 Carmen Rubio
Carmen Rubio Rudy Luna
Rudy Luna Artemio Rosiles
Artemio Rosiles Moisés Rubio-Osornio
Moisés Rubio-Osornio- 1Neurophysiology Department, National Institute of Neurology and Neurosurgery, Manuel Velasco Suárez, Mexico City, Mexico
- 2Experimental Laboratory of Neurodegenerative Diseases, National Institute of Neurology and Neurosurgery, Manuel Velasco Suárez, Mexico City, Mexico
Epilepsy is a neurological disorder in which, in many cases, there is poor pharmacological control of seizures. Nevertheless, it may respond beneficially to alternative treatments such as dietary therapy, like the ketogenic diet or caloric restriction. One of the mechanisms of these diets is to produce a hyperpolarization mediated by the adenosine triphosphate (ATP)-sensitive potassium (KATP) channels (KATP channels). An extracellular increase of K+ prevents the release of Ca2+ by inhibiting the signaling of the Wnt pathway and the translocation of β-catenin to the cell nucleus. Wnt ligands hyperpolarize the cells by activating K+ current by Ca2+. Each of the diets described in this paper has in common a lower use of carbohydrates, which leads to biochemical, genetic processes presumed to be involved in the reduction of epileptic seizures. Currently, there is not much information about the genetic processes implicated as well as the possible beneficial effects of diet therapy on epilepsy. In this review, we aim to describe some of the possible genes involved in Wnt pathways, their regulation through the KATP channels which are implicated in each one of the diets, and how they can reduce epileptic seizures at the molecular level.
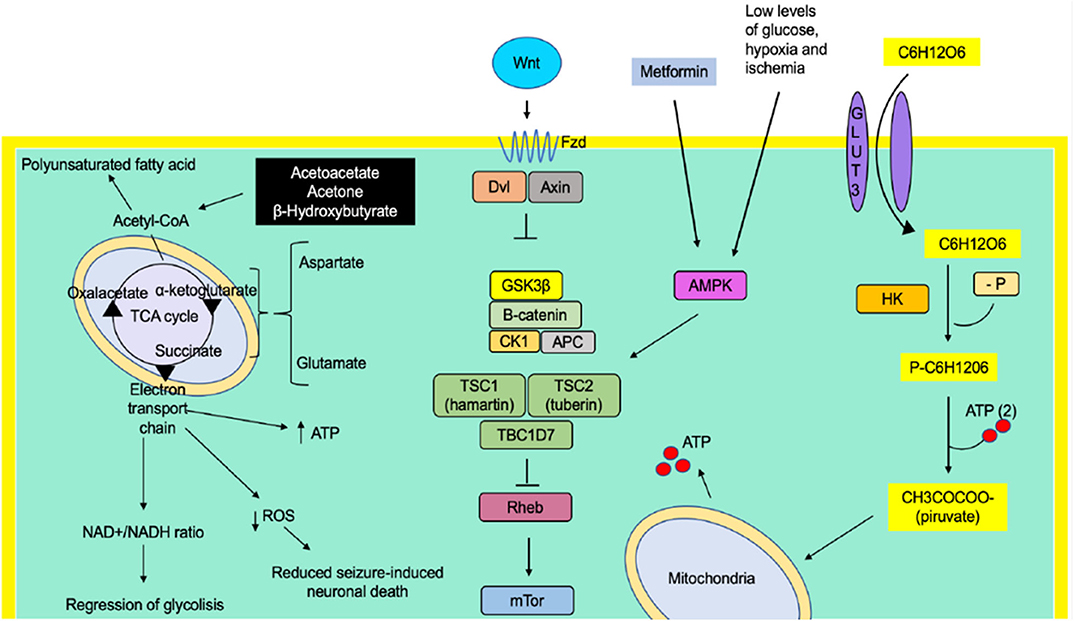
Graphical Abstract. The ketogenic diet has shown a decrease in seizures through the production of ketone bodies that cause an effect on neuronal function and the release of neurotransmitters, such as a decrease of glutamate, and adenosine, mitochondrial function, increase of GABA synthesis, and KATP channels. Caloric restriction may inhibit glycolysis and, just like the ketogenic diet, is involved in mitochondrial biogenesis, decreased oxidative stress, and decreasing inflammation, but it appears that it does not have an ionic gradient effect on KATP channels. The ketogenic diet and caloric restriction per se down-regulated and up-regulated some genes involved in seizure activity.
Introduction
Epilepsy, despite having a large number of different treatments, is actually one of the diseases with poor control of epileptic seizures. This is because of the lack of understanding of epileptogenesis, seizure genesis, seizure spread, and seizure suppression (1). Approximately 30% of patients have refractory epilepsy, which means that adequate trials of at least two drug programs have failed (2, 3). Drug treatment is usually aimed at having a neuronal anti-glutamatergic effect or demands the actions of GABA; some anti-epileptics act on ion channels of calcium, sodium, or potassium, stabilizing the neuronal membranes and thus avoiding the synchronization and the propagation of abnormal discharges. An alternative to counteract epileptic seizures is surgery, but just a selected number of patients are candidates for this type of treatment. These are the many reasons why it is necessary to look for alternative therapies for seizure control (4).
Neuronal activity is closely related to the metabolism of carbohydrates that influence neuronal excitability, which can become abnormal as it happens in epilepsy. Carbohydrates promote the production of fatty acids for the generation of ketone bodies. Animal experiments have provided evidence that the anticonvulsant effect of the ketogenic diet (KD) is mediated by acetone, which has shown efficacy in the treatment of epilepsy, though it is not the only mechanism described (5). Glucose is the main substrate for obtaining energy in adult brains. Approximately 63% of glucose supplies is estimated to be used to obtain energy in a 7-days-old rat's brain; this amount increases with age (6).
Since 1921, dietary therapies have been suggested to have an important role in epileptic seizure control, including infantile spasms, Doose syndrome, Rett syndrome, tuberous sclerosis, Dravet syndrome (7, 8), and other neurological diseases. Seizure control is obtained by different mechanisms. What they all have in common is an overall reduction in glucose levels, elevated free fatty acids, and ketone levels (9). Thus, dietary therapy may play a role in slowing down the progression of a specific disorder (10).
The Wnt signaling pathway is actively involved in glucose regulation pathways, especially by increasing glucose absorption in cortical neurons independently of the activation of target genes and the synaptic effect of Wnt (11). In recent years, attempts have been made to describe the participation of adenosine triphosphate (ATP)-sensitive potassium (KATP) channels (KATP channels) as a convergent mechanism of the effect that restrictive diets provide, because of a reduction in glycolytic pathways and the increase of ketone bodies, and therefore a cellular excitability reduction by lowering cytoplasmic ATP and activating the KATP channels. Therefore, the KATP channels rather than closed by ketone bodies could be activated, generating an output of positive ions and decreasing neuronal excitability (12). The association of the neuronal excitability is gaining importance, which depends mostly on these KATP channels and the Wnt pathways, though a clear relationship with glycolytic pathways and their metabolic regulation remain to be clarified.
Therefore, the objective throughout this review is to describe the Wnt pathway as a regulator of the KATP channels involved in the ketogenic diet and caloric restriction as well as to point out the possible genes involved in each of the diets and how they can molecularly achieve a reduction in epileptic seizures (Tables 1 and 2). To accomplish our objective, we have looked into articles concerning restrictive diet therapy, their multiple and different mechanisms of action involving cellular metabolism, molecular signaling, and gene interaction, all associated with epileptogenesis in experimental and clinical research. In the gene tables below, we looked for the authors who firstly reported these genes in literature and for the authors that associated them with diet therapy, Wnt signaling pathway, epileptogenesis, and KATP channels.

Table 1. Down and up regulated genes implicated in the Wnt and mTor signaling pathway after caloric restriction therapy.

Table 2. Down and up regulated genes implicated in the Wnt signaling pathway and pxidative stress after ketogenic diet therapy.
Caloric Restriction
Caloric restriction (CR) is described to be a natural dietary therapy that improves health, prolongs longevity, and reduces the effects of new inflammatory diseases in rodents and humans (77–79). CR occurs by partial dietary restriction and differs from acute fasting or starvation. CR reduces caloric energy consumption without causing eating disorders or any specific nutrient deficiency (80). In addition to improving health status, CR has anticonvulsant effects in murine and other epilepsy models (81, 82).
Moreover, it is proposed that the anti-epileptic action of CR may require a reduction in glucose or insulin concentrations and an increase in the β-hydroxybutyrate (β-HB) ketone body (83), along with increased fatty acid oxidation and reduced lipid accumulation in some tissues, as CR has been strongly related with improving insulin sensitivity and increasing adiponectin levels (84). Electrographically, CR has shown an increase in the neuron discharge threshold, providing an anti-epileptic effect (85). Also, CR in rats with epilepsy induction by the Kindling model increased the threshold after a stimulus applied to the amygdala and showed a decreased duration of epileptic activity (86). The abovementioned finding suggests that CR carries an anti-seizure profile and is completely independent of changes in glucose, insulin, or β-HB (82).
Talevi and Rocha (9) mention that CR may inhibit glycolysis and, just like the KD, is involved in mitochondrial biogenesis and decreases oxidative stress and inflammation, but it appears that it does not have an ionic gradient effect on the KATP channels. However, according to Kawamura et al. (87) in 2010, in neurons from CA3 of the hippocampus after CR and the ketogenic diet, the glucose levels decreased and provoked an increase of ATP after opening K+ channels, lowering neural excitability and stabilizing the membrane. After CR in rats, a reduction in the degeneration of GABAergic neurons in the hippocampus and the entorhinal cortex was shown before the administration of kainic acid (88).
Physiological Effects of Caloric Restriction on Wnt/B-Catenin on the Brain
New signaling pathways have been postulated on the effects of caloric restriction in the brain, one of them involving glycolytic metabolism in neurons, but little is known about the intervention of the Wnt/β-catenin pathway (89, 90). The brain is an organ that consumes large amounts of glucose. In brain tissue, glucose is oxidized by glycolysis and oxidative phosphorylation to produce ATP, the product with the highest consumption by the neuron during the formation of the ionic gradient after synaptic transmission (91, 92).
