
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 12 January 2021
Sec. Neuroepidemiology
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.525933
This article is part of the Research TopicEpidemiology of Atypical Demyelinating DiseasesView all 15 articles
 Helen Cross1
Helen Cross1 Farahna Sabiq1
Farahna Sabiq1 Nathalie Ackermans2
Nathalie Ackermans2 Andrew Mattar1Shelly Au1Mark Woodhall3
Andrew Mattar1Shelly Au1Mark Woodhall3 Bo Sun3Virginia Devonshire1
Bo Sun3Virginia Devonshire1 Robert Carruthers1Ana Luiza Sayao1
Robert Carruthers1Ana Luiza Sayao1 Virender Bhan1Alice Schabas1Jillian Chan1
Virender Bhan1Alice Schabas1Jillian Chan1 Marvin Fritzler4
Marvin Fritzler4 Patrick Waters3
Patrick Waters3 Anthony Traboulsee1*
Anthony Traboulsee1*Introduction: Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease is a recently described central nervous system (CNS) inflammatory disorder with phenotypic overlap with Neuromyelitis Optica Spectrum Disorder (NMOSD). NMOSD seronegative patients, and those with limited forms of the disorder, become suspects for MOG antibody-associated disease. We describe a multi-ethnic population with MOG antibody seropositivity from the University of British Columbia MS/NMO clinic.
Methods: AQP4-antibody seronegative patients presenting 2005–2016 with CNS inflammatory disease suspicious for NMOSD, as well as 20 MS controls, were retrospectively tested for MOG-IgG1 antibodies by live cell-based assay at Oxford Autoimmune Neurology Diagnostic Laboratory (UK) and by a commercial fixed cell-based assay at MitogenDx (Calgary, Canada). Additional MOG seropositive cases were identified through routine clinical interaction (2016–2018) using one of these laboratories. Clinical data was reviewed retrospectively.
Results: Retrospective testing identified 21 MOG seropositives (14 by live assay only, 3 by fixed assay only and 4 by both) representing 14% of the “NMOSD suspects” cohort. One multiple sclerosis (MS) control serum was MOG seropositive. Twenty additional MOG positive cases were identified prospectively. Of 42 patients (27 female), median disease onset age was 29 years (range 3–62; 9 pediatric cases), 20 (47%) were non-Caucasian, and 3 (7%) had comorbid autoimmune disease. Most common onset phenotypes were optic neuritis (23, 55%; 8 bilateral) and myelitis (9, 21%; 6 longitudinally extensive) Three of the patients in our cohort experienced cortical encephalitis; two presented with seizures. Onset was moderate-severe in 64%, but 74% had good response to initial steroid therapy. Cumulative relapse probability for the MOG positive group at 1 year was 0.428 and at 4 years was 0.628. Most had abnormal brain imaging, including cortical encephalitis and poorly demarcated subcortical and infratentorial lesions. Few “classic MS” lesions were seen. Optic nerve lesions (frequently bilateral) were long and predominantly anterior, but 5 extended to the chiasm. Spinal cord lesions were long and short, with involvement of multiple spinal regions simultaneously, including the conus medullaris.
Conclusions: Our MOG seropositive patients display phenotypes similar to previous descriptions, including cortical lesions with seizures and conus medullaris involvement. Many patients relapsed, predominantly in a different CNS location from onset. Serologic data from two different cell-based antibody assays highlight the discrepancies between live and fixed testing for MOG antibodies.
Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease is a recently described central nervous system (CNS) inflammatory disorder. There are a few large published adult case series (1–5), however the full clinical and radiological spectrum, and optimal management are not yet clear. The majority of published studies are based on Caucasian populations.
MOG antibody-associated disease has similarity to Neuromyelitis Optica Spectrum Disorders (NMOSD) in terms of clinical and imaging phenotypes (6), suggesting that patients within the aquaporin 4 (AQP4) antibody seronegative cohort become suspects for MOG antibody-associated disease. Previously published literature suggests approximately 40% of NMOSD AQP4-negative cohorts are MOG antibody positive (7). Additionally, MOG antibodies can be present in 10–20% of idiopathic atypical demyelinating diseases not meeting full NMOSD criteria (7, 8). MOG antibody-associated disease is however a distinctly different disorder from NMOSD, both immunologically and pathologically (9, 10). This distinction means recognition of these patients is important.
The University of British Columbia (UBC) MS/NMO referral clinic is the largest in British Columbia for CNS inflammatory disorders. NMOSD are known to be more prevalent in non-Caucasian populations. Given the multi-ethnicity of the British Columbian population, this clinic serves a NMOSD cohort of over 200 patients. Whilst primarily an adult clinic, some pediatric cases are also referred.
The primary aim of the study was to describe the population of MOG antibody seropositive patients at the UBC MS/NMO referral clinic, both clinically and radiologically, with comparison to other published MOG antibody-associated disease cohorts, as well as to the rest of our “NMO-suspects” AQP4 negative cohort.
A secondary aim was to systematically examine for autoantibody comorbidity [MOG, AQP4, and N-methyl-D-aspartate receptor subunit 1 (NMDAR) antibodies] within patients with CNS inflammatory disorders.
We identified a cohort of MOG antibody patients within our clinic from two sources: retrospectively via batch testing of stored serum samples and prospectively via routine clinical testing. Two different laboratories were utilized for the testing.
We searched our database for AQP4 antibody seronegative patients who were seen at the UBC MS/NMO clinic between 2005 and 2016. We included those who were NMOSD criteria positive (11) or had acute disseminated encephalomyelitis (ADEM) (12), longitudinally extensive or severe transverse myelitis (LETM), severe or recurrent optic neuritis (ON), tumefactive brain lesions, and patients with encephalopathy with white matter lesions and/or cortical lesions, with no clear diagnosis. Patients with neurosarcoidosis, lymphoma, stroke, or vasculitis were excluded. Additionally, we included 20 randomly selected patients with clinically definite multiple sclerosis (CDMS) (13) as controls.
Stored serum samples were tested by live cell-based assay at the Oxford Autoimmune Neurology Diagnostic Laboratory, UK, and on a fixed commercial cell-based assay (Euroimmun AG, Lübeck, Germany) by MitogenDx in Calgary, Canada.
Additional MOG antibody positive cases, in most cases tested only at a single center, were identified (2016–2019) through routine clinical testing at MitogenDx or Oxford Autoimmune Neurology Diagnostic Laboratory. Testing for MOG antibodies was at the discretion of the attending clinician, in most cases being sent due to demyelinating presentations atypical for MS or suggestive of NMOSD.
The systematic testing for AQP4-antibodies and NMDAR antibodies was performed for the retrospective cohort at Oxford Autoimmune Neurology Diagnostic Laboratory via cell-based assay. All prospective cases had AQP4-antibodies tested at MitogenDx via cell-based assay.
