepsy Curr. (2018) 18:318–22. doi: 10.5698/1535-7597.18.5.318
3. Lord LD, Expert P, Huckins JF, Turkheimer FE. Cerebral energy metabolism and the brain's functional network architecture: an integrative review. J Cereb Blood Flow Metab. (2013) 33:1347–54. doi: 10.1038/jcbfm.2013.94
4. Wu Y, Liu D, Song Z. Neuronal networks and energy bursts in epilepsy. Neuroscience. (2015) 287:175–86. doi: 10.1016/j.neuroscience.2014.06.046
5. Waters D, Adeloye D, Woolham D, Wastnedge E, Patel S, Rudan I. Global birth prevalence and mortality from inborn errors of metabolism: a systematic analysis of the evidence. J Glob Health. (2018) 8:021102. doi: 10.7189/jogh.08.021102
6. Juweid ME, Cheson BD. Positron-emission tomography and assessment of cancer therapy. N Engl J Med. (2006) 354:496–507. doi: 10.1056/NEJMra050276
7. Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab. (2010) 7:7. doi: 10.1186/1743-7075-7-7
8. Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. (2017) 31:5–19. doi: 10.1016/j.ccell.2016.12.006
9. Wheless JW. History of the ketogenic diet. Epilepsia. (2008) 49:3–5. doi: 10.1111/j.1528-1167.2008.01821.x
10. Giordano C, Marchio M, Timofeeva E, Biagini G. Neuroactive peptides as putative mediators of antiepileptic ketogenic diets. Front Neurol. (2014) 5:63. doi: 10.3389/fneur.2014.00063
11. Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, et al. 2-deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. (2006) 9:1382–7. doi: 10.1038/nn1791
12. Gimenez-Cassina A, Martinez-Francois JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, et al. BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron. (2012) 74:719–30. doi: 10.1016/j.neuron.2012.03.032
13. Sada N, Lee S, Katsu T, Otsuki T, Inoue T. Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science. (2015) 347:1362–7. doi: 10.1126/science.aaa1299
14. Duncan JSP, Winston GPP, Koepp MJP, Ourselin SP. Brain imaging in the assessment for epilepsy surgery. Lancet Neurol. (2016) 15:420–33. doi: 10.1016/S1474-4422(15)00383-X
15. Dhamija R, Patterson MC, Wirrell EC. Epilepsy in children—when should we think neurometabolic disease? J Child Neurol. (2012) 27:663–71. doi: 10.1177/0883073811435829
16. Rho JM. How does the ketogenic diet induce anti-seizure effects? Neurosci Lett. (2017) 637:4–10. doi: 10.1016/j.neulet.2015.07.034
17. Dienel GA. Brain glucose metabolism: integration of energetics with function. Physiol Rev. (2019) 99:949–1045. doi: 10.1152/physrev.00062.2017
18. Williams LR, Leggett RW. Reference values for resting blood flow to organs of man. Clin Phys Physiol Meas. (1989) 10:187–217. doi: 10.1088/0143-0815/10/3/001
19. Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. (2001) 21:1133–45. doi: 10.1097/00004647-200110000-00001
20. van Hall G, Stromstad M, Rasmussen P, Jans O, Zaar M, Gam C, et al. Blood lactate is an important energy source for the human brain. J Cereb Blood Flow Metab. (2009) 29:1121–9. doi: 10.1038/jcbfm.2009.35
21. Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. (2017) 25:262–84. doi: 10.1016/j.cmet.2016.12.022
22. Li X, Yokono K, Okada Y. Phosphofructokinase, a glycolytic regulatory enzyme has a crucial role for maintenance of synaptic activity in guinea pig hippocampal slices. Neurosci Lett. (2000) 294:81–4. doi: 10.1016/S0304-3940(00)01535-4
