 Elisa Gouvea Bogossian
Elisa Gouvea Bogossian Lorenzo Peluso
Lorenzo Peluso Jacques Creteur
Jacques Creteur Fabio Silvio Taccone
Fabio Silvio Taccone- Intensive Care Department, Erasmus Hospital, Université Libre de Bruxelles, Brussels, Belgium
Hyperventilation is a commonly used therapy to treat intracranial hypertension (ICTH) in traumatic brain injury patients (TBI). Hyperventilation promotes hypocapnia, which causes vasoconstriction in the cerebral arterioles and thus reduces cerebral blood flow and, to a lesser extent, cerebral blood volume effectively, decreasing temporarily intracranial pressure. However, hyperventilation can have serious systemic and cerebral deleterious effects, such as ventilator-induced lung injury or cerebral ischemia. The routine use of this therapy is therefore not recommended. Conversely, in specific conditions, such as refractory ICHT and imminent brain herniation, it can be an effective life-saving rescue therapy. The aim of this review is to describe the impact of hyperventilation on extra-cerebral organs and cerebral hemodynamics or metabolism, as well as to discuss the side effects and how to implement it to manage TBI patients.
Introduction
Intracranial hypertension (ICHT) is the most critical and potentially devastating complication in traumatic brain injury (TBI) patients (1). Since the skull is a rigid compartment, the total volume of the intracranial contents, i.e., brain tissue, blood, and cerebral spinal fluid, will remain constant over time. An increase in the volume of one of these components is initially compensated by shifting parts of the others (i.e., compression of the cerebral venous system can decrease global cerebral blood volume; increased CSF reabsorption and CSF displacement toward the basal cisterns and spinal compartment can decrease the CSF volume); when these mechanisms can no longer compensate for further volume changes intracranial pressure (ICP) will rapidly rise (2). Both the duration of ICHT and the absolute maximum value of ICP have an impact on patients' outcome (3); therefore, therapies aimed at controlling ICP and minimizing the ICHT burden are the cornerstone of TBI management (4).
Although different therapeutic interventions are available, none of them has shown a significant impact on patients' outcome and some potential side effects may limit their use. Modulation of arterial carbon dioxide pressure (PaCO2) has been used since decades in neuro-anesthesia and in neuro-intensive care, because lowering PaCO2 (i.e., hypocapnia) through increased minute volume ventilation (i.e., hyperventilation) can rapidly contribute to reduce the volume of the swollen brain and help control ICP (5); these effects are mediated by cerebral vasoconstriction and reduction in cerebral blood flow (CBF) and cerebral blood volume (CBV) (1, 2).
Although commonly used, hyperventilation has not been extensively supported by robust evidence, its effects might be transient and may not improve the probability of neurological recovery (3). Moreover, by decreasing CBF, hyperventilation may trigger or enhance brain ischemia (4, 5). In addition, hyperventilation has some extra-cerebral effects that may negatively impact patients' outcome (6). As such, if hyperventilation is frequently employed in TBI patients (7), the potential risks associated with this therapy require an optimal understanding on how to manage PaCO2 in TBI patients.
The aim of this review is to describe the effects of hyperventilation on brain physiology and to discuss its use in the management of TBI patients. Only studies focusing on controlled hyperventilation (i.e., modification of minute ventilation in TBI patients treated with mechanical ventilation and controlled modes) have been evaluated, while pre-hospital hyperventilation or spontaneous hyperventilation will not be discussed.
Hyperventilation, Hypocapnia and Systemic Effects
Hyperventilation is characterized by elevated minute alveolar ventilation, which can be secondary to an increase of tidal volume and/or respiratory rate, if the dead space remains constant. This condition is typically observed as a physiological response to hypoxemia, systemic inflammation, chest trauma, or pain; however, in the setting of TBI management, “controlled hyperventilation” is a modification of minute ventilation to obtain hypocapnia (i.e., PaCO2 <38 mmHg) in order to manipulate cerebral hemodynamics and compliance (8). In this setting, acceptable ranges of PaCO2 in clinical practice are considered between 35 and 45 mmHg at sea level (8); when hyperventilation is applied, it can classified into moderate (PaCO2 31–35 mmHg), forced (PaCO2 26–30 mmHg), or intensified forced (PaCO2 <26 mmHg), according to PaCO2 levels (9).
There are several non-cerebral effects related to this therapeutic strategy (Table 1); as TBI patients often have lung injury (7, 10, 11) due to micro-aspiration, pneumonia or lung contusions (12), promoting hyperventilation by increasing tidal volume can induce ventilator-induced lung injury (VILI) (13) and potentially delay pulmonary healing or worsen outcome (14–16). Moreover, hyperventilation may increase intra-thoracic pressure, which would favor right ventricular dysfunction or, in hypovolemic patients, cause an impairment of the venous return and decrease cardiac output (17). Moreover, hypocapnia compromises coronary blood flow and is associated with an increased risk of myocardial ischemia (18) and the development of arrhythmias (19). Prolonged hyperventilation is associated with respiratory alkalosis (20); alkalemia would shift the oxygen dissociation curve of hemoglobin toward the left, increasing the hemoglobin affinity for oxygen and compromising tissue oxygen delivery (18). Hypocapnia and respiratory alkalosis also lead to pulmonary vasodilation (19) and bronchoconstriction (21), which result in ventilation to perfusion (V/Q) mismatch and secondary hypoxemia in TBI patients with pre-existing lung injury. In animal studies, hypocapnia also decreased surfactant production (22) and increases the permeability of the alveolo-capillary barrier (23), although this has not well-demonstrated in humans. Hyperventilation may also increase intra-abdominal pressure, which can secondarily increase ICP (24); hypocapnia decreases blood flow to the kidneys, skin and muscles tissues and increases platelet adhesion and aggregation (12). Finally, hyperventilation is often associated with electrolytes disturbances, such as hypokalemia, hypocalcemia, and hypophosphatemia (12). Taken all together, these findings suggest that controlled hyperventilation and hypocapnia should be applied with extreme caution to all critically ill patients, because of several negative effects on targets organs.