The stimulation of cortical neurons with Wnt3a stimulates glucose absorption without significant changes in the expression and the functionality of glucose transporter 3 (GLUT3), but with an increase in its affinity. Furthermore, by stimulating the same ligand, the activity of two glycolysis regulatory enzymes (hexokinase enzyme and 6-phosphofruct-2-kinase/fructose-2,3-bisphosphatase) increases, as they are correlated directly with the increase in glucose absorption (11) (Figure 1). Besides the Wnt pathway, AKT participates in the glucose increase by inducing glycogen synthesis due to the inhibition of 3-kinase-glycogen synthase in rapamycin proteins (93, 94). Alterations in the Wnt/β-catenin signaling pathways have also been identified, generating mutations in genes involved in gluconeogenesis and glutamine metabolism in hepatocytes. Many of these proteins involved are associated with mitochondrial dysfunction and carbohydrate metabolism, suggesting that having defects in Wnt signaling may also determine a metabolic change in the energy utilization of cells toward glycolysis and staying away from fatty acid oxidation (95). The intervention of Wnt/β-catenin signaling pathways in glycolytic pathways and their indirect role in the cell membrane activity of epileptic neurons are still not fully understood. Wnt signaling pathways are essential for the development and the function of the central nervous system because it modulates key processes such as hippocampal neurogenesis, synaptic cleft formation, and mitochondrial regulation (96, 97). The insulin receptor substrate 1 signaling protein (IRS-1) induces a transcriptional effect of Wnt. The higher level of IRS-1 drives the activation of mitochondrial biogenesis. Moreover, in insulin-sensitive cell types, it improves insulin signaling, again raising the possibility that Wnt proteins can be utilized to modulate glucose homeostasis (65).
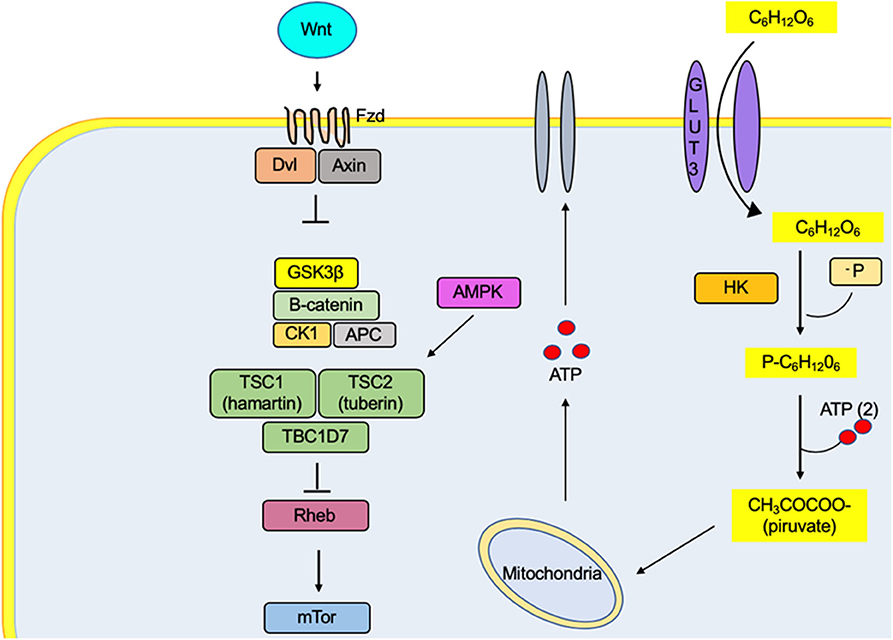
Figure 1. The metabolic effect of Wnt promotes the absorption of glucose in the neuron by increasing the affinity of the GLUT3 transporter for its substrate. In the same way, it increases the activity of the enzyme hexokinase, one of the proteins regulating the glycolytic rate. Therefore, there is an increase in the ATP concentrations, a metabolic substrate for the regulation of channels responsible for the excitability of the cell membrane. The activation of the Fzd receptors by its Wnt ligand causes the decoupling of β-catenin degradation complex (GSK3β, CK1, APC, and β-catenin proteins), preventing their degradation by the proteasome and generating an accumulation in the cytosol and its translocation to the nucleus, where it binds to T-cell factor/lymphoid-enhancer binding factor family transcription factors, thus promoting the transcription of genes related to lipid metabolism and cell differentiation, among others.
Wnt Pathways
Wnt signals can be divided into two main pathways: the canonical pathway (β-catenin-dependent signaling pathway) and the non-canonical pathway (β-catenin-independent). The first has been implicated in leading the regulation of transcriptional activity and the activation of genes such as c-Myc, cyclin D, and the TCF/LEF (T-cell factor/lymphoid promoter-binding factor) pathway that determines cell fate such as determination, proliferation, and differentiation of stem cells. The binding of a specific Wnt (such as Wnt1, Wnt10b, or Wnt3A) to their Fzd (Frizzled)-LRP5 or Fzd-LRP6 receptors results in the activation of intracellular heterotrimeric G proteins and Dvl (Disheveled) proteins, which in the dentate gyrus leads to phosphorylation and recruitment of Axin to LRP5 (or LRP6) and participating as a co-receptor (18, 19, 98). The abovementioned event leads to the dissociation of β-catenin and the degradation complex by promoting its stabilization and the accumulation of the molecule in the cytosol. Thus, it is translocated toward the nucleus and activates the transcription factors of the TCF/LEF families (21, 99), c-Myc, and cyclin D. Certain intracellular molecules expressed by Wnt-dependent activation of β-catenin jointly participate in the pyramidal cells of the region hippocampal CA1 and can increase the sensitivity to pentylenetetrazole and kainate by inducing seizures and cell death (100). The opened KATP channels produce ROS due to gene triggering, which increases mitochondrial production. The block of KATP channels induces the release of Ca2+ in the nucleus, leading to the expression of the c-myc gene (101). In 2008, Jeon et al. (102) reported that 10 repeated electroconvulsive seizures in the rat's brain exert a neuroprotective effect on the inhibition of c-Myc protein levels and subsequent inactivation of the Bad proapoptotic protein while allowing the activation of BclXL. However, the overexpression effect of β-catenin on c-Myc and cyclin D causes neuronal death in Purkinje cells and the granule layer of the cerebellum of rats with repeated generalized seizures. Activation of β-catenin can induce the overexpression of c-Myc and cyclin D that exert an apoptotic effect through positive regulation and activation of proapoptotic proteins (103). The voltage-gated calcium channel β4 subunit inhibits cyclin-D1, which prevents the activity by β-catenin due to an interaction with TCF4 and controlling different transcription factors (104).
The second Wnt-dependent or non-canonical pathway has been preferentially implicated in cell movement (105, 106). This pathway alters intracellular Ca2+ activated by Wnt5a and simultaneously decreases potassium currents, leading to an increase in neuronal excitability, specifically in CA1 pyramidal neurons (107). The activation of Fzd receptors by its Wnt ligand induces the inhibition of the enzyme GSK3β, which generates the activation of tuberin protein (TSC2) (15). Without these signals, TSC2 inhibits the brain protein-enriched homologous Ras protein (Rheb), an activator of mTORC1 (108). Thus, inactivation of Rheb by TSC2 leads to the inhibition of mTORC1, and inhibition of TSC2 leads to the indirect activation of mTORC1.
Recently, our research group reported that mild CR (15%) significantly increased the phosphorylation of protein kinase activated by adenosine monophosphate (AMPK) (Figure 2). Furthermore, it reduces the phosphorylation of ribosomal protein (S6) and PKB/Akt in the neocortex and the hippocampus, suggesting that mild CR inhibits mTORC1 signaling pathways. Therefore, an effect of raising the discharge threshold potential has been pointed out, suggesting an anticonvulsant action (82).
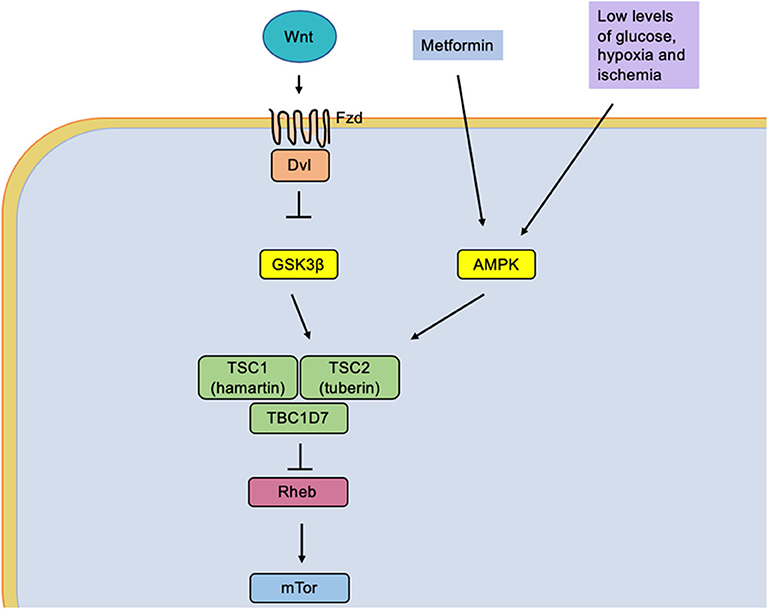
Figure 2. The low glucose levels caused by caloric restriction induce an activation of the protein AMPK (adenosine monophosphate-activated by protein kinase). It simultaneously causes activation of the tuberin complex that causes an indirect inhibition of the mTOR molecular complex by the Rheb protein (homologous Ras protein-enriched brain), a direct activator of mTOR.
It is a fact that due to the glycolytic regulation of CR, this diet gets involved in the Wnt signaling pathway. We suggest that the resulting proteins of this pathway are part of seizure pathogenesis in experimental models, proposing that CR could help in seizure control through the proteins of the Wnt signaling pathway. Nevertheless, further research needs to be done to understand this mechanism and suggest a specific molecular/genetic target for therapeutic intervention.
The Effects of Caloric Restriction in the mTOR Signaling Pathway
One of the Wnt functions is to increase gene expression through the transcriptional activity of β-catenin, as well as protein synthesis through the mammalian target of rapamycin (mTOR). However, their interaction is not completely understood. Caloric restriction reduces the epileptic activity inhibiting the mTOR signaling pathway. This is a main pathophysiology component of epileptic seizure (109). The hyperactivation of mTOR participates in epilepsy; the inhibition on this kinase results in beneficial anti-convulsive effects. It is related to cell proliferation, protein synthesis, cell survival, angiogenesis, apoptosis, and cell motility.
mTOR is divided into two complexes, mTORC1, implicated in cell growth and proliferation as well as metabolism, and mTORC2, involved in the regulation of protein kinases and the cytoskeletal elements of the cell (110). Mutations in the mTOR pathway are associated with dysplasias, epilepsy, and neurodevelopmental disorders like tuberous sclerosis complex, Cowden's syndrome, polyhydramnios, megalencephaly, and neurofibromatosis type 1 (111).
