 John Michael S. Sanchez1
John Michael S. Sanchez1 J. Scott McNally2
J. Scott McNally2 Melissa M. Cortez3
Melissa M. Cortez3 James Hemp4Laura A. Pace4
James Hemp4Laura A. Pace4 Stacey L. Clardy3,5*
Stacey L. Clardy3,5*- 1Division of Microbiology and Immunology, Department of Pathology, University of Utah, Salt Lake City, UT, United States
- 2Department of Radiology, Utah Center for Advanced Imaging Research, University of Utah, Salt Lake City, UT, United States
- 3Department of Neurology, Imaging and Neurosciences Center, University of Utah, Salt Lake City, UT, United States
- 4Division of Gastroenterology, Department of Internal Medicine, University of Utah, Salt Lake City, UT, United States
- 5George E. Whalen Veterans Affairs Medical Center, Salt Lake City, UT, United States
The central nervous system (CNS) is an important regulator of the gastrointestinal tract, and CNS dysfunction can result in significant and disabling gastrointestinal symptom manifestation. For patients with neuroimmunologic and neuroinflammatory conditions, the recognition of gastrointestinal symptoms is under-appreciated, yet the gastrointestinal manifestations have a dramatic impact on quality of life. The current treatment strategies, often employed independently by the neurologist and gastroenterologist, raise the question of whether such patients are being treated optimally when siloed in one specialty. Neuroimmunogastroenterology lies at the borderlands of medical specialties, and there are few resources to guide neurologists in this area. Here, we provide an overview highlighting the potential mechanisms of crosstalk between immune-mediated neurological disorders and gastrointestinal dysfunction.
Introduction
The central nervous system (CNS) is an important regulator of the gastrointestinal tract, and CNS dysfunction can result in gastrointestinal symptom manifestation (1). Examples can be found in lesions of the hindbrain, such as in the medullary raphe nuclei that controls gastric motility and secretion through secretion of 5HT (5-hydroxytryptophan) and substance P (2, 3); midbrain, as seen in cerebellum-associated nausea and emesis related to motion sickness (4); and forebrain, including amygdala control of gastric secretion (5). While such conditions may be familiar to the practicing neurologist, neuroinflammatory-associated gastrointestinal disorders are less recognized.
Patients with inflammatory and autoimmune neurological disorders often have accompanying gastrointestinal symptoms, although the etiology of these gastrointestinal symptoms can vary. For example, a majority of multiple sclerosis patients report symptoms suggestive of a gastrointestinal motility disorder (6), likely related to involvement of the autonomic nervous system, while intractable nausea and vomiting in neuromyelitis optica spectrum disorder is directly attributable to central nervous system (CNS) lesions of the area postrema (7). For patients with other systemic neuroimmunologic and neuroinflammatory conditions, the recognition of gastrointestinal symptoms is likely under-appreciated, perhaps owing to more dramatic symptoms in other organ systems—yet the gastrointestinal manifestations of these disease processes can have a dramatic impact on quality of life.
Likewise, for patients presenting primarily with gastrointestinal symptoms, there is often an underappreciation of the neurologic, neuroimmune, and inflammatory nature of the condition. For example, transcriptomic analysis of patients with idiopathic gastroparesis reveal a clear role of the immune system in the disease process (8), and similarly, in patients with esophageal dysmotility disorders, there is a high rate of autoantibody detection (9, 10). Yet, most patients are treated surgically (11), rather than with immune-modulating therapy. The current treatment strategies, often employed independently by the neurologist and gastroenterologist, raise the question of whether such patients are being treated optimally when siloed in one specialty—especially if there is not a recognition of the potential neurologic, neuroimmune, immunologic and inflammatory underpinnings of the symptoms. Indeed, neuroimmunogastroenterology lies at the borderlands of medical specialties, and there are few currently available resources to guide neurologists and gastroenterologists in this area. Here, we provide an overview of recent literature highlighting the potential mechanisms of crosstalk between immune-mediated neurological disorders and gastrointestinal dysfunction. In addition, we report three clinical cases to emphasize the diagnostic and therapeutic avenues to evaluate and treat gastrointestinal complaints in patients with inflammatory and autoimmune neurological disorders.