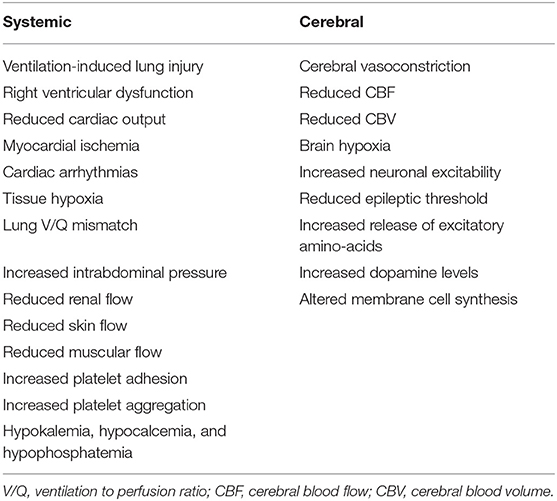
Table 1. Potential side effects associated with hyperventilation in the human setting.
Effects of Hyperventilation and Hypocapnia on Brain Physiology and Metabolism
The brain has a high energy requirement, being responsible for 20% of total body oxygen consumption (25). Since the brain is incapable of storing energy, rapid adjustments of CBF are essential to maintain an adequate supply of oxygen and nutrients to brain tissue (26). Several mechanisms, collectively called “cerebral autoregulation,” are effective to keep CBF within the necessary values to meet the cerebral energetical demand (26). As such, CBF is heterogenous and varies according to the metabolic activity of each cerebral region (27); resistance arterioles contract and dilate to regulate CBF in response to different stimuli, such as blood pressure, blood viscosity, transmural pressure, metabolic demand, tissue pH, and electrolytes or PaCO2 (28).
In particular, these resistance arterioles respond to variations in PaCO2 between 20 and 60 mmHg by contracting (i.e., hypocapnia) or dilating (i.e., hypercapnia) (2), a phenomenon called “cerebro-vascular CO2 reactivity.” This response to PaCO2 variations is probably pH-mediated (18) (i.e., low pH or high H+ concentrations will promote vasodilation, while high pH and low H+ vasoconstriction), and is proportionally more relevant with hypercapnia than with hypocapnia (29). Around 70% of the CBV is located within the venous system and is not affected by changes in PaCO2; therefore, changes in CBV following hyperventilation are restricted to the arterial component and are associated with a decrease in CBF (18). In particular, for each mmHg-decrease in PaCO2, there is an approximate decrease of 3% in CBF (2), although the impact of hypocapnia on ICP is less pronounced (5).
Hyperventilation can also result in brain hypoxia (Table 1). The main mechanism suggested is the reduction of oxygen supply due global and/or regional hypoperfusion caused by the reduction in CBF (30, 31). Moreover, due to the above-mentioned systemic effects, hypocapnia can lead to additional VILI, with impaired gas exchanges and hypoxemia, and can alter the oxygen hemoglobin dissociation curve, with reduced oxygen delivery (18).
Finally, alkalemia and hypocapnia increase neuronal excitability (32), reduce the epileptic threshold and/or prolong convulsive activities (33). In animal studies, hypocapnia led to an increased cerebral consumption and depletion of local glucose (34, 35). Hypocapnia has also been associated with neurotoxicity (12), by inducing the release of cytotoxic excitatory amino-acid (36), increasing dopamine levels in the basal ganglia (37) and by promoting the inappropriate incorporation of choline into the phospholipids of cell membranes (38).
Controlled Hyperventilation in TBI Patients
Hyperventilation has been reported to effectively control ICHT in TBI patients (39, 40); in Table 2, a summary of most relevant studies reporting data on hypocapnia, ICP and outcome in this patients' population has been provided.
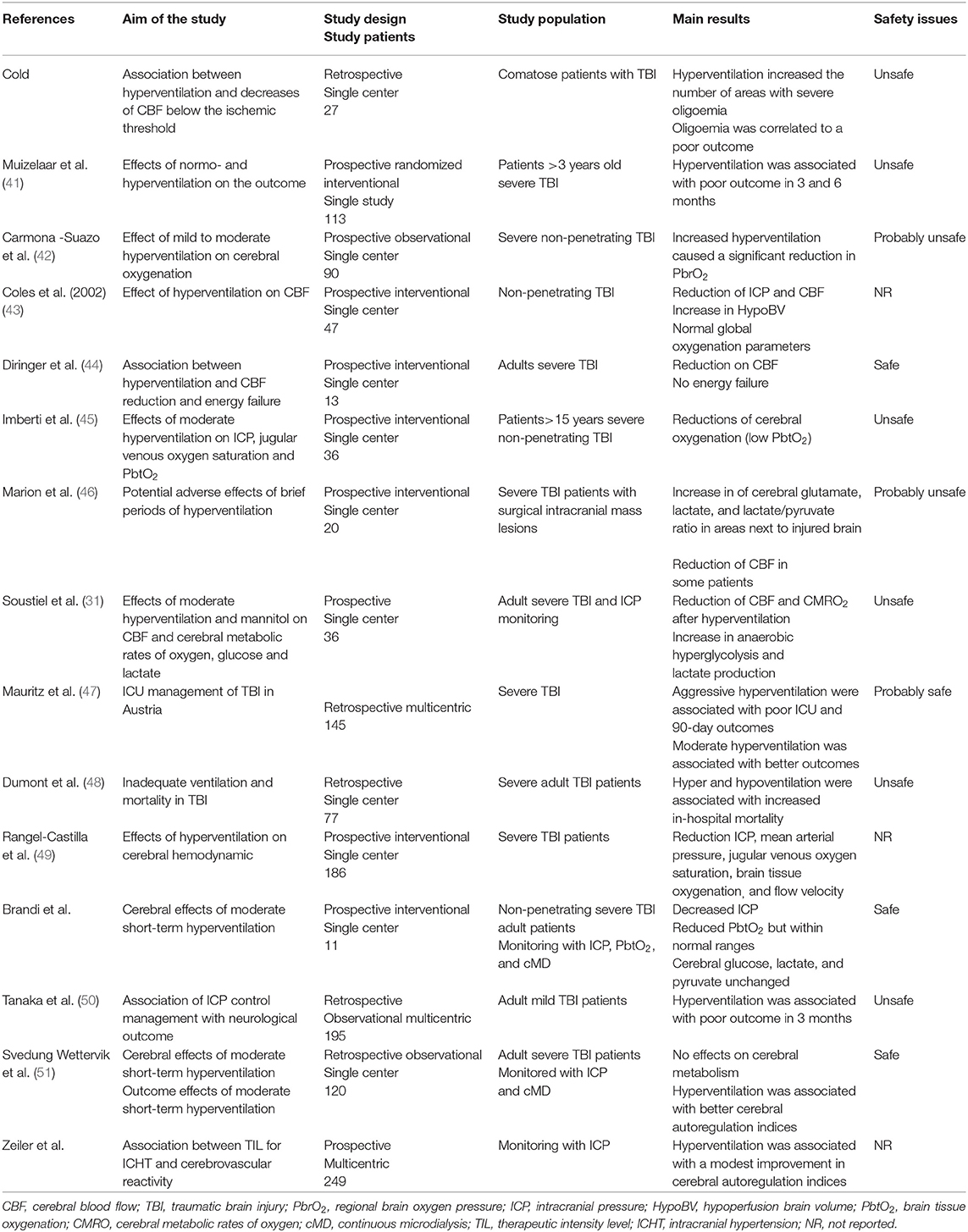
Table 2. List of most relevant clinical studies dealing with controlled hyperventilation in traumatic brain injury (TBI) patients.