Prospective testing of NMDAR antibodies (MitogenDx, via cell-based assay) was not uniformly performed for prospective cases. The timing of serum sampling for MOG antibody testing in relation to clinical disease activity was not standardized. The majority of samples were taken at routine clinic visits, which may or may not have been at the time of a relapse. MOG titers were unfortunately not available, nor were serial test results.
We compared the phenotypic features of our MOG antibody positive patients with the published literature, as well as with the AQP4-antibody and MOG-antibody seronegative patients (from our retrospective cohort) who remained in the idiopathic CNS inflammatory disorders category.
Clinical data pertaining to demographics, disease onset and course, clinical syndromes and response to treatment, was collected by 3 clinicians (HC, NA, and AM) via retrospective chart review of the clinic electronic medical records system (established 2015), as well as provincial health databases for earlier clinical interaction records. Detailed ophthalmic examinations were mostly not documented. Cerebrospinal fluid studies were not performed for many patients and this data was therefore not captured.
Radiological review for the MOG positive patients only was performed by a neuroradiologist (FS), by reviewing the magnetic resonance (MR) imaging available for these patients from the time of first disease presentation, or the first MRI within 5 years of this time. T2 lesions were counted and lesion locations and characteristics were noted. Where possible brain, spinal cord, and orbital imaging was reviewed. Where serial imaging was available, only the first scan was analyzed to maintain consistency.
Ethnicity was captured as Caucasian (European ancestry), Asian (Chinese, Japanese, Korean, Vietnamese or Filipino ancestry), South Asian (Indian Subcontinent), or Other (First Nations or other non-Caucasian ancestry). Clinical severity of disease at onset was classified as mild [VA < 20/100, sensory only (excluding marked neuropathic pain), non-disabling motor], moderate (VA ≥ 20/100, ≤ 20/800, marked neuropathic pain, disabling motor, bladder and bowel involvement) or severe (VA > 200/800, inability to walk, incontinence). Where unable to find specific information, we were guided by clinical impression of severity. Response to initial steroid therapy was graded as good (full recovery or minor residual disability), or poor (minimal change in clinical picture or significant ongoing disability) based on the assessment at the subsequent clinic visit (≥1 month later, but the timing of this was not standardized). Recovery on follow-up was assessed at the patient's last recorded clinic visit, and graded as full/very good (no or minimal disability on clinical impression at last follow-up or EDSS ≤ 2.0), predominantly moderate-severe residual visual disability (VA > 20/100 in worse affected eye), predominantly moderate-severe residual motor disability (motor disability affecting function), predominantly moderate-severe residual bladder, bowel or sexual disability (disability impacting lifestyle), or combination disability when more than one moderate-severe category was present.
No new clinical or radiological data, or blood samples were collected specifically for the study.
Statistical procedures were performed using IBM SPSS Statistics, Version 25.0 (IBM Corp., Armonk, NY). An independent samples t-test was used to determine if there were significant differences in numerical variables such as age at onset and duration of follow-up.Chi-square tests and Fisher's exact tests were used to compare the clinical data between the MOG seropositive and the double seronegative groups of patients, with p < 0.05 considered to be significant. A Kaplan-Meier analysis was used to determine the cumulative relapse curves and cumulative relapse probabilities. Four patients had to be excluded for this analysis due to lack of specific time data for first relapse. A formal comparison of relapse probability at 1 year between the MOG positive and seronegative group was performed with a z-test.
All our clinic patients are offered the option of voluntary participation in research conducted at our center. If they provide written consent, their clinical and imaging information is included in our research database. Additional specific ethics approval for this sub-study was obtained (H19-01146) and it was carried out in compliance with the Helsinki Declaration of 1975 for human studies as revised in 2013.
Retrospective Cohort
We identified a total of 146 patients (4% pediatric, 70% female) who fulfilled our search criteria. Twenty-one of these patients (14% pediatric, 52% female) were found to have MOG antibody positivity on testing: 14 by live assay only, 3 by fixed assay only and 4 by both, representing 14% of patients who were clinically suspicious for NMO and seronegative for AQP4 antibodies. One of the CDMS patients was MOG antibody positive by the live assay test. AQP4 antibodies were detected by live cell-based assay in eight patients previously considered seronegative by a fixed test. Two otherwise seronegative patients and one CDMS patient were found to have NMDAR antibodies by the live test (see Table 1). No patient was found to have dual antibody seropositivity.
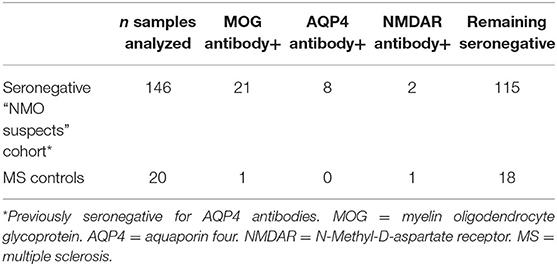
Table 1. Results of antibody testing in retrospective cohort.
Prospective Cohort
An additional 20 MOG positive cases were identified through routine clinical practice (19 via fixed assay at Mitogen and 1 via live assay at Oxford). One of these patients was also found to have positive NMDAR antibodies.
Across the two cohorts, the median timing of test serum sample collection in relation to disease onset or last relapse was 1 month (range 0–132) (see Supplementary Table 2).
The clinical features of patients identified by the two different tests are presented in Supplementary Table 1, in comparison to the cohort as a whole. The text below refers to the entire MOG antibody positive cohort.
Of the 42 patients (27 female) with a median disease onset age of 29 years (range 3–62; 9 pediatric cases), 20 (47%) were non-Caucasian (9 Asian, 7 South Asian, 4 other), and 3 (7%) had comorbid autoimmune disease (thyroid disease and psoriasis).
Most common phenotypes at onset were isolated optic neuritis in 23 patients (55%; bilateral in eight) and isolated myelitis in nine (21%; longitudinally extensive in six). Other onset phenotypes included brainstem presentations, combinations of optic neuritis, and myelitis and cerebral syndromes [which included focal deficits due to tumefactive lesions (3), seizures due to cortical lesions (1), or ADEM (2)]. As the majority of our cohort was adult, the proportion of cases with ADEM at onset was low.
Severity of disease at onset was moderate-severe in 64%, but the majority (74%) had good response to initial steroid therapy. Twenty-one percent of patients received other acute treatments in addition to high dose steroids, including plasma exchange, mitoxantrone, and intravenous immunoglobulins.
Using a formal Kaplan-Meier assessment, 3 patients who had relapsed had to be excluded due to lack of clarity on exact time to relapse. The cumulative relapse probability for the MOG positive group at 1 year was 0.428 (95% CI 0.244–0.567), at 4 years was 0.628 (95% CI 0.431–0.757), and at 10 years was 0.81 (95% CI 0.602–0.909) (see Figure 1).