The TOR protein family plays a role in regulating mRNA transcription initiation and protein translation in response to intracellular concentrations of amino acids and other essential nutrients. The mTOR pathway is activated by ingestion, rising from blood glucose levels, which consequently increases insulin. Excess protein in the diet stimulates the mTOR pathway, phosphorylating the insulin receptor, among others, while generating more insulin, which can aggravate the Ser/Thr phosphorylation of the insulin receptor through mTORC1 activation. mTORC1 activation suppresses Wnt canonical pathway expression and regulates Fzd and Dvl for stem cell functioning and cellular homeostasis (112). As mentioned before, Wnt protein inhibits GSK3β through Fzd ligands, activating TSC2. When TSC2 is inhibited, it indirectly activates mTORC1 by supporting the relation between mTORC1 and Wnt pathways (15, 108).
This pathway responds to different extra- and intracellular cues. It also affects mitochondrial function. When mTOR is hyperactivated, it induces the production of ROS and eventually causes neuronal damage. A high level of AMPK leads to mTOR pathway regulation. AMPK is stimulated at low energy levels that later activate tuberin protein (encoded by tuberous sclerosis 2 gene, TSC2). Tuberin plus hamartin (encoded by TSC1) inhibits complex mTORC1 (113). Additionally, AMPK removes insulin and growth factors, activating protein kinase B (PKB/Akt), which inhibits tuberin and activates mTORC1 (114). If there is an increase in mTOR cascade function, epilepsy also increases. Therefore, the inhibition of mTOR has shown anti-convulsive effects (115).
Diets such as the ketogenic diet and caloric restriction have shown anti-epileptic actions in several different animal models due to mTOR pathway inhibition. Among the anti-epileptic effects of CR found in epileptic inbred mice, delayed seizure onset and decreased seizure incidence are found (78), as well as increased paired-pulse inhibition, an increased threshold of maximal dentate gyrus activation, no spreading of depression-like events (116), delayed kindling rate in seizure-prone rats (85), and a maximization after-discharge threshold in electric kindling epilepsy model (82).
There is undoubtedly a physiologic connection of the Wnt and the mTOR signaling pathways that gives us a more profound understanding of the interplay between mTORC1 and Wnt/β-catenin signaling pathway. However, more research needs to be done to conclude a novel therapeutic target.
Ketogenic Diet
Although the different mechanisms of action in diets are not fully understood yet, the KD has shown a decrease in seizures in about 50% of the patients (50). About 50% of individuals treated with KD have reduced seizure frequency (117), even stopping seizures at all in 10–15% (118). Studies suggest that this relies on the production of ketone bodies that cause an effect on neuronal function and release of neurotransmitters, such as a decrease of glutamate and an increase of GABA synthesis.
On the other hand, it has not been set specifically which epileptic syndromes the KD could benefit, but its effectiveness has been proven in glucose transporter 1 deficiency syndrome, Angelman syndrome, and tuberous sclerosis complex, and a potential action in autoimmune epilepsy and encephalitis has been suggested due to the KD anti-inflammatory actions (119). Pyruvate dehydrogenase deficiency has been reported to have a beneficial response to the KD. However, the KD is not appropriate for some patients with conditions of primary carnitine deficiencies and β-oxidation defects due to the inability to metabolize ketone bodies (120). A small percentage of patients have adverse effects like gastrointestinal symptoms and high serum lipids in short-term studies, which do not allow them to follow the diet as a treatment, which suggests that genetic changes are involved (50, 117). Currently, there are different approaches to reduce the adverse effects of the KD (121).
Moreover, 15 years ago, research found 42 genes to be involved in intracellular metabolism and signal transduction pathways, specifically oxidative phosphorylation and mitochondrial protein metabolism expressed in different ways after KD treatment (122). Additionally, a reduction in seizures was demonstrated in 50% of a group of patients with epilepsy, with the potassium sodium-activated channel subfamily T after KD treatment (45).
Neurotransmitters Involved in the Ketogenic Diet
GABA
For epilepsy, numerous pathologic pathways produce neuronal excitability resulting in seizures, including insufficient synaptic inhibition, and changes in the receptors like low GABAA (GABAA-Rs) levels in the molecular layer of the dentate gyrus shown in kindled rats (123). Ketone bodies have multiple direct and indirect effects that modify neuronal excitability, intracellular metabolic changes, and modification in cell gene expression. Furthermore, it has been confirmed that the KD and CR enhance GABAA-Rs by increasing inhibition specifically in the dentate gyrus (116), a structure highly related to epileptic activity. The inhibition of glycolysis by 2-deoxy-D-glucose (2DG) has been observed to provide neuroprotective effects against seizures (124). Perhaps this inhibition provides a decrease in the ATP/ADP ratio. In 2013, Kossoff and Wang (125) mentioned that 2DG inhibition could prevent epileptic seizures by a different mechanism than the one presented by the KD since 2DG does not produce ketosis; however, it also increases GABAA-Rs at synaptic or membrane levels due to glyceraldehyde-3-phosphate dehydrogenase (126).
Aspartate; Glutamate to GABA
It is suggested that aspartate plays a significant role in hippocampal epileptogenesis since it is synthesized by glutamate when glucose is not available and functions as the excitatory amino acid that is released after depolarization, increasing its levels in the intra-synaptic space. The KD increases blood branched-chain amino acids, specifically high levels of leucine in the brain, which block the transamination of glutamate to aspartate (127). Another mechanism that supports the function of the KD in decreasing aspartate is the reduction of oxaloacetate, which is converted to aspartate through transamination reaction (128).
Ketosis induces glutamate decarboxylase, causing fewer levels of glutamate and high levels of GABA (129), by some mechanism not yet understood. The KD response alters the glutamate–glutamine cycle by intensifying the astrocytic glutamine pathway extracting glutamate from the synaptic clefts and returning it to the presynaptic neuron as a GABA precursor (Figure 3). In 2010, Juge et al. (130) showed that ketone bodies, especially acetoacetate, competed reversibly for the chlorine (Cl−) binding site in the vesicular glutamate transporter, decreasing glutamate release, which had been available to glutamic acid decarboxylase for GABA synthesis in GABAergic neurons only (131).
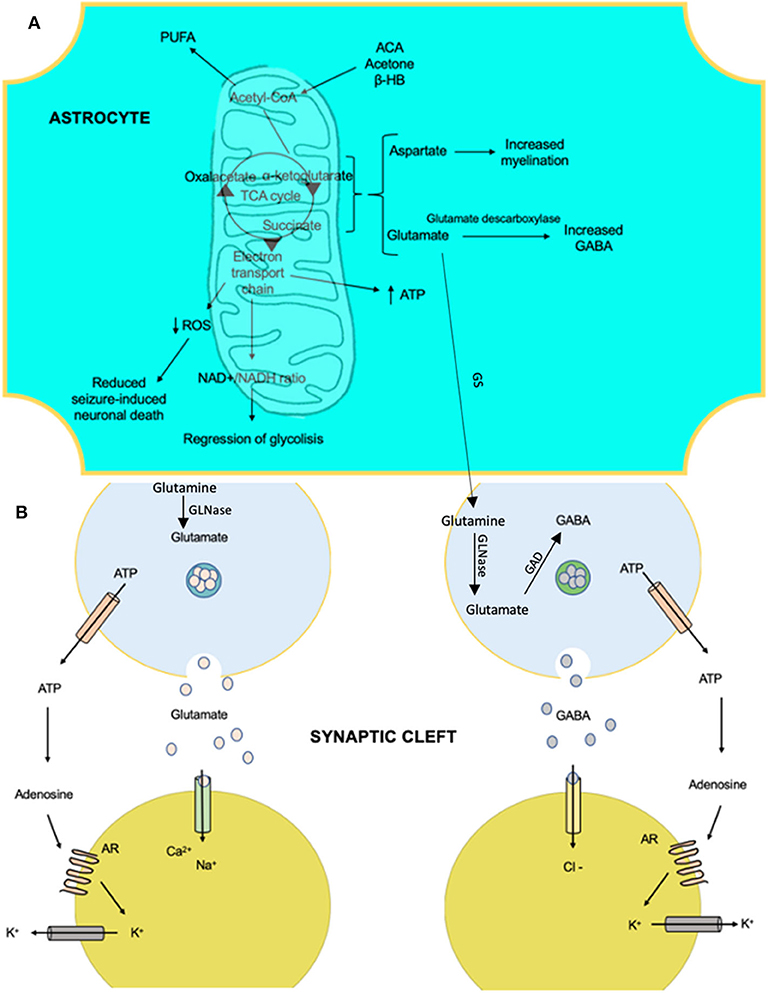
Figure 3. The anti-epileptic effects that involve the metabolism of ketone bodies and the activity of neurotransmitters with the ketogenic diet. (A) Products of the mitochondria in the metabolism of ketone bodies in brain cells, which confer anti-epileptic effects. These effects are linked to changes in the amounts of neurotransmitters and influence membrane polarity. (B) The effects of certain products on ion transfer at the synapse. AR, adenosine receptor A1; ACA, acetoacetate; β-HB, beta-hydroxybutyrate; GAD, glutamate decarboxylase; GS, glutamine synthetase; GLNase, glutaminase; PUFA, polyunsaturated fatty acid; ROS, reactive oxygen species.
Another possible mechanism may be due to an inhibition of the voltage-sensitive calcium channels enhanced by GABAergic function through receptors after KD (132), causing greater stability among neurons in the hippocampus and control over transport proteins and Na+/K+-ATPase pump, causing ionic homeostasis for a more prolonged period than expected. It was demonstrated that, after KD therapy, there is a higher resistance to metabolic stress, raising the threshold to release larger amounts of ATP (10).
GABA, Glutamate, and Wnt
Wnt 5a in the non-canonical calcium pathway causes higher levels of GABAA-Rs in the neuron surface by facilitating membrane insertion and increasing neuronal inhibition by regulating the synaptic plasticity of inhibitory circuits in the hippocampus (31, 133). When tyrosine-protein kinase receptor (RTK) is expressed, the non-canonical pathway of Wnt increases intracellular calcium (Wnt/Ca2+ pathway), causing high levels of PKC and leading to regulate channel opening and NMDAR, a receptor for glutamate. Specifically, Wnt5a maintains a basal NMDAR synaptic transmission in calcium-dependent neurons, as shown in the CA1 of the hippocampus, and it has been described that SNAP25 peptide may play a role in this mechanism as well (107, 134).