Obrist et al. showed that hyperventilation could rapidly reduce ICP in half of TBI patients, although this was associated to a reduction in CBF in almost all of them (4). The relationship between PaCO2 and ICP is not linear and the most important effects are observed between PaCO2 values of 30 and 50 mmHg (52). Moreover, prolonged hyperventilation (53) will be associated with a progressive reduction of its vasoconstrictive effects, because of the perivascular normalization of pH due to local buffering. As reduced CBF (i.e., oligemia) is frequently observed in the early phase after TBI (54), prolonged hyperventilation should not be initiated in these patients without CBF monitoring. Cerebral blood flow can be measured directly, using Xenon computed tomography (CT) scan, CT perfusion (CTP) scan, or positron emission tomography (PET) scan, but these techniques involve injection of radioactive tracers or contrast media and require patients' transportation, which is not always feasible in severe TBI cases with ICHT (55). Indirect CBF velocities assessment using transcranial Doppler (TCD) ultrasonography does not directly correspond to absolute CBF values (56), although elevated pulsatility index (PI >1.2), low diastolic velocities in the middle cerebral artery (<20 cm/s) and estimated ICP using validated formulas might be helpful to identify TBI patients at risk of hypoperfusion. Different studies have shown a reduction in CBF levels during hyperventilation (5, 57–59); also, the most relevant reduction in CBF was observed in the peri-contusional areas, which are more vulnerable to secondary injuries (59).
If the reduction in CBF is quite consistent, controlled hyperventilation (i.e., mean PaCO2 from 37 to 30 mmHg) could improve indices of cerebral autoregulation function in TBI patients with disturbed pressure-reactivity at baseline, whereas those with intact pressure-reactivity at baseline would have no effect of such intervention (60). Another large cohort study also showed that mild hyperventilation was associated with lower pressure reactivity index (i.e., better autoregulatory function), in particular on day 2 after injury (61). One hypothesis is that hypocapnia and related vasoconstriction could reestablish endothelial reactivity in cerebral vessels, which were previously dilated in order to compensate for reduced cerebral oxygen delivery in the presence of ICHT.
Nevertheless, if a reduction in CBF is observed during hyperventilation, it remains unclear whether this phenomenon is associated with signs of cellular hypoxic injury and anaerobic metabolism. In severe TBI patients, Diringer et al. (62) observed that short and moderate hyperventilation significantly decreased CBF but did not impair global cerebral metabolism and oxygen extraction. As such, the use of neuromonitoring, in particular of cerebral oxygenation and/or metabolism, could provide important findings about the brain tolerance to controlled hyperventilation. Forced hyperventilation has been associated with reduced cerebral oxygenation, which was measured by the jugular bulb oximetry (SjO2, i.e., the threshold for cerebral hypoxia being <55%), although these results were not consistent in all studies (63–65). However, SjO2 reflect hemispheric global oxygenation and tissue hypoxia may occur even within normal SjO2 values (66). Brain tissue oxygen tension (PbtO2) is a regional technique, is well-correlated with local CBF and can directly monitor the areas at higher-risk of secondary ischemia (67). The effects of hyperventilation on PbtO2 are variable, with some studies reported a significant reduction in brain oxygenation (68–71) while others showing no major changes (72, 73) and some reporting an increase in PbtO2, in particular due to the large reduction in ICP with previous cerebral vasodilation (i.e., hyperemia) (45, 74). Recent studies reported unchanged PbtO2 values in adult severe TBI patients undergoing moderate hyperventilation and with a median PbtO2 value at baseline within normal values (i.e., >30 mmHg) (39, 40).
Cerebral metabolic function can be assessed at bedside using the microdialysis technique, or performing PET and magnetic resonance imaging (MRI) spectroscopy studies. In one study, early hyperventilation (i.e., 24–36 h after injury) was associated with a significant increase in tissue lactate and lactate/pyruvate ratio, suggesting anaerobic metabolism and tissue hypoxia (46); these metabolic effects were less pronounced at a late phase (i.e., 3–4 days after TBI). However, two recent studies showed no effect of moderate hyperventilation on cerebral metabolites in adult TBI patients (39, 40). Using PET scan, one study (n = 9) showed that moderate and intense hyperventilation resulted in reduced CBF and increased oxygen extraction, but a constant oxygen metabolism (i.e., no energy failure) (44). In two larger studies (43, 75), hyperventilation (i.e., PaCO2 <30 mmHg) increased the volume of hypoperfused cerebral areas within the injured brain, which, in the absence of increased oxygen extraction, would result in tissue hypoxia.
With all these potential side effects, which is the effect of controlled hyperventilation on the outcome of TBI patients? In one large retrospective study (n = 251), Gordon et al. (76) reported a lower mortality in TBI patients undergoing hyperventilation (i.e., PaCO2 between 25 and 30 mmHg for 6 to 41 days); however, more severe neurological sequelae were observed among survivors in the hyperventilation group when compared to the other. Only one prospective randomized clinical trial has investigated the effects of hyperventilation in this setting; Muizelaar et al. (41) compared the 3- and 6-month neurological outcome of patients who were kept at a median PaCO2 of 25 mm Hg to those kept at a median of 35 mmHg for 5 days: forced hyperventilation was associated with a higher proportion of patients with poor outcome. Interestingly, among patients being treated with controlled hyperventilation and tromethamine (THAM, i.e., a buffer that prevents pH changes within the extracellular cerebral fluid and excessive vasoconstriction), there was a higher proportion of patients with long-term favorable neurological outcome when compared to the others.