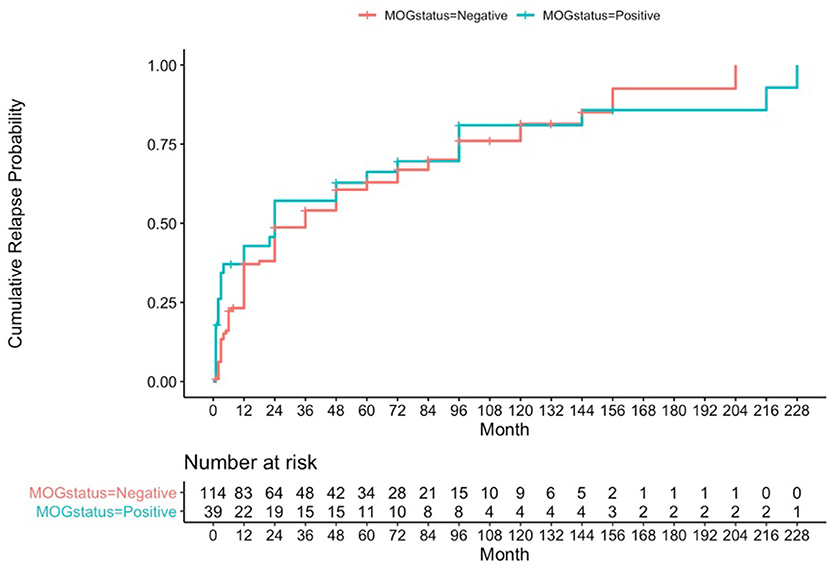
Figure 1. Kaplan-Meier analysis for cumulative relapse probabilities in the MOG-antibody positive and AQP4- and MOG-antibody seronegative groups.
Upon examining correlations between onset phenotype and future relapses, it was shown that 68% of patients with isolated optic neuritis and 78% of isolated transverse myelitis relapsed, whilst 100% of those who had presented with cerebral or combination presentations relapsed. Relapses most frequently affected the optic nerves or spinal cord, but the majority experienced different phenotypes on relapse to that experienced at onset (26/34 patients), including brainstem and cerebral or cortical phenotypes. Patients experiencing restricted phenotypes with recurrent disease were most likely to present with optic neuritis (6/7); one patient had pure recurrent transverse myelitis.
No patients had a progressive disease course, although patients with this phenotype are not routinely tested for MOG antibodies at our center.
In total 57% had no or minimal disability at last follow-up. Significant ongoing disability affected predominantly bladder/bowel in 12%, vision in 7%, motor in 2%, and a combination poor outcome was seen in 19%. Chronic steroid-sparing therapies were used in 25/42 patients (59.5%). Azathioprine was the most common first line agent (15/25), but mycophenolate mofetil (MMF) (4/25), and B-cell depleting monoclonal antibody therapies (6/25) were also used. Nine patients required a second line therapy, and one required a third line of treatment. (These constituted mostly MMF and B cell depleting therapies for those not previously using, as well as maintenance IVIG and an interleukin 6 receptor inhibiting monoclonal antibody). Supplementary Tables 2, 3 provide further details on the individual patient presentations.
In comparison to the MOG-antibody positive patients, the AQP4- and MOG-antibody negative patients showed a higher female predominance 2.5:1 vs. 1.8:1, and a slightly older age at onset (median 39 vs. 29 years).
The seronegative patients were more likely to present with isolated myelitis (57%) at onset than isolated optic neuritis (20%). Onset with neurological deficits in more than one location was more common in the seronegative group (12%) compared to the MOG-antibody positive group (5%). Disability at onset was similarly moderate to severe in both groups, but more patients in the MOG-antibody positive group showed a good response to initial steroid therapy (74 vs. 32%).
Cumulative relapse probabilities in the AQP4- and MOG-antibody seronegative group was 0.371 at 1 year (95% CI 0.274–0.455), 0.606 at 4 years (95% CI 0.498–0.691), and 0.814 at 10 years (95% CI 0.682–0.891) (see Figure 1). A formal z-test comparison was performed for estimated relapse probability at 12 months vs. the MOG positive group using their two standard error estimates of 0.04605 and 0.08148, and this was found to be non-significant (z = −0.609, 95% CI −0.240 to 0.126, p-value = 0.54).
In terms of clinical syndromes experienced at any point during the disease course, MOG-antibody positive patients were more likely to have experienced optic neuritis (74 vs. 40%) or brainstem (38 vs. 21%) presentations than the seronegative group. Myelitis was more common in the seronegative group (80 vs. 52%).
Tumefactive cerebral lesions presenting with the expected focal neurological deficits were common in both groups. Other symptomatic cerebral presentations also occurred, with some difference between the two groups. In the MOG-antibody group, two patients presented with seizures due to cortical lesions. In the seronegative group one patient presented with chorea and another with psychosis.
Both groups had a significant proportion of patients with residual disability affecting function at last follow-up −43% in the MOG-antibody positive group and 51% in the seronegative group (see Table 2).
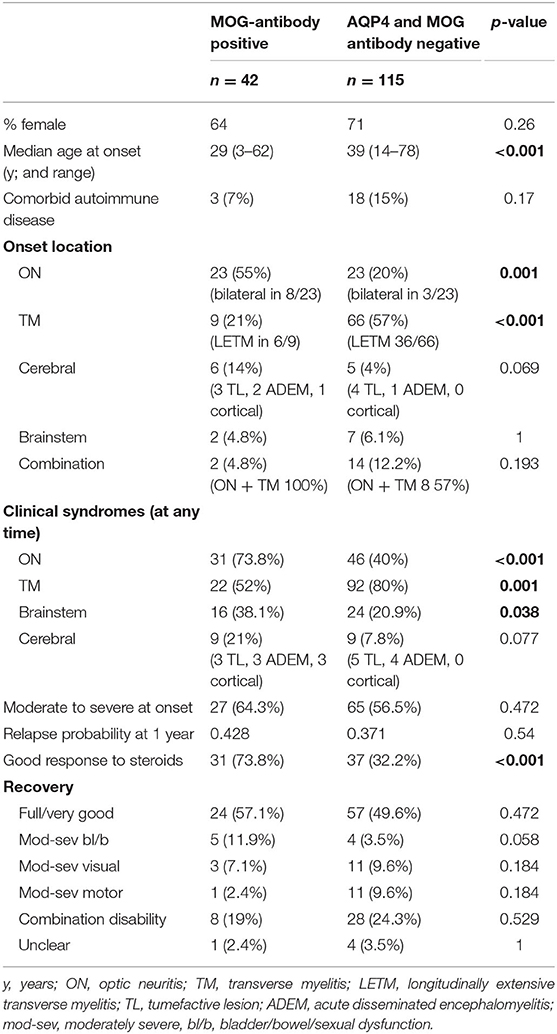
Table 2. Comparison of MOG-antibody positive and AQP4, MOG-antibody seronegative cohorts.