The Ketogenic Diet Modulates the Mitochondria, so Does Wnt
Seizures and epileptogenesis cause the mitochondria to release reactive oxygen species (ROS), causing oxidative stress and cellular macromolecule dysfunction, affecting the production of ATP and mtDNA stability. Overall, this damage increases neuronal excitability. For instance, in temporal lobe epilepsy (TLE), mitochondrial dysfunction after oxidative stress is associated with excitotoxicity and apoptosis in the hippocampus (135, 136). How histological and functional modifications occur in the hippocampus in TLE have been shown; some of these changes include neuronal loss in CA1 and CA3. In other types of epilepsy, gliosis and granule cell dispersion in dentate gyrus have been also demonstrated (137, 138).
Studies have shown that the KD decreases the reactive oxygen species of the mitochondria of the hippocampus (139), one of the most vulnerable structures to the epileptic phenomena (140). In the hippocampus, the KD up-regulates genes involved in metabolic pathways and down-regulates genes implicated in intracellular signal transduction, resulting in an increase of mitochondrial ATP synthase beta subunit and its precursor and enhancing the brain ATP production while maintaining the neuronal membranes in continued depolarization (42). Ketone bodies prevent cell death by activating the KATP channels in the mitochondria (mitoKATP channel) by glycolytic reduction (141). Mitochondrial biogenesis has been proposed after a chronic ketosis state. The mitochondrial biogenesis allows an increase in the storage of phosphates, glutamate, and glutamine, resulting in an ATP increase. The Wnt signals activate mitochondrial biogenesis and reactive oxygen species generation, leading to cell damage (65). The Armcx groups of proteins have a bimodal subcellular localization, associated with the mitochondrial outer membrane; its overexpression produces mitochondrial alterations, demonstrating that this family of proteins plays an important role in the regulation of mitochondrial dynamics and aggregation. These genes are expressed highly in the developing and adult nervous systems. Furthermore, higher levels of expression of Armcx3, which is part of the exclusive mammalian gene family, interacts with the Kinesin/Miro/Trak2 complex in a Ca2+-dependent manner, regulating mitochondrial dynamics and neuron trafficking. When the intracellular Ca2+ levels are depleted, there is also a degradation of Armcx3 through the Wnt signaling pathways, generating a mitochondrial aggregation effect reversed by the overexpression of Armcx3 and evaded by the activation of PKC. Therefore, the Wnt pathway can control mitochondrial dynamics by regulating Armcx proteins (142). The canonical pathway of Wnt did not alter the pattern of Armcx3-induced mitochondrial aggregation; it was also observed that the non-canonical pathway Wnt/PKC regulated both mitochondrial aggregation and Armcx3 protein levels; it was reduced with the activation of the non-canonical Wnt/Ca2+ pathway, suggesting that Wnt regulates mitochondrial distribution and dynamics through degradation of the Armcx3 protein (40).
Many controversies are documented about complex enzymes; there is a general agreement on the decrease of complex I, which has shown mitochondrial dysfunction. However, others differ in the alteration of complexes II, III, and IV (136). After induced seizure, complexes I and IV of the mitochondria were decreased, and an increase of complex II provoked mitochondrial dysfunction due to oxidative stress, associated to a down-regulation in the encoded proteins for the electron transport chain, essential to membrane permeability, neurotransmitter biosynthesis, and the production of ATP, among other functions, resulting in a ROS increase, which continues a vicious cycle of producing oxidative stress and mitochondrial dysfunction. The KD could prevent mitochondrial complexes I, II, and III inhibition and increase uncoupling proteins (UCP2), reducing oxidative stress in the brain due to a decrease of free radicals and expressing antioxidant proteins that make the brain more resilient to oxidative stress (143).
After the KD, an enhancement of the mitochondria increases the glutathione (GSH) levels and biosynthesis in the hippocampus, contributing to a lower level of ROS as hydrogen peroxide (H2O2) and improvement of mtDNA (144). The abovementioned condition is maintained by an increase of low levels of 4-hydroxy-2 non-enal, a product of lipid peroxidation, which leads to a protective transcription factor pathway involving Nrf2 protein, increasing GSH biosynthesis by gene transcription after the KD treatment (74).
Mitochondrial permeability transition (mPT) is suggested to be the reason for cell death prevention due to the KD treatment after having been inhibited by oxidative stress. Ketone bodies act in a similar way to cyclosporine A, which can increase the threshold of calcium-induced mPT opening (145). It also decreases brain pH, inhibiting proton-sensitive ion channels, while increasing monocarboxylic transporters (MCT1, a protein that transports ketone bodies to the brain) (42).
Mechanisms of Action of the Ketogenic Diet and the Participation of KATP Channels in Neuronal Excitability
In recent years, more and more research has been carried out on ion channels as responsible for the ketogenic effect of restrictive diets on neuronal membrane potential. The latest studies indicate that the KD modifies the KATP channels' function. These channels are part of the metabolic state of the cell and the membrane potential due to their response and sensitivity to variations in concentrations of ATP; therefore, the channels are inhibited by high concentrations of intracellular ATP and activated by ADP (146), causing variations in the membrane potential of the neuron by allowing variations of K+ ion conductance (Figure 4). In experimental models, evidence still suggests that the KD restores and increases ATP levels (147–149). We suggest that the activation of KATP channels leads to this increase of ATP to provide its neural stability. These KATP channels are present in multiple neural areas. Regardless of being areas with glucose-sensitive function, it presents a high expressiveness specifically in the hippocampus (150–152). KATP channels of the neuronal membrane are inhibited in a basal state, except in states of severe metabolic deprivation (anoxia and ischemia), with both situations leading to cellular stress (153, 154).
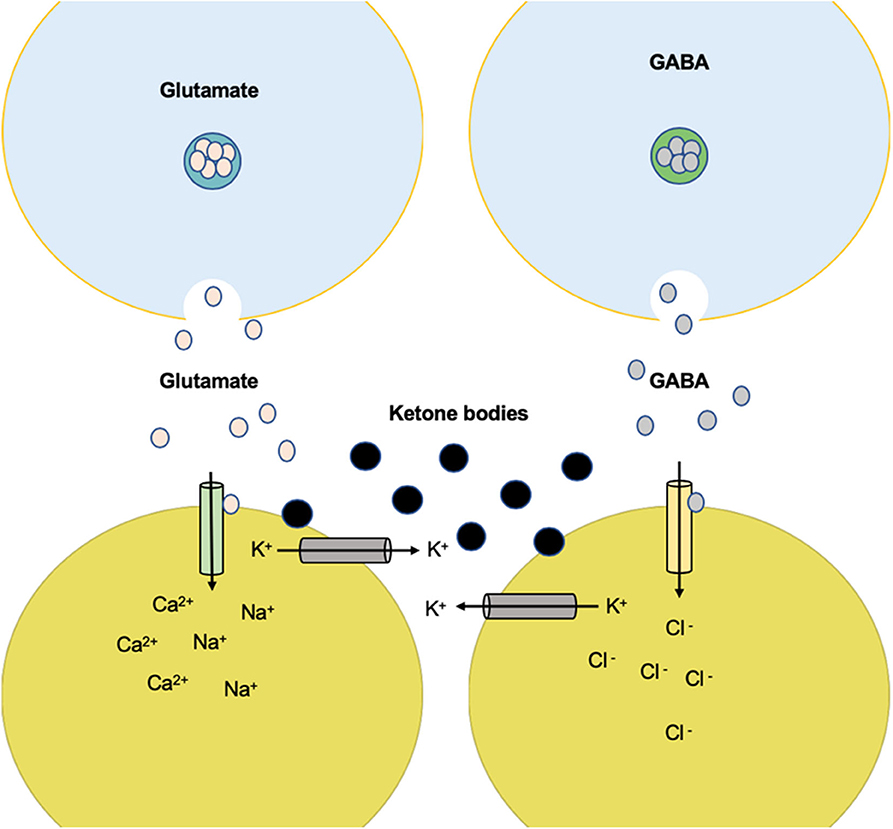
Figure 4. Anticonvulsant effect of K+ channel-mediated ketone body metabolism. Ketone body metabolism increases overall ATP but reduces glycolysis and glycolytic ATP synthesis. The reduction of ATP near the plasma membrane can disinhibit the KATP channels and therefore reduce electrical activity. High electrical activity (as in a seizure) increases Na+ input and ATP utilization near the plasma membrane. This produces negative changes in activity through the KATP channels. The set point at which this negative feedback safety mechanism is activated is determined by the level of glycolytic ATP synthesis.
A study by Shen et al. in 2016, using dopaminergic neurons of the substantia nigra, carried out a patch-clamp type registry, which showed the activation of AMPK during the “whole cell” that caused a gradual increase of the KATP channel functionality and vice versa; the inhibition of AMPK activity caused a decrease in KATP channels. In 2011, Ikematsu et al. (155) showed that AMPK activation generated the function of voltage-regulated K+ channels (Kv2.1) by the phosphorylation of two channel sites (s440 and s537), inducing membrane hyperpolarization. This is important because AMPK is essential for neuronal energy consumed after an epileptic seizure. Besides that, a high level of AMPK has been shown to up-regulate the Bcl2 modifying factor gene, preventing neuronal death in CA1 and CA3 of the hippocampus induced by seizures (61).
The participation of Bad (BCL-2 cell death agonist) in neuronal metabolism has been mentioned, proven by its genetic deletion that generates a protective effect against epileptic seizures. Bad's genetic modifications decrease glycolytic metabolism, inducing a marked increase in activity in the KATP channels (57). Bad ablation reduces the epileptiform activity of the hippocampal-entorhinal circuit in picrotoxin-induced epileptiform neurons (partial inhibitor of GABAA-Rs). Therefore, it confers a protective effect against seizures dependent on the KATP channels' activity (58).
Tanner et al. (156) showed that bursts of stimulation in dentate nuclei neurons increase the probability of opening the KATP channels through the administration of the ketone body R-β-hydroxybutyrate. They also found that neuron over-stimulation by triggering five action potentials by applying a current of 20 Hz in the same area caused a post-stimulation hyperpolarization mediated by the KATP channels, suggesting the excitation limitation of seizure spread through the activation of KATP channels.