23. Díaz-García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G. Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metabolism. (2017) 26:361–74. doi: 10.1016/j.cmet.2017.06.021
24. Pellerin L. Food for thought: the importance of glucose and other energy substrates for sustaining brain function under varying levels of activity. Diabetes Metab. (2010) 36:S59–63. doi: 10.1016/S1262-3636(10)70469-9
25. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. (1994) 91:10625–9. doi: 10.1073/pnas.91.22.10625
26. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell. (2017) 171:358–71. doi: 10.1016/j.cell.2017.09.019
27. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. (2017) 551:115–8. doi: 10.1038/nature24057
28. Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. J Cereb Blood Flow Metab. (2012) 32:1152–66. doi: 10.1038/jcbfm.2011.149
29. Rasmussen P, Wyss MT, Lundby C. Cerebral glucose and lactate consumption during cerebral activation by physical activity in humans. FASEB J. (2011) 25:2865–73. doi: 10.1096/fj.11-183822
30. Matsui T, Omuro H, Liu Y, Soya M, Shima T, McEwen BS, et al. Astrocytic glycogen-derived lactate fuels the brain during exhaustive exercise to maintain endurance capacity. Proc Natl Acad Sci USA. (2017) 114:6358–63. doi: 10.1073/pnas.1702739114
31. Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metabolism. (2016) 23:94–102. doi: 10.1016/j.cmet.2015.10.010
32. Patel DC, Tewari BP, Chaunsali L, Sontheimer H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat Rev Neurosci. (2019) 20:282–97. doi: 10.1038/s41583-019-0126-4
33. Nelson SR, Schulz DW, Passonneau JV, Lowry OH. Control of glycogen levels in brain. J Neurochem. (1968) 15:1271–9. doi: 10.1111/j.1471-4159.1968.tb05904.x
34. Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. (2011) 144:810–23. doi: 10.1016/j.cell.2011.02.018
35. Clasadonte J, Scemes E, Wang Z, Boison D, Haydon PG. Connexin 43-mediated astroglial metabolic networks contribute to the regulation of the sleep-wake cycle. Neuron. (2017) 95:1365–80. doi: 10.1016/j.neuron.2017.08.022
36. Hertz L, Chen Y. Glycogenolysis, an astrocyte-specific reaction, is essential for both astrocytic and neuronal activities involved in learning. Neuroscience. (2018) 370:27–36. doi: 10.1016/j.neuroscience.2017.06.025
37. Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. II Choroid plexus and ependymal epithelia, endothelia and pericytes. J Neurocytol. (1986) 15:511–24. doi: 10.1002/jemt.1060030406
38. Osorio I, Frei MG, Sornette D, Milton J, Lai Y. Epileptic seizures: quakes of the brain? Phys Rev E Stat Nonlin Soft Matter Phys. (2010) 82:21919. doi: 10.1103/PhysRevE.82.021919
39. Rathore C, Dickson JC, Teotonio R, Ell P, Duncan JS. The utility of 18F-fluorodeoxyglucose PET (FDG PET) in epilepsy surgery. Epilepsy Res. (2014) 108:1306–14. doi: 10.1016/j.eplepsyres.2014.06.012
40. Engel JJ, Lubens P, Kuhl DE, Phelps ME. Local cerebral metabolic rate for glucose during petit mal absences. Ann Neurol. (1985) 17:121–8. doi: 10.1002/ana.410170204
41. Prevett MC, Duncan JS, Jones T, Fish DR, Brooks DJ. Demonstration of thalamic activation during typical absence seizures using H2(15)O and PET. Neurology. (1995) 45:1396–402. doi: 10.1212/WNL.45.7.1396
42. Bilo L, Meo R, de Leva MF, Vicidomini C, Salvatore M, Pappata S. Thalamic activation and cortical deactivation during typical absence status monitored using [18F]FDG-PET: a case report. Seizure. (2010) 19:198–201. doi: 10.1016/j.seizure.2010.01.009