Discussion: A Practical Approach
The initial PaCO2 targets in TBI patients with normal ICP values undergoing mechanical ventilation should be within normal values (i.e., 38–42 mmHg—Figure 1); although Brain Trauma Foundation guidelines could not identify an optimal threshold for PaCO2 values in the initial phase of TBI management [(66), international consensus recommended for these “physiological” values as oligemia is frequent in the first 24–48 h after injury and could be aggravated by hypocapnia (49, 50). Prophylactic (i.e., in the absence of ICHT) and prolonged hyperventilation is not recommended and should not be used, (61) as it would provide no benefits and could result in tissue hypoxia and cerebral metabolic disturbances. In order to detect cerebral oligemia in these patients, an initial CTP scan could be helpful to identify very low CBF values, which would result in secondary ischemia in case PaCO2 would decrease below physiological values. In the absence of CTP, cerebral ultrasound, using a combination of PI, estimated ICP and diastolic CBF velocity, could identify patients at risk of cerebral hypoperfusion. A close attention on gas analyses monitoring, requiring repeated sampling and end-tidal CO2 (etCO2) monitoring, is necessary in this phase, as hyperventilation in the absence of elevated ICP is frequently observed in adult TBI patients (61). Importantly, etCO2 might have some limitations in case of concomitant severe chest trauma and hemodynamic instability (i.e., low cardiac output) (77). Whether targeting normal PaCO2 or pH values would be the most appropriate approach remains unknown in these patients. However, as cerebral perivascular pH could be influenced by other factors than PaCO2 and systemic pH (i.e., local metabolism, K+, isolated cerebral hypoxia) and therefore be less accurately predicted, using PaCO2 levels and quantifying changes in cerebral hemodynamics and physiology to PaCO2 changes at bedside is more feasible for physicians. If ICP remains within acceptable values after 48 h from the injury (i.e., <15 mmHg), lower PaCO2 values (i.e., 33–36 mmHg) could result in improved cerebral autoregulation (66), however it is hard to recommend this approach routinely in all severe TBI patients. TBI patients at the highest risk of impaired cerebral autoregulation are those with diffuse brain injury (51); as such, if TCD assessment shows normal CBF velocities in these patients, lower PaCO2 values (i.e., 33–36 mmHg) could be tolerated, although a more comprehensive neuromonitoring would be the only effective solution to detect the potential occurrence of tissue hypoxia.
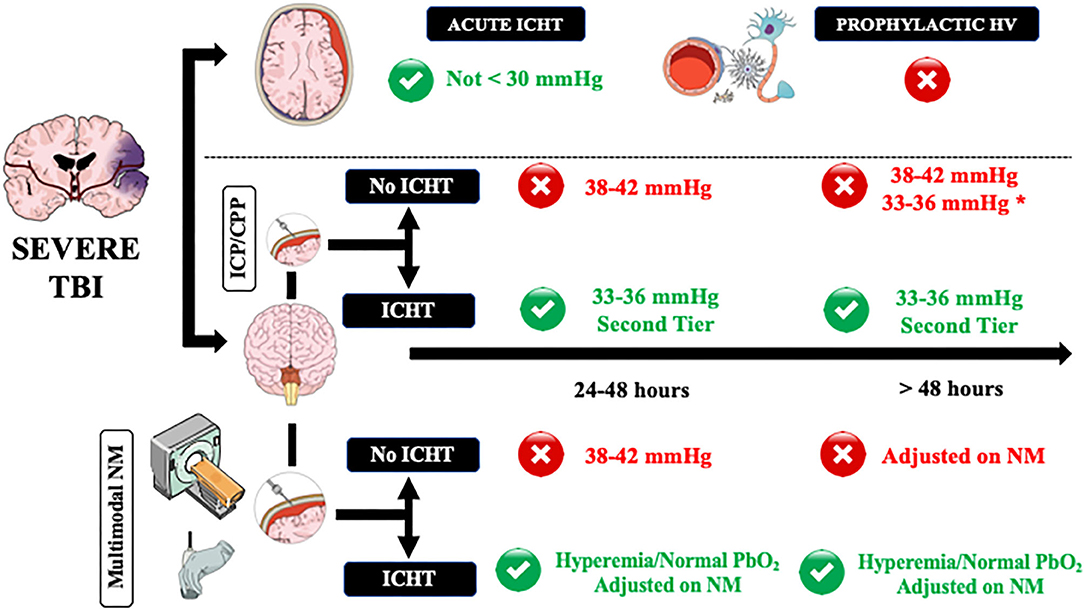
Figure 1. A practical approach on how to manage controlled hyperventilation and hypocapnia in traumatic brain injury (TBI) patients. Acute intracranial hypertension (ICHT) = life-threatening elevation in ICP, in particular when signs of herniation (i.e., anisocoria, apnea, hypertension, bradycardia) are present. ICHT, intracranial pressure close to the critical threshold for therapy without signs of herniation; HV, controlled hyperventilation and hypocapnia. Green and red circles refers to the potential use (green) or contraindication (red) to the use of HV. NM, neuromonitoring. *In case of diffuse brain injury but with high potential risk of tissue hypoxia. **Adjusted on neuromonitoring.
As hyperventilation combined with hypocapnia is the most rapidly available method to reduce ICP, moderate and brief hyperventilation should be used to treat life-threatening elevation in ICP, in particular when signs of herniation (i.e., anisocoria, apnea, hypertension, bradycardia) are present (61) and could be used as a bridge toward additional interventions (i.e., repeated CT-scan; osmotic therapy; surgery). In this setting, hyperventilation should be of short duration and, if possible, should never decrease below PaCO2 values of 30 mmHg because: (a) the most important effects of PaCO2 on ICP are observed between 30 and 50 mmHg and (b) most of the relevant cerebral side effects were reported for forced hyperventilation. If possible, the use of hyperventilation should be minimized for these patients during the first 24 h after injury, when CBF often is the most reduced (61), unless CBF could be measured.