Chronic maintenance therapies were also used by some patients in the persistently seronegative group (62/115, 54%). Azathioprine was again most common (43/62 patients), but MMF (5) and antiCD20 monoclonal antibodies (5) were also used for some patients, as were traditional MS therapies (10). Second line therapy (predominantly MMF) was used by 22 (19%), and third line (predominantly antiCD20 monoclonal antibodies) by 11 (9.5%).
MR imaging was available for 35/42 of the MOG-antibody positive patients (Table 3); 32/35 were performed on 1.5 tesla MRI and three on 3 tesla MRI. Gadolinium contrast was administered in 18/35 cases. The majority of scans (22/35) were performed at the time of first disease presentation.

Table 3. MRI results summary: combined MOG cohort.
All 35 patients had brain imaging available for review. In nine, no lesions were detected, whilst 26 had abnormal scans. Number of lesions varied widely from ≤3 lesions in 10 patients, four to nine lesions in six patients and more than nine lesions in the remaining 10 patients. Supratentorial lesions were seen in 24/26 patients, bilateral in 18, and “fluffy” or poorly demarcated in 11. Seven of the 24 had lesions larger than two centimeters and five (of these seven) demonstrated contrast enhancement. In the majority of patients (23/26) the lesions were located in the subcortical white matter, but in 14 of these there were no lesions adjacent to the lateral ventricles. Seven patients had cortical lesions. Six patients had lesions in the corpus callosum, but none displayed diffuse splenial involvement. Four patients had lesions in the basal ganglia. One had lesions adjacent to the third ventricle. Whilst five patients had one or two of the components of previously described “classic MS” lesion findings (14) of Dawson's fingers, inferior temporal lobe lesions and S- or U-shaped juxtacortical lesions, no patients had all three characteristics.
8/26 patients had infratentorial brain involvement. In all eight the brainstem was involved, three had additional cerebellar involvement. In terms of location in the brainstem, seven patients had pontine lesions, six cerebellar peduncular lesions and five had lesions in the periaqueductal gray matter. Four patients had medullary lesions (two in the area postrema) and six had lesions adjacent to the fourth ventricle. Only one patient had diffuse brainstem involvement.
Orbital imaging (with minimum coronal T2 views with fat suppression) was available for 14 patients. Thirteen patients showed abnormality in their optic nerves, with bilateral involvement in eight patients. All 13 patients had long lesions, eight of these were exclusively anteriorly situated in the optic nerve, but five extended posteriorly to involve the chiasm.
Spinal cord imaging was available for 27 patients (however five of these had only had cervical cord imaging). 15/27 scans were normal. The 12 abnormal scans showed mixed patterns of involvement: three had only longitudinally extensive lesions (more than or equal to three vertebral segments), three had only short lesions and five had both longitudinally extensive and short lesions. In terms of lesion location, all patients had involvement of more than one spinal region. 7/12 patients had involvement of the conus medullaris. No patients displayed significant cord atrophy.
See attached Figures 2–7 for sample images from our patient cohort, and Table 3 and Figure 8 for a summary of MRI features.
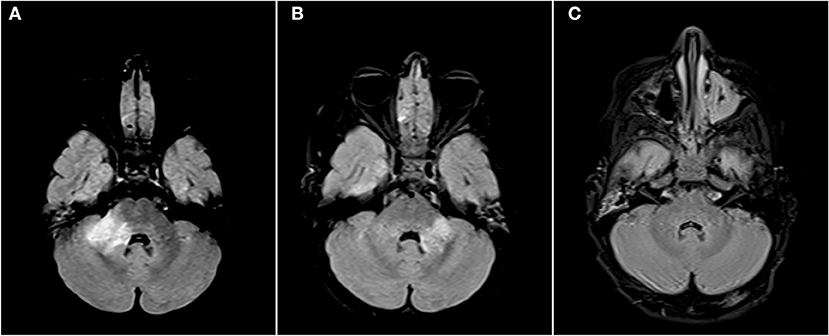
Figure 2. Axial FLAIR images of the brain from the same patient across serial examinations. (A) At the time of disease presentation: a FLAIR hyperintense lesion involves the right middle cerebellar peduncle. (B) Approximately 2 months later: the lesion in the right middle cerebellar peduncle has nearly completely resolved and there has been interval development of a new lesion in left middle cerebellar peduncle. (C) More than 1 year later: The posterior fossa lesions as well as other supratentorial lesions (not shown) have completely resolved.
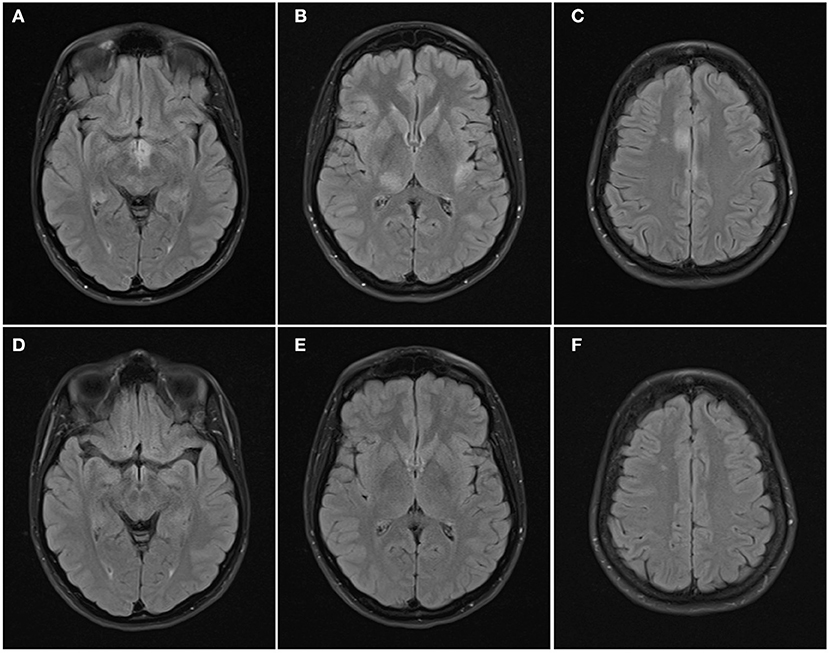
Figure 3. Axial FLAIR images of the brain from the same patient across serial examinations. (A–C) At the time of disease presentation: FLAIR hyperintense lesions involve the hypothalamus and optic tracts (A), right thalamus and left insular cortex (B), and parasagittal frontal cortex bilaterally and right centrum semiovale (C). (D–F) 5 months later: the lesions have all resolved except for the lesion in the right centrum semiovale.