43. Mirrione MM, Schiffer WK, Siddiq M, Dewey SL, Tsirka SE. PET imaging of glucose metabolism in a mouse model of temporal lobe epilepsy. Synapse. (2006) 59:119–21. doi: 10.1002/syn.20216
44. Van Paesschen W, Dupont P, Van Driel G, Van Billoen H, Maes A. SPECT perfusion changes during complex partial seizures in patients with hippocampal sclerosis. Brain. (2003) 126(Pt 5):1103–11. doi: 10.1093/brain/awg108
45. Brooks GA. The science and translation of lactate shuttle theory. Cell Metab. (2018) 27:757–85. doi: 10.1016/j.cmet.2018.03.008
46. Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. (2005) 57:226–35. doi: 10.1002/ana.20380
47. Pan JW, Williamson A, Cavus I, Hetherington HP, Zaveri H, Petroff OAC, et al. Neurometabolism in human epilepsy. Epilepsia. (2008) 49:31–41. doi: 10.1111/j.1528-1167.2008.01508.x
48. Chatzikonstantinou A, Ebert AD, Hennerici MG. Cerebrospinal fluid findings after epileptic seizures. Epileptic Disord. (2015) 17:453–9. doi: 10.1684/epd.2015.0779
49. Calabrese VP, Gruemer HD, James K, Hranowsky N, DeLorenzo RJ. Cerebrospinal fluid lactate levels and prognosis in status epilepticus. Epilepsia. (1991) 32:816–21. doi: 10.1111/j.1528-1157.1991.tb05538.x
50. Patching SG. Glucose transporters at the blood-brain barrier: function, regulation and gateways for drug delivery. Mol Neurobiol. (2017) 54:1046–77. doi: 10.1007/s12035-015-9672-6
51. Szablewski L. Glucose transporters in brain: in health and in alzheimer's disease. J Alzheimers Dis. (2017) 55:1307–20. doi: 10.3233/JAD-160841
52. Hildebrand MS, Damiano JA, Mullen SA, Bellows ST, Oliver KL, Dahl HH, et al. Glucose metabolism transporters and epilepsy: only GLUT1 has an established role. Epilepsia. (2014) 55:e18–21. doi: 10.1111/epi.12519
53. De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. (1991) 325:703–9. doi: 10.1056/NEJM199109053251006
54. Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE. Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology. (2010) 75:432–40. doi: 10.1212/WNL.0b013e3181eb58b4
55. Klepper J, Wang D, Fischbarg J, Vera JC, Jarjour IT, O'Driscoll KR. Defective glucose transport across brain tissue barriers: a newly recognized neurological syndrome. Neurochem Res. (1999) 24:587–94. doi: 10.1023/A:1022544131826
56. Wolking S, Becker F, Bast T, Wiemer-Kruel A, Mayer T, Lerche H, et al. Focal epilepsy in glucose transporter type 1 (Glut1) defects: case reports and a review of literature. J Neurol. (2014) 261:1881–6. doi: 10.1007/s00415-014-7433-5
57. Pong AW, Geary BR, Engelstad KM, Natarajan A, Yang H, De Vivo DC. Glucose transporter type I deficiency syndrome: epilepsy phenotypes and outcomes. Epilepsia. (2012) 53:1503–10. doi: 10.1111/j.1528-1167.2012.03592.x
58. Klepper J, Akman C, Armeno M, Auvin S, Cervenka M, Cross HJ, et al. Glut1 deficiency syndrome (Glut1DS): state of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open. (2020) 5:354–65. doi: 10.1002/epi4.12414
59. Soltesz G, Acsadi G. Association between diabetes, severe hypoglycaemia, and electroencephalographic abnormalities. Arch Dis Child. (1989) 64:992–6. doi: 10.1136/adc.64.7.992
60. Reid CA, Kim TH, Berkovic SF, Petrou S. Low blood glucose precipitates spike-and-wave activity in genetically predisposed animals. Epilepsia. (2011) 52:115–20. doi: 10.1111/j.1528-1167.2010.02911.x