Literature Review
General Mechanisms of Neuroimmune-Gastrointestinal Crosstalk
The brain-gut axis incorporates bi-directional signals from the central nervous system, autonomic nervous system, enteric nervous system, gut microbiota, and immune system to respond to the dynamic needs of human physiology. The sympathetic and parasympathetic divisions of the autonomic nervous system provide efferent cues to modulate gastrointestinal motility, secretion, and blood flow to the gastrointestinal tract (12). In contrast, afferent neurons of the enteric nervous system relay information to the CNS regarding the enteric milieu, including mechanical distension and the fed/fast state of the gastrointestinal tract (13). Within the gastrointestinal tract itself resides the densest collection of commensal microorganisms, the gut microbiota, which produce neuroactive molecules and have substantial influence on mucosal immunity (Figure 1A) (14). In fact, the majority of the human body's immune cells are found within gut-associated lymphoid tissue and are key mediators of the brain-gut axis.
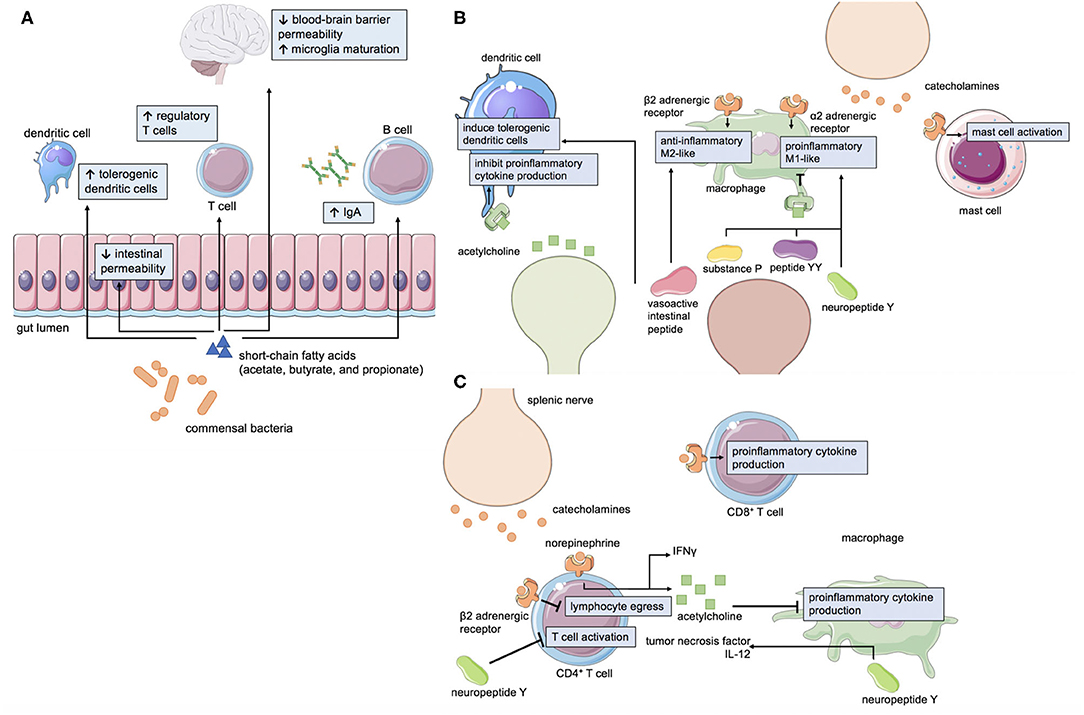
Figure 1. Mechanisms of neuroimmune-gastrointestinal crosstalk. (A) Short-chain fatty acids (SCFAs) such as acetate, butyrate, and propionate are metabolites generated by commensal gut bacteria that affect intestinal permeability, immunity, and CNS physiology. Neurotransmitters and neuropeptides can influence the function of (B) innate and (C) adaptive immune cells.
Cells of the innate immune system are classically understood to mount the initial immune response to pathogens, however the crosstalk between cells of the immune and nervous systems are functionally significant in the gastrointestinal tract. Catecholamines, a major class of sympathetic neurotransmitters, have been shown to differentially induce pro- (M1-like) and anti- (M2-like) inflammatory macrophages in the intestine (15). The effect of catecholamine signaling appears to be context-dependent as activation of α2 adrenoreceptors promotes the production of proinflammatory cytokines (16, 17) whereas activation of β2 adrenoreceptors promotes the production of anti-inflammatory cytokines (18). Mast cells express adrenergic receptors, and signaling through these receptors can modulate the secretion of mast cell products (19). In contrast, the major parasympathetic neurotransmitter, acetylcholine, has been shown to decrease pro-inflammatory cytokine production in macrophages and dendritic cells, among other immune cell types (18, 20, 21). Serotonin, 95% of which is synthesized in the gut (22), modulates the polarization of macrophages (23) and phagocytosis (24). Mast cells themselves have the ability to produce serotonin (25), which can directly affect gastrointestinal motility, and histamine, which can activate sensory neurons in the gastrointestinal tract (26). Neuropeptides, including substance P, neuropeptide Y and peptide YY, can enhance the production of pro-inflammatory cytokines in monocytes (27) and enhance phagocytosis in macrophages (28). Vasoactive intestinal peptide (VIP), another neuropeptide, induces regulatory dendritic cells (29, 30) which can then go on to induce regulatory T cells. VIP can also reduce TGF-β1 (31), which can drive pro-inflammatory T-helper-17 cells, and increase IL-10 production in macrophages (32). Thus, neurons of the enteric nervous system and intestine-resident innate immune cells are intimately associated through bidirectional signaling (Figure 1B).