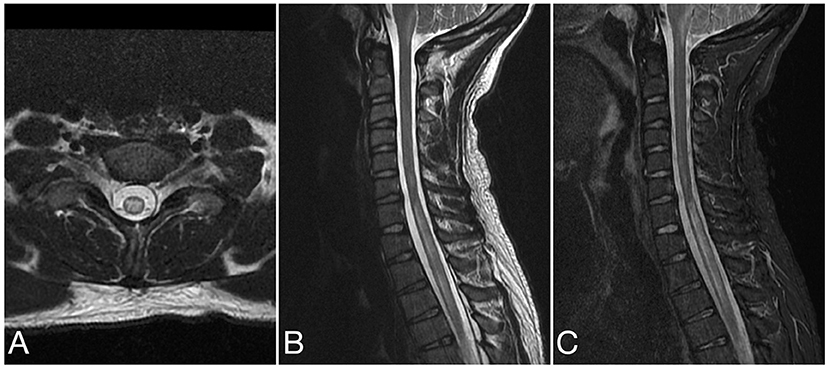
Figure 4. Axial T2 (A), sagittal T2 (B), and sagittal STIR (C) images of the cervical spine from the same patient at the time of disease presentation. A longitudinally extensive T2/STIR hyperintense lesion (≥3 contiguous vertebral segments) involve the spinal cord at the cervicothoracic junction.
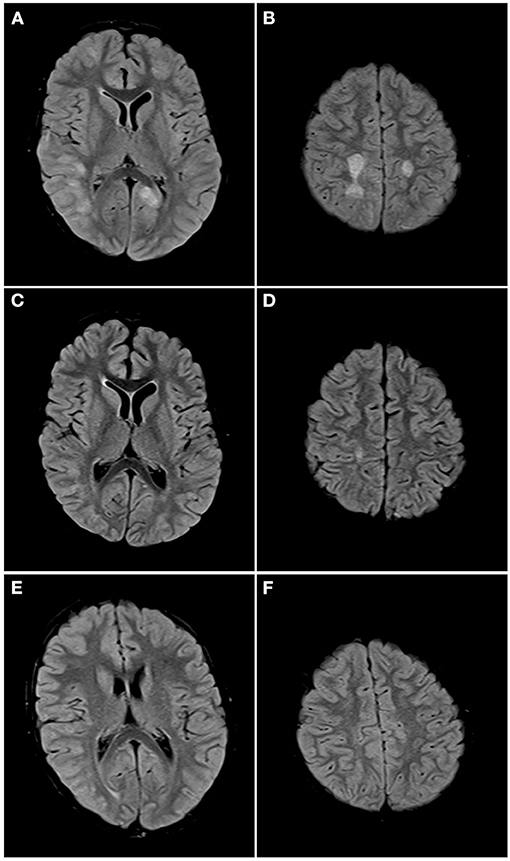
Figure 5. Axial FLAIR images of the brain from the same patient across serial examinations. (A,B) At the time of disease presentation: FLAIR hyperintense lesions involve the cortex and subcortical white matter of the right posterior temporal, right lateral occipital and left parasagittal occipital lobes (A) and the centrum semiovale bilaterally (B). (C,D) Approximately 1 month later: The lesions in both cerebral hemispheres have nearly completely resolved with a small residual lesion in the right centrum semiovale. (E,F) One year later: All the lesions have completely resolved.
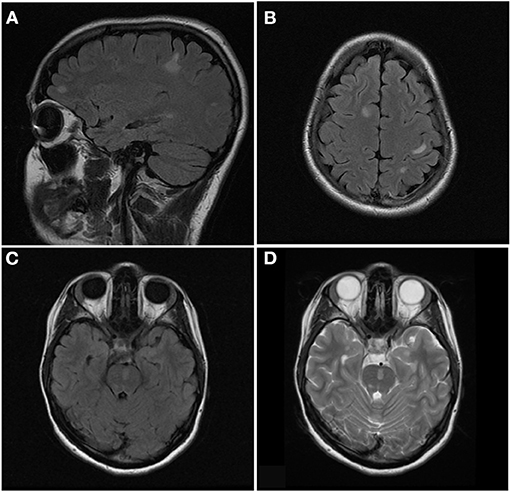
Figure 6. Sagittal FLAIR (A), axial FLAIR (B,C), and axial T2 (D) images of the brain from the same patient at the time of disease presentation. (A,B) Multiple FLAIR hyperintense lesions involve the juxtacortical and subcortical white matter of both cerebral hemispheres. (C,D) Additional T2/FLAIR hyperintense lesions involves the juxtacortical white matter of the left temporal lobe and pons.
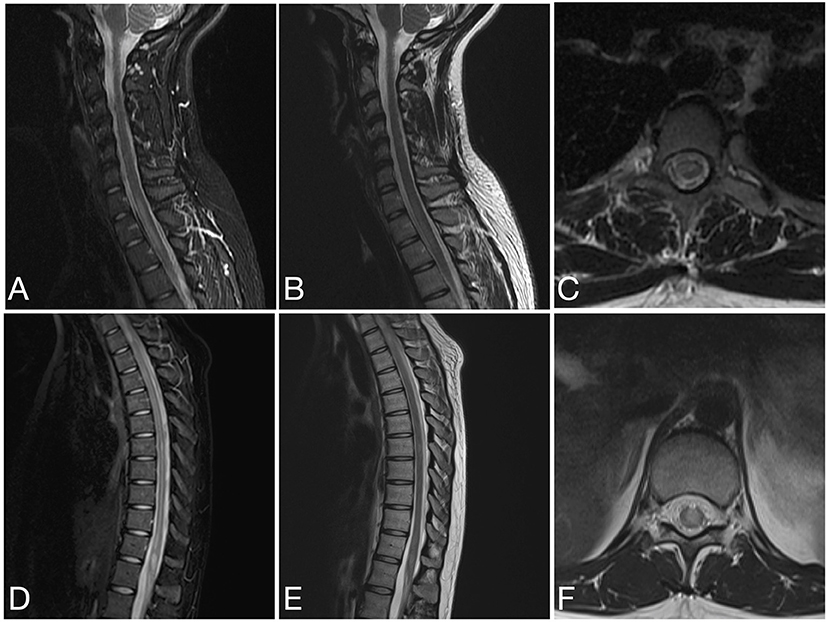
Figure 7. Sagittal STIR (A), sagittal T2 (B) and axial T2 (C) images of the cervical spine and sagittal STIR (D), sagittal T2 (E), and axial T2 (F) images of the thoracic spine from the same patient as in Figure 6 at the time of disease presentation. (A–C) A short segment T2/STIR hyperintense lesion (2 contiguous vertebral segments) involve the spinal cord at the cervicothoracic junction. (D–F) Other short segment T2/STIR hyperintense lesions involve the thoracic spinal cord and conus medullaris.