In patients with ICP values remaining close to the critical threshold for therapy (i.e., 20–22 mmHg), international guidelines recommended the use of controlled hyperventilation only as “tiers 2” therapy, after the failure of increased sedation and osmotics infusion (78); indeed, controlled hyperventilation produced similar effects on ICP but more metabolic disturbances of cerebral metabolism than mannitol in TBI patients (61). In these patients, it is recommended to set PaCO2 around 33–36 mmHg and avoid values <30 mmHg (49). In the absence of other neuromonitoring than ICP and cerebral perfusion pressure (CPP), CTP scan and TCD are helpful to suggest normal or high (i.e., hyperemia) CBF values, which would logically respond to hyperventilation. However, in case of oligemia, physicians could decide to avoid hypocapnia and move to “tiers 3” therapy, such as barbiturates, hypothermia, or decompressive craniectomy, to treat ICHT as hypocapnia would result in additional cerebral hypoperfusion.
In our opinion, invasive neuromonitoring should be also considered in severe TBI patients and ICHT to optimize overall management and, in particular, to assess the effects of low PaCO2 values on cerebral oxygenation and metabolism. As such, PaCO2 values and hyperventilation could be adjusted to the brain tolerance and therapeutic targets individualized on the patients' need. In case of the need for low PaCO2 values and concomitant lung injury, other interventions aiming at reducing CO2 production, such as increasing sedation or hypothermia, could be considered to induce hypocapnia and avoid lung stress and VILI. However, this strategy should be further evaluated in clinical studies.
If controlled PaCO2 values are mandatory in severe TBI patients because of the significant effects on brain hemodynamics and compliance, high doses of sedatives, often in association with neuromuscular blocking agents (NMBAs), are required to adjust ventilatory parameters to obtain desired PaCO2 targets. As such, in the absence of aggressive therapies for ICHT, it remains unknown when it would be safe to discontinue sedatives and eventually tolerate spontaneous hyperventilation in these patients. If this occurs after several (i.e., >7–10) days from injury, the risk of oligemia would be probably limited. Also, spontaneous hyperventilation after acute brain injury is often triggered by local acidosis (i.e., low pH surrounding the respiratory center located in the brainstem), which result in vascular dilation and would probably compensate from vasoconstriction induced by hyperventilation. In these cases, non-invasive (i.e., TCD) and invasive (i.e., PbtO2) monitoring would again be helpful to individualize therapeutic decisions.
In conclusions, controlled hyperventilation is effective in reducing ICP but it also reduces CBF and might have both cerebral and systemic serious side effects. As such, normal PaCO2 values should be maintained in the early phase after TBI if ICP remains within acceptable values. Controlled hyperventilation (i.e., never below PaCO2 of 30 mmHg) should be used as a temporary life-saving intervention in case of severe intracranial hypertension; PaCO2 levels should be also adjusted and individualized in each patient using CTP and cerebral ultrasound of, whenever possible, advanced multimodal neuromonitoring.
Author Contributions
EG, LP, JC, and FT contribute equally to the elaboration of this manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Raichle ME, Plum F. Hyperventilation and cerebral blood flow. Stroke. (1972) 3:566–75. doi: 10.1161/01.STR.3.5.566
2. Cold GE. Cerebral blood flow in acute head injury. The regulation of cerebral blood flow and metabolism during the acute phase of head injury, and its significance for therapy. Acta Neurochir Suppl. (1990) 49:1–64. doi: 10.1007/978-3-7091-9101-9_1
3. Raichle ME, Posner JB, Plum F. Cerebral blood flow during and after hyperventilation. Arch Neurol. (1970) 23:394–403. doi: 10.1001/archneur.1970.00480290014002
4. Obrist WD, Langfitt TW, Jaggi JL, Cruz J, Gennarelli TA. Cerebral blood flow and metabolism in comatose patients with acute head injury. Relationship to intracranial hypertension. J Neurosurg. (1984) 61:241–53. doi: 10.3171/jns.1984.61.2.0241
5. Fortune JB, Feustel PJ, deLuna C, Graca L, Hasselbarth J, Kupinski AM. Cerebral blood flow and blood volume in response to O2 and CO2 changes in normal humans. J Trauma. (1995) 39:463–471; discussion: 471–2. doi: 10.1097/00005373-199509000-00012
6. Stocchetti N, Maas AI, Chieregato A, van der Plas AA. Hyperventilation in head injury: a review. Chest. (2005) 127:1812–27. doi: 10.1378/chest.127.5.1812
7. Hendrickson CM, Howard BM, Kornblith LZ, Conroy AS, Nelson MF, Zhuo H, et al. The acute respiratory distress syndrome following isolated severe traumatic brain injury. J Trauma Acute Care Surg. (2016) 80:989–97. doi: 10.1097/TA.0000000000000982
9. Neumann JO, Chambers IR, Citerio G, Enblad P, Gregson BA, Howells T, et al. The use of hyperventilation therapy after traumatic brain injury in Europe: an analysis of the BrainIT database. Intensive Care Med. (2008) 34:1676–82. doi: 10.1007/s00134-008-1123-7
10. Bratton SL, Davis RL. Acute lung injury in isolated traumatic brain injury. Neurosurgery. (1997) 40:707–12; discussion: 712. doi: 10.1097/00006123-199704000-00009