A study targeting substantia nigra pars reticularis (an area that participates in seizure activity) and ketone bodies observed reduction in neural excitability through KATP channels and GABA. Ketone bodies keep KATP channels open, which might increase the expression of the GABAB subunit (157).
An extracellular increase of K+ prevents the release of Ca2+, inhibiting the signaling of the Wnt pathway and the translocation of β-catenin to the cell nucleus. Wnt ligands hyperpolarized the cells by activating K+ current by Ca2+ (157). The Wnt response also depends on the activation of Bcl9 that activates genes that have response sequences to Wnt (158, 159).
Adenosine may play a relevant mechanism of the KD as a neuroprotective and anticonvulsant effect. Masino et al. (160) proposed the KD as metabolic stress that would cause a decrease in the enzyme adenosine kinase, an inhibitory regulator of adenosine, inducing its increase in the cell and activating its receptors, causing a tonic inhibition of presynaptic and postsynaptic neurons through the adenosine receptor 1 (RA1) and conferring a protective effect against epileptic seizures. Moreover, adenosine has shown to increase seizures in experimental models and decrease DNA methylation, even after discontinuing the diet (161). In experimental models, the KD has demonstrated to improve cerebral infarcts because of its association to increase extracellular adenosine levels, reduce volume infarcts, and boost regional cerebral blood flow, again by expressing the neuroprotective effect of the KD (162).
Conclusion
The KD has shown a decrease in seizures through the production of ketone bodies that cause an effect on neuronal function and neurotransmitter release, such as a decrease of glutamate, enhanced mitochondrial function, an increase of GABA synthesis, and KATP channels' activation. On the other hand, CR may inhibit glycolysis and, like the KD, is involved in mitochondrial biogenesis, a decrease of oxidative stress, and inflammation reduction. The KD and CR per se down-regulates and up-regulates certain genes involved in seizure activity. There is still not enough evidence, because of the limited number of studies and small sample sizes, to suggest a specific treatment related to these genes and the association with KATP channels to control epileptic seizures. In Figure 5, we aim to show the relationship between genes found to be regulated by restrictive diets in epileptogenesis; we observed that almost all genes interact.
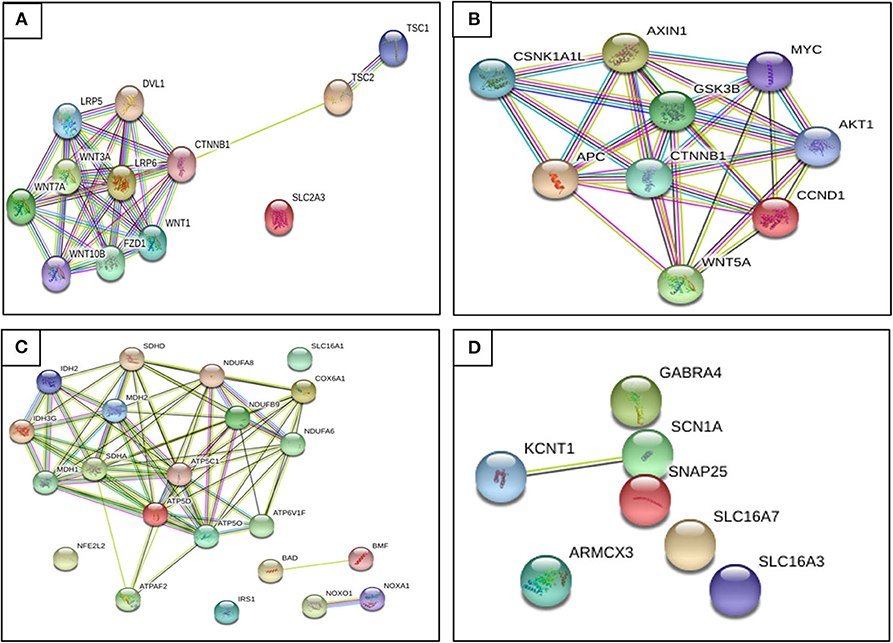
Figure 5. Relationship between genes found regulated by restrictive diets in epileptogenesis. (A) Genes up-regulated on caloric restriction (CR). (B) Genes down-regulated on CR. (C) Genes up-regulated on KD. (D) Genes up-regulated on KD. We observe that almost all genes interact. Until recently, there has not been evidence described to prove some of these genes as a potential pharmacotherapy, though we hope to provide a better understanding of epileptogenesis and seizure pathophysiology and future ideas and research to find a possible diet therapy focused on gene expression (retrieved from https://string-db.org/).
The molecular changes that intervene in the glycolytic metabolism of neurons are crucial in modulating the electrophysiological response to epileptic seizures. Understanding the metabolic pathways interfering with the functionality of the KATP channels in neurons with epileptiform crises is of great importance because the evidence shows that it could be introduced as a new pharmacological target for the new therapeutic generations being formulated for the modification of these metabolic pathways. Nevertheless, there has not been current evidence described to prove that some of these genes has a potential anticonvulsant effect. We hope to provide a better understanding of epileptogenesis and seizure pathophysiology and future ideas and research to find a possible diet therapy focused on gene expression in the regulation in Wnt pathways and their regulation through the KATP channels to reduce epileptic seizures.
Author Contributions
All the authors contributed in equal ways to the writing and making of this manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The present work was possible thanks to grants provided by CONACYT (287959) and the National Institute of Neurology and Neurosurgery.
References
1. Billakota S, Devinsky O, Kim KW. Why we urgently need improved epilepsy therapies for adult patients. Neuropharmacology. (2020) 170:107855. doi: 10.1016/j.neuropharm.2019.107855
2. Kwan P, Brodie MJ. Refractory epilepsy: mechanisms and solutions. Expert Rev Neurother. (2006) 6:397–406. doi: 10.1586/14737175.6.3.397
3. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad-hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. (2009) 51:1069–77. doi: 10.1111/j.1528-1167.2009.02397.x
4. Rubio C, Custodio V, Juárez F, Paz C. Stimulation of the superior cerebellar peduncle during the development of amygdaloid kindling in rats. Brain Res. (2004) 1010:151–5. doi: 10.1016/j.brainres.2004.03.015
5. Seymour KJ, Bluml S, Sutherling J, Sutherling W, Ross BD. Identification of cerebral acetone by 1H-MRS in patients with epilepsy controlled by ketogenic diet. MAGMA. (1999) 8:33–42. doi: 10.1016/S1352-8661(99)00006-X
6. Vannucci RC, Yager JY, Vannucci SJ. Cerebral glucose and energy utilization during the evolution of hypoxic- ischemic brain damage in the immature rat. J Cereb Blood Flow Metab. (1994) 14:279–88. doi: 10.1038/jcbfm.1994.35
8. Wang HS, Lin KL. Ketogenic diet: an early option for epilepsy treatment, instead of a last choice only. Biomed J. (2013) 36:16–7. doi: 10.4103/2319-4170.107155
9. Talevi A, Rocha L. Antiepileptic Drug Discovery. 1st edn. New York, NY: Springer Science; Business Media (2016). p. 163–74.
10. Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. (2006) 60:223–35. doi: 10.1002/ana.20899
11. Cisternas P, Salazar P, Silva-Álvarez C, Barros LF, Inestrosa NC. Activation of Wnt signaling in cortical neurons enhances glucose utilization through glycolysis. J Biol Chem. (2016) 291:25950–64. doi: 10.1074/jbc.M116.735373
12. Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J Neurosci. (2007) 27:3618–25. doi: 10.1523/JNEUROSCI.0132-07.2007
13. Manning BD, Toker A. AKT/PK signaling: navigating the network. Cell. (2017) 169:381–405. doi: 10.1016/j.cell.2017.04.001
14. Kuhn I, Bartholdi MF, Salamon H, Feldman RI, Roth RA, Johnson PH. Identification of AKT-regulated genes in inducible MERAkt cells. Physiol Genomics. (2001) 7:105–14. doi: 10.1152/physiolgenomics.00052.2001
15. Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. (2006) 126:955–68. doi: 10.1016/j.cell.2006.06.055
16. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. (1991) 66:589–600. doi: 10.1016/0092-8674(81)90021-0
17. Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, et al. Identification of FAP locus genes from chromosome 5q21. Science. (1991) 253:661–5. doi: 10.1126/science.1651562
18. Tamai K, Zeng X, Liu C, Zhang X, Harada Y, Chang Z, et al. A mechanism for Wnt coreceptor activation. Mol Cell. (2004) 13:149–56. doi: 10.1016/S1097-2765(03)00484-2
20. Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. Embo J. (1998) 17:1371–84. doi: 10.1093/emboj/17.5.1371
21. Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, et al. Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3β. Science. (1988) 280:596–9. doi: 10.1126/science.280.5363.596
22. Rubio C, Taddei E, Acosta J, Custodio V, Paz C. Neuronal excitability in epileptogenic zones regulated by the Wnt/B-Catenin pathway. CNS Neurol Disord. (2020) 19:2–11. doi: 10.2174/1871527319666200120143133
23. Desjardins PR, Lue PF, Liew CC, Gornal AG. Purification and properties of rat liver nuclear protein kinases. Can. J. Biochem. (1972) 50:1249–59. doi: 10.1139/o72-170
24. Ozawa M, Baribault H, Kemler R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J. (1989) 8:1711–7. doi: 10.1002/j.1460-2075.1989.tb03563.x
25. Hodges SL, Lugo JN. Wnt/β-catenin signaling as a potential target for novel epilepsy therapies. Epilepsy Res. (2018) 146:9–16. doi: 10.1016/j.eplepsyres.2018.07.002
26. Xiong Y, Connolly T, Futcher B, Beach D, Human. D-type cyclin. Cell Press. (1991) 65:691–9. doi: 10.1016/0092-8674(91)90100-D
27. Siegfried E, Chou TB, Perrimon N. Wingless signaling acts through zeste-white 3, the Drosophila homolog of glycogen synthase kinase-3, to regulate engrailed and establish cell fate. Cell. (1992) 71:1167–79. doi: 10.1016/S0092-8674(05)80065-0
28. Bishop JM, Varmus H. Functions and origins of retroviral transforming genes. In: Weis R, Teich N, Varmus H, Coffin JM, editors. RNA Tumor Viruses. Cold Spring Harbor: Cold Spring Harbor Laboratory. (1985). p. 249–356.