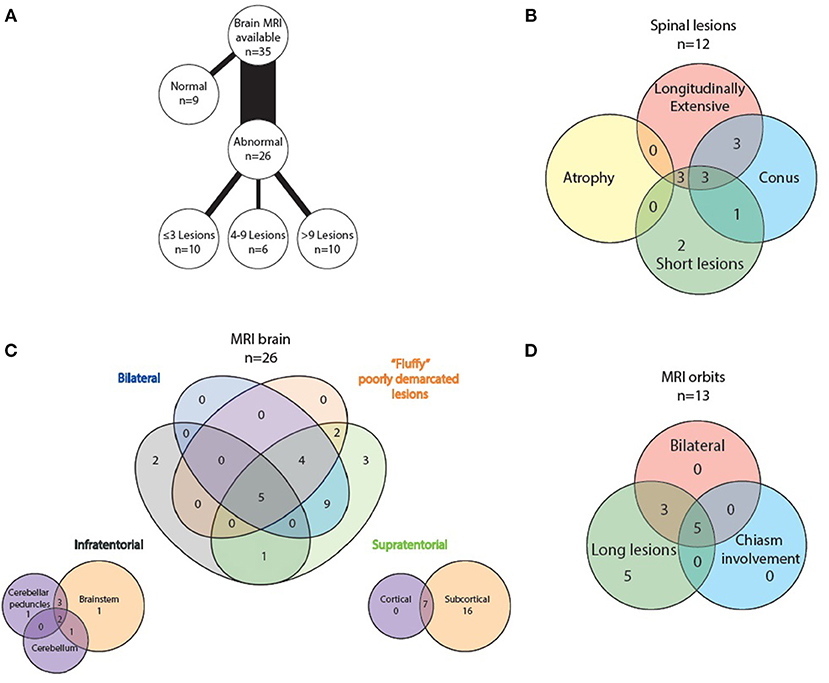
Figure 8. Radiological features of MOG positive MRI scans: (A) lesion load on abnormal brain scans, (B) spinal lesion characteristics, (C) MRI brain topography, and (D) optic nerve lesion characteristics.
We report a mixed pediatric (9) and adult (33) cohort of 42 MOG antibody positive patients from our multi-ethnic clinic in British Columbia, Canada. Retrospectively we identified 21/146 MOG antibody positives representing a frequency of 14% within the seronegative NMOSD “suspects”/idiopathic atypical demyelinating syndrome cohort which is similar to previous reports (8–20%) (7, 15, 16).
Demographically our patients are similar to other MOG cohorts (1–4, 8) in terms of a balanced gender distribution (64% female vs. 44–68%), onset age (29 vs. 27–37 years) and rate of comorbid autoimmune disorders (7 vs. 7–11%), but our cohort had a higher proportion of non-Caucasian patients (47 vs. 8–27%). Additionally, the onset phenotype was characteristic: 55% with optic neuritis, 21% transverse myelitis (including 7 patients with radiologically confirmed lesions in the conus medullaris, a feature frequently identified in MOG antibody mediated myelitis). With only 9 pediatric patients (median age 7, range 3–14 years) we did not see a high number of ADEM cases, which is typically seen in children under the age of 11 years. This finding was confirmed in a Chinese study examining differences in presentation between adult and pediatric MOG antibody disease (17).
More recently seizures and neuropsychiatric change related to cerebral cortical encephalitic lesions (5, 18–20) have been added to the MOG-antibody clinical phenotype (5, 18–20). Three of the patients in our cohort experienced cortical encephalitis, two of whom presented with seizures. This is not a rare presentation, in fact in a large Chinese series (5), 20.7% of the MOG cohort presented with cortical encephalitis. One of our patients with this presentation on relapse was also found to have NMDAR receptor antibodies. This coexistence of NMDAR and MOG antibodies has been previously described (21, 22). MOG-associated demyelinating episodes can occur simultaneously to NMDAR encephalitis, or can precede or follow (21).
One of our “clinically definite multiple sclerosis” cases was found to be MOG-antibody positive. This patient had presented with recurrent optic neuritis and a mild brainstem relapse, and had minimal MRI lesions. The clinical course of this patient has been atypical for MS, and the attending physician had recently been questioning the diagnosis. MOG antibody disease is felt to be distinct from MS, however as there is not yet a biomarker for MS, we are bound by clinical diagnostic criteria (McDonald criteria) which are limited in their sensitivity and specificity. One caveat of the McDonald criteria is that other potential disorders need to be excluded. A study from Germany (23) examined different groups of MS patients for MOG antibodies and found a rate of 5% MOG-antibody positivity in a group selected for “NMOSD-type presentations” with severe optic neuritis, myelitis and brainstem presentations. The authors suspect that the rate of MOG-antibody positivity in an unselected MS cohort is likely closer to 1%. Overall MOG antibodies are considered a marker of a non-MS disease (23, 24).
Radiologically, MOG antibody disease has some phenotypic overlap with NMOSD but can usually be distinguished from MS (13, 25). Brain imaging can be normal in MOG antibody disease (26, 27), but when lesions are seen they tend to be few, poorly demarcated or tumefactive and most often infratentorial (2, 25, 26). On follow-up imaging, lesions frequently show marked improvement or even resolution (2, 28). Seventy-four percent of our analyzed MRI brain scans were abnormal. The lesion frequency appeared bimodal with 42% (10/24) having <4 lesions and an equal number (10/24) with more than 9 lesions. Infratentorial involvement was relatively less common in our population, however as we only formally examined the first set of MR imaging performed for each patient, this may reflect what we found in our clinical onset phenotypes—that only five percent had brainstem involvement at first clinical presentation. We did not systematically analyse follow-up scans, but lesion resolution was noted in some patients anecdotally which is highlighted with the representative images displayed (see Figures 2–7).
In MOG antibody-associated disease, optic nerve lesions tend to be longer, but more anteriorly situated, sparing the optic chiasm with frequent optic disc edema at presentation (29). Our cohort demonstrated predominantly long lesions, but optic chiasm involvement was seen (5/13), previously reported as rare in this disorder (30, 31).
Spinal cord lesions are most frequently longitudinally extensive, but can also be short (32, 33). This was seen in our cohort with 3/12 abnormal spinal MRIs showing LETM only, 3/12 short lesions only, and both short and long lesions were present in 6/12 scans. Involvement of the conus medullaris is particularly characteristic (32), and was seen in 7/12 of our abnormal spinal MRIs.
Due to the retrospective nature of the study, follow-up times were not standardized so a simple relapse proportion could not be calculated, however a cumulative relapse probability at 10 years was 0.81 (although for this duration n = 4). This is a higher proportion of relapsing patients than some other series (1–3), however groups from Germany (33) and Spain (34) report similar high relapse rates of 80 and 78%, particularly optic neuritis relapses. Our cohort is from a referral center for CNS autoimmune disorders and could therefore have a referral bias for more severe or relapsing patients. In addition, due to the selection criteria of the AQP4 seronegative cohort for retrospective testing, it is likely that more severe cases are included in our cohort. Length of initial steroid treatment was not standardized and this data were not available for all patients. It is therefore possible, that early relapses occurred in those with short steroid exposure.