The adaptive immune system is also involved in neuro-immune crosstalk in the gastrointestinal tract (Figure 1C). Interestingly, some T cells are intrinsically capable of producing acetylcholine via choline acetyltransferase (21). Acetylcholine-producing T cells can be activated by efferent signals from the vagus nerve to the splenic nerve (21). Conversely, afferent vagal signaling can result in anti-inflammatory effects (33). Catecholamine signaling via β2-adrenergic receptors interact with chemokine receptors to control T and B cell migration (34) and can differentially modulate cytokine production in different T cell subsets (35, 36). Similarly, dopamine can modulate T cell migration and cytokine secretion (37). Neuropeptides can also modulate the production of cytokines by T cells (38, 39). VIP is one such example that increases inflammatory cytokine production in T cells (40). Overall, the large collection of innate and adaptive immune cells that reside in the gut are linked to cues from the nervous system and the microbiota to maintain homeostasis in a constantly changing environment.
The Role of the Gut Microbiota and Gut Permeability in Neuroinflammatory Disease
From birth, and likely even in utero, the community of bacteria, archaea, viruses, and fungi that colonize the human gut play an important role in the physiology of the gastrointestinal, immune, and nervous systems (41). Commensal microorganisms and their metabolites can influence gastric secretion and motility by directing serotonin production; modulate the differentiation and function of immune cells; and increase neurogenesis (42). The epithelium of the gastrointestinal tract is critical to maintaining a barrier between the luminal contents of the intestines and the rest of the body, however this barrier has selective permeability that allows for the absorption of nutrients as well as certain microbial products (43, 44). In some neuroinflammatory conditions, the permeability of the intestinal barrier is increased that can allow for pathogenic translocation of gut microorganisms and microbiota-derived products into circulation that promote inflammation (45). Conversely, certain commensal microorganisms including certain strains of Lactobacillus and Bifidobacterium have the ability to upregulate the expression of tight junction proteins between epithelial cells and strengthen the gut barrier (46). Evidence for the role of the gut microbiota and the intestinal barrier in neuroinflammation has been found in a number of disease states including autism spectrum disorder, multiple sclerosis, and Parkinson's disease. Understanding the mechanisms by which the gut microbiota and intestinal permeability influences neuroinflammation is an intense area of research.
Autism spectrum disorder is a continuum of neurodevelopmental conditions characterized by repetitive behaviors, altered sociability, and language difficulties (47). Gastrointestinal dysfunction and increased permeability of the gastrointestinal barrier is a common comorbidity of autism (48). Patients with autism spectrum disorder also display alterations in gut bacterial communities, including a lower abundance of Akkermansia, Bacteroides, Escherichia coli, and Enterococcus and a higher abundance of Faecalibacterium and Lactobacillus, compared to typically developing controls (49). Gnotobiotic mice that receive fecal microbiota from autism spectrum disorder patients exhibit exacerbated autism-like behaviors compared to mice that receive fecal microbiota from typically developing controls (50). Specific bacteria, such as Bacteroides fragilis and Lactobacillus reuteri, have been shown to modulate mouse models of autism spectrum disorder by reducing gut inflammation, permeability and targeting oxytocinergic and dopaminergic networks (51–53). Importantly, microbiota-based therapies including fecal microbiota transplant, antibiotics, and probiotics have shown benefit in small clinical studies (54–56).
Multiple sclerosis is an immune-mediated demyelinating disease of the CNS and the most common cause of non-traumatic physical disability in young adults (57). Gastrointestinal dysmotility and increased permeability is prevalent in patients with multiple sclerosis and in experimental autoimmune encephalomyelitis (EAE), the most commonly used mouse model of multiple sclerosis (58–61). A number of studies have reported changes in specific strains of bacteria such as elevated Methanobrevibacter and Porphyromonas