29. Zhou J, Wu M, Taniyasu S, Besso H, Tanaka O, Saruwatari Y, et al. Dammarane- saponins of sanchi-ginseng, roots of panax notoginseng. Structures of new saponins, notoginsenosides-R1 and -R2, and identification of ginsenosides-Rg2 And -Rh1. Chem Pharm Bull. (1981) 29:2844–50. doi: 10.1248/cpb.29.2844
30. Adamson M, Dennis C, Delaney S, Christiansen J, Monkley S, Kozak C, et al. Isolation and genetic mapping of two novel members of the murine Wnt gene family, Wnt11 and Wnt12, and the mapping of Wnt5a and Wnt7a. Genomics. (1994) 24:9–13. doi: 10.1006/geno.1994.1575
31. Cuitino L, Godoy JA, Farías GG, Couve A, Bonansco C, Fuenzalida M, et al. Wnt-5a modulates recycling of functional GABAA receptors on hippocampal neurons. J Neurosci. (2010) 30:8411–20. doi: 10.1523/JNEUROSCI.5736-09.2010
32. Peifer M, Rauskolb C, Williams M, Riggleman B, Wieschaus E. The segment polarity gene armadillo interacts with the wingless signaling pathway in both embryonic and adult pattern formation. Development. (1991) 111:1029–43.
33. Perrimon N, Mahowald AP. Multiple functions of segment polarity genes in Drosophila. Dev Biol. (1987) 119:587–600. doi: 10.1016/0012-1606(87)90061-3
34. Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, et al. Arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. (2000) 407:527–30. doi: 10.1038/35035110
35. Kayano T, Fukumoto H, Eddy RL, et al. Evidence for a family of human glucose transporter-like proteins. Sequence and gene localization of a protein expressed in fetal skeletal muscle and other tissues. J Biol Chem. (1988) 263:15245–8.
36. Janssen L, Sandkuyl L, Merkens E, Maat-Kievit J, Sampson J, Fleury P, et al. Genetic heterogeneity in tuberous sclerosis. Genomics. (1990) 8:237–42. doi: 10.1016/0888-7543(90)90277-2
38. Lee F, Lane T, Kuo A, Shackleford G, Leder P. Insertional mutagenesis identifies a member of the wnt gene family as a candidate oncogene in the mammary epithelium of Int-2/Fgf-3 transgenic mice. Proc Natl Acad Sci USA. (1995) 92:2268–72. doi: 10.1073/pnas.92.6.2268
39. Roelink H, Wagenaar E, Lopes da Silva S, Nusse R. Wnt-3, a gene activated by proviral insertion in mouse mammary tumors, is homologous to Int-1/Wnt-1 and is normally expressed in mouse embryos and adult brain. Proc Natl Acad Sci USA. (1990) 87:4519–23. doi: 10.1073/pnas.87.12.4519
40. Serrat R, López-Doménech G, Mirra S, Quevedo M, Garcia-Fernàndez J, Ulloa F, et al. (2013). The non-canonical Wnt/PKC pathway regulates mitochondrial dynamics through degradation of the arm-like domain-containing protein Alex3. PLoS ONE. (2013) 8:e67773. doi: 10.1371/journal.pone.0067773
41. Kurochkin I, Yonemitsu N, Funahashi S, Nomura H. ALEX1, a novel human armadillo repeat protein that is expressed differentially in normal tissues and carcinomas. Biochem Biophys Res Commun. (2000) 280:340–7. doi: 10.1006/bbrc.2000.4125
42. Sook Noh H, Po Lee H, Wook Kim D, Soo Kang S, Jae Cho G, Rho JM, et al. A cDNA microarray analysis of gene expression profiles in rat hippocampus following a ketogenic diet. Mol Brain Res. (2004) 129:80–7. doi: 10.1016/j.molbrainres.2004.06.020
43. Khrestchatisky M, MacLennan A, Chiang M, Xu W, Jackson M, Brecha N, et al. A novel α subunit in rat brain GABAA receptors. Neuron. (1989) 3:745–53. doi: 10.1016/0896-6273(89)90243-2
44. Paulais M, Lachheb S, Teulon J. A Na+- and Cl—activated K+ channel in the thick ascending limb of mouse kidney. J Gen Physiol. (2006) 127:205–15. doi: 10.1085/jgp.200509360
45. Borlot F, Abushama A, Morrison-Levy N, Jain P, Puthenveettil VK, Abukhalid M, et al. KCNT1-related epilepsy: an international multicenter cohort of 27 pediatric cases. Epilepsia. (2020) 61:679–92. doi: 10.1111/epi.16480
46. Garcia C, Brown M, Pathak R, Goldstein J. cDNA Cloning of MCT2, a second monocarboxylate transporter expressed in different cells than MCT1. J Biol Chem. (1995) 270:1843–9. doi: 10.1074/jbc.270.4.1843
47. Cho CH. Commentary: targeting LDH enzymes with a stiripentol analog to treat epilepsy. Front Cell Neurosci. (2015) 9:264. doi: 10.3389/fncel.2015.00264
48. Price NT, Jackson VN, Halestrap AP. Cloning and sequencing of four new mammalian monocarboxylate transporter (MCT) homologues confirms the existence of a transporter family with an ancient past. Biochem J. (1998) 329:321–28. doi: 10.1042/bj3290321
49. Noda M, Ikeda T, Kayano T, Suzuki H, Takeshima H, Kurasaki M, et al. Existence of distinct sodium channel messenger rnas in rat brain. Nature. (1986) 320:188–92. doi: 10.1038/320188a0
50. Dutton SBB, Escayg A. Genetic influences on ketogenic diet efficacy. Epilepsia. (2008) 49:67–9. doi: 10.1111/j.1528-1167.2008.01839.x
51. Oyler G, Higgins G, Hart R, Battenberg E, Billingsley M, Bloom F, et al. The identification of a novel synaptosomal-associated protein, SNAP-25, differentially expressed by neuronal subpopulations. J Cel Biol. (1989) 109:3039–52. doi: 10.1083/jcb.109.6.3039
52. Nonneman D, Rohrer G. Comparative mapping of a region on chromosome 10 containing qtl for reproduction in swine. Animal Genetics. (2002) 34:42–6. doi: 10.1046/j.1365-2052.2003.00928.x
53. Jordan MEAM, Breen G. Molecular cloning of an import precursor of the -subunit of the human mitochondrial ATP synthase complex. Biochim Biophys Acta. (1992) 1130:123–6. doi: 10.1016/0167-4781(92)90477-H
54. Devenish R, Prescott M, Boyle G, Nagley P. The oligomycin axis of mitochondrial ATP synthase: OSCP and the proton channel. J Bioenerg Biomembranes. (2000) 32:507–15. doi: 10.1023/A:1005621125812
55. Yokoyama K, Ohkuma S, Taguchi H, Yasunaga T, Wakabayashi T, Yoshida M. V-type H+ - ATPase/Synthase from a termophillic eubacterium, thermus thermophilus. J Biol Chem. (2000) 275:13955–61. doi: 10.1074/jbc.275.18.13955
56. Wang Z, White P, Ackerman S. Atp11p And Atp12p are assembly factors for the F1-Atpase in human mitochondria. J Biol Chem. (2001) 276:30773–8. doi: 10.1074/jbc.M104133200
57. Giménez-Cassina A, Martínez-François JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, et al. BAD-Dependent regulation of fuel metabolism and KATP channel activity confers resistance to epileptic seizures. Neuron. (2012) 74:719–30. doi: 10.1016/j.neuron.2012.03.032
58. MartÍnez-François JR, Fernández-Agüera MC, Nathwani N, Lahmann C, Burnham VL, Danial NN, et al. BAD and KATP channels regulate neuron excitability and epileptiform activity. Elife. (2018) 7:e32721. doi: 10.7554/eLife.32721
59. Yang E, Zha J, Jockel J, Boise L, Thompson C, Korsmeyer S. Bad, a heterodimeric partner for Bcl-Xl and Bcl-2, displaces Bax and promotes cell death. Cell Press. (1995) 80:285–91. doi: 10.1016/0092-8674(95)90411-5
60. Puthalakath H, Villunger A, O'Reilly L, Beaumont J, Coultas L, Cheney R, et al. Bmf: A proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. (2001) 293:1829–32. doi: 10.1126/science.1062257
61. Yang Y, Zhu B, Zheng F, Li Y, Zhang Y, Hu Y, et al. Chronic metformin treatment facilitates seizure termination. Biochem Biophys Res Commun. (2017) 484:450–5. doi: 10.1016/j.bbrc.2017.01.157
62. Smith EO, Lomax MI. Structural organization of the bovine gene for the heart/muscle isoform of cytochrome c oxidase subunit via. Biochim Biophys Acta. (1993) 1174:63–71. doi: 10.1016/0167-4781(93)90092-R
63. Shiga K. lsoenzyme Pattern of NADP Dependent Isocitrate Dehydrogenase in Human Tissue and the Effect of Manganese and Magnesium on Tetrazolium Method. Japanese J Clin Chem. (1978) 7:63-71. doi: 10.14921/jscc1971b.6.1_63
64. Okamoto K, Matsuzaka Y, Yoshikawa Y, Takaki A, Kulski J, Tamiya G, et al. Identification of nad+-dependent isocitrate dehydrogenase 3 γ-like (IDH3GL) gene and its genetic polymorphisms. GENE. (2003) 323:141–8. doi: 10.1016/j.gene.2003.09.014
65. Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ, Elledge SJ. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. (2010) 24:1507–18. doi: 10.1101/gad.1924910
66. Sun X, Miralpeix M, Myers M, Glasheen E, Backer J, Kahn C, et al. Expression and function of IRS-1 in insulin signal transmission. J Biol Chem. (1992) 267:22662–22612.