61. Badawy RAB, Vogrin SJ, Lai A, Cook MJ. Cortical excitability changes correlate with fluctuations in glucose levels in patients with epilepsy. Epilepsy Behav. (2013) 27:455–60. doi: 10.1016/j.yebeh.2013.03.015
62. Stockler-Ipsiroglu S, van Karnebeek C. Cerebral creatine deficiencies: a group of treatable intellectual developmental disorders. Semin Neurol. (2014) 34:350–6. doi: 10.1055/s-0034-1386772
63. Leuzzi V, Mastrangelo M, Battini R, Cioni G. Inborn errors of creatine metabolism and epilepsy. Epilepsia. (2013) 54:217–27. doi: 10.1111/epi.12020
64. Dhar SU, Scaglia F, Li FY, Smith L, Barshop BA, Eng CM, et al. Expanded clinical and molecular spectrum of guanidinoacetate methyltransferase (GAMT) deficiency. Mol Genet Metab. (2009) 96:38–43. doi: 10.1016/j.ymgme.2008.10.008
65. Cheillan D, Joncquel-Chevalier CM, Briand G, Salomons GS, Mention-Mulliez K, Dobbelaere D, et al. Screening for primary creatine deficiencies in French patients with unexplained neurological symptoms. Orphanet J Rare Dis. (2012) 7:96. doi: 10.1186/1750-1172-7-96
66. Battini R, Alessandrì MG, Leuzzi V, Moro F, Tosetti M, Bianchi MC, et al. Arginine: glycine amidinotransferase (AGAT) deficiency in a newborn: early treatment can prevent phenotypic expression of the disease. J Pediatrics. (2006) 148:828–30. doi: 10.1016/j.jpeds.2006.01.043
67. Bianchi MC, Tosetti M, Battini R, Leuzzi V, Alessandri MG, Carducci C, et al. Treatment monitoring of brain creatine deficiency syndromes: a 1H- and 31P-MR spectroscopy study. AJNR Am J Neuroradiol. (2007) 28:548–54. Available online at: http://www.ajnr.org/content/28/3/548
68. Cecil KM, Salomons GS, Ball WJ, Wong B, Chuck G, Verhoeven NM, et al. Irreversible brain creatine deficiency with elevated serum and urine creatine: a creatine transporter defect? Ann Neurol. (2001) 49:401–4.
69. DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. (2008) 31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302
70. Bindoff LA, Engelsen BA. Mitochondrial diseases and epilepsy. Epilepsia. (2012) 53:92–7. doi: 10.1111/j.1528-1167.2012.03618.x
71. Berkovic SF, Carpenter S, Evans A, Karpati G, Shoubridge EA, Andermann F, et al. Myoclonus epilepsy and ragged-red fibres (MERRF). 1. A clinical, pathological, biochemical, magnetic resonance spectrographic and positron emission tomographic study. Brain. (1989) 112(Pt 5):1231–60. doi: 10.1093/brain/112.5.1231
72. Tzoulis C, Tran GT, Coxhead J, Bertelsen B, Lilleng PK, Balafkan N, et al. Molecular pathogenesis of polymerase gamma-related neurodegeneration. Ann Neurol. (2014) 76:66–81. doi: 10.1002/ana.24185
73. Zsurka GP, Kunz WSP. Mitochondrial dysfunction and seizures: the neuronal energy crisis. Lancet Neurol. (2015) 14:956–66. doi: 10.1016/S1474-4422(15)00148-9
74. Benarroch EE. Na+, K+-ATPase: functions in the nervous system and involvement in neurologic disease. Neurology. (2011) 76:287–93. doi: 10.1212/WNL.0b013e3182074c2f
75. Bowser DN, Khakh BS. ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci. (2004) 24:8606–20. doi: 10.1523/JNEUROSCI.2660-04.2004
76. Kawamura M, Gachet C, Inoue K, Kato F. Direct excitation of inhibitory interneurons by extracellular ATP mediated by P2Y1 receptors in the hippocampal slice. J Neurosci. (2004) 24:10835–45. doi: 10.1523/JNEUROSCI.3028-04.2004