11. Zygun DA, Kortbeek JB, Fick GH, Laupland KB, Doig CJ. Non-neurologic organ dysfunction in severe traumatic brain injury. Crit Care Med. (2005) 33:654–60. doi: 10.1097/01.CCM.0000155911.01844.54
12. Godoy DA, Seifi A, Garza D, Lubillo-Montenegro S, Murillo-Cabezas F. Hyperventilation therapy for control of posttraumatic intracranial hypertension. Front Neurol. (2017) 8:250. doi: 10.3389/fneur.2017.00250
13. Mascia L, Zavala E, Bosma K, Pasero D, Decaroli D, Andrews P, et al. High tidal volume is associated with the development of acute lung injury after severe brain injury: an international observational study. Crit Care Med. (2007) 35:1815–20. doi: 10.1097/01.CCM.0000275269.77467.DF
14. Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. (2000) 342:1301–8. doi: 10.1056/NEJM200005043421801
15. Holland MC, Mackersie RC, Morabito D, Campbell AR, Kivett VA, Patel R, et al. The development of acute lung injury is associated with worse neurologic outcome in patients with severe traumatic brain injury. J Trauma. (2003) 55:106–11. doi: 10.1097/01.TA.0000071620.27375.BE
16. Rincon F, Ghosh S, Dey S, Maltenfort M, Vibbert M, Urtecho J, et al. Impact of acute lung injury and acute respiratory distress syndrome after traumatic brain injury in the United States. Neurosurgery. (2012) 71:795–803. doi: 10.1227/NEU.0b013e3182672ae5
17. Imai T, Uchiyama M, Maruyama N, Yoshikawa D, Fujita T. Influence of constant sustained positive airway pressure on right ventricular performance. Intensive Care Med. (1993) 19:8–12. doi: 10.1007/BF01709271
18. Curley G, Kavanagh BP, Laffey JG. Hypocapnia and the injured brain: more harm than benefit. Crit Care Med. (2010) 38:1348–59. doi: 10.1097/CCM.0b013e3181d8cf2b
19. Mazzara JT, Ayres SM, Grace WJ. Extreme hypocapnia in the critically ill patient. Am J Med. (1974) 56:450–6. doi: 10.1016/0002-9343(74)90475-6
20. Seifter JL. Integration of acid-base and electrolyte disorders. N Engl J Med. (2015) 372:391–2. doi: 10.1056/NEJMc1414731
21. O'Cain CF, Hensley MJ, McFadden ER Jr, Ingram RH Jr. Pattern and mechanism of airway response to hypocapnia in normal subjects. J Appl Physiol Respir Environ Exerc Physiol. (1979) 47:8–12. doi: 10.1152/jappl.1979.47.1.8
22. Oyarzun MJ, Donoso P, Quijada D. Role of hypocapnia in the alveolar surfactant increase induced by free fatty acid intravenous infusion in the rabbit. Respiration. (1986) 49:187–94. doi: 10.1159/000194878
23. Reynolds AM, Zadow SP, Scicchitano R, McEvoy RD. Airway hypocapnia increases microvascular leakage in the guinea pig trachea. Am Rev Respir Dis. (1992) 145:80–4. doi: 10.1164/ajrccm/145.1.80
24. Citerio G, Vascotto E, Villa F, Celotti S, Pesenti A. Induced abdominal compartment syndrome increases intracranial pressure in neurotrauma patients: a prospective study. Crit Care Med. (2001) 29:1466–71. doi: 10.1097/00003246-200107000-00027
25. Mink JW, Blumenschine RJ, Adams DB. Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Physiol. (1981) 241:R203–12. doi: 10.1152/ajpregu.1981.241.3.R203
26. Aaslid R, Lindegaard KF, Sorteberg W, Nornes H. Cerebral autoregulation dynamics in humans. Stroke. (1989) 20:45–52. doi: 10.1161/01.STR.20.1.45
27. Ingvar DH, Cronqvist S, Ekberg R, Risberg J, Hoedt-Rasmussen K. Normal values of regional cerebral blood flow in man, including flow and weight estimates of gray and white matter. A preliminary summary. Acta Neurol Scand Suppl. (1965) 14:72–8. doi: 10.1111/j.1600-0404.1965.tb01958.x
28. Czosnyka M, Brady K, Reinhard M, Smielewski P, Steiner LA. Monitoring of cerebrovascular autoregulation: facts, myths, and missing links. Neurocrit Care. (2009) 10:373–86. doi: 10.1007/s12028-008-9175-7
29. Brian JE Jr. Carbon dioxide and the cerebral circulation. Anesthesiology. (1998) 88:1365–86. doi: 10.1097/00000542-199805000-00029
30. Weckesser M, Posse S, Olthoff U, Kemna L, Dager S, Muller-Gartner HW. Functional imaging of the visual cortex with bold-contrast MRI: hyperventilation decreases signal response. Magn Reson Med. (1999) 41:213–6. doi: 10.1002/(SICI)1522-2594(199901)41:1<213::AID-MRM31>3.0.CO;2-S
31. Soustiel JF, Mahamid E, Chistyakov A, Shik V, Benenson R, Zaaroor M. Comparison of moderate hyperventilation and mannitol for control of intracranial pressure control in patients with severe traumatic brain injury–a study of cerebral blood flow and metabolism. Acta Neurochir. (2006) 148:845–51; discussion: 851. doi: 10.1007/s00701-006-0792-7
32. Huttunen J, Tolvanen H, Heinonen E, Voipio J, Wikstrom H, Ilmoniemi RJ, et al. Effects of voluntary hyperventilation on cortical sensory responses. Electroencephalographic and magnetoencephalographic studies. Exp Brain Res. (1999) 125:248–54. doi: 10.1007/s002210050680
33. Bergsholm P, Gran L, Bleie H. Seizure duration in unilateral electroconvulsive therapy. The effect of hypocapnia induced by hyperventilation and the effect of ventilation with oxygen. Acta Psychiatr Scand. (1984) 69:121–8. doi: 10.1111/j.1600-0447.1984.tb02475.x
34. Berson FG, Spatz M, Klatzo I. Effects of oxygen saturation and pCO2 on brain uptake of glucose analogues in rabbits. Stroke. (1975) 6:691–6. doi: 10.1161/01.STR.6.6.691
35. Samra SK, Turk P, Arens JF. Effect of hypocapnia on local cerebral glucose utilization in rats. Anesthesiology. (1989) 70:523–6. doi: 10.1097/00000542-198903000-00024
36. Graham EM, Apostolou M, Mishra OP, Delivoria-Papadopoulos M. Modification of the N-methyl-D-aspartate (NMDA) receptor in the brain of newborn piglets following hyperventilation induced ischemia. Neurosci Lett. (1996) 218:29–32. doi: 10.1016/0304-3940(96)13114-1