67. Mowbray J. Evidence for the role of a specific monocarboxylate transporter in the control of pyruvate oxidation by rat liver mitochondria. North Holland Publishing Company. (1974) 44:344–7. doi: 10.1016/0014-5793(74)81174-9
68. Conde-del Pino E, Pérez-Vilar M, Cintrón-Rivera A, Señeriz R. Studies in schistosoma mansoni. I. malic and lactic dehydrogenase of adult worms and cercariae. Exp Parasitol. (1966) 18:320–6. doi: 10.1016/0014-4894(66)90033-6
69. Dunbar D, Shibasaki Y, Dobbie L, Andersson B, Brookes B. In situ hybridization mapping of genomic clones for five human respiratory chain complex I genes. Cytogenet Cell Genet. (1997) 78:21–4. doi: 10.1159/000134618
70. Triepels R, van den Heuvel L, Loeffen J, Smeets R, Trijbels F, Smeitink J. The nuclear-encoded human NADH:ubiquinone oxidoreductase NDUFA8 subunit: cdna cloning, chromosomal localization, tissue distribution, and mutation detection in complex-I-deficient patients. Human Genetics. (1998) 103:557–63. doi: 10.1007/s004390050869
71. Walker JE, Arizmendi JM, Dupuis A, et al. Sequences of 20 subunits of NADH:ubiquinone oxidoreductase from bovine heart mitochondria. Application of a novel strategy for sequencing proteins using the polymerase chain reaction. J Mol Biol. (1992) 226:1051–72. doi: 10.1016/0022-2836(92)91052-Q
72. Bánfi B, Clark R, Steger K, Krause K. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. (2002) 278:3510–3. doi: 10.1074/jbc.C200613200
73. Scheck AC, Abdelwahab MG, Fenton KE, Stafford P. The ketogenic diet for the treatment of glioma: insights from genetic profiling. Epilepsy Res. (2012) 100:327–37. doi: 10.1016/j.eplepsyres.2011.09.022
74. Milder J, Patel M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. (2012) 100:295–303. doi: 10.1016/j.eplepsyres.2011.09.021
75. Moi P, Chan K, Asunis I, Cao A, Kan Y. Isolation of NF-E2-Related Factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Genetics. (1994) 91:9926–30. doi: 10.1073/pnas.91.21.9926
76. Wood D, Darlison M, Wilde R, Guest J. Nucleotide sequence encoding the flavoprotein and hydrophobic subunits of the succinate dehydrogenase of Escherichia Coli. Biochem J. (1984) 222:519–34. doi: 10.1042/bj2220519
77. Weindruch R, Walford RL, Fligiel S, Guthrie D. The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. J Nutr. (1986) 116:641–54. doi: 10.1093/jn/116.4.641
78. Greene AE, Todorova MT, McGowan R, Seyfried TN. Caloric restriction inhibits seizure susceptibility in epileptic EL mice by reducing blood glucose. Epilepsia. (2001) 42:1371–8. doi: 10.1046/j.1528-1157.2001.17601.x
79. Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci USA. (2003) 100:2911–6. doi: 10.1073/pnas.0536856100
80. Stafstrom C, Rho J. Epilepsy And The Ketogenic Diet. 1st edn. Totowa, NJ: Humana Press (2010). p. 247–64.
81. Bough KJ, Valiyil R, Han FT, Eagles DA. Seizure resistance is dependent upon age and calorie restriction in rats fed a ketogenic diet. Epilepsy Res. (1999) 35:21–8. doi: 10.1016/S0920-1211(98)00125-9
82. Phillips-Farfán BV, Rubio MC, Custodio V, Paz C, Carvajal KG. Caloric restriction protects against electrical kindling of the amygdala by inhibiting the mTOR signaling pathway. Front Cell Neurosci. (2015) 9:90. doi: 10.3389/fncel.2015.00090
83. Thio LL, Erbayat-Altay E, Rensing N, Yamada KA. Leptin contributes to slower weight gain in juvenile rodents on a ketogenic diet. Pediatr Res. (2006) 60:413–7. doi: 10.1203/01.pdr.0000238244.54610.27
84. Plank M, Wuttke D, Van Dam S, Clarke SA, De Magalhães JP. A meta-analysis of caloric restriction gene expression profiles to infer common signatures and regulatory mechanisms. Mol Biosyst. (2012) 8:1339–49. doi: 10.1039/c2mb05255e
85. Azarbar A, McIntyre DC, Gilby KL. Caloric restriction alters seizure disposition and behavioral profiles in seizure-prone (fast) versus seizure-resistant (slow) rats. Behav Neurosci. (2010) 124:106–14. doi: 10.1037/a0018307
86. Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. (1969) 25:295–330. doi: 10.1016/0014-4886(69)90128-9
87. Kawamura MJr, Ruskin DN, Masino SA. Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J Neurosci. (2010) 30:3886–95. doi: 10.1523/JNEUROSCI.0055-10.2010
88. Contestabile A, Ciani E, Contestabile A. Dietary restriction differentially protects from neurodegeneration in animal models of excitotoxicity. Brain Res. (2004) 1002:162–6. doi: 10.1016/j.brainres.2004.01.005
89. Benzler J, Ganjam GK, Krüger M, Pinkenburg O, Kutschke M, Stöhr S, et al. Hypothalamic glycogen synthase kinase 3β has a central role in the regulation of food intake and glucose metabolism. Biochem J. (2012) 447:175–84. doi: 10.1042/BJ20120834
90. Benzler J, Andrews ZB, Pracht C, Stöhr S, Shepherd PR, Grattan DR, et al. Hypothalamic Wnt signalling is impaired during obesity and reinstated by leptin treatment in male mice. Endocrinology. (2013) 154:4737–45. doi: 10.1210/en.2013-1746
91. Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. (2001) 21:1133–45. doi: 10.1097/00004647-200110000-00001
92. Howarth C, Gleeson P, Attwell D. Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab. (2012) 32:1222–32. doi: 10.1038/jcbfm.2012.35
93. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol Cell. (2002) 10:151–62. doi: 10.1016/S1097-2765(02)00568-3
94. Patel P, Woodgett JR. Glycogen Synthase Kinase 3: A Kinase for All Pathways?. Curr Top Dev Biol. (2017) 123:277–302. doi: 10.1016/bs.ctdb.2016.11.011
95. Chafey P, Finzi L, Boisgard R, Caüzac M, Clary G, Broussard C, et al. Proteomic analysis of β-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics. (2009) 9:3889–900. doi: 10.1002/pmic.200800609
96. Budnik V, Salinas PC. Wnt signaling during synaptic development and plasticity. Curr Opin Neurobiol. (2011) 21:151–9. doi: 10.1016/j.conb.2010.12.002
97. Inestrosa NC, Varela-Nallar L. Wnt signaling in the nervous system and in Alzheimer's disease. J Mol Cell Biol. (2014) 6:64–74. doi: 10.1093/jmcb/mjt051
98. Grumolato L, Liu G, Mong P, Mudbhary R, Biswas R, Arroyave R, et al. Canonical and non-canonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev. (2010) 24:2517–30. doi: 10.1101/gad.1957710
99. Chien AJ, Conrad WH, Moon RT. A Wnt survival guide: from flies to human disease. J Invest Dermatol. (2009) 129:1614–27. doi: 10.1038/jid.2008.445
100. Koeller HB, Ross ME, Glickstein SB. Cyclin D1 in excitatory neurons of the adult brain enhances kainate-induced neurotoxicity. Neurobiol Dis. (2008) 31:230–41. doi: 10.1016/j.nbd.2008.04.010
101. Quesada I, Rovira JM, Martin F, Roche E, Nadal A, Soria B. Nuclear KATP channels trigger nuclear Ca(2+) transients that modulate nuclear function. Proc Natl Acad Sci USA. (2002) 99:9544–9. doi: 10.1073/pnas.142039299
102. Jeon WJ, Kim SH, Seo MS, Kim Y, Kang UG, Juhnn YS, et al. Repeated electroconvulsive seizure induces c-Myc down-regulation and Bad inactivation in the rat frontal cortex. Exp Mol Med. (2008) 40:435–44. doi: 10.3858/emm.2008.40.4.435
103. Rubio C, Rosiles-Abonce A, Trejo-Solís C, Rubio-Osornio M, Mendoza C, Custodio V, et al. Increase signaling of Wnt/β-catenin pathway and presence of apoptosis in cerebellum of kindled rats. CNS Neurol Disord. (2017) 16:772–80. doi: 10.2174/1871527316666170117114513
104. Rima M, Daghsni M, Lopez A, Fajloun Z, Lefrancois L, Dunach M, et al. Down- regulation of the Wnt/β-catenin signaling pathway by Cacnb4. Mol Biol Cell. (2017) 28:3699–708. doi: 10.1091/mbc.e17-01-0076
105. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. (2004) 20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126
106. Kilian B, Mansukoski H, Barbosa FC, Ulrich F, Tada M, Heisenberg CP. The role of Ppt/Wnt5 in regulating cell shape and movement during zebrafish gastrulation. Mech Dev. (2003) 120:467–76. doi: 10.1016/s0925-4773(03)00004-2
107. McQuate A, Latorre-Esteves E, Barria A. A Wnt/calcium signaling cascade regulates neuronal excitability and trafficking of NMDARs. Cell Rep. (2017) 21:60–9. doi: 10.1016/j.celrep.2017.09.023
108. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. (2002) 4:648–57. doi: 10.1038/ncb839
109. Liu J, Reeves C, Michalak Z. Evidence for mTOR pathway activation in a spectrum of epilepsy-associated pathologies. Acta Neuropathol Commun. (2014) 2:71. doi: 10.1186/2051-5960-2-71
110. Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. (2002) 10:457–68. doi: 10.1016/S1097-2765(02)00636-6
111. Galanopoulou AS, Gorter JA, Cepeda C. Finding a better drug for epilepsy: the mTOR pathway as an antiepileptogenic target. Epilepsia. (2012) 53:1119–30. doi: 10.1111/j.1528-1167.2012.03506.x
112. Zeng H, Lu B, Zamponi R, et al. mTORC1 signaling suppresses Wnt/β-catenin signaling through DVL-dependent regulation of Wnt receptor FZD level. Proc Natl Acad Sci USA. (2018) 115:E10362–9. doi: 10.1073/pnas.1808575115
113. Potter WB, O'Riordan KJ, Barnett D, Osting SMK, Wagoner M, Burger C, et al. Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS ONE. (2010) 5:e8996. doi: 10.1371/journal.pone.0008996
114. Wong M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J. (2013) 36:40–50. doi: 10.4103/2319-4170.110365
115. Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol. (2016) 12:379–92. doi: 10.1038/nrneurol.2016.81
116. Bough KJ, Schwartzkroin PA, Rho JM. Calorie restriction and ketogenic diet diminish neuronal excitability in rat dentate gyrus in vivo. Epilepsia. (2003) 44:752–60. doi: 10.1046/j.1528-1157.2003.55502.x