77. Vaillend C, Mason SE, Cuttle MF, Alger BE. Mechanisms of neuronal hyperexcitability caused by partial inhibition of Na+-K+-ATPases in the rat CA1 hippocampal region. J Neurophysiol. (2002) 88:2963–78. doi: 10.1152/jn.00244.2002
78. Mosienko V, Teschemacher AG, Kasparov S. Is L-lactate a novel signaling molecule in the brain? J Cereb Blood Flow Metab. (2015) 35:1069–75. doi: 10.1038/jcbfm.2015.77
79. Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J, et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem. (2009) 284:2811–22. doi: 10.1074/jbc.M806409200
80. Tang F, Lane S, Korsak A, Paton JFR, Gourine AV, Kasparov S, et al. Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat Commun. (2014) 5:3284. doi: 10.1038/ncomms4284
81. Bozzo L, Puyal J, Chatton J. Lactate modulates the activity of primary cortical neurons through a receptor-mediated pathway. PLoS ONE. (2013) 8:e71721. doi: 10.1371/journal.pone.0071721
82. Jourdain P, Allaman I, Rothenfusser K, Fiumelli H, Marquet P, Magistretti PJ. L-lactate protects neurons against excitotoxicity: implication of an ATP-mediated signaling cascade. Sci Rep. (2016) 6:21250. doi: 10.1038/srep21250
83. de Castro AH, Briquet M, Schmuziger C, Restivo L, Puyal J, Rosenberg N, et al. The lactate receptor HCAR1 modulates neuronal network activity through the activation of galpha and gbetagamma subunits. J Neurosci. (2019) 39:4422–33. doi: 10.1523/JNEUROSCI.2092-18.2019
84. Jorwal P, Sikdar SK. Lactate reduces epileptiform activity through HCA1 and GIRK channel activation in rat subicular neurons in an in vitro model. Epilepsia. (2019) 60:2370–85. doi: 10.1111/epi.16389
85. Ziemann AE, Schnizler MK, Albert GW, Severson MA, Howard MR, Welsh MJ, et al. Seizure termination by acidosis depends on ASIC1a. Nat Neurosci. (2008) 11:816–22. doi: 10.1038/nn.2132
86. Ghatak S, Sikdar SK. Lactate modulates the intracellular pH sensitivity of human TREK1 channels. Pflügers Arch Eur J Physiol. (2016) 468:825–36. doi: 10.1007/s00424-016-1795-8
87. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature. (2010) 467:863–7. doi: 10.1038/nature09413
88. Wang Y, Martins I, Ma Y, Kepp O, Galluzzi L, Kroemer G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy. (2013) 9:1624–5. doi: 10.4161/auto.25873
89. Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov. (2008) 7:575–90. doi: 10.1038/nrd2605
90. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. (2010) 90:291–366. doi: 10.1152/physrev.00021.2009
91. Forte N, Medrihan L, Cappetti B, Baldelli P, Benfenati F. 2-Deoxy-d-glucose enhances tonic inhibition through the neurosteroid-mediated activation of extrasynaptic GABAA receptors. Epilepsia. (2016) 57:1987–2000. doi: 10.1111/epi.13578
92. von Kugelgen I, Hoffmann K. Pharmacology and structure of P2Y receptors. Neuropharmacology. (2016) 104:50–61. doi: 10.1016/j.neuropharm.2015.10.030
93. Lovatt D, Xu Q, Liu W, Takano T, Smith NA, Schnermann J, et al. Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc Natl Acad Sci USA. (2012) 109:6265–70. doi: 10.1073/pnas.1120997109
94. Hartman AL, Gasior M, Vining EP, Rogawski MA. The neuropharmacology of the ketogenic diet. Pediatr Neurol. (2007) 36:281–92. doi: 10.1016/j.pediatrneurol.2007.02.008
95. Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. (2013) 36:32–40. doi: 10.1016/j.tins.2012.11.005