Whilst 57% of patients in our cohort had a good functional outcome at last assessment, 43% were left with significant residual disability after repeated relapses. This is in contrast to earlier reports of MOG-antibody associated disease being relatively benign in comparison to NMOSD, however as our experience with the disease grows, more reports are emerging that indicate that significant (especially visual) disability accrual is in fact occurring in many patients with repeated relapses (4, 33, 35). These data are important for ongoing treatment decisions where lesion resolution is often dramatic and short-term treatment over months is routine.
Our study was not able to incorporate MOG antibody titers or serial test results. We were therefore unable to determine if these factors had influence on disease severity or likelihood of relapse. It has been reported that higher antibody titers at onset may be associated with a more severe disease presentation, but may not be predictive of future relapses (17, 36). Longitudinal persistence of MOG seropositivity may be associated with increased relapse risk (2, 37). The serologic data in this study are from two different cell-based antibody assays, one presenting native human MOG as the substrate (live test) and the other one presenting MOG overexpressed in cells that have been chemically stabilized (fixed test). These tests were discordant. Of the 22 sera retrospectively tested as positive for MOG antibodies, only 4 were positive on both assays, 15 patients were only identified by live testing while 3 were uniquely positive on the fixed test. This highlights the significant discrepancies between live and fixed testing for MOG antibodies which, although not the focus of this study, requires formal investigation (38). In a similar fashion 8/146 serum samples, identified as seronegative AQP4 by stabilized commercial testing (Euroimmun AG), were identified as AQP4 antibody seropositive by live testing on sera that had been stored for many years (range 1–12). These data are not trivial and have important implications for clinical decisions in managing patient care in this severely disabling disease. These data recapitulate multiple other studies (38–40).
Our multi-ethnic clinic population from British Columbia, Canada display similar demographic and phenotypic features to those previously described. We confirm rarer presenting features or “red flags” suggesting MOG-antibody positivity in patients, such as seizures with cortical lesions and conus medullaris involvement in patients with myelitis. Positive anti-MOG antibodies can rule out MS in patients with an atypical clinical MS disease course. Importantly, many of our MOG-antibody positive patients relapsed and were left with significant disability. International collaborative research efforts could address the clear need for a biomarker to identify patients likely to relapse as well as to establish formal treatment guidelines.
All datasets generated for this study are included in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by University of British Columbia Clinical Research Ethics Board. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
HC drafted and coordinated the manuscript, she was also involved in study design, data collection, and analysis. FS collected the neuroradiology data. NA was involved in study design and initial data collection. AM assisted with data collection. SA performed some of the statistical analysis. MW performed laboratory analyses at Oxford. BS created the venn diagrams. VD, RC, ALS, VB, ASc, and JC contributed patients and assisted with manuscript review. MF coordinated laboratory assessments at MitogenDx and assisted with manuscript review. PW coordinated laboratory assessments at Oxford and assisted with manuscript review. AT was involved in study design, manuscript review, and coordination of the team. All authors contributed to the article and approved the submitted version.
MF is the director of MitogenDx. He is a paid consultant, has received honoraria or gifts in kind from Inova Diagnostics (San Diego, CA). PW is the director of Oxford Autoimmune Neurology Diagnostic Laboratory. He holds patents for antibody testing and has received consulting honoraria from Biogen Idec, Euroimmun AG, Mereo Biopharma, UBC, and retrogenix.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank Haiyan Hou and Meifeng Zhang (MitogenDx, Calgary, AB) for laboratory analyses, as well as Dr. Carolyn Taylor (Department of Statistics UBC) for assistance with the Kaplan-Meier analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.525933/full#supplementary-material
1. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
2. Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology. (2018) 90:e1858–69. doi: 10.1212/WNL.0000000000005560
3. de Mol C, Wong YYM, van Pelt ED, Wokke BH, Siepman TAM, Neuteboom RF, et al. The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. Multiple Sclerosis J. (2019) 2019:1352458519845112. doi: 10.1177/1352458519845112
4. Ramanathan S, Mohammed S, Tantsis E, Nguyen TK, Merheb V, Fung V, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. (2018) 89:127–37. doi: 10.1136/jnnp-2017-316880
5. Wang L, Zhangbou J, Zhou L, Zhang Y, Li H, Li Y, et al. Encephalitis is an important clinical component of myelin oligodendrocyte glycoprotein antibody associated demyelination: a single-center cohort study in Shanghai, China. Eur J Neurol. (2019) 26:168–74. doi: 10.1111/ene.13790
6. Kitley J, Waters P, Woodhall M, Leite M, Murchison A, George J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. (2014) 71:276–83. doi: 10.1001/jamaneurol.2013.5857
7. Hamid SHM, Whittam D, Mutch K, Linaker S, Solomon T, Das K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. (2017) 264:2088–94. doi: 10.1007/s00415-017-8596-7
8. Hoftberger R, Sepulveda M, Armangue T, Blanco Y, Rostasy K, Calvo AC, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler. (2015) 21:866–74. doi: 10.1177/1352458514555785
9. Narayan R, Simpson A, Fritsche K, Salama S, Pardo S, Mealy M, et al. MOG antibody disease: a review of MOG antibody seropositive neuromyelitis optica spectrum disorder. Multiple Sclerosis Related Disord. (2018) 25:66–72. doi: 10.1016/j.msard.2018.07.025
10. Levy M, Giovanni G, Hawkes C, Waubant E. Refining the nosology of antigen-specific diseases within the spectrum of neuromyelitis optica. Multiple Sclerosis Related Disord. (2018) 25:A1–2. doi: 10.1016/j.msard.2018.09.027
11. Wingerchuk D, Banwell B, Bennett B, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
12. Pohl D, Alper G, Van Haren K, Kornberg A, Lucchinetti C, Tenembaum S, et al. Acute disseminated encephalomyelitis: updates on an inflammatory CNS syndrome. Neurology. (2016) 87:S38–45. doi: 10.1212/WNL.0000000000002825