37. Pastuszko P, Wilson DF. Activation of tyrosine hydroxylase in striatum of newborn piglets in response to hypocapnic ischemia and recovery. Adv Exp Med Biol. (1997) 411:65–73. doi: 10.1007/978-1-4615-5865-1_8
38. Mykita S, Golly F, Dreyfus H, Freysz L, Massarelli R. Effect of CDP-choline on hypocapnic neurons in culture. J Neurochem. (1986) 47:223–31. doi: 10.1111/j.1471-4159.1986.tb02853.x
39. Ghajar J, Hariri RJ, Narayan RK, Iacono LA, Firlik K, Patterson RH. Survey of critical care management of comatose, head-injured patients in the United States. Crit Care Med. (1995) 23:560–7. doi: 10.1097/00003246-199503000-00023
40. Matta B, Menon D. Severe head injury in the United Kingdom and Ireland: a survey of practice and implications for management. Crit Care Med. (1996) 24:1743–8. doi: 10.1097/00003246-199610000-00023
41. Muizelaar JP, Marmarou A, Ward JD, Kontos HA, Choi SC, Becker DP, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. (1991) 75:731–9. doi: 10.3171/jns.1991.75.5.0731
42. Carmona Suazo JA, Maas AI, van den Brink WA, van Santbrink H, Steyerberg EW, Avezaat CJ. CO2 reactivity and brain oxygen pressure monitoring in severe head injury. Crit Care Med. (2000) 28:3268–74. doi: 10.1097/00003246-200009000-00024
43. Coles JP, Minhas PS, Fryer TD, Smielewski P, Aigbirihio F, Donovan T, et al. Effect of hyperventilation on cerebral blood flow in traumatic head injury: clinical relevance and monitoring correlates. Crit Care Med. (2002) 30:1950–9. doi: 10.1097/00003246-200209000-00002
44. Diringer MN, Videen TO, Yundt K, Zazulia AR, Aiyagari V, Dacey RG Jr, et al. Regional cerebrovascular and metabolic effects of hyperventilation after severe traumatic brain injury. J Neurosurg. (2002) 96:103–8. doi: 10.3171/jns.2002.96.1.0103
45. Imberti R, Bellinzona G, Langer M. Cerebral tissue PO2 and SjvO2 changes during moderate hyperventilation in patients with severe traumatic brain injury. J Neurosurg. (2002) 96:97–102. doi: 10.3171/jns.2002.96.1.0097
46. Marion DW, Puccio A, Wisniewski SR, Kochanek P, Dixon CE, Bullian L, et al. Effect of hyperventilation on extracellular concentrations of glutamate, lactate, pyruvate, and local cerebral blood flow in patients with severe traumatic brain injury. Crit Care Med. (2002) 30:2619–25. doi: 10.1097/00003246-200212000-00001
47. Mauritz W, Janciak I, Wilbacher I, Rusnak M. Australian severe TBISI: severe traumatic brain injury in Austria IV: intensive care management. Wien Klin Wochenschr. (2007) 119:46–55. doi: 10.1007/s00508-006-0763-2
48. Dumont TM, Visioni AJ, Rughani AI, Tranmer BI, Crookes B. Inappropriate prehospital ventilation in severe traumatic brain injury increases in-hospital mortality. J Neurotrauma. (2010) 27:1233–41. doi: 10.1089/neu.2009.1216
49. Rangel-Castilla L, Lara LR, Gopinath S, Swank PR, Valadka A, Robertson C. Cerebral hemodynamic effects of acute hyperoxia and hyperventilation after severe traumatic brain injury. J Neurotrauma. (2010) 27:1853–63. doi: 10.1089/neu.2010.1339
50. Tanaka C, Tagami T, Unemoto K, Kudo S, Takehara A, Kaneko J, et al. Intracranial pressure management and neurological outcome for patients with mild traumatic brain injury who required neurosurgical intervention: a Japanese database study. Brain Inj. (2019) 33:869–74. doi: 10.1080/02699052.2019.1614667
51. Svedung Wettervik T, Howells T, Hillered L, Nilsson P, Engquist H, Lewen A, et al. Mild hyperventilation in traumatic brain injury-relation to cerebral energy metabolism, pressure autoregulation, and clinical outcome. World Neurosurg. (2020) 133:e567–75. doi: 10.1016/j.wneu.2019.09.099
52. Paul RL, Polanco O, Turney SZ, McAslan TC, Cowley RA. Intracranial pressure responses to alterations in arterial carbon dioxide pressure in patients with head injuries. J Neurosurg. (1972) 36:714–20. doi: 10.3171/jns.1972.36.6.0714
53. Muizelaar JP, van der Poel HG, Li ZC, Kontos HA, Levasseur JE. Pial arteriolar vessel diameter and CO2 reactivity during prolonged hyperventilation in the rabbit. J Neurosurg. (1988) 69:923–7. doi: 10.3171/jns.1988.69.6.0923
54. Fieschi C, Battistini N, Beduschi A, Boselli L, Rossanda M. Regional cerebral blood flow and intraventricular pressure in acute head injuries. J Neurol Neurosurg Psychiatry. (1974) 37:1378–88. doi: 10.1136/jnnp.37.12.1378
55. Fantini S, Sassaroli A, Tgavalekos KT, Kornbluth J. Cerebral blood flow and autoregulation: current measurement techniques and prospects for noninvasive optical methods. Neurophotonics. (2016) 3:031411. doi: 10.1117/1.NPh.3.3.031411
56. Naqvi J, Yap KH, Ahmad G, Ghosh J. Transcranial Doppler ultrasound: a review of the physical principles and major applications in critical care. Int J Vasc Med. (2013) 2013:629378. doi: 10.1155/2013/629378
57. Skippen P, Seear M, Poskitt K, Kestle J, Cochrane D, Annich G, et al. Effect of hyperventilation on regional cerebral blood flow in head-injured children. Crit Care Med. (1997) 25:1402–9. doi: 10.1097/00003246-199708000-00031
58. Dahl B, Bergholt B, Cold GE, Astrup J, Mosdal B, Jensen K, et al. CO(2) and indomethacin vasoreactivity in patients with head injury. Acta Neurochir. (1996) 138:265–73. doi: 10.1007/BF01411736
59. Sioutos PJ, Orozco JA, Carter LP, Weinand ME, Hamilton AJ, Williams FC. Continuous regional cerebral cortical blood flow monitoring in head-injured patients. Neurosurgery. (1995) 36:943–9; discussion: 949–50. doi: 10.1097/00006123-199505000-00009
60. Steiner LA, Balestreri M, Johnston AJ, Coles JP, Chatfield DA, Pickard JD, et al. Effects of moderate hyperventilation on cerebrovascular pressure-reactivity after head injury. Acta Neurochir Suppl. (2005) 95:17–20. doi: 10.1007/3-211-32318-X_4
61. Carney N, Totten AM, O'Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery. (2017) 80:6–15. doi: 10.1227/NEU.0000000000001432
62. Diringer MN, Yundt K, Videen TO, Adams RE, Zazulia AR, Deibert E, et al. No reduction in cerebral metabolism as a result of early moderate hyperventilation following severe traumatic brain injury. J Neurosurg. (2000) 92:7–13. doi: 10.3171/jns.2000.92.1.0007
63. Unterberg AW, Kiening KL, Hartl R, Bardt T, Sarrafzadeh AS, Lanksch WR. Multimodal monitoring in patients with head injury: evaluation of the effects of treatment on cerebral oxygenation. J Trauma. (1997) 42(5 Suppl):S32–7. doi: 10.1097/00005373-199705001-00006
64. von Helden A, Schneider GH, Unterberg A, Lanksch WR. Monitoring of jugular venous oxygen saturation in comatose patients with subarachnoid haemorrhage and intracerebral haematomas. Acta Neurochir Suppl. (1993) 59:102–6. doi: 10.1007/978-3-7091-9302-0_18
65. Oertel M, Kelly DF, Lee JH, Glenn TC, Vespa PM, Martin NA. Is CPP therapy beneficial for all patients with high ICP? Acta Neurochir Suppl. (2002) 81:67–8. doi: 10.1007/978-3-7091-6738-0_16
66. Oddo M, Bosel J. Participants in the international multidisciplinary consensus conference on multimodality M: monitoring of brain and systemic oxygenation in neurocritical care patients. Neurocrit Care. (2014) 21(Suppl. 2):S103–20. doi: 10.1007/s12028-014-0024-6
67. Hemphill JC 3rd, Knudson MM, Derugin N, Morabito D, Manley GT. Carbon dioxide reactivity and pressure autoregulation of brain tissue oxygen. Neurosurgery. (2001) 48:377–83; discussion: 383–74. doi: 10.1227/00006123-200102000-00028
68. van Santbrink H, Maas AI, Avezaat CJ. Continuous monitoring of partial pressure of brain tissue oxygen in patients with severe head injury. Neurosurgery. (1996) 38:21–31. doi: 10.1097/00006123-199601000-00007
69. Meixensberger J, Jager A, Dings J, Baunach S, Roosen K. Multimodal hemodynamic neuromonitoring–quality and consequences for therapy of severely head injured patients. Acta Neurochir Suppl. (1998) 71:260–2. doi: 10.1007/978-3-7091-6475-4_75
70. Fandino J, Stocker R, Prokop S, Imhof HG. Correlation between jugular bulb oxygen saturation and partial pressure of brain tissue oxygen during CO2 and O2 reactivity tests in severely head-injured patients. Acta Neurochir. (1999) 141:825–34. doi: 10.1007/s007010050383
71. Gopinath SP, Valadka AB, Uzura M, Robertson CS. Comparison of jugular venous oxygen saturation and brain tissue PO2 as monitors of cerebral ischemia after head injury. Crit Care Med. (1999) 27:2337–45. doi: 10.1097/00003246-199911000-00003
72. Schneider GH, Sarrafzadeh AS, Kiening KL, Bardt TF, Unterberg AW, Lanksch WR. Influence of hyperventilation on brain tissue-PO2, PCO2, and pH in patients with intracranial hypertension. Acta Neurochir Suppl. (1998) 71:62–5. doi: 10.1007/978-3-7091-6475-4_20
73. Gupta AK, Hutchinson PJ, Al-Rawi P, Gupta S, Swart M, Kirkpatrick PJ, et al. Measuring brain tissue oxygenation compared with jugular venous oxygen saturation for monitoring cerebral oxygenation after traumatic brain injury. Anesth Analg. (1999) 88:549–53. doi: 10.1097/00000539-199903000-00016
74. Imberti R, Ciceri M, Bellinzona G, Pugliese R. The use of hyperventilation in the treatment of plateau waves in two patients with severe traumatic brain injury: contrasting effects on cerebral oxygenation. J Neurosurg Anesthesiol. (2000) 12:124–7. doi: 10.1097/00008506-200004000-00011
75. Menon DK, Coles JP, Gupta AK, Fryer TD, Smielewski P, Chatfield DA, et al. Diffusion limited oxygen delivery following head injury. Crit Care Med. (2004) 32:1384–90. doi: 10.1097/01.CCM.0000127777.16609.08
76. Gordon E. Controlled respiration in the management of patients with traumatic brain injuries. Acta Anaesthesiol Scand. (1971) 15:193–208. doi: 10.1111/j.1399-6576.1971.tb05461.x
77. Lee SW, Hong YS, Han C, Kim SJ, Moon SW, Shin JH, et al. Concordance of end-tidal carbon dioxide and arterial carbon dioxide in severe traumatic brain injury. J Trauma. (2009) 67:526–30. doi: 10.1097/TA.0b013e3181866432
78. Hawryluk GWJ, Aguilera S, Buki A, Bulger E, Citerio G, Cooper DJ, et al. A management algorithm for patients with intracranial pressure monitoring: the Seattle International Severe Traumatic Brain Injury Consensus Conference (SIBICC). Intensive Care Med. (2019) 45:1783–94. doi: 10.1007/s00134-019-05805-9
Keywords: traumatic brain injury, hyperventilation, hypocapnia, intracranial hypertension, cerebral ischemia
Citation: Gouvea Bogossian E, Peluso L, Creteur J and Taccone FS (2021) Hyperventilation in Adult TBI Patients: How to Approach It? Front. Neurol. 11:580859. doi: 10.3389/fneur.2020.580859
Received: 07 July 2020; Accepted: 21 December 2020;
Published: 28 January 2021.
Edited by:
Rafael Badenes, University of Valencia, SpainReviewed by:
Marcel Aries, Maastricht University, NetherlandsRajaNandini Muralidharan, Winthrop University Hospital, United States
Copyright © 2021 Gouvea Bogossian, Peluso, Creteur and Taccone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio Silvio Taccone, ZnRhY2NvbmVAdWxiLmFjLmJl