96. Dwarkanath BS, Zolzer F, Chandana S, Bauch T, Adhikari JS, Muller WU, et al. Heterogeneity in 2-deoxy-D-glucose-induced modifications in energetics and radiation responses of human tumor cell lines. Int J Radiat Oncol Biol Phys. (2001) 50:1051–61. doi: 10.1016/S0360-3016(01)01534-6
97. Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. (2006) 440:470–6. doi: 10.1038/nature04711
98. He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. (2004) 43:31–42. doi: 10.1016/j.neuron.2004.06.019
99. Lahteinen S, Pitkanen A, Saarelainen T, Nissinen J, Koponen E, Castren E. Decreased BDNF signalling in transgenic mice reduces epileptogenesis. Eur J Neurosci. (2002) 15:721–34. doi: 10.1046/j.1460-9568.2002.01897.x
100. Elmariah SB, Crumling MA, Parsons TD, Balice-Gordon RJ. Postsynaptic TrkB-mediated signaling modulates excitatory and inhibitory neurotransmitter receptor clustering at hippocampal synapses. J Neurosci. (2004) 24:2380–93. doi: 10.1523/JNEUROSCI.4112-03.2004
101. Reddy DS. Role of hormones and neurosteroids in epileptogenesis. Front Cell Neurosci. (2013) 7:115. doi: 10.3389/fncel.2013.00115
102. Martínez-François JR, Fernández-Agüera MC, Nathwani N, Lahmann C, Burnham VL, Danial NN, et al. BAD and KATP channels regulate neuron excitability and epileptiform activity. eLife. (2018) 7:e32721. doi: 10.7554/eLife.32721
103. Lian XY, Khan FA, Stringer JL. Fructose-1,6-bisphosphate has anticonvulsant activity in models of acute seizures in adult rats. J Neurosci. (2007) 27:12007–11. doi: 10.1523/JNEUROSCI.3163-07.2007
104. Ding Y, Wang S, Zhang MM, Guo Y, Yang Y, Weng SQ, et al. Fructose-1,6-diphosphate inhibits seizure acquisition in fast hippocampal kindling. Neurosci Lett. (2010) 477:33–6. doi: 10.1016/j.neulet.2010.04.030
105. Ding Y, Wang S, Jiang Y, Yang Y, Zhang M, Guo Y, et al. Fructose-1,6-diphosphate protects against epileptogenesis by modifying cation-chloride co-transporters in a model of amygdaloid-kindling temporal epilepticus. Brain Res. (2013) 1539:87–94. doi: 10.1016/j.brainres.2013.09.042
106. Shao LR, Wang G, Stafstrom CE. The glycolytic metabolite, fructose-1,6-bisphosphate, blocks epileptiform bursts by attenuating voltage-activated calcium currents in hippocampal slices. Front Cell Neurosci. (2018) 12:168. doi: 10.3389/fncel.2018.00168
107. Shao LR, Rho JM, Stafstrom CE. Glycolytic inhibition: a novel approach toward controlling neuronal excitability and seizures. Epilepsia Open. (2018) 3:191–7. doi: 10.1002/epi4.12251
108. Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Tomson T, et al. Progress report on new antiepileptic drugs: a summary of the fourteenth eilat conference on new antiepileptic drugs and devices (EILAT XIV). I Drugs in preclinical and early clinical development. Epilepsia. (2018) 59:1811–41. doi: 10.1111/epi.14557
109. Zandbergen E, Haan R, Hijdra A. Systematic review of prediction of poor outcome in anoxic-ischaemic coma with biochemical markers of brain damage. Intensive Care Med. (2001) 27:1661–7. doi: 10.1007/s001340101076
110. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. (2013) 123:3685–92. doi: 10.1172/JCI69741
111. Erakovic V, Zupan G, Varljen J, Laginja J, Simonic A. Altered activities of rat brain metabolic enzymes in electroconvulsive shock-induced seizures. Epilepsia. (2001) 42:181–9. doi: 10.1046/j.1528-1157.2001.30499.x
112. Cho CH. Frontier of epilepsy research-mTOR signaling pathway. Exp Mol Med. (2011) 43:231–74. doi: 10.3858/emm.2011.43.5.032