13. Polman CH, Reingold S, Banwell B, Clanet M, Cohen J, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. (2011) 69:292–302. doi: 10.1002/ana.22366
14. Juryńczyk M, Tackley G, Kong Y, Geraldes R, Matthews L, Woodhall M, et al. Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry. (2017) 88:132–6. doi: 10.1136/jnnp-2016-314005
15. Siritho S, Sato D, Kaneko K, Fujihara K, Prayoonwiwat N. The clinical spectrum associated with myelin oligodendrocyte glycoprotein antibodies (anti-MOG-Ab) in Thai patients. Multiple Sclerosis J. (2016) 22:964–8. doi: 10.1177/1352458515614093
16. Tanaka M, Tanaka K. Anti-MOG antibodies in adult patients with demyelinating disorders of the central nervous system. J Neuroimmunol. (2014) 270:98–9. doi: 10.1016/j.jneuroim.2014.03.001
17. Chen L, Chen C, Zhong X, Sun X, Zhu H, Haixia Z, et al. Different features between pediatric-onset and adult-onset patients who are seropositive for MOG-IgG: a multicenter study in South China. J Neuroimmunol. (2018) 321:83–91. doi: 10.1016/j.jneuroim.2018.05.014
18. Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflammation. (2017) 4:e322. doi: 10.1212/NXI.0000000000000322
19. Fujimori J, Takai Y, Nakashima I, Sato D, Takahashi T, Kaneko K, et al. Bilateral frontal cortex encephalitis and paraparesis in a patient with anti-MOG antibodies. J Neurol Neurosurg Psychiatry. (2017) 88:534–6. doi: 10.1136/jnnp-2016-315094
20. Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and encephalitis in myelin oligodendrocyte glycoprotein IgG disease vs aquaporin 4 IgG disease. JAMA Neurol. (2018) 75:65–71. doi: 10.1001/jamaneurol.2017.3196
21. Titulaer MJ, Hoftberger R, Iizuka T, Leypoldt F, McCracken L, Cellucci T, et al. Overlapping demyelinating syndromes and anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. (2014) 75:411–28. doi: 10.1002/ana.24117
22. Fan S, Xu Y, Ren H, Guan H, Feng F, Gao X, et al. Comparison of myelin oligodendrocyte glycoprotein (MOG)-antibody disease and AQP4-IgG-positive neuromyelitis optica spectrum disorder (NMOSD) when they co-exist with anti-NMDA (N-methyl-D-aspartate) receptor encephalitis. Multiple Sclerosis Related Disord. (2018) 20:144–52. doi: 10.1016/j.msard.2018.01.007
23. Spadaro M, Gerdes L, Krumbholz M, Ertl-Wagner B, Thaler F, Schuh E, et al. Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol Neuroimmunol Neuroinflammation. (2016) 3:e257. doi: 10.1212/NXI.0000000000000257
24. Hacohen Y, Absoud M, Deiva K, Hemingway C, Nytrova P, Woodhall M, et al. Myelin oligodendrocyte glycoprotein antibodies are associated with a non-MS course in children. Neurol Neuroimmunol Neuroinflammation. (2015) 2:e81. doi: 10.1212/NXI.0000000000000081
25. Jurynczyk M, Geraldes R, Probert F, Woodhall M, Waters P, Tackley G, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain. (2017) 140:617–27. doi: 10.1093/brain/aww350
26. Kim SM, Woodhall M, Kim J, Kim S, Park K, Vincent A, et al. Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol Neuroimmunol Neuroinflammation. (2015) 2:e163. doi: 10.1212/NXI.0000000000000163
27. Sato DK, Callegaro D, Lana-Peixoto M, Waters P, de Haidar Jorge F, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. (2014) 82:474–81. doi: 10.1212/WNL.0000000000000101
28. Waters P, Fadda G, Woodhall M, O'Mahoney J, Brown R, Castro D, et al. Serial anti-myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol. (2019) 77:82–93. doi: 10.1001/jamaneurol.2019.2940
29. Ramanathan S, Prelog K, Barnes E, Tansis E, Reddel S, Henderson A, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Multiple Sclerosis J. (2016) 22:470–82. doi: 10.1177/1352458515593406
30. Biotti D, Bonneville F, Tournaire E, Ayrignac X, Dalliere C, Mahieu L, et al. Optic neuritis in patients with anti-MOG antibodies spectrum disorder: MRI and clinical features from a large multicentric cohort in France. J Neurol. (2017) 264:2173–5. doi: 10.1007/s00415-017-8615-8
31. Zhao Y, Tan S, Chan T, Xu Q, Zhao J, Teng D, et al. Clinical features of demyelinating optic neuritis with seropositive myelin oligodendrocyte glycoprotein antibody in Chinese patients. Br J Ophthalmol. (2018) 102:1372–7. doi: 10.1136/bjophthalmol-2017-311177
32. Dubey D, Pittock S, Krecke K, Morris P, Sechi E, Zalewski N, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. (2019) 76:301–9. doi: 10.1001/jamaneurol.2018.4053
33. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. (2016) 13:280. doi: 10.1186/s12974-016-0718-0
34. Sepúlveda M, Armangue T, Martinez-Hernandez E, Arrambide G, Sola-Valls N, Sabatar L, et al. Clinical spectrum associated with MOG autoimmunity in adults: significance of sharing rodent MOG epitopes. J Neurol. (2016) 263:1349–60. doi: 10.1007/s00415-016-8147-7
35. Piccolo L, Woodhall M, Tackley G, Jurynczyk M, Kong Y, Domingos J, et al. Isolated new onset 'atypical'optic neuritis in the NMO clinic: serum antibodies, prognoses and diagnoses at follow-up. J Neurol. (2016) 263:370–9. doi: 10.1007/s00415-015-7983-1
36. Cobo-Calvo A, Sepulveda M, d'Indy H, Armangue T, Ruiz A, Maillart E, et al. Usefulness of MOG-antibody titres at first episode to predict the future clinical course in adults. J Neurol. (2019) 266:806–15. doi: 10.1007/s00415-018-9160-9
37. Oliveira LM, Apostolos-Pereira SL, Pitombeira MS, Bruel Torretta PH, Callegaro D, Sato DK. Persistent MOG-IgG positivity is a predictor of recurrence in MOG-IgG-associated optic neuritis, encephalitis and myelitis. Multiple Sclerosis J. (2019) 25:1907–14. doi: 10.1177/1352458518811597
38. Waters PJ, Komorowski L, Woodhall M, Lederer S, Majed M, Fryer J, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology. (2019) 92:e1250–5. doi: 10.1212/WNL.0000000000007096
39. Waters P, Reindl M, Saiz A, Schanda K, Tuller F, Kral V, et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry. (2016) 87:1005–15. doi: 10.1136/jnnp-2015-312601
Keywords: myelin oligodendrocyte glycoprotein (MOG) antibodies, aquaporin 4 antibodies, multiple sclerosis (MS), demyelination, neuroinflammation, neuromyelitis optica
Citation: Cross H, Sabiq F, Ackermans N, Mattar A, Au S, Woodhall M, Sun B, Devonshire V, Carruthers R, Sayao AL, Bhan V, Schabas A, Chan J, Fritzler M, Waters P and Traboulsee A (2021) Myelin Oligodendrocyte Glycoprotein (MOG) Antibody Positive Patients in a Multi-Ethnic Canadian Cohort. Front. Neurol. 11:525933. doi: 10.3389/fneur.2020.525933
Received: 10 January 2020; Accepted: 07 December 2020;
Published: 12 January 2021.
Edited by:
Michael Levy, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Kristen Marie Krysko, University of California, San Francisco, United StatesCopyright © 2021 Cross, Sabiq, Ackermans, Mattar, Au, Woodhall, Sun, Devonshire, Carruthers, Sayao, Bhan, Schabas, Chan, Fritzler, Waters and Traboulsee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony Traboulsee, dC50cmFib3Vsc2VlQHViYy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.