117. Martin-McGill KJ, Jackson CF, Bresnahan R, Levy RG, Cooper PN. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst Rev. (2018) 11:CD001903. doi: 10.1002/14651858.CD001903.pub4
118. Freeman JM, Kossoff EH, Hartman AL. The ketogenic diet: one decade later. Pediatrics. (2007) 119:535–43. doi: 10.1542/peds.2006-2447
119. Husari KS, Cervenka MC. The ketogenic diet all grown up—Ketogenic diet therapies for adults. Epilepsy Res. (2020) 162:106319. doi: 10.1016/j.eplepsyres.2020.106319
120. Schoeler NE, Simpson Z, Whiteley VJ, Nguyen P, Meskell R, Lightfoot K, et al. Biochemical assessment of patients following ketogenic diets for epilepsy: Current practice in the UK and Ireland. Epilepsia Open. (2020) 5:73–9. doi: 10.1002/epi4.12371
121. Blackford R. Not your parents' ketogenic diet and flexibility in 2020. Epilepsy Res. (2020) 162:106307. doi: 10.1016/j.eplepsyres.2020.106307
122. Stafford P, Abdelwahab MG, Kim DY, Preul MC, Rho JM, Scheck AC. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr Metab. (2010) 7:74. doi: 10.1186/1743-7075-7-74
123. O'Donnell-Luria AH, Pais LS, Faundes V, Wood JC, Sveden A, Luria V, et al. Heterozygous variants in kmt2e cause a spectrum of neurodevelopmental disorders and epilepsy. Am J Hum Genet. (2019) 104:1210–22. doi: 10.1016/j.ajhg.2019.03.021
124. Garriga-Canut M, Schoenike B, Qaz R, Bergendahl K, Daley TJ, Pfender RM, et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. (2006) 9:1382–7. doi: 10.1038/nn1791
125. Kossoff E, Wang HS. Dietary therapies for epilepsy. Biomed J. (2013) 36:2–8. doi: 10.4103/2319-4170.107152
126. Stafstrom CE, Ockuly JC, Murphree L, Valley MT, Roopra A, Sutula TP. Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Ann Neurol. (2009) 65:435–47. doi: 10.1002/ana.21603
127. Yudkoff M, Daikhin Y, Nissim I, Lazarow A, Nissim I. Ketogenic diet, amino acid metabolism, and seizure control. J Neurosci Res. (2001) 66:931–40. doi: 10.1002/jnr.10083
128. Yudkoff M, Daikhin Y, Nissim I, Horyn O, Lazarow A, Luhovyy B, et al. Response of brain amino acid metabolism to ketosis. Neurochem Int. (2005) 47:119–28. doi: 10.1016/j.neuint.2005.04.014
129. Yudkoff M, Daikhin Y, Horyn O, Nissim I, Nissim I. Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia. (2008) 49:73–5. doi: 10.1111/j.1528-1167.2008.01841.x
130. Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara C, et al. Metabolic control of vesicular glutamate transport and release. Neuron. (2010) 68:99–112. doi: 10.1016/j.neuron.2010.09.002
131. Kaminski RM, Livingood MR, Rogawski MA. Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice. Epilepsia. (2004) 45:864–67. doi: 10.1111/j.0013-9580.2004.04504.x
132. Nishimura T, Schwarzer C, Gasser E, Kato N, Vezzani A, Sperk G. Altered expression of GABAA and GABAB receptor subunit mRNAs in the hippocampus after kindling and electrically induced status epilepticus. Neuroscience. (2005) 134:691–704. doi: 10.1016/j.neuroscience.2005.04.013
133. Fuenzalida M, Espinoza C, Pérez MÁ, Tapia-Rojas C, Cuitino L, Brandan E, et al. Wnt signaling pathway improves central inhibitory synaptic transmission in a mouse model of Duchenne muscular dystrophy. Neurobiol Dis. (2016) 86:109–20. doi: 10.1016/j.nbd.2015.11.018
134. Cerpa W, Gambrill A, Inestrosa NC, Barria A. Regulation of NMDA-receptor synaptic transmission by Wnt signaling. J Neurosci. (2011) 31:9466–71. doi: 10.1523/JNEUROSCI.6311-10.2011
135. Chuang YC, Chang AY, Lin JW, Hsu SP, Chan SH. Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia. (2004) 45:1202–9. doi: 10.1111/j.0013-9580.2004.18204.x
136. Waldbaum S, Patel M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. (2010) 88:23–45. doi: 10.1016/j.eplepsyres.2009.09.020
137. Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. (1990) 535:195–204. doi: 10.1016/0006-8993(90)91601-C
138. El Bahh B, Lespinet V, Lurton D, Coussemacq M, Le Gal La Salle G, Rougier A. Correlations between granule cell dispersion, mossy fiber sprouting, and hippocampal cell loss in temporal lobe epilepsy. Epilepsia. (1999) 40:1393–401. doi: 10.1111/j.1528-1157.1999.tb02011.x
139. Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK, Rho JM. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol. (2004) 55:576–80. doi: 10.1002/ana.20062
140. Rubio C, Rubio-Osornio M, Retana-Marquez S, Lopez M, Custodio V, Paz C. In vivo experimental models of epilepsy. Cent Nerv Syst Agents Med Chem. (2010) 10:298–309. doi: 10.2174/187152410793429746
141. Palmer S, Towne MC, Pearl PL, Pelletier RC, Genetti CA, Shi J, et al. SLC6A1 mutation and ketogenic diet in epilepsy with myoclonic-atonic seizures. Pediatr Neurol. (2016) 64:77–9. doi: 10.1016/j.pediatrneurol.2016.07.012
142. López-Doménech G, Serrat R, Mirra S, D'Aniello S, Somorjai I, Abad A, et al. The Eutherian Armcx genes regulate mitochondrial trafficking in neurons and interact with Miro and Trak2. Nat Commun. (2012) 3:1–12. doi: 10.1038/ncomms1829
143. Yang H, Shan W, Zhu F, Wu J, Wang Q. Ketone bodies in neurological diseases: focus on neuroprotection and underlying mechanisms. Front Neurol. (2019) 10:585. doi: 10.3389/fneur.2019.00585
144. Jarrett SG, Milder JB, Liang LP, Patel M. The ketogenic diet increases mitochondrial glutathione levels. J Neurochem. (2008) 106:1044–51. doi: 10.1111/j.1471-4159.2008.05460.x
145. Kim DY, Davis LM, Sullivan PG, Maalouf M, Simeone TA, van Brederode J, et al. Ketone bodies are protective against oxidative stress in neocortical neurons. J Neurochem. (2007) 101:1316–26. doi: 10.1111/j.1471-4159.2007.04483.x
146. Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. (1983) 305:147–8. doi: 10.1038/305147a0
147. Nakazawa M, Kodama S, Matsuo T. Effects of ketogenic diet on electroconvulsive threshold and brain contents of adenosine nucleotides. Brain Dev. (1983) 5:375–80. doi: 10.1016/S0387-7604(83)80042-4
148. Nylen K, Velazquez JL, Sayed V, Gibson KM, Burnham WM, Snead OC 3rd. The effects of a ketogenic diet on ATP concentrations and the number of hippocampal mitochondria in Aldh5a1(-/-) mice. Biochim Biophys Acta. (2009) 1790:208–12. doi: 10.1016/j.bbagen.2008.12.005
149. DeVivo DC, Leckie MP, Ferrendelli JS, McDougal DBJr. Chronic ketosis and cerebral metabolism. Ann, Neurol. (1978) 3:331–7. doi: 10.1002/ana.410030410
150. Karschin C, Ecke C, Ashcroft FM, Karschin A. Overlapping distribution of K(ATP) channel-forming Kir6.2 subunit and the sulfonylurea receptor SUR1 in rodent brain. FEBS Lett. (1997) 401:59–64. doi: 10.1016/S0014-5793(96)01438-X
151. Dunn-Meynell AA, Rawson NE, Levin BE. Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res. (1998) 814:41–54. doi: 10.1016/S0006-8993(98)00956-1
152. Zawar C, Plant TD, Schirra C, Konnerth A, Neumcke B. Cell-type specific expression of ATP-sensitive potassium channels in the rat hippocampus. J Physiol. (1999) 514:327–41.
153. Pierrefiche O, Bischoff AM, Richter DW. ATP-sensitive K+ channels are functional in expiratory neurones of normoxic cats. J Physiol. (1996) 494:399–409. doi: 10.1113/jphysiol.1996.sp021501
154. Allen TGJ, Brown DA. Modulation of the excitability of cholinergic basal forebrain neurones by KATP channels. J Physiol. (2004) 554:353–70. doi: 10.1113/jphysiol.2003.055889
155. Ikematsu N, Dallas ML, Ross FA, Lewis RW, Rafferty JN, David JA, et al. Phosphorylation of the voltage-gated potassium channel Kv2.1 by AMP-activated protein kinase regulates membrane excitability. Proc Natl Acad Sci USA. (2011) 108:18132–7. doi: 10.1073/pnas.1106201108
156. Tanner GR, Lutas A, Martínez-François JR, Yellen G. Single KATP channel opening in response to action potential firing in mouse dentate granule neurons. J Neurosci. (2011) 31:8689–96. doi: 10.1523/JNEUROSCI.5951-10.2011
157. Ashmore J, Olsen H, Sørensen N, Thrasivoulou C, Ahmed A. Wnts control membrane potential in mammalian cancer cells. J Physiol. (2019) 597:5899–914. doi: 10.1113/JP278661
158. Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. (1999) 398:422–6. doi: 10.1038/18884
159. Kramps T, Peter O, Brunner E, et al. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell. (2002) 109:47–60. doi: 10.1016/S0092-8674(02)00679-7
160. Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, et al. A ketogenic diet suppresses seizures in mice through adenosine A 1 receptors. J Clin Invest. (2011) 121:2679–83. doi: 10.1172/JCI57813
161. Lusardi TA, Akula KK, Coffman SQ, Ruskin DN, Masino SA, Boison D. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology. (2015) 99:500–9. doi: 10.1016/j.neuropharm.2015.08.007
Keywords: epilepsy, ketogenic diet, caloric restriction, KATP channels, Wnt
Citation: Rubio C, Luna R, Rosiles A and Rubio-Osornio M (2020) Caloric Restriction and Ketogenic Diet Therapy for Epilepsy: A Molecular Approach Involving Wnt Pathway and KATP Channels. Front. Neurol. 11:584298. doi: 10.3389/fneur.2020.584298
Received: 19 July 2020; Accepted: 28 September 2020;
Published: 05 November 2020.
Edited by:
Kette D. Valente, Universidade de São Paulo, BrazilReviewed by:
David Ruskin, Trinity College, United StatesKatia Lin, Federal University of Santa Catarina, Brazil
Copyright © 2020 Rubio, Luna, Rosiles and Rubio-Osornio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Moisés Rubio-Osornio, cnVvbW9uJiN4MDAwNDA7Z21haWwuY29t