113. Zhao Z, Han F, Yang S, Wu J, Zhan W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett. (2015) 358:17–26. doi: 10.1016/j.canlet.2014.11.046
114. Lund IV, Hu Y, Raol YH, Benham RS, Faris R, Russek SJ, et al. BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci Signal. (2008) 1:a9. doi: 10.1126/scisignal.1162396
115. Zha X, Wang F, Wang Y, He S, Jing Y, Wu X, et al. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res. (2011) 71:13–8. doi: 10.1158/0008-5472.CAN-10-1668
116. Venkatesh HS, Chaumeil MM, Ward CS, Haas-Kogan DA, James CD, Ronen SM. Reduced phosphocholine and hyperpolarized lactate provide magnetic resonance biomarkers of PI3K/Akt/mTOR inhibition in glioblastoma. Neuro Oncol. (2012) 14:315–25. doi: 10.1093/neuonc/nor209
117. Lee SC, Marzec M, Liu X, Wehrli S, Kantekure K, Ragunath PN, et al. Decreased lactate concentration and glycolytic enzyme expression reflect inhibition of mTOR signal transduction pathway in B-cell lymphoma. NMR Biomed. (2013) 26:106–14. doi: 10.1002/nbm.2825
118. Lim JS, Kim W, Kang H, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. (2015) 21:395–400. doi: 10.1038/nm.3824
119. Vamecq J, Vallée L, Lesage F, Gressens P, Stables JP. Antiepileptic popular ketogenic diet: emerging twists in an ancient story. Prog Neurobiol. (2005) 75:1–28. doi: 10.1016/j.pneurobio.2004.11.003
120. Martin-McGill KJ, Jackson CF, Bresnahan R, Levy RG, Cooper PN. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst Rev. (2018) 11:D1903. doi: 10.1002/14651858.CD001903.pub4
121. Martin-McGill KJ, Bresnahan R, Levy RG, Cooper PN. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst Rev. (2020) 6:D1903. doi: 10.1002/14651858.CD001903.pub5
122. Miranda MJ, Turner Z, Magrath G. Alternative diets to the classical ketogenic diet–can we be more liberal? Epilepsy Res. (2012) 100:278–85. doi: 10.1016/j.eplepsyres.2012.06.007
123. Yancy WJ, Olsen MK, Guyton JR, Bakst RP, Westman EC. A low-carbohydrate, ketogenic diet versus a low-fat diet to treat obesity and hyperlipidemia: a randomized, controlled trial. Ann Intern Med. (2004) 140:769–77. doi: 10.7326/0003-4819-140-10-200405180-00006
124. Accurso A, Bernstein RK, Dahlqvist A, Draznin B, Feinman RD, Fine EJ, et al. Dietary carbohydrate restriction in type 2 diabetes mellitus and metabolic syndrome: time for a critical appraisal. Nutr Metab. (2008) 5:9. doi: 10.1186/1743-7075-5-9
125. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. (2010) 10:267–77. doi: 10.1038/nrc2817
126. Stafstrom CE, Rho JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol. (2012) 3:59. doi: 10.3389/fphar.2012.00059
127. Simeone TA, Simeone KA, Stafstrom CE, Rho JM. Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology. (2018) 133:233–41. doi: 10.1016/j.neuropharm.2018.01.011
128. Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara C, et al. Metabolic control of vesicular glutamate transport and release. Neuron. (2010) 68:99–112. doi: 10.1016/j.neuron.2010.09.002
129. Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, et al. A ketogenic diet suppresses seizures in mice through adenosine A(1) receptors. J Clin Invest. (2011) 121:2679–83. doi: 10.1172/JCI57813
130. Dunwiddie TV, Worth T. Sedative and anticonvulsant effects of adenosine analogs in mouse and rat. J Pharmacol Exp Ther. (1982) 220:70–6.
