 Elisa Canepa
Elisa Canepa Silvia Fossati
Silvia Fossati- Alzheimer's Center at Temple (ACT), Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
Alzheimer's disease (AD) is a chronic neurodegenerative disorder and the most prevalent cause of dementia. The main cerebral histological hallmarks are represented by parenchymal insoluble deposits of amyloid beta (Aβ plaques) and neurofibrillary tangles (NFT), intracellular filamentous inclusions of tau, a microtubule-associated protein. It is well-established that cerebrovascular dysfunction is an early feature of AD pathology, but the detrimental mechanisms leading to blood vessel impairment and the associated neurovascular deregulation are not fully understood. In 90% of AD cases, Aβ deposition around the brain vasculature, known as cerebral amyloid angiopathy (CAA), alters blood brain barrier (BBB) essential functions. While the effects of vascular Aβ accumulation are better documented, the scientific community has only recently started to consider the impact of tau on neurovascular pathology in AD. Emerging compelling evidence points to transmission of neuronal tau to different brain cells, including astrocytes, as well as to the release of tau into brain interstitial fluids, which may lead to perivascular neurofibrillar tau accumulation and toxicity, affecting vessel architecture, cerebral blood flow (CBF), and vascular permeability. BBB integrity and functionality may therefore be impacted by pathological tau, consequentially accelerating the progression of the disease. Tau aggregates have also been shown to induce mitochondrial damage: it is known that tau impairs mitochondrial localization, distribution and dynamics, alters ATP and reactive oxygen species production, and compromises oxidative phosphorylation systems. In light of this previous knowledge, we postulate that tau can initiate neurovascular pathology in AD through mitochondrial dysregulation. In this review, we will explore the literature investigating tau pathology contribution to the malfunction of the brain vasculature and neurovascular unit, and its association with mitochondrial alterations and caspase activation, in cellular, animal, and human studies of AD and tauopathies.
Introduction
Alzheimer's Disease, Neurovascular Unit and Blood-Brain Barrier Dysfunction
Alzheimer's disease (AD) is a progressive and deadly neurodegenerative disorder recognized by the World Health Organization as the most prevalent form of dementia (1). AD initial symptoms typically manifest as mild cognitive impairment (MCI). Although MCI does not always convert to AD or dementia, a percentage of MCI cases will worsen over the years, eventually progressing to the severe cognitive decline, emotional and behavioral changes, visuospatial and motor deficits, characteristics of late-stage AD (2). These devastating cognitive and physical aspects are correlated with neuronal loss and extensive brain atrophy, predominantly in the hippocampus and cortex (3). The most studied and scientifically confirmed neuropathological hallmarks of AD are senile plaques (SP) and neurofibrillary tangles (NFT) in association with neuronal degeneration, loss of synapses, neuroinflammation, and oxidative stress (4). Furthermore, conspicuous studies indicate that brain vascular dysregulation is an early feature of AD, contributing to the progression of the pathology, and suggesting a tight link between cerebrovascular alterations and neurodegeneration (5–8). SP are abnormal parenchymal insoluble deposits of amyloid-beta (Aβ), which derives from amyloid precursor protein (APP) processing. Aβ monomers aggregate into more complex species (oligomers and protofibrils) which represent the toxic species for most brain cells (9, 10) and eventually form fibrils (11). Interestingly, Aβ accumulation also takes place around the brain micro- and macro-vasculature, in a well-described AD pathological feature known as cerebral amyloid angiopathy (CAA) (12), which leads to severe cerebral vascular dysfunction (CVD) (10, 13–16). NFT are intracellular inclusions of paired helical filaments (PHF) of the microtubule-associated protein (MAP) tau. Physiologically, specific phosphorylation and dephosphorylation events regulate tau microtubule (MT)-binding property, thus modulating tau functions, including promoting MT assembly and dynamics, and maintaining neuronal spatial organization and stability (17, 18). In AD brains, tau phosphorylation levels are 3–4-fold higher compared to non-demented adult brains (19, 20), and phosphorylation facilitates tau aggregation, causing cell morphology, functionality, and viability disruption. Besides the documented effects of Aβ deposition around cerebral blood vessels (BVs), recent evidence indicates that tau pathology induces CVD (21–23), and that the presence of hippocampal perivascular tau strictly correlates with blood brain barrier (BBB) permeability and loss of integrity (24), prompting new questions to elucidate the molecular mechanisms through which tau toxicity affects the cerebral vasculature, in both AD and other tauopathies.
The BBB is a highly specialized and selective semipermeable interface between the central nervous system (CNS) and the peripheral circulation, regulating the entrance of blood specific components into the brain and the clearance of potentially neurotoxic substances from the CNS to the blood. The morpho-functional unit, which includes the BBB and is responsible for maintaining its unique properties, is the neurovascular unit (NVU). The NVU is constituted by endothelial cells (ECs) that form the BVs and are strongly attached to each other via a complex network of sealing proteins named tight junctions (TJs) (25), by the surrounding smooth muscle cells (SMC) or pericytes, together with neurons, astrocytes, and microglia cells (8, 26–28). BV-surrounding cells influence and co-operate in maintaining the structural and functional BBB phenotype (26). Astrocytes are the major glial cell type (~98%) at the parenchymal basal membrane of the microvasculature (29), with microglial cells occupying the remaining surface. In addition to their contribution to BBB maintenance and permeability, astrocytes are considered immunocompetent cells, playing a central role in detecting detrimental signals following which they start secreting cytokines and chemokines, boosting innate and adaptive immune cell activation and trafficking, and thus affecting BBB function (30). Microglia are brain innate resident immune cells, involved in the active surveillance of CNS. When exposed to pathogens, tissue damage or toxic substances, microglial cells become activated, releasing pro-inflammatory cytokines, chemotactic factors, and mediate adaptive immunity, acting as antigen-presenting cells (31, 32). If activated, they can interact with cerebral microvasculature, and increase capillary permeability by producing reactive oxygen species (ROS), and promoting monocyte and lymphocyte migration through the BBB (33, 34). Importantly, the extensive astrogliosis and microgliosis present in AD brains (35–38), may further exacerbate vascular dysfunction. Morphologically, AD brains display BBB changes such as reduced microvascular density, increased capillary tortuosity and fragmentation with fewer intact branches, atrophic string vessels, and changes in vessel diameter (8, 39, 40), that impair vascular fitness causing decreased cerebral blood flow (CBF), hypoxia, dysregulated nutrient and oxygen transport to the CNS, and disrupted cerebral clearance. As a consequence, amplification of cellular stress, accumulation of toxic metabolic waste, uncontrolled detrimental inflammatory response and infiltration of blood-borne molecules and cells occur, eliciting neurodegenerative processes and progressive decrease in cognitive functions (8, 41, 42). All together, these data provide a strong evidence of the connection between BBB malfunction and AD, although the leading molecular mechanisms triggering CVD have not been fully elucidated.
While Aβ aggregation and deposition have long been associated with cerebrovascular alterations in AD, the scientific community has recently started to consider the role of tau in BBB dysfunction, supported by a rapidly growing literature that confirms CVD in other tauopathies.
Here, we will review what is known about tau and neurovascular dysregulation, exploring and providing perspective on some of the possible molecular events through which tau may exert its toxicity on the NVU.
Tau and Its Transmission
The MAPs family includes three classes of polypeptides predominantly expressed in neurons: MAP1, MAP2, and tau. While MAP1 and MAP2 have been largely found in dendrites, tau has been detected mainly in axons (43, 44), where it is involved in primary functions, including neuronal development control (45–47), vesicular and axonal transport (48, 49), and neuronal polarity maintenance (50), amongst others. Structurally, tau presents a basic proline-rich region (aa155–242) which contains serine (S), threonine (T), and tyrosine (Y) potential phosphorylation sites (51). In physiological state, tau is phosphorylated or dephosphorylated based on the balance of kinase (e.g., GSK3β and CDK5) and phosphatase (e.g., PP1, PP2A, B and C) activities (47, 52–55). This equilibrium confers tau the ability to bind and stabilize tubulin polymerization, requisite condition for maintaining axonal and dendritic shape, and thus functionality (56, 57). Due to hyperphosphorylation, tau MT-binding property is lost, inducing its oligomerization and the formation of PHF which progressively aggregate into NFT (58, 59). As a consequence, alterations of cytoskeleton architecture occur, leading to axonal transport disruption, synaptic dysfunction, and eventually neuronal cell death (47, 60–62). Indeed, a characteristic brain accumulation of highly phosphorylated tau is found in AD, and other tauopathies, such as progressive supranuclear palsy (PSP), Pick's disease (PiD), corticobasal degeneration (CBD), and frontotemporal dementia FTD (63).
Although research in neurodegenerative disorders focuses mainly on tau hyperphosphorylation, it is relevant to mention that tau can also be subject to concurrent or alternative post-translational modifications (PTMs), including N- and C-terminal proteolytic cleavage (truncation), nitration, glycosylation, acetylation, glycation, ubiquitination, or polyamination (64), that exacerbate tau pathology. For instance, in AD human brains, C-terminal tau fragments generated by caspase-3 cleavage at aspartic acid residue 421 (D421) have been detected (65). Considerable evidence shows that caspase-3-truncated tau species are particularly prone to phosphorylation (65), and that caspase-dependent cleavage process increases tau propensity to self-aggregate, boosting the rate of tau polymerization and NFT assembly (45, 47, 59, 66), and fostering dendritic spine loss, synaptic impairment and memory deficits (67, 68). Similarly, caspase-6 activity derived N-terminal truncated tau segments have been found in AD cerebral tissue (69).
Collectively, these data indicate that misfolded conformations of tau facilitate the development of aggregates that appear to be crucial for neuronal demise in AD and other tauopathies.
In the last decade, a wealth of studies has suggested that misfolded tau spreads in a prion-like manner in tauopathy brains (70–72). The hypothesis of the prion-like propagation has been proposed from the observation that the progressive accumulation of tau appeared to spread in a foreseeable pattern, along known anatomically connected neuronal networks (73), analogously to that reported for prion proteins (72).
Albeit tau is an intracellular protein, both tau and P-tau can also be detected in brain interstitial fluid (74), cerebrospinal fluid (CSF) (75), and, as shown more recently, in blood (76–79). In AD patients, CSF and plasma tau and phosphorylated tau (P-tau) concentrations are significantly higher compared to healthy controls. Since tau in biofluids is considered to be for the most part neuron-derived, CSF and plasma tau concentrations typically represent the intensity of neurodegeneration (76, 78, 80, 81). CSF P-tau, on the other hand, is an established AD biomarker also used in clinical trials, reflecting more specifically the progression of tau pathology and correlating with cognitive dysfunction. Recently, plasma P-tau181 and P-tau217 have also been proposed as novel biomarkers for AD (82–84). Although blood tau and P-tau fragments are of great interest as biomarkers, these peptides are present at very low concentrations in the circulation (in the low pg/ml), and it is still unknown if circulating tau species may be able to induce toxic effects on the vasculature, both in the brain and in the periphery. In particular, more studies are needed to clarify any possible toxic effects of blood P-tau, especially in conditions in which cerebral vessels are already dysfunctional. On the other hand, CSF P-tau, present at higher concentrations in the perivascular spaces, may be a likely contributor to neurovascular pathology, especially in situations in which perivascular clearance is compromised, which may facilitate its accumulation around brain vessels (85–87). Interestingly, it has been reported that, besides prompting intracellular toxicity, tau can be secreted and taken up by healthy neurons (88–90), indicating that extracellular or CSF P-tau may be critical for the progression of tau pathology. Recent studies in the AD field have demonstrated that seed-competent tau spreads along neural connections, is detected in synaptosomes and white matter axons, and that tau seeding precedes the presence of hyperphosphorylated tau in synaptically connected regions (91–94). To further elucidate the propagating properties of tau, many studies have investigated the potential molecular processes involved. Numerous neuronal mechanisms have been identified as non-mutually exclusive sources of extracellular tau, including synaptic secretion (95), direct unconventional translocation across the plasma-membrane (96, 97), release in extracellular vesicles such as exosomes (98–100) and ectosomes (101), or tunneling nanotubes (TNTs) (102, 103). In pathological conditions, neuronal deterioration may also account for extracellular tau which is internalized by neighboring cells via bulk endocytosis (88, 89, 104, 105), clathrin- (106, 107) or low-density lipoprotein receptor related protein 1 (LRP1)-mediated endocytosis (108), micropinocytosis by heparin sulfate proteoglycans (90, 109–111) or TNTs (102, 103) (Figure 1). Following cellular entry of tau, neurons can seed physiological monomers, perpetuating the pathological process (112). It has also been demonstrated that neuronal activity can induce tau release (95), enhancing intercellular spread of tau (113).
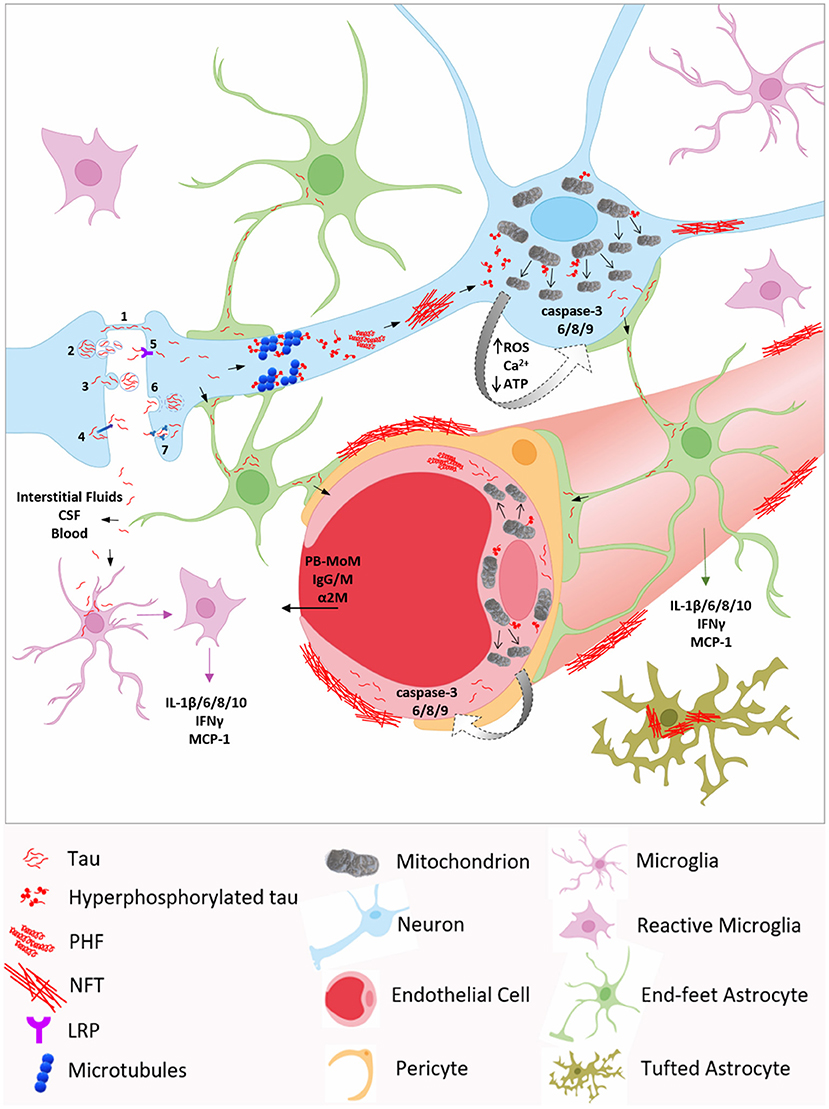
Figure 1. Molecular mechanisms underlying neurovascular tau toxicity in AD and other tauopathies. Neuronal transmission through (1) tunneling nanotubes (TNTs), (2) exosomes, (3) ectosomes, or (4) plasma membrane may account for extracellular tau, which can be internalized by neighboring neurons via (5) low-density lipoprotein receptor (LRP), (6) clathrin-mediated endocytosis, (7) micropinocytosis by heparin sulfate proteoglycans, or (1) TNTs. Tau is also detected in brain interstitial fluids, cerebrospinal fluid (CSF) and in blood, reflecting the intensity of neurodegeneration. Inside the neuron, hyperphosphorylated tau promotes microtubules disassembly, aggregates into oligomers and paired helical filaments (PHF) which, in turn, accumulate, leading to neurofibrillary tangles (NFT) deposition. Phosphorylated tau can interact with mitochondrial proteins, such as Drp1, triggering excessive mitochondrial fission and mitochondrial dysfunction, including elevated ROS production, Ca2+ homeostasis dysregulation, decreased ATP production, and ultimately caspase activation. Caspase-3 cleaved tau facilitates tau phosphorylation and self-aggregation, further exacerbating tau pathology. Tau can propagate to astrocytes and microglia, and be internalized via micropinocytosis and CX3CR1, respectively. In astrocytes, the accumulation of tau fibrils around the nucleus confers the characteristic tufted phenotype. Tau pathological species, through astrocytic end-feet and interstitial fluids, may spread to endothelial cells and pericytes, inducing blood brain barrier (BBB) disruption and permeability to blood-borne components, including peripheral blood monocyte-derived macrophages (PB-MoM), immunoglubulins (IgGs and IgMs), and α2-macroglobulin (α2M). Moreover, the presence of tau within glial cells induces the secretion of several pro-inflammatory cytokines and chemokines, such as IL-1β,−6, - 8,−10, IFNγ, and MCP-1, initiating a high inflammatory state, which contributes to BBB integrity loss.
As mentioned above, AD and tauopathy brains display widespread astrogliosis and microgliosis, which closely correlate with the areas of neurodegeneration (114, 115). Intriguingly, several studies have reported the accumulation of toxic tau in astrocytes and microglia in different tauopathies (116–121). Endogenous glial tau expression has been reported (122, 123), although at much lower levels compared to neurons (124). Therefore, it has been postulated that the intracellular presence of tau in these cell types may be, at least in part, explained by its internalization from the extracellular milieu. Recent evidence has demonstrated that astrocytic tau uptake occurs via micropinocytosis, followed by enhanced lysosomal pathways (125). It is likely that, in pathological conditions, tau engulfed astrocytes exhibit dysregulated clearance mechanisms, further intensifying tau propagation and toxicity. Additionally, it is known that microglia have the competence to phagocytize tau (126–132), and the presence of microglial aggregated or hyperphosphorylated tau has been observed in mice ad humans with tau pathology (119, 120, 126–128, 131, 133). The microglial process of tau internalization seems to be mediated by CX3CR1 receptor (120, 133). More importantly, at the site of tau colocalization with astrocytes and microglia, several pro-inflammatory cytokines have been detected (134), indicating a high neuroinflammatory state. During inflammation, reactive glial cells continuously secrete cytokines and chemokines, which further recruit and activate innate and adaptive immune cells (30–32), initiating a feed-forward detrimental inflammatory response, feature of AD and tauopathies (4, 134), which may also affects neurovascular and BBB function. Another hypothesis proposes that tau toxicity might be mediated by the brain vasculature, considering that vascular dysfunction, dendritic deterioration and inflammation take place before neuronal loss in a mouse model of tau-induced neurodegeneration (135) or before tau phosphorylation in a salt-rich diet model (136). In turn, tau toxicity may also be in part responsible for the vascular dysfunction present in AD and related disorders. In confirmation of this, considerable studies have shown that tau pathology triggers alterations of BVs and vascular inflammation (23, 135, 137–139), which may be induced by tau oligomers and fibrils deposition around the microvasculature (23, 24, 140). Moreover, tau-induced neuroinflammation can additionally damage BBB, leading to the infiltration of peripheral immune cells and endothelial signaling molecules expression (141, 142), which keep glial cells active (22, 143), worsening inflammation, neurovascular pathology, and consequentially neuronal activity.
All together, these results suggest how tau toxicity may propagate from neurons to NVU components, and potentially activate harmful mechanisms that interfere with neurovascular function and inflammation, exacerbating neurodegeneration.
TAU and Neurovascular Dysfunction
Cell Studies
As previously described, the BBB is a dynamic and extremely specialized barrier which physically separates CNS and systemic circulation. The NVU is a complex functional unit including the BBB and is responsible for its properties and health. ECs, mural cells (including vascular smooth muscle cells and pericytes), astrocytes, microglia and neurons (26, 144) compose the NVU and contribute to its functions. Multiple studies have confirmed NVU abnormalities in AD, describing CVD as an early event in AD pathology, and establishing a direct correlation between brain vasculature dysregulation and neurodegeneration (5–8). The potential molecular pathways linking tau toxicity and neurovascular dysfunction have recently started to be evaluated, both in vitro and in vivo (Table 1). As reported below, a growing body of literature shows that other tauopathies, in addition to AD, also display severe cerebrovascular changes (22, 24, 28, 142, 145–149) (Table 2).
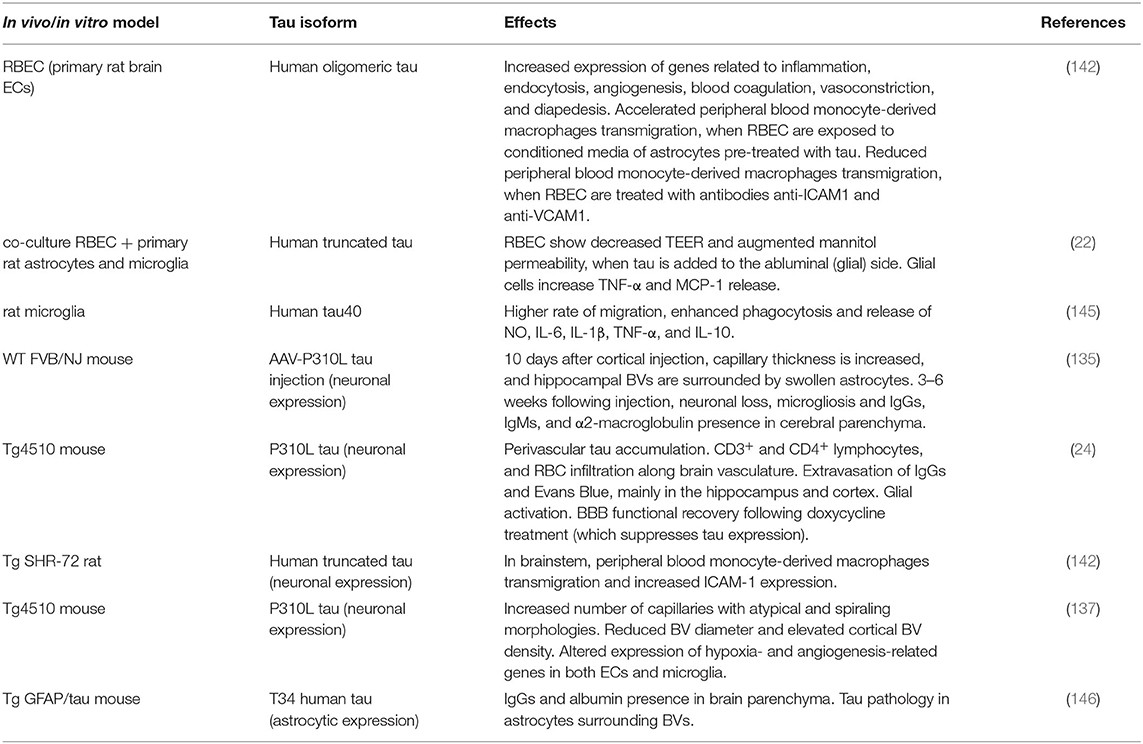
Table 1. Effects of tau on the NVU in vitro and in vivo models.
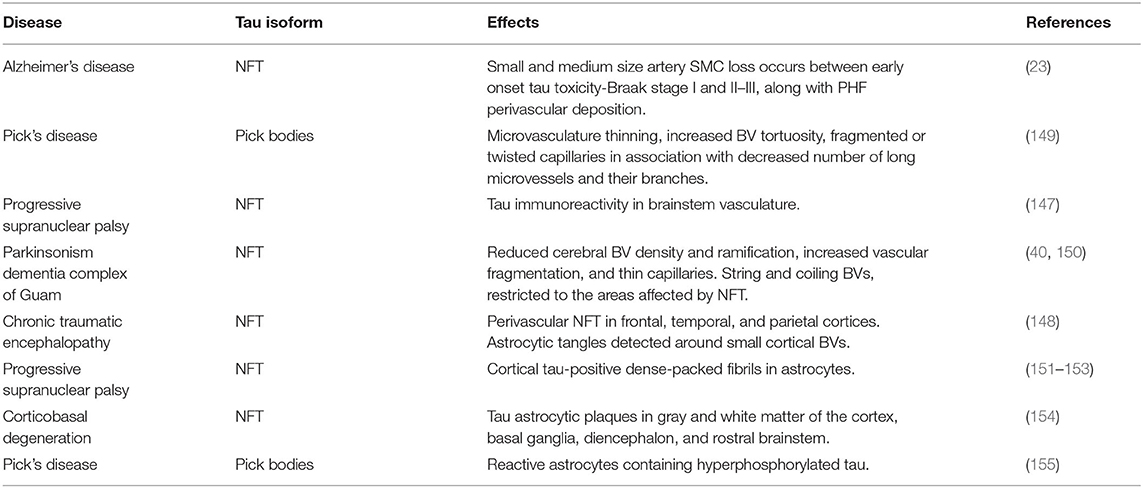
Table 2. Effects of tau on the NVU in AD and other tauopathies.
A recent study has demonstrated that primary rat brain ECs (RBEC) exposed to human oligomeric tau dramatically increase the expression of genes related to inflammation, endocytosis, angiogenesis, blood coagulation, and vasoconstriction (142). The authors have also observed upregulated expression of genes involved in diapedesis, the process of immune cell migration across the endothelial wall from the blood circulation to brain parenchyma. Using RBEC monolayers as an in vitro BBB model, they have analyzed tau-induced transmigration of peripheral blood monocyte-derived macrophages (PB-MoM) isolated from rat. When RBEC are incubated in conditioned media (CM) of astrocytes previously challenged with tau, a 3-fold accelerated transmigration of PB-MoM has been detected. In the same conditions, EC pre-treatment with monoclonal antibodies anti-ICAM-1 and anti-VCAM-1 reduces PB-MoM transmigration, indicating that these specific endothelial adhesion molecules may play a role in tau-induced trafficking of blood cells across BBB (142). The effects of human truncated tau have been evaluated in a similar in vitro model of BBB, consisting of a co-culture of primary RBEC and mixed rat primary astrocyte (85–90%)-microglia (10–15%) cultures (22). When tau is added to the abluminal compartment of the co-culture chamber, where astrocytes and microglia are seeded, a significant decrease of trans-endothelial electrical resistance (TEER) has been observed, concomitantly with an augmented mannitol permeability through the BBB, in contrast to controls. The measurement of pro-inflammatory molecules indicates that BBB breakdown is likely mediated by TNF-α and the chemokine MCP-1, which are abundantly released by glial cells, following tau treatment (22). A different in vitro study confirmed the harmful effect of tau on NVU components, showing that cultured rat microglia, transfected with human tau40, were significantly activated compared to control. Activation was confirmed by higher ability of migration, enhanced phagocytosis and increased CM levels of nitric oxide (NO), and IL-1β, IL-6, TNF-α, and IL-10 inflammatory cytokines (145), which may act as mediators of tau-induced BBB dysfunction. Indeed, if on one hand IL-1β and TNF-α regulate the expression of endothelial TJ proteins (156, 157), on the other hand, stimulation of ECs with both TNF-α and IFN-γ increases the level of adhesion molecules, such as ICAM-1 and VCAM-1 (158–161), facilitating immune cell paracellular migration to cerebral parenchyma and amplifying tau-triggered toxicity.
Overall, these in vitro studies point to deleterious effects of tau on vascular and immune cellular and molecular pathways responsible for NVU function. These effects and the main respective references are summarized in Table 1.
Animal Studies
Several in vivo studies have contributed to corroborate the impact of tau pathology on the brain vasculature and NVU (Table 1). In one of these studies, as a model of tauopathy, wild-type mice have been injected with recombinant adeno-associated virus (AAV) vector to express mutant tau P301L (encoding for familial FTD tau protein), specifically in neurons (135). Compared to control littermates, AAV-tauP301L–injected animals displayed significantly increased capillary thickness as early as 10 days after injection, with many BVs within the CA1 region of the hippocampus surrounded by swollen astrocytes, most likely participating in the substantial constriction of the capillaries. Interestingly, in these animals, neurodegeneration coincided with microgliosis, which occurred 3–6 weeks following injection, demonstrating that tau-mediated vascular defects preceded neuronal loss. In concomitance with the onset of neurodegeneration, BBB integrity was further compromised, as parenchymal IgGs, IgMs, and α2-macroglobulin were detected (135). In aged (12–15-month-old) tetracycline inducible rTg4510 mice, which overexpress tau P301L protein, a variety of vascular abnormalities have been described, mainly in hippocampus and cortex. Progressively with age, rTg4510 animals exhibited tau perivascular accumulation together with BBB breakdown, measured as significant CD3+ and CD4+ lymphocyte, and red blood cell (RBC) infiltration along the vasculature, and extravasation of IgGs and Evans Blue, a dye that binds serum albumin, which does not cross the BBB, unless it is injured (24). In this model, tau pathology triggered glial activation, identified as high expression of glial fibrillary acidic protein (GFAP), astrocytic intermediate filament protein, and heat shock protein 27 (Hsp27), marker of reactive astrocytes (162) and involved in BBB regulation through its role in actin stabilization (163). Noteworthy, in comparison to WT or younger Tg animals, treatment with doxycycline, which suppressed tau expression, led to BBB functional recovery in old rTg4510 mice, with reduced T cell and RBC infiltration, and decreased Hsp27 and GFAP levels (24), indicating tau direct involvement in CVD. Transmigration of blood-borne cells, such as PB-MoM, has also been reported in cerebral areas with increased ICAM-1 expression, associated with neurofibrillary pathology, in Tg SHR-72 rat model which stably expresses human tau protein truncated at amino acids 151–391 (aa 151-391/4R) (142), confirming the role of tau in triggering detrimental changes in BBB. Using the Tg4510 murine model, another study has documented multiple brain vascular changes, including increased number of capillaries characterized by atypical and spiraling morphologies, reduced vessel diameter, and elevated cortical BV density, in 15-month-old Tg animals. RNA analysis has revealed an altered expression of hypoxia- and angiogenesis-related genes specifically in ECs and microglia. The greatest fold change has been found in endothelial Serpine1, a gene that encodes for plasminogen activator inhibitor (PAI-1) protein (137), known for stimulating migration of ECs (164), modulating proteolytic activity (165), fundamental for extracellular matrix remodeling during angiogenesis, and for regulating microglia motility and phagocytosis (166). Intriguingly, also Tg mice expressing the T34 human tau isoform specifically in astrocytes, have been shown to develop BBB disruption, verified as IgG and albumin presence in the brain parenchyma, in association with prominent tau pathology in astrocytic foot processes surrounding BVs (146). In summary, tau has been shown to trigger multiple detrimental brain vascular and glial changes in animal models, which are detailed in Table 1.
Human Studies
Over the last few decades, the AD cerebral microvasculature has been described as atrophic, thin, fragmented, twisted or tortuous, and with glomerular loop formations (40). Ultrastructural analysis has reported atrophic ECs, and remarkably decreased (167) and compromised morphology (168) of TJs. In addition, AD brains present anomalous focal constrictions and general degeneration of SMC (169–171), swollen astrocytic end-feet (172, 173), and atrophic pericytes (169). As a consequence, AD subjects display disrupted CBF and brain hypoperfusion (28, 174–176). Interestingly, a 2016 study has demonstrated that vascular changes are correlated with Braak stages, which classify the degree of tau pathology, showing that small and medium size artery SMC loss occurs between early onset tau toxicity-Braak stage I and II–III, along with PHF perivascular deposition (23).
These results bolster a direct connection of tau pathology with vascular abnormalities. As a confirmation, in PiD, where cerebral aggregates of hyperphosphorylated tau (Pick bodies) are present (149), hallmarks like microvasculature thinning, increased BV tortuosity, fragmented or twisted capillaries in association with decreased number of long microvessels and their branches, are as severe as in AD (40). Moreover, atrophic brain areas display a considerable disorganization of the vascular basal lamina distribution (150). A recent paper has described oligomeric tau immunoreactivity in brain vasculature of other tauopathies, such as PSP (147). It is also known that Parkinsonism dementia complex of Guam, characterized by abundant tau neurofibrillary pathology, shows reduced cerebral BV density and ramification, increased vascular fragmentation, and thin capillaries (40). Furthermore, string and coiling vessels are typical, and restricted to the areas affected by NFT (150). Repetitive mild or moderate traumatic brain injury (TBI) seems to be associated with higher risk of AD development (177), and may lead to chronic traumatic encephalopathy (CTE) (148, 178, 179). Strikingly, perivascular NFT of hyperphosphorylated tau are one of the most common pathological hallmarks of CTE, starting at very early stages of the disease, mainly in frontal, temporal, and parietal cortices (particularly in the depth of the sulci) (180). With the progression of the disorder, also fibrillar astrocytic tangles are detected around small cortical BVs (148), plausibly exacerbating vascular dysfunction. Similarly, other tauopathies display high phosphorylated tau accumulation in astrocytes. For instance, in PSP, astroglial phenotype is often referred to as tufted (151–153), showing tau-positive dense-packed fibrils forming tufts around single or double nuclei, mainly in the frontal cortex, striatum, and thalamus (181, 182). Additionally, CBD is characterized by tau astrocytic plaques (154), defined as punctate or spindle-shaped aggregates, forming irregular rounded structures (181, 182). Reactive astrocytes containing hyperphosphorylated tau have been found in PiD as well (155).
Associations between tau pathology and white matter hyperintensities (WMH) have also been reported, showing that increasing cortical P-tau burden independently predicted the severity of WMH, indicating a potentially important role of tau in the pathogenesis of WM damage (183, 184).
Collectively, these human studies provide strong evidence for an association between tau toxicity and vascular dysregulation in multiple tauopathies (summarized in Table 2).
To better dissect how tau pathology may affect cerebrovascular function, we propose below some of the molecular and cellular mechanisms which have been shown to be affected by tau, and may also mediate its effects on the vessel walls.
TAU and Mitochondrial Dysregulation
Tau and Mitochondrial Dynamics
Mitochondria are essential organelles for cell survival and death, playing a primary role in energy metabolism and apoptotic processes (185). Balanced mitochondrial fission and fusion dynamics are pivotal events in regulating their shape, size, and number, enabling a correct morphology and distribution, and thus their capacity to meet high energy cellular demands, such as those of brain cells (186–188). A cytosolic guanosine triphosphatase (GTPase), named dynamin-like protein 1 (Drp1), assembling into spiral filaments around mitochondria, interacts with outer membrane proteins, including mitochondrial fission factor (Mff) and fission protein-1 (Fis1), to regulate the division process (189). On the other hand, the interaction of mitochondrial outer membrane proteins, such as the GTPases mitofusin 1 and 2 (Mfn1 and Mfn2), with optic dominant atrophy 1 (Opa1) inner mitochondrial membrane protein, mediates fusion (190). It has been reported that mitochondrial activity and phenotype are directly and tightly modulated by cellular and environmental stimuli (191). Therefore, it is not surprising that mitochondrial dysfunction and aberrant morphology are predominant pathological early features of AD brains (192–194).
Although with some discrepancies, most likely due to different tau isoforms and cell types used, multiple in vitro studies have pointed to direct effects of tau on mitochondrial dynamics (Table 3). Both human WT full-length (hTau) and P301L mutated tau isoforms have been shown to promote mitochondrial perinuclear accumulation, a feature of AD brains (203), in animal and cellular models (195, 203). Moreover, in HEK293 cells, hTau enhanced Mfn1, Mfn2, and Opa1 protein expression. Specifically, the decrease of hTau-mediated Mfn2 polyubiquitination underlaid Mfn2 accumulation, and the resulting increased mitochondrial fusion (195). Conversely, primary cortical neurons from tau KO mice, transfected with caspase-3 truncated tau presented mitochondrial fragmentation, together with a significant reduction of Opa1 levels, compared to control neurons (196). Another study has reported that, following treatment with okadaic acid, a well-known PP2A inhibitor, rat brain ECs (RBE4) displayed elevated tau phosphorylation and increased mitochondrial fission (Drp1 and Fis1) and fusion (Mfn1, Mfn2, and Opa1) protein levels, suggesting tau-dependent mitochondrial dynamics alterations, and confirming the expression of tau in ECs (197). Mfn1, Mfn2, and Opa1 accumulation have also been detected in brains of 6-month-old hTau Tg mice, in comparison with age-matched WT littermates (195). Additionally, in tau P301L Tg mice, another in vivo model of tauopathy, the partial genetic ablation of Drp1 reduced mitochondrial dysfunction and rescued mitochondrial dynamics (198). Excessive mitochondrial fission triggered by an atypical interaction between hyperphosphorylated tau and Drp1 has been described in multiple AD mouse models (199). Importantly, similar findings have been reported in AD human brains, where phosphorylated tau has been found to physically interact with Drp1, with little or no physical interaction occurring in control subjects (199). Moreover, a disrupted balance of fission and fusion, both at mRNA and protein levels (200, 201), has been found in AD patients, with most studies reporting a shift toward fission (200, 202). Overall, these data point to direct effects of tau on mitochondrial dynamics in multiple cellular (including ECs), animal models of tauopathies, and in human AD brains. These effects and their specific references are summarized in Table 3.
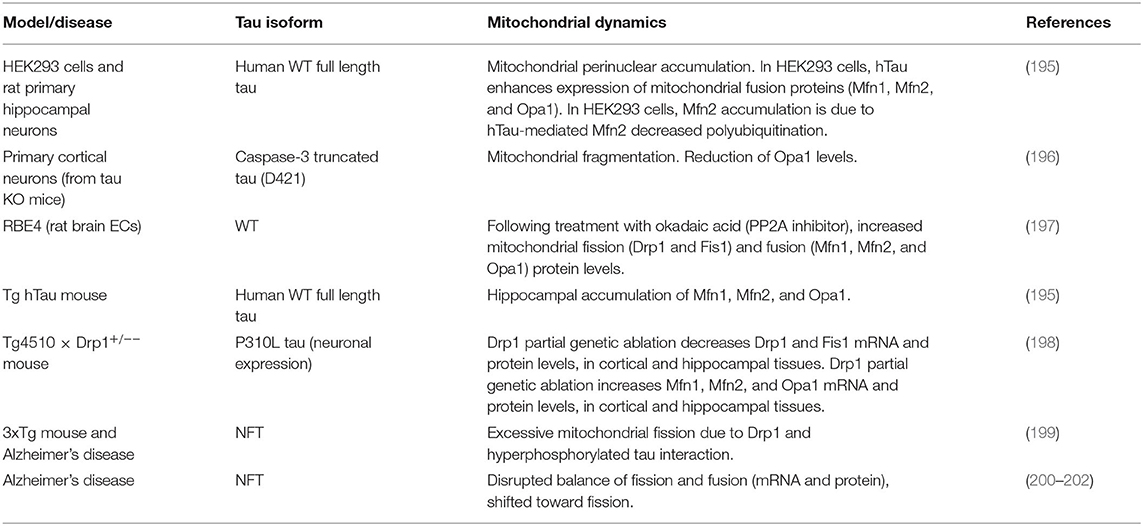
Table 3. Tau and mitochondrial dynamics.
Tau and Mitochondrial Dysfunction
Mitochondria are important for multiple cell functions in healthy and diseased brains, including energy production, intracellular Ca2+ homeostasis control, cell cycle regulation, ROS generation, apoptosis, and, in neurons, synaptic plasticity maintenance (10, 15, 185, 204–209). Amongst these, a pivotal role of the mitochondria is to provide energy to the cell from nutrient sources, through adenosine trisphosphate (ATP) production, accomplished via tricarboxylic acid cycle (TCA or Krebs cycle) and oxidative phosphorylation (OxPhos). Mitochondrial bioenergetics dysfunction and elevated mitochondrial ROS production have been reported to cause neuronal degeneration, and eventually cell death, in AD and other neuropathological conditions (194, 205, 206, 210). Mitochondrial dysfunction is also considered one of the earliest and probable causative steps in the AD pathogenesis (211–215). Tau has been recognized as a mediator of mitochondrial dysfunction, in both in vitro and in vivo models (216–219), as well as in human tauopathies, including AD (Table 4).
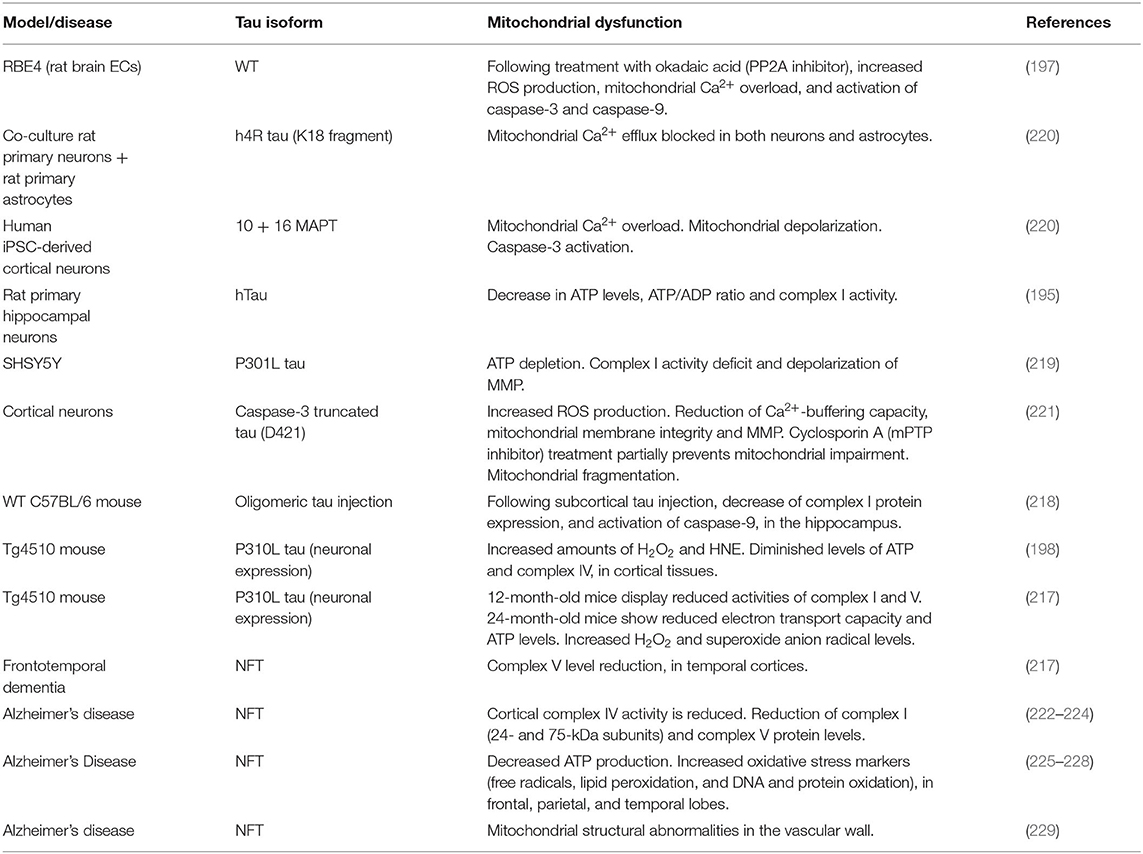
Table 4. Tau and mitochondrial dysfunction.
Cell experiments showed that rat brain ECs challenged with okadaic acid, prompted tau hyperphosphorylation in concomitance with ROS production, mitochondrial Ca2+ overload, and activation of caspase-3 and caspase-9, markers of apoptosis (197). The treatment of rat primary neuronal-astrocytic co-culture with the repeat domain of tau (K18) blocked mitochondrial Ca2+ efflux via impairment of NCLX, mitochondrial Na+/Ca2+ exchanger, in both neurons and astrocytes. Mitochondrial Ca2+ overload occurred also in human iPSC-derived cortical neurons expressing 10 + 16 MAPT mutation (linked to FTD), along with mitochondrial depolarization and caspase-3 activation (220). Rat primary hippocampal neurons stably expressing hTau displayed a significant decrease in ATP levels, ATP/ADP ratio, complex I activity, and cell viability (195). Similarly, overexpression of P301L tau in neuronal SHSY5Y cells led to ATP depletion and pronounced complex I activity deficit, along with depolarization of mitochondrial membrane potential (MMP) (219), which is physiologically generated by complexes I, III, and IV. In cortical neurons, the inducible expression of D421-cleaved caspase-3 truncated tau triggered mitochondrial fragmentation, increased ROS production, and a significant reduction of Ca2+-buffering capacity, mitochondrial membrane integrity and MMP. Interestingly, the treatment with cyclosporin A, which inhibits mitochondrial permeability transition pore (mPTP), partially prevented tau-induced mitochondrial impairment (221). mPTP is a mitochondrial channel that opens in pathological circumstances, enhancing mitochondrial permeability to ions and small molecules (230–232), therefore inducing MMP failure, decrease of ATP production, release of mitochondrial content, and cell death (233–237). One of the components of mPTP is voltage-dependent anion channel (VDAC) protein (238). Remarkably, VDAC protein levels have been described to progressively increase in correlation with Braak stages in AD brains, where phosphorylated tau has been found to interact with VDAC (239). WT mice subjected to subcortical injection of tau oligomers exhibited decrease of NADH-ubiquinone oxidoreductase (complex I) protein expression and a considerable activation of caspase-9, when the hemispheres were compared with the ones injected with fibrillar or monomeric tau. Validating the tau propagation hypothesis, these changes have been found in the hippocampus, where IHC analysis revealed the co-localization between tau oligomers and mitochondria, specifically in CA1 cells (218). In another study, P301L mice exhibited increased amounts of hydrogen peroxide (H2O2) and 4-hydroxy-2-nonenol (HNE), indicator of lipid peroxidation, when compared to WT animals. Moreover, Tg animals showed markedly diminished levels of ATP and cytochrome C oxidase (complex IV) (198), further corroborating the role of tau toxicity in mitochondrial bioenergetics failure. Using the same murine model of tauopathy, it has been demonstrated that brains of 12-month-old Tg mice displayed significantly reduced activities of complex I and V. In these animals, tau-induced mitochondrial dysregulation worsened with aging, since 24-month-old mice showed increasingly reduced electron transport capacity and ATP levels, together with incremented ROS levels (H2O2 and superoxide anion radicals) (217). Of note, complex V level reduction has been also found in human familial FTD brains (217). In AD, several studies have shown significant decrease of mitochondrial proteins and activity (222–224). Additional studies have observed decreased ATP production and increased oxidative stress markers, such as free radicals, lipid peroxidation, and DNA and protein oxidation (225–228). Strikingly, mitochondrial structural abnormalities have been reported to occur also in the vascular wall of AD subjects, compared to age-matched controls (229). The literature summarized above points to a contribution of tau to mitochondrial dysfunction. Although more studies specifically targeted to understand the mitochondrial effects of tau in ECs or the NVU are still needed, the available data (197) suggest that mitochondrial dysfunction may be one of the mechanisms by which tau impairs cerebrovascular health.
TAU and Caspases
Caspases are cysteine-dependent proteases which cleave multiple intracellular substrate proteins after an aspartic acid residue, playing a primary role in apoptosis (240–244). Based on their structure and their hierarchical position in the apoptotic signaling cascade, caspases are categorized into upstream initiators (caspase-2,−8,−9, and−10) and downstream effectors (caspase-3,−6, and−7) (245–247). In brain cells, including neurons and glial cells, caspase activation does not always result in apoptosis (248, 249).
In neurodegenerative disorders, including AD, several stressors, such as deficits of oxygen and growth factors, excitotoxicity, inflammation, dysregulation of Ca2+ homeostasis and oxidative stress, may induce caspase activation, contributing to the etiopathogenesis of the disease (250–252). The associations between tau toxicity and caspase activation, and the relative references are listed in Table 5.

Table 5. Tau and caspases.
Multiple studies have provided evidence that human AD brains display significant caspase-3,−6,−7,−8, and−9 upregulation, compared to controls (69, 255–258). A large body of literature has focused on caspase-3, recognized as the main effector of the apoptotic process (262, 263). The presence of active caspase-3 within NFT is considered one of the earliest biomarkers of AD (259), with a high degree of colocalization in neurons, astrocytes and BVs, in subjects with overt pathology (256). This underpins a tight correlation between tau toxicity and neurovascular dysfunction, which is possibly caspase-mediated. To further corroborate this hypothesis, another study has demonstrated the activation of caspase-3 in CD68+ cells, identified by the authors as reactive microglia, in the frontal cortex of AD brains, relative to age- and gender-matched healthy controls (248). Moreover, neurons and a subset of degenerating astrocytes positive for active caspase-3 have been described in FTD cerebral samples (260). The activation of caspase-3 has been described to precede and to lead NFT formation in 7-month-old rTg4510 mice. Remarkably, when animals have been treated with doxycycline to suppress tau expression, the levels of cleaved caspase-3 in tangle-bearing neurons were diminished, while tangles remained, suggesting that soluble tau is the upstream initiator of caspase-3 activation, which in turn leads to the fibrillary progression (253).
Interestingly, among more than 400 proteins that can be cleaved by caspases (264–266), tau has been detected as substrate of caspase-2 (267),−3 (65), and−6 (69). Tau fragments derived from caspase-3 and−6 cleavage have been found in AD subjects (65, 69). Specifically, D421-cleaved caspase-3 truncated tau is accounted as an early feature of AD (268), and it has also been observed in other tauopathies, including PiD, PSP and CBD (269). Considering that caspase-3 cleaved tau shows an enhanced propensity to phosphorylation (65), and therefore to self-aggregation, besides the canonical role in apoptosis, caspase-3 activation may also function as a trigger for tau polymerization and NFT assembly (45, 47, 59, 66), ultimately causing dendritic spine loss and synaptic dysfunction (67, 68), as well as possible toxic effects for neurovascular cells. Accordingly, the injection of human tau-4R in WT mice led to the neuronal activation of caspase-3, tau truncation and aggregation. Moreover, when animals were injected with truncated tau, D421+ neurons showed colocalization of cleaved tau with the endogenous WT isoform, together with an accumulation of misfolded tau, further confirming that D421 tau fragment precedes and is sufficient to induce tau conformational and phosphorylation changes, and that the process is likely caspase-3-mediated (and initiated by soluble tau) (253). Active caspase-3 and D421-cleaved caspase-3 truncated tau have also been reported to occur within GFAP+, CD68+, and EBA+ (endothelial barrier antigen) cells in the white matter of corpus callosum, in a rat model of TBI, corroborating the involvement of caspase-3 activation in tau-mediated neurovascular damage (254). In addition, caspase-3 derived tau fragments have been found in NFT in the hippocampus, DG and frontal cortex, in human vascular dementia brains. Importantly, active caspase-3 has been detected in BVs and pre-tangle (but not NFT) neurons that colocalized with cleaved tau, supporting that caspase-3 activation is a required step for the cleavage of tau and the consequential NFT development at the NVU level (261).
Since Aβ oligomeric aggregates have also been shown to induce caspase-3 activation in neurovascular cells, including endothelial, glial, and neuronal cells (10, 13–15, 206–208), the presence of Aβ deposits surrounding cerebral vessels in AD and CAA, promoting caspase-3 activation, may also prompt an increase in the levels of caspase-3 truncated tau, which is more prone to phosphorylation and aggregation, and may therefore enhance tau toxicity at the NVU (270). In addition, AD subjects display a 2–3-fold increase of active caspase-6 in the temporal and frontal cortex, where caspase-6 cleaved tau is found into intracellular, extracellular, and even immature tangles (257).
Overall, this evidence supports the hypothesis that both tau and Aβ can initiate caspase activation, which may lead to apoptosis and, concomitantly, promote tau aggregation and NFT formation, further exacerbating tau-induced neurovascular pathology. In particular, the presence of active caspases in the brain vasculature (254, 256, 261), accompanied by the discussed evidence of tau transmission within neurovascular cells, supports the hypothesis that caspase activation may induce accumulation of toxic caspase-3 cleaved tau in cerebrovascular and glial cells, thus precipitating neurovascular pathology in AD, CAA, and tauopathies.
Discussion
The impact of tau on neurovascular pathology, although previously understudied, has recently become an active topic of research for the AD and dementia scientific community. Although more specific mechanistic studies in vessel wall cells are needed, here we postulate that tau may propagate from neurons to NVU cellular components such as ECs, astrocytes and microglia, and that inflammatory and mitochondrial alterations induced by tau in these cells may underlie its toxicity at the NVU (Figure 1). We further hypothesize that caspase activation, and in particular active caspase-3 tau cleavage, may also play a primary role in pathological tau-induced vascular dysfunction. Whether oligomeric Aβ aggregates, possibly through caspase-mediated mechanisms, contribute to perivascular tau oligomerization and NFT deposition is an intriguing hypothesis in need of additional exploration. Contextually, the lack of knowledge of the cell-specific detrimental molecular mechanisms initiated by tau arises new questions. Further studies will be necessary to elucidate tau-driven endothelial, pericyte, astrocytic, and microglial harmful cellular events. Amongst these, it will be critical to dissect the possible link between tau dysregulated clearance, altered inflammatory response and development of tau aggregates around cerebral BVs, in neurodegenerative diseases, including AD and other tauopathies, such as PiD, PSP, and CTE. Although tau toxicity seems to be responsible for neurovascular compromise, other studies suggest that early CVD can also cause tau pathological accumulation (136). Both these mechanisms may induce a vicious cycle between CVD and tau pathology, which still remains largely understudied. Establishing causality of these effects when studying a disease like AD, presenting with different pathologies, such as amyloidosis, tauopathy, cerebrovascular dysfunction, and neuroinflammation, often overlapping at the same time in the same brains, will require multiple and properly-designed studies.
Moreover, developing strategies to promote perivascular clearance, such as immunotherapy (271) and to target neurovascular cell-specific mechanisms, including mitochondrial dysfunction (207, 208, 272), to counteract toxicity of both amyloid and tau, will be essential research efforts to ameliorate CVD in AD and other tauopathies.
Author Contributions
EC and SF designed and conceptualized the review. EC wrote the review draft and did the literature search. SF critically revised and edited the manuscript, provided relevant insights, additional literature search, and acquired funding. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH R01NS104127 and R01AG062572 grants and the Edward N. and Della L. Thome Memorial Foundation, Awards Program in Alzheimer's Disease Drug Discovery Research, awarded to SF.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Nicole Lemon for editing the manuscript and Thaira Torielli for drafting the figure.
References
1. World Health Organization. Dementia Fact Sheet. (2019). Available online at: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed September 19, 2019).
3. Caselli RJ, Beach TG, Yaari R, Reiman EM. Alzheimer's disease a century later. J Clin Psychiatry. (2006) 67:1784–800. doi: 10.4088/JCP.v67n1118
4. Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. (2010) 362:329–44. doi: 10.1056/NEJMra0909142
5. Kalaria RN. The role of cerebral ischemia in Alzheimer's disease. Neurobiol Aging. (2000) 21:321–30. doi: 10.1016/S0197-4580(00)00125-1
6. Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol. (2001) 64:575–611. doi: 10.1016/S0301-0082(00)00068-X
7. de la Torre JC. Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. (2004) 3:184–90. doi: 10.1016/S1474-4422(04)00683-0
8. Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. (2005) 28:202–8. doi: 10.1016/j.tins.2005.02.001
9. Finder VH, Glockshuber R. Amyloid-beta aggregation. Neurodegener Dis. (2007) 4:13–27. doi: 10.1159/000100355
10. Fossati S, Cam J, Meyerson J, Mezhericher E, Romero IA, Couraud PO, et al. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-beta variants in cells composing the cerebral vessel walls. FASEB J. (2010) 24:229–41. doi: 10.1096/fj.09-139584
11. Muller-Hill B, Beyreuther K. Molecular biology of Alzheimer's disease. Annu Rev Biochem. (1989) 58:287–307. doi: 10.1146/annurev.bi.58.070189.001443
12. Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, part XV. Neurology. (1996) 46:1592–6. doi: 10.1212/WNL.46.6.1592
13. Fossati S, Ghiso J, Rostagno A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer's abeta. Cell Death Dis. (2012) 3:e321. doi: 10.1038/cddis.2012.55
14. Fossati S, Ghiso J, Rostagno A. Insights into caspase-mediated apoptotic pathways induced by amyloid-beta in cerebral microvascular endothelial cells. Neurodegener Dis. (2012) 10:324–8. doi: 10.1159/000332821
15. Ghiso J, Fossati S, Rostagno A. Amyloidosis associated with cerebral amyloid angiopathy: cell signaling pathways elicited in cerebral endothelial cells. J Alzheimers Dis. (2014) 42(Suppl. 3):S167–76. doi: 10.3233/JAD-140027
16. Fossati S, Todd K, Sotolongo K, Ghiso J, Rostagno A. Differential contribution of isoaspartate post-translational modifications to the fibrillization and toxic properties of amyloid beta and the Asn23 Iowa mutation. Biochem J. (2013) 456:347–60. doi: 10.1042/BJ20130652
17. Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol. (1986) 103(6 Pt 2):2739–46. doi: 10.1083/jcb.103.6.2739
18. Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. (1992) 3:1141–54. doi: 10.1091/mbc.3.10.1141
19. Khatoon S, Grundke-Iqbal I, Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem. (1992) 59:750–3. doi: 10.1111/j.1471-4159.1992.tb09432.x
20. Khatoon S, Grundke-Iqbal I, Iqbal K. Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett. (1994) 351:80–4. doi: 10.1016/0014-5793(94)00829-9
21. Vidal R, Calero M, Piccardo P, Farlow MR, Unverzagt FW, Mendez E, et al. Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE-epsilon4 allele. Acta Neuropathol. (2000) 100:1–12. doi: 10.1007/s004010051186
22. Kovac A, Zilkova M, Deli MA, Zilka N, Novak M. Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J Alzheimers Dis. (2009) 18:897–906. doi: 10.3233/JAD-2009-1197
23. Merlini M, Wanner D, Nitsch RM. Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer's disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol. (2016) 131:737–52. doi: 10.1007/s00401-016-1560-2
24. Blair LJ, Frauen HD, Zhang B, Nordhues BA, Bijan S, Lin YC, et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol Commun. (2015) 3:8. doi: 10.1186/s40478-015-0186-2
25. Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. (1963) 17:375–412. doi: 10.1083/jcb.17.2.375
26. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. (2010) 37:13–25. doi: 10.1016/j.nbd.2009.07.030
27. Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. (2005) 57:173–85. doi: 10.1124/pr.57.2.4
28. Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. (2004) 5:347–60. doi: 10.1038/nrn1387
29. Oksanen M, Lehtonen S, Jaronen M, Goldsteins G, Hamalainen RH, Koistinaho J. Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell Mol Life Sci. (2019) 76:2739–60. doi: 10.1007/s00018-019-03111-7
30. Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. (2007) 28:138–45. doi: 10.1016/j.it.2007.01.005
31. Streit WJ, Conde JR, Fendrick SE, Flanary BE, Mariani CL. Role of microglia in the central nervous system's immune response. Neurol Res. (2005) 27:685–91. doi: 10.1179/016164105X49463
32. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. (2007) 10:1538–43. doi: 10.1038/nn2014
33. Persidsky Y, Ghorpade A, Rasmussen J, Limoges J, Liu XJ, Stins M, et al. Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am J Pathol. (1999) 155:1599–611. doi: 10.1016/S0002-9440(10)65476-4
34. Hudson LC, Bragg DC, Tompkins MB, Meeker RB. Astrocytes and microglia differentially regulate trafficking of lymphocyte subsets across brain endothelial cells. Brain Res. (2005) 1058:148–60. doi: 10.1016/j.brainres.2005.07.071
35. Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging. (1988) 9:339–49. doi: 10.1016/S0197-4580(88)80079-4
36. McGeer PL, Akiyama H, Itagaki S, McGeer EG. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett. (1989) 107:341–6. doi: 10.1016/0304-3940(89)90843-4
37. Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, et al. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochem Int. (2001) 39:341–8. doi: 10.1016/S0197-0186(01)00041-9
38. Hayes A, Thaker U, Iwatsubo T, Pickering-Brown SM, Mann DM. Pathological relationships between microglial cell activity and tau and amyloid beta protein in patients with Alzheimer's disease. Neurosci Lett. (2002) 331:171–4. doi: 10.1016/S0304-3940(02)00888-1
39. Ostergaard L, Aamand R, Gutierrez-Jimenez E, Ho YC, Blicher JU, Madsen SM, et al. The capillary dysfunction hypothesis of Alzheimer's disease. Neurobiol Aging. (2013) 34:1018–31. doi: 10.1016/j.neurobiolaging.2012.09.011
40. Buee L, Hof PR, Delacourte A. Brain microvascular changes in Alzheimer's disease and other dementias. Ann N Y Acad Sci. (1997) 826:7–24. doi: 10.1111/j.1749-6632.1997.tb48457.x
41. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. (2011) 12:723–38. doi: 10.1038/nrn3114
42. Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. (2009) 118:103–13. doi: 10.1007/s00401-009-0522-3
43. Dehmelt L, Halpain S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. (2005) 6:204. doi: 10.1186/gb-2004-6-1-204
44. Halpain S, Dehmelt L. The MAP1 family of microtubule-associated proteins. Genome Biol. (2006) 7:224. doi: 10.1186/gb-2006-7-6-224
45. Ding H, Matthews TA, Johnson GV. Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. J Biol Chem. (2006) 281:19107–14. doi: 10.1074/jbc.M511697200
46. McMillan PJ, Kraemer BC, Robinson L, Leverenz JB, Raskind M, Schellenberg G. Truncation of tau at E391 promotes early pathologic changes in transgenic mice. J Neuropathol Exp Neurol. (2011) 70:1006–19. doi: 10.1097/NEN.0b013e31823557fb
47. Kolarova M, Garcia-Sierra F, Bartos A, Ricny J, Ripova D. Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis. (2012) 2012:731526. doi: 10.1155/2012/731526
48. Dolan PJ, Johnson GV. The role of tau kinases in Alzheimer's disease. Curr Opin Drug Discov Devel. (2010) 13:595–603.
49. Rodriguez-Martin T, Cuchillo-Ibanez I, Noble W, Nyenya F, Anderton BH, Hanger DP. Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging. (2013) 34:2146–57. doi: 10.1016/j.neurobiolaging.2013.03.015
50. Avila J, Jimenez JS, Sayas CL, Bolos M, Zabala JC, Rivas G, et al. Tau structures. Front Aging Neurosci. (2016) 8:262. doi: 10.3389/fnagi.2016.00262
51. Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. (1985) 101:1371–8. doi: 10.1083/jcb.101.4.1371
52. Arrasate M, Perez M, Avila J. Tau dephosphorylation at tau-1 site correlates with its association to cell membrane. Neurochem Res. (2000) 25:43–50. doi: 10.1023/A:1007583214722
53. Maas T, Eidenmuller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem. (2000) 275:15733–40. doi: 10.1074/jbc.M000389200
54. Maccioni RB, Otth C, Concha II, Munoz JP. The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. Eur J Biochem. (2001) 268:1518–27. doi: 10.1046/j.1432-1327.2001.02024.x
55. Hernandez F, Lucas JJ, Avila J. GSK3 and tau: two convergence points in Alzheimer's disease. J Alzheimers Dis. (2013) 33(Suppl. 1):S141–4. doi: 10.3233/JAD-2012-129025
56. Wolfe MS. The role of tau in neurodegenerative diseases and its potential as a therapeutic target. Scientifica. (2012) 2012:796024. doi: 10.6064/2012/796024
57. Liu SL, Wang C, Jiang T, Tan L, Xing A, Yu JT. The role of Cdk5 in Alzheimer's disease. Mol Neurobiol. (2016) 53:4328–42. doi: 10.1007/s12035-015-9369-x
58. Crespo-Biel N, Theunis C, Van Leuven F. Protein tau: prime cause of synaptic and neuronal degeneration in Alzheimer's disease. Int J Alzheimers Dis. (2012) 2012:251426. doi: 10.1155/2012/251426
59. de Calignon A, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to neurodegeneration in a murine model of Alzheimer's disease. Med Sci. (2010) 26:787–9. doi: 10.1051/medsci/20102610787
60. Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci USA. (2001) 98:6923–8. doi: 10.1073/pnas.121119298
61. Dorostkar MM, Zou C, Blazquez-Llorca L, Herms J. Analyzing dendritic spine pathology in Alzheimer's disease: problems and opportunities. Acta Neuropathol. (2015) 130:1–19. doi: 10.1007/s00401-015-1449-5
62. Liu F, Li B, Tung EJ, Grundke-Iqbal I, Iqbal K, Gong CX. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci. (2007) 26:3429–36. doi: 10.1111/j.1460-9568.2007.05955.x
63. Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener. (2008) 3:8. doi: 10.1186/1750-1326-3-8
64. Beharry C, Cohen LS, Di J, Ibrahim K, Briffa-Mirabella S, Alonso Adel C. Tau-induced neurodegeneration: mechanisms and targets. Neurosci Bull. (2014) 30:346–58. doi: 10.1007/s12264-013-1414-z
65. Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci. (2005) 25:5365–75. doi: 10.1523/JNEUROSCI.1125-05.2005
66. Hanger DP, Wray S. Tau cleavage and tau aggregation in neurodegenerative disease. Biochem Soc Trans. (2010) 38:1016–20. doi: 10.1042/BST0381016
67. D'Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci. (2011) 14:69–76. doi: 10.1038/nn.2709
68. Zhao Y, Tseng IC, Heyser CJ, Rockenstein E, Mante M, Adame A, et al. Appoptosin-mediated caspase cleavage of tau contributes to progressive supranuclear palsy pathogenesis. Neuron. (2015) 87:963–75. doi: 10.1016/j.neuron.2015.08.020
69. Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer's disease brain. Neurobiol Dis. (2002) 11:341–54. doi: 10.1006/nbdi.2002.0549
70. Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. (2010) 33:317–25. doi: 10.1016/j.tins.2010.04.003
71. Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. (2010) 11:301–7. doi: 10.1038/nrm2873
72. Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. (2013) 501:45–51. doi: 10.1038/nature12481
73. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/BF00308809
74. Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. (2011) 31:13110–7. doi: 10.1523/JNEUROSCI.2569-11.2011
75. Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D. The tau protein in human cerebrospinal fluid in Alzheimer's disease consists of proteolytically derived fragments. J Neurochem. (1997) 68:430–3. doi: 10.1046/j.1471-4159.1997.68010430.x
76. Dage JL, Wennberg AMV, Airey DC, Hagen CE, Knopman DS, Machulda MM, et al. Levels of tau protein in plasma are associated with neurodegeneration and cognitive function in a population-based elderly cohort. Alzheimers Dement. (2016) 12:1226–34. doi: 10.1016/j.jalz.2016.06.001
77. Deters KD, Risacher SL, Kim S, Nho K, West JD, Blennow K, et al. Plasma tau association with brain atrophy in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. (2017) 58:1245–54. doi: 10.3233/JAD-161114
78. Fossati S, Ramos Cejudo J, Debure L, Pirraglia E, Sone JY, Li Y, et al. Plasma tau complements CSF tau and P-tau in the diagnosis of Alzheimer's disease. Alzheimers Dement. (2019) 11:483–92. doi: 10.1016/j.dadm.2019.05.001
79. Zetterberg H, Wilson D, Andreasson U, Minthon L, Blennow K, Randall J, et al. Plasma tau levels in Alzheimer's disease. Alzheimers Res Ther. (2013) 5:9. doi: 10.1186/alzrt163
80. Blennow K, Zetterberg H. The past and the future of Alzheimer's disease fluid biomarkers. J Alzheimers Dis. (2018) 62:1125–40. doi: 10.3233/JAD-170773
81. Mielke MM, Hagen CE, Wennberg AMV, Airey DC, Savica R, Knopman DS, et al. Association of plasma total tau level with cognitive decline and risk of mild cognitive impairment or dementia in the mayo clinic study on aging. JAMA Neurol. (2017) 74:1073–80. doi: 10.1001/jamaneurol.2017.1359
82. Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. (2020) 19:422–33. doi: 10.1016/S1474-4422(20)30071-5
83. Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. (2020) 26:379–86. doi: 10.1038/s41591-020-0755-1
84. Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. (2020) 324:772–81. doi: 10.1001/jama.2020.12134
85. de Leon MJ, Li Y, Okamura N, Tsui WH, Saint-Louis LA, Glodzik L, et al. Cerebrospinal fluid clearance in Alzheimer disease measured with dynamic PET. J Nucl Med. (2017) 58:1471–6. doi: 10.2967/jnumed.116.187211
86. Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. (2015) 11:457–70. doi: 10.1038/nrneurol.2015.119
87. Carare RO, Aldea R, Agarwal N, Bacskai BJ, Bechman I, Boche D, et al. Clearance of interstitial fluid (ISF) and CSF (CLIC) group-part of vascular professional interest area (PIA): cerebrovascular disease and the failure of elimination of amyloid-beta from the brain and retina with age and Alzheimer's disease-opportunities for therapy. Alzheimers Dement. (2020) 12:e12053. doi: 10.1002/dad2.12053
88. Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. (2009) 284:12845–52. doi: 10.1074/jbc.M808759200
89. Guo JL, Lee VM. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. (2011) 286:15317–31. doi: 10.1074/jbc.M110.209296
90. Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of tau aggregation by fibrillar species. J Biol Chem. (2012) 287:19440–51. doi: 10.1074/jbc.M112.346072
91. DeVos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, et al. Synaptic tau seeding precedes tau pathology in human Alzheimer's disease brain. Front Neurosci. (2018) 12:267. doi: 10.3389/fnins.2018.00267
92. Furman JL, Vaquer-Alicea J, White CL 3rd, Cairns NJ, Nelson PT, Diamond MI. Widespread tau seeding activity at early braak stages. Acta Neuropathol. (2017) 133:91–100. doi: 10.1007/s00401-016-1644-z
93. Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer's disease and PART. Acta Neuropathol. (2018) 136:57–67. doi: 10.1007/s00401-018-1855-6
94. Kaufman SK, Thomas TL, Del Tredici K, Braak H, Diamond MI. Characterization of tau prion seeding activity and strains from formaldehyde-fixed tissue. Acta Neuropathol Commun. (2017) 5:41. doi: 10.1186/s40478-017-0442-8
95. Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. (2013) 14:389–94. doi: 10.1038/embor.2013.15
96. Katsinelos T, Zeitler M, Dimou E, Karakatsani A, Muller HM, Nachman E, et al. Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. (2018) 23:2039–55. doi: 10.1016/j.celrep.2018.04.056
97. Merezhko M, Brunello CA, Yan X, Vihinen H, Jokitalo E, Uronen RL, et al. Secretion of tau via an unconventional non-vesicular mechanism. Cell Rep. (2018) 25:2027–35.e4. doi: 10.1016/j.celrep.2018.10.078
98. Polanco JC, Scicluna BJ, Hill AF, Gotz J. Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J Biol Chem. (2016) 291:12445–66. doi: 10.1074/jbc.M115.709485
99. Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. (2012) 287:3842–9. doi: 10.1074/jbc.M111.277061
100. Wang Y, Balaji V, Kaniyappan S, Kruger L, Irsen S, Tepper K, et al. The release and trans-synaptic transmission of tau via exosomes. Mol Neurodegener. (2017) 12:5. doi: 10.1186/s13024-016-0143-y
101. Dujardin S, Begard S, Caillierez R, Lachaud C, Delattre L, Carrier S, et al. Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS ONE. (2014) 9:e100760. doi: 10.1371/journal.pone.0100760
102. Abounit S, Wu JW, Duff K, Victoria GS, Zurzolo C. Tunneling nanotubes: a possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion. (2016) 10:344–51. doi: 10.1080/19336896.2016.1223003
103. Tardivel M, Begard S, Bousset L, Dujardin S, Coens A, Melki R, et al. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological tau protein assemblies. Acta Neuropathol Commun. (2016) 4:117. doi: 10.1186/s40478-016-0386-4
104. Santa-Maria I, Varghese M, Ksiezak-Reding H, Dzhun A, Wang J, Pasinetti GM. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of tau protein in aggresomes. J Biol Chem. (2012) 287:20522–33. doi: 10.1074/jbc.M111.323279
105. Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem. (2013) 288:1856–70. doi: 10.1074/jbc.M112.394528
106. Calafate S, Flavin W, Verstreken P, Moechars D. Loss of bin1 promotes the propagation of tau pathology. Cell Rep. (2016) 17:931–40. doi: 10.1016/j.celrep.2016.09.063
107. Evans LD, Wassmer T, Fraser G, Smith J, Perkinton M, Billinton A, et al. Extracellular monomeric and aggregated tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep. (2018) 22:3612–24. doi: 10.1016/j.celrep.2018.03.021
108. Rauch JN, Luna G, Guzman E, Audouard M, Challis C, Sibih YE, et al. LRP1 is a master regulator of tau uptake and spread. Nature. (2020) 580:381–5. doi: 10.1038/s41586-020-2156-5
109. Falcon B, Cavallini A, Angers R, Glover S, Murray TK, Barnham L, et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J Biol Chem. (2015) 290:1049–65. doi: 10.1074/jbc.M114.589309
110. Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci USA. (2013) 110:E3138–47. doi: 10.1073/pnas.1301440110
111. Rauch JN, Chen JJ, Sorum AW, Miller GM, Sharf T, See SK, et al. Tau internalization is regulated by 6-O sulfation on heparan sulfate proteoglycans (HSPGs). Sci Rep. (2018) 8:6382. doi: 10.1038/s41598-018-24904-z
112. Falcon B, Noad J, McMahon H, Randow F, Goedert M. Galectin-8-mediated selective autophagy protects against seeded tau aggregation. J Biol Chem. (2018) 293:2438–51. doi: 10.1074/jbc.M117.809293
113. Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. (2016) 19:1085–92. doi: 10.1038/nn.4328
114. Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol. (1995) 52:81–8. doi: 10.1001/archneur.1995.00540250089017
115. Togo T, Dickson DW. Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol. (2002) 104:398–402. doi: 10.1007/s00401-002-0569-x
116. Buee L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and pick's disease. Brain Pathol. (1999) 9:681–93. doi: 10.1111/j.1750-3639.1999.tb00550.x
117. Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. (2016) 131:87–102. doi: 10.1007/s00401-015-1509-x
118. Ferrer I, Garcia MA, Gonzalez IL, Lucena DD, Villalonga AR, Tech MC, et al. Aging-related tau astrogliopathy (ARTAG): not only tau phosphorylation in astrocytes. Brain Pathol. (2018) 28:965–85. doi: 10.1111/bpa.12593
119. Odawara T, Iseki E, Kosaka K, Akiyama H, Ikeda K, Yamamoto T. Investigation of tau-2 positive microglia-like cells in the subcortical nuclei of human neurodegenerative disorders. Neurosci Lett. (1995) 192:145–8. doi: 10.1016/0304-3940(95)11595-N
120. Bolos M, Llorens-Martin M, Jurado-Arjona J, Hernandez F, Rabano A, Avila J. Direct evidence of internalization of tau by microglia in vitro and in vivo. J Alzheimers Dis. (2016) 50:77–87. doi: 10.3233/JAD-150704
121. Leyns CEG, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. (2017) 12:50. doi: 10.1186/s13024-017-0192-x
122. Hallmann AL, Arauzo-Bravo MJ, Mavrommatis L, Ehrlich M, Ropke A, Brockhaus J, et al. Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci Rep. (2017) 7:42991. doi: 10.1038/srep42991
123. Walker LC. Glial tauopathy: neurons optional? J Exp Med. (2020) 217:e20191915. doi: 10.1084/jem.20191915
124. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
125. Martini-Stoica H, Cole AL, Swartzlander DB, Chen F, Wan YW, Bajaj L, et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J Exp Med. (2018) 215:2355–77. doi: 10.1084/jem.20172158
126. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. (2015) 18:1584–93. doi: 10.1038/nn.4132
127. Brelstaff J, Tolkovsky AM, Ghetti B, Goedert M, Spillantini MG. Living neurons with tau filaments aberrantly expose phosphatidylserine and are phagocytosed by microglia. Cell Rep. (2018) 24:1939–48.e4. doi: 10.1016/j.celrep.2018.07.072
128. de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. (2012) 73:685–97. doi: 10.1016/j.neuron.2011.11.033
129. De Marco D, Taggenbrock R, Crespo R, Koudstaal W, Ramsburg E, Apetri A. Cell-based assay to study antibody-mediated tau clearance by microglia. J Vis Exp. 2018:58576. doi: 10.3791/58576
130. Funk KE, Mirbaha H, Jiang H, Holtzman DM, Diamond MI. Distinct therapeutic mechanisms of tau antibodies: promoting microglial clearance versus blocking neuronal uptake. J Biol Chem. (2015) 290:21652–62. doi: 10.1074/jbc.M115.657924
131. Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D, et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. J Neuroinflammation. (2018) 15:269. doi: 10.1186/s12974-018-1309-z
132. Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep. (2015) 5:11161. doi: 10.1038/srep11161
133. Bolos M, Llorens-Martin M, Perea JR, Jurado-Arjona J, Rabano A, Hernandez F, et al. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol Neurodegener. (2017) 12:59. doi: 10.1186/s13024-017-0200-1
134. Nilson AN, English KC, Gerson JE, Barton Whittle T, Nicolas Crain C, Xue J, et al. Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J Alzheimers Dis. (2017) 55:1083–99. doi: 10.3233/JAD-160912
135. Jaworski T, Lechat B, Demedts D, Gielis L, Devijver H, Borghgraef P, et al. Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am J Pathol. (2011) 179:2001–15. doi: 10.1016/j.ajpath.2011.06.025
136. Faraco G, Hochrainer K, Segarra SG, Schaeffer S, Santisteban MM, Menon A, et al. Dietary salt promotes cognitive impairment through tau phosphorylation. Nature. (2019) 574:686–90. doi: 10.1038/s41586-019-1688-z
137. Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, et al. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer's disease. Proc Natl Acad Sci USA. (2018) 115:E1289–E98. doi: 10.1073/pnas.1710329115
138. Kim HJ, Park S, Cho H, Jang YK, San Lee J, Jang H, et al. Assessment of extent and role of tau in subcortical vascular cognitive impairment using 18F-AV1451 positron emission tomography imaging. JAMA Neurol. (2018) 75:999–1007. doi: 10.1001/jamaneurol.2018.0975
139. Majerova P, Garruto RM, Kovac A. Cerebrovascular inflammation is associated with tau pathology in guam parkinsonism dementia. J Neural Transm. (2018) 125:1013–25. doi: 10.1007/s00702-018-1883-3
140. Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Hernandez C, Barrett AD, Dineley K, et al. Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer's disease mouse model. J Neurosci. (2015) 35:4857–68. doi: 10.1523/JNEUROSCI.4989-14.2015
141. Laurent C, Dorothee G, Hunot S, Martin E, Monnet Y, Duchamp M, et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. (2017) 140:184–200. doi: 10.1093/brain/aww270
142. Majerova P, Michalicova A, Cente M, Hanes J, Vegh J, Kittel A, et al. Trafficking of immune cells across the blood-brain barrier is modulated by neurofibrillary pathology in tauopathies. PLoS ONE. (2019) 14:e0217216. doi: 10.1371/journal.pone.0217216
143. Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. (2012) 3:1227. doi: 10.1038/ncomms2230
144. Lecrux C, Hamel E. The neurovascular unit in brain function and disease. Acta Physiol. (2011) 203:47–59. doi: 10.1111/j.1748-1716.2011.02256.x
145. Wang L, Jiang Q, Chu J, Lin L, Li XG, Chai GS, et al. Expression of Tau40 induces activation of cultured rat microglial cells. PLoS ONE. (2013) 8:e76057. doi: 10.1371/journal.pone.0076057
146. Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM, et al. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci. (2005) 25:3539–50. doi: 10.1523/JNEUROSCI.0081-05.2005
147. Castillo-Carranza DL, Nilson AN, Van Skike CE, Jahrling JB, Patel K, Garach P, et al. Cerebral microvascular accumulation of tau oligomers in Alzheimer's disease and related tauopathies. Aging Dis. (2017) 8:257–66. doi: 10.14336/AD.2017.0112
148. McKee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. (2015) 25:350–64. doi: 10.1111/bpa.12248
149. Rohn TT, Day RJ, Catlin LW, Brown RJ, Rajic AJ, Poon WW. Immunolocalization of an amino-terminal fragment of apolipoprotein E in the pick's disease brain. PLoS ONE. (2013) 8:e80180. doi: 10.1371/journal.pone.0080180
150. Buee L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, et al. Pathological alterations of the cerebral microvasculature in Alzheimer's disease and related dementing disorders. Acta Neuropathol. (1994) 87:469–80. doi: 10.1007/BF00294173
151. Arima K. Ultrastructural characteristics of tau filaments in tauopathies: immuno-electron microscopic demonstration of tau filaments in tauopathies. Neuropathology. (2006) 26:475–83. doi: 10.1111/j.1440-1789.2006.00669.x
152. Arima K, Nakamura M, Sunohara N, Ogawa M, Anno M, Izumiyama Y, et al. Ultrastructural characterization of the tau-immunoreactive tubules in the oligodendroglial perikarya and their inner loop processes in progressive supranuclear palsy. Acta Neuropathol. (1997) 93:558–66. doi: 10.1007/s004010050652
153. Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. (2010) 23:394–400. doi: 10.1097/WCO.0b013e32833be924
154. Kouri N, Whitwell JL, Josephs KA, Rademakers R, Dickson DW. Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol. (2011) 7:263–72. doi: 10.1038/nrneurol.2011.43
155. Takeda N, Kishimoto Y, Yokota O. Pick's disease. Adv Exp Med Biol. (2012) 724:300–16. doi: 10.1007/978-1-4614-0653-2_23
156. Trickler WJ, Mayhan WG, Miller DW. Brain microvessel endothelial cell responses to tumor necrosis factor-alpha involve a nuclear factor kappa B (NF-kappaB) signal transduction pathway. Brain Res. (2005) 1048:24–31. doi: 10.1016/j.brainres.2005.04.028
157. Wu B, Ma Q, Khatibi N, Chen W, Sozen T, Cheng O, et al. Ac-YVAD-CMK decreases blood-brain barrier degradation by inhibiting caspase-1 activation of interleukin-1beta in intracerebral hemorrhage mouse model. Transl Stroke Res. (2010) 1:57–64. doi: 10.1007/s12975-009-0002-z
158. Ohara Y, McCarron RM, Hoffman TT, Sugano H, Bembry J, Lenz FA, et al. Adrenergic mediation of TNF alpha-stimulated ICAM-1 expression on human brain microvascular endothelial cells. Acta Neurochir Suppl. (2000) 76:117–20. doi: 10.1007/978-3-7091-6346-7_24
159. Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. (2011) 585:3770–80. doi: 10.1016/j.febslet.2011.04.066
160. Ruprecht K, Kuhlmann T, Seif F, Hummel V, Kruse N, Bruck W, et al. Effects of oncostatin M on human cerebral endothelial cells and expression in inflammatory brain lesions. J Neuropathol Exp Neurol. (2001) 60:1087–98. doi: 10.1093/jnen/60.11.1087
161. Sonar SA, Lal G. Differentiation and transmigration of CD4 T cells in neuroinflammation and autoimmunity. Front Immunol. (2017) 8:1695. doi: 10.3389/fimmu.2017.01695
162. Koren J 3rd, Jinwal UK, Lee DC, Jones JR, Shults CL, Johnson AG, et al. Chaperone signalling complexes in Alzheimer's disease. J Cell Mol Med. (2009) 13:619–30. doi: 10.1111/j.1582-4934.2008.00557.x
163. Leszczynski D, Joenvaara S, Reivinen J, Kuokka R. Non-thermal activation of the hsp27/p38MAPK stress pathway by mobile phone radiation in human endothelial cells: molecular mechanism for cancer- and blood-brain barrier-related effects. Differentiation. (2002) 70:120–9. doi: 10.1046/j.1432-0436.2002.700207.x
164. Isogai C, Laug WE, Shimada H, Declerck PJ, Stins MF, Durden DL, et al. Plasminogen activator inhibitor-1 promotes angiogenesis by stimulating endothelial cell migration toward fibronectin. Cancer Res. (2001) 61:5587–94.
165. Czekay RP, Wilkins-Port CE, Higgins SP, Freytag J, Overstreet JM, Klein RM, et al. PAI-1: an integrator of cell signaling and migration. Int J Cell Biol. (2011) 2011:562481. doi: 10.1155/2011/562481
166. Jeon H, Kim JH, Kim JH, Lee WH, Lee MS, Suk K. Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J Neuroinflammation. (2012) 9:149. doi: 10.1186/1742-2094-9-149
167. Stewart PA, Hayakawa K, Akers MA, Vinters HV. A morphometric study of the blood-brain barrier in Alzheimer's disease. Lab Invest. (1992) 67:734–42.
168. Claudio L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer's disease patients. Acta Neuropathol. (1996) 91:6–14. doi: 10.1007/s004010050386
169. Miyakawa T, Uehara Y, Desaki J, Kimura T, Kuramoto R. Morphological changes of microvessels in the brain with Alzheimer's disease. Jpn J Psychiatry Neurol. (1988) 42:819–24. doi: 10.1111/j.1440-1819.1988.tb01171.x
170. Hashimura T, Kimura T, Miyakawa T. Morphological changes of blood vessels in the brain with Alzheimer's disease. Jpn J Psychiatry Neurol. (1991) 45:661–5. doi: 10.1111/j.1440-1819.1991.tb01187.x
171. Kimura T, Hashimura T, Miyakawa T. Observations of microvessels in the brain with Alzheimer's disease by the scanning electron microscopy. Jpn J Psychiatry Neurol. (1991) 45:671–6. doi: 10.1111/j.1440-1819.1991.tb01189.x
172. Higuchi Y, Miyakawa T, Shimoji A, Katsuragi S. Ultrastructural changes of blood vessels in the cerebral cortex in Alzheimer's disease. Jpn J Psychiatry Neurol. (1987) 41:283–90. doi: 10.1111/j.1440-1819.1987.tb00414.x
173. Yamashita K, Miyakawa T, Katsuragi S. Vascular changes in the brains with Alzheimer's disease. Jpn J Psychiatry Neurol. (1991) 45:79–84. doi: 10.1111/j.1440-1819.1991.tb00509.x
174. Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, et al. Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med. (2000) 343:450–6. doi: 10.1056/NEJM200008173430701
175. Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. (2004) 35(11 Suppl. 1):2616–9. doi: 10.1161/01.STR.0000143224.36527.44
177. Fann JR, Ribe AR, Pedersen HS, Fenger-Gron M, Christensen J, Benros ME, et al. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry. (2018) 5:424–31. doi: 10.1016/S2215-0366(18)30065-8
178. Katsumoto A, Takeuchi H, Tanaka F. Tau pathology in chronic traumatic encephalopathy and Alzheimer's disease: similarities and differences. Front Neurol. (2019) 10:980. doi: 10.3389/fneur.2019.00980
179. McKee AC. The neuropathology of chronic traumatic encephalopathy: the status of the literature. Semin Neurol. (2020) 40:359–69. doi: 10.1055/s-0040-1713632
180. Stein TD, Alvarez VE, McKee AC. Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther. (2014) 6:4. doi: 10.1186/alzrt234
181. Ikeda K, Akiyama H, Kondo H, Haga C, Tanno E, Tokuda T, et al. Thorn-shaped astrocytes: possibly secondarily induced tau-positive glial fibrillary tangles. Acta Neuropathol. (1995) 90:620–5. doi: 10.1007/BF00318575
182. Schultz C, Ghebremedhin E, Del Tredici K, Rub U, Braak H. High prevalence of thorn-shaped astrocytes in the aged human medial temporal lobe. Neurobiol Aging. (2004) 25:397–405. doi: 10.1016/S0197-4580(03)00113-1
183. McAleese KE, Firbank M, Dey M, Colloby SJ, Walker L, Johnson M, et al. Cortical tau load is associated with white matter hyperintensities. Acta Neuropathol Commun. (2015) 3:60. doi: 10.1186/s40478-015-0240-0
184. Strain JF, Smith RX, Beaumont H, Roe CM, Gordon BA, Mishra S, et al. Loss of white matter integrity reflects tau accumulation in Alzheimer disease defined regions. Neurology. (2018) 91:e313–e8. doi: 10.1212/WNL.0000000000005864
185. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. (2012) 148:1145–59. doi: 10.1016/j.cell.2012.02.035
186. Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet. (2009) 18:R169–76. doi: 10.1093/hmg/ddp326
187. Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, et al. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer's disease. J Alzheimers Dis. (2010) 20(Suppl. 2):S401–12. doi: 10.3233/JAD-2010-100666
188. Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. (2013) 23:64–71. doi: 10.1016/j.tcb.2012.10.006
189. Kageyama Y, Zhang Z, Sesaki H. Mitochondrial division: molecular machinery and physiological functions. Curr Opin Cell Biol. (2011) 23:427–34. doi: 10.1016/j.ceb.2011.04.009
190. Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. (2009) 20:3525–32. doi: 10.1091/mbc.e09-03-0252
191. Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol. (2013) 304:R393–406. doi: 10.1152/ajpregu.00584.2012
192. Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta. (2014) 1842:1240–7. doi: 10.1016/j.bbadis.2013.10.015
193. Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants. (2017) 6:25. doi: 10.3390/antiox6020025
194. Guo L, Tian J, Du H. Mitochondrial dysfunction and synaptic transmission failure in Alzheimer's disease. J Alzheimers Dis. (2017) 57:1071–86. doi: 10.3233/JAD-160702
195. Li XC, Hu Y, Wang ZH, Luo Y, Zhang Y, Liu XP, et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep. (2016) 6:24756. doi: 10.1038/srep24756
196. Perez MJ, Vergara-Pulgar K, Jara C, Cabezas-Opazo F, Quintanilla RA. Caspase-cleaved tau impairs mitochondrial dynamics in Alzheimer's disease. Mol Neurobiol. (2018) 55:1004–18. doi: 10.1007/s12035-017-0385-x
197. Placido AI, Pereira CMF, Correira SC, Carvalho C, Oliveira CR, Moreira PI. Phosphatase 2A inhibition affects endoplasmic reticulum and mitochondria homeostasis via cytoskeletal alterations in brain endothelial cells. Mol Neurobiol. (2017) 54:154–68. doi: 10.1007/s12035-015-9640-1
198. Kandimalla R, Manczak M, Fry D, Suneetha Y, Sesaki H, Reddy PH. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer's disease. Hum Mol Genet. (2016) 25:4881–97. doi: 10.1093/hmg/ddw312
199. Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet. (2012) 21:2538–47. doi: 10.1093/hmg/dds072
200. Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. (2009) 29:9090–103. doi: 10.1523/JNEUROSCI.1357-09.2009
201. Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. (2011) 20:2495–509. doi: 10.1093/hmg/ddr139
202. Bonda DJ, Wang X, Perry G, Smith MA, Zhu X. Mitochondrial dynamics in Alzheimer's disease: opportunities for future treatment strategies. Drugs Aging. (2010) 27:181–92. doi: 10.2165/11532140-000000000-00000
203. Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, et al. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. Am J Pathol. (2011) 179:2071–82. doi: 10.1016/j.ajpath.2011.07.004
204. Jacobson J, Duchen MR. Interplay between mitochondria and cellular calcium signalling. Mol Cell Biochem. (2004) 256–7:209–18. doi: 10.1023/B:MCBI.0000009869.29827.df
205. Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. (2008) 60:748–66. doi: 10.1016/j.neuron.2008.10.010
206. Parodi-Rullan R, Sone JY, Fossati S. Endothelial mitochondrial dysfunction in cerebral amyloid angiopathy and Alzheimer's disease. J Alzheimers Dis. (2019) 72:1019–39. doi: 10.3233/JAD-190357
207. Fossati S, Giannoni P, Solesio ME, Cocklin SL, Cabrera E, Ghiso J, et al. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol Dis. (2016) 86:29–40. doi: 10.1016/j.nbd.2015.11.006
208. Solesio ME, Peixoto PM, Debure L, Madamba SM, de Leon MJ, Wisniewski T, et al. Carbonic anhydrase inhibition selectively prevents amyloid beta neurovascular mitochondrial toxicity. Aging Cell. (2018) 17:e12787. doi: 10.1111/acel.12787
209. Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis. (2010) 20(Suppl. 2):S265–79. doi: 10.3233/JAD-2010-100339
210. Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. (2011) 20:4515–29. doi: 10.1093/hmg/ddr381
211. Balietti M, Giorgetti B, Casoli T, Solazzi M, Tamagnini F, Burattini C, et al. Early selective vulnerability of synapses and synaptic mitochondria in the hippocampal CA1 region of the Tg2576 mouse model of Alzheimer's disease. J Alzheimers Dis. (2013) 34:887–96. doi: 10.3233/JAD-121711
212. Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA. (2010) 107:18670–5. doi: 10.1073/pnas.1006586107
213. Gibson GE, Shi Q. A mitocentric view of Alzheimer's disease suggests multi-faceted treatments. J Alzheimers Dis. (2010) 20(Suppl. 2):S591–607. doi: 10.3233/JAD-2010-100336
214. Xie H, Guan J, Borrelli LA, Xu J, Serrano-Pozo A, Bacskai BJ. Mitochondrial alterations near amyloid plaques in an Alzheimer's disease mouse model. J Neurosci. (2013) 33:17042–51. doi: 10.1523/JNEUROSCI.1836-13.2013
215. Varghese M, Zhao W, Wang J, Cheng A, Qian X, Chaudhry A, et al. Mitochondrial bioenergetics is defective in presymptomatic Tg2576 AD mice. Transl Neurosci. (2011) 2:1–5. doi: 10.2478/s13380-011-0011-8
216. Amadoro G, Corsetti V, Stringaro A, Colone M, D'Aguanno S, Meli G, et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J Alzheimers Dis. (2010) 21:445–70. doi: 10.3233/JAD-2010-100120
217. David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem. (2005) 280:23802–14. doi: 10.1074/jbc.M500356200
218. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. (2011) 6:39. doi: 10.1186/1750-1326-6-39
219. Schulz KL, Eckert A, Rhein V, Mai S, Haase W, Reichert AS, et al. A new link to mitochondrial impairment in tauopathies. Mol Neurobiol. (2012) 46:205–16. doi: 10.1007/s12035-012-8308-3
220. Britti E, Ros J, Esteras N, Abramov AY. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium. (2020) 86:102150. doi: 10.1016/j.ceca.2019.102150
221. Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem. (2009) 284:18754–66. doi: 10.1074/jbc.M808908200
222. Mutisya EM, Bowling AC, Beal MF. Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J Neurochem. (1994) 63:2179–84. doi: 10.1046/j.1471-4159.1994.63062179.x
223. Schagger H, Ohm TG. Human diseases with defects in oxidative phosphorylation. 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur J Biochem. (1995) 227:916–21. doi: 10.1111/j.1432-1033.1995.tb20219.x
224. Kim SH, Vlkolinsky R, Cairns N, Fountoulakis M, Lubec G. The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer's disease. Life Sci. (2001) 68:2741–50. doi: 10.1016/S0024-3205(01)01074-8
225. Gibson GE, Sheu KF, Blass JP. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm. (1998) 105:855–70. doi: 10.1007/s007020050099
226. Parker WD Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology. (1990) 40:1302–3. doi: 10.1212/WNL.40.8.1302
227. Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. (2000) 21:455–62. doi: 10.1016/S0197-4580(00)00112-3
228. Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. (2005) 93:953–62. doi: 10.1111/j.1471-4159.2005.03053.x
229. Aliyev A, Chen SG, Seyidova D, Smith MA, Perry G, de la Torre J, et al. Mitochondria DNA deletions in atherosclerotic hypoperfused brain microvessels as a primary target for the development of Alzheimer's disease. J Neurol Sci. (2005) 229–30:285–92. doi: 10.1016/j.jns.2004.11.040
230. Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. (1979) 195:460–7. doi: 10.1016/0003-9861(79)90372-2
231. Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. (1979) 195:453–9. doi: 10.1016/0003-9861(79)90371-0
232. Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys. (1979) 195:468–77. doi: 10.1016/0003-9861(79)90373-4
233. Kroemer G, Blomgren K. Mitochondrial cell death control in familial parkinson disease. PLoS Biol. (2007) 5:e206. doi: 10.1371/journal.pbio.0050206
234. Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. (2013) 12:674–83. doi: 10.4161/cc.23599
235. Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol. (2015) 78:100–6. doi: 10.1016/j.yjmcc.2014.09.023
236. Bernardi P, Di Lisa F, Fogolari F, Lippe G. From ATP to PTP and back: a dual function for the mitochondrial ATP synthase. Circ Res. (2015) 116:1850–62. doi: 10.1161/CIRCRESAHA.115.306557
237. Jonas EA, Porter GA Jr, Beutner G, Mnatsakanyan N, Alavian KN. Cell death disguised: the mitochondrial permeability transition pore as the c-subunit of the F(1)F(O) ATP synthase. Pharmacol Res. (2015) 99:382–92. doi: 10.1016/j.phrs.2015.04.013
238. Rao VK, Carlson EA, Yan SS. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim Biophys Acta. (2014) 1842:1267–72. doi: 10.1016/j.bbadis.2013.09.003
239. Manczak M, Reddy PH. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer's disease. Hum Mol Genet. (2012) 21:5131–46. doi: 10.1093/hmg/dds360
240. Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science. (1992) 256:97–100. doi: 10.1126/science.1373520
241. Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. (1995) 376:37–43. doi: 10.1038/376037a0
242. Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, et al. Human ICE/CED-3 protease nomenclature. Cell. (1996) 87:171. doi: 10.1016/S0092-8674(00)81334-3
243. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. (1996) 86:147–57. doi: 10.1016/S0092-8674(00)80085-9
244. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. (1998) 281:1312–6. doi: 10.1126/science.281.5381.1312
246. Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell. (2002) 9:459–70. doi: 10.1016/S1097-2765(02)00482-3
247. Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. (2004) 5:897–907. doi: 10.1038/nrm1496
248. Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. (2011) 472:319–24. doi: 10.1038/nature09788
249. Hyman BT, Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci. (2012) 13:395–406. doi: 10.1038/nrn3228
250. Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer's disease. Neuroscientist. (2002) 8:276–83. doi: 10.1177/1073858402008003013
251. Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium. (2010) 47:183–9. doi: 10.1016/j.ceca.2009.12.014
252. Chen X, Guo C, Kong J. Oxidative stress in neurodegenerative diseases. Neural Regen Res. (2012) 7:376–85. doi: 10.3969/j.issn.1673-5374.2012.05.009
253. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, et al. Caspase activation precedes and leads to tangles. Nature. (2010) 464:1201–4. doi: 10.1038/nature08890
254. Glushakova OY, Glushakov AO, Borlongan CV, Valadka AB, Hayes RL, Glushakov AV. Role of caspase-3-mediated apoptosis in chronic caspase-3-cleaved tau accumulation and blood-brain barrier damage in the corpus callosum after traumatic brain injury in rats. J Neurotrauma. (2018) 35:157–73. doi: 10.1089/neu.2017.4999
255. Selznick LA, Holtzman DM, Han BH, Gokden M, Srinivasan AN, Johnson EM, et al. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J Neuropathol Exp Neurol. (1999) 58:1020–6. doi: 10.1097/00005072-199909000-00012
256. Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW. Activated caspase-3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res. (2001) 898:350–7. doi: 10.1016/S0006-8993(01)02018-2
257. Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer's disease. Am J Pathol. (2004) 165:523–31. doi: 10.1016/S0002-9440(10)63317-2
258. Matsui T, Ramasamy K, Ingelsson M, Fukumoto H, Conrad C, Frosch MP, et al. Coordinated expression of caspase 8, 3 and 7 mRNA in temporal cortex of Alzheimer disease: relationship to formic acid extractable abeta42 levels. J Neuropathol Exp Neurol. (2006) 65:508–15. doi: 10.1097/01.jnen.0000229238.05748.12
259. Gastard MC, Troncoso JC, Koliatsos VE. Caspase activation in the limbic cortex of subjects with early Alzheimer's disease. Ann Neurol. (2003) 54:393–8. doi: 10.1002/ana.10680
260. Su JH, Nichol KE, Sitch T, Sheu P, Chubb C, Miller BL, et al. DNA damage and activated caspase-3 expression in neurons and astrocytes: evidence for apoptosis in frontotemporal dementia. Exp Neurol. (2000) 163:9–19. doi: 10.1006/exnr.2000.7340
261. Day RJ, Mason MJ, Thomas C, Poon WW, Rohn TT. Caspase-cleaved tau co-localizes with early tangle markers in the human vascular dementia brain. PLoS ONE. (2015) 10:e0132637. doi: 10.1371/journal.pone.0132637
262. Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. (2000) 407:802–9. doi: 10.1038/35037739
263. Shimohama S. Apoptosis in Alzheimer's disease–an update. Apoptosis. (2000) 5:9–16. doi: 10.1023/A:1009625323388
264. Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. (1999) 68:383–424. doi: 10.1146/annurev.biochem.68.1.383
265. Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. (2003) 10:76–100. doi: 10.1038/sj.cdd.4401160
266. Luthi AU, Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ. (2007) 14:641–50. doi: 10.1038/sj.cdd.4402103
267. Zhao X, Kotilinek LA, Smith B, Hlynialuk C, Zahs K, Ramsden M, et al. Caspase-2 cleavage of tau reversibly impairs memory. Nat Med. (2016) 22:1268–76. doi: 10.1038/nm.4199
268. Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. (2004) 114:121–30. doi: 10.1172/JCI200420640
269. Newman J, Rissman RA, Sarsoza F, Kim RC, Dick M, Bennett DA, et al. Caspase-cleaved tau accumulation in neurodegenerative diseases associated with tau and alpha-synuclein pathology. Acta Neuropathol. (2005) 110:135–44. doi: 10.1007/s00401-005-1027-3
270. You Y, Perkins A, Cisternas P, Munoz B, Taylor X, You Y, et al. Tau as a mediator of neurotoxicity associated to cerebral amyloid angiopathy. Acta Neuropathol Commun. (2019) 7:26. doi: 10.1186/s40478-019-0680-z
271. Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer's disease. Trends Mol Med. (2015) 21:394–402. doi: 10.1016/j.molmed.2015.03.003
Keywords: tau, neurovascular unit, mitochondria, caspases, Alzheimer's disease, tauopathies, vascular dysfunction
Citation: Canepa E and Fossati S (2021) Impact of Tau on Neurovascular Pathology in Alzheimer's Disease. Front. Neurol. 11:573324. doi: 10.3389/fneur.2020.573324
Received: 16 June 2020; Accepted: 24 November 2020;
Published: 07 January 2021.
Edited by:
Sonia Do Carmo, McGill University, CanadaReviewed by:
Boon Lead Tee, Trinity College Dublin, IrelandSudeshna Das, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2021 Canepa and Fossati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Fossati, c2lsdmlhLmZvc3NhdGlAdGVtcGxlLmVkdQ==