131. DeVivo DC, Leckie MP, Ferrendelli JS, McDougal DJ. Chronic ketosis and cerebral metabolism. Ann Neurol. (1978) 3:331–7. doi: 10.1002/ana.410030410
132. Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci. (2007) 27:3618–25. doi: 10.1523/JNEUROSCI.0132-07.2007
133. Lund TM, Ploug KB, Iversen A, Jensen AA, Jansen-Olesen I. The metabolic impact of beta-hydroxybutyrate on neurotransmission: reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. J Neurochem. (2015) 132:520–31. doi: 10.1111/jnc.12975
134. Huttenlocher PR, Wilbourn AJ, Signore JM. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology. (1971) 21:1097–103. doi: 10.1212/WNL.21.11.1097
135. Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. (2008) 7:500–6. doi: 10.1016/S1474-4422(08)70092-9
136. Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, et al. A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia. (2009) 50:1109–17. doi: 10.1111/j.1528-1167.2008.01870.x
137. Chang P, Augustin K, Boddum K, Williams S, Sun M, Terschak JA, et al. Seizure control by decanoic acid through direct AMPA receptor inhibition. Brain. (2016) 139:431–43. doi: 10.1093/brain/awv325
138. Rogawski MA. A fatty acid in the MCT ketogenic diet for epilepsy treatment blocks AMPA receptors. Brain. (2016) 139:306–9. doi: 10.1093/brain/awv369
139. Hanada T, Hashizume Y, Tokuhara N, Takenaka O, Kohmura N, Ogasawara A, et al. Perampanel: a novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia. (2011) 52:1331–40. doi: 10.1111/j.1528-1167.2011.03109.x
140. Thijs RD, Surges R, O'Brien TJ, Sander JW. Epilepsy in adults. Lancet. (2019) 393:689–701. doi: 10.1016/S0140-6736(18)32596-0
141. Chen Z, Brodie MJ, Liew D, Kwan P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30-year longitudinal cohort study. JAMA Neurol. (2018) 75:279–86. doi: 10.1001/jamaneurol.2017.3949
142. Löscher W, Klitgaard H, Twyman RE, Schmidt D. New avenues for anti-epileptic drug discovery and development. Nat Rev Drug Discov. (2013) 12:757–76. doi: 10.1038/nrd4126
143. Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H. Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A. Br J Pharmacol. (2008) 154:1662–71. doi: 10.1038/bjp.2008.198
144. Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci USA. (2004) 101:9861–6. doi: 10.1073/pnas.0308208101
145. Fisher JL. The anti-convulsant stiripentol acts directly on the GABA(A) receptor as a positive allosteric modulator. Neuropharmacology. (2009) 56:190–7. doi: 10.1016/j.neuropharm.2008.06.004
146. Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Tomson T, et al. Progress report on new antiepileptic drugs: a summary of the fourteenth eilat conference on new antiepileptic drugs and devices (EILAT XIV). II Drugs in more advanced clinical development. Epilepsia. (2018) 59:1842–66. doi: 10.1111/epi.14555
Keywords: epilepsy, energy metabolism, brain, ATP-sensitive potassium channel, anticonvulsants
Citation: Fei Y, Shi R, Song Z and Wu J (2020) Metabolic Control of Epilepsy: A Promising Therapeutic Target for Epilepsy. Front. Neurol. 11:592514. doi: 10.3389/fneur.2020.592514
Received: 10 August 2020; Accepted: 20 November 2020;
Published: 08 December 2020.
Edited by:
Sherifa Ahmed Hamed, Assiut University Hospitals, EgyptReviewed by:
David Ruskin, Trinity College, United StatesMario Tombini, Campus Bio-Medico University, Italy
Copyright © 2020 Fei, Shi, Song and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi Song, ZG9jc29uZ0AxMjYuY29t; Jinze Wu, d3VqaW56ZTExMTdAaG90bWFpbC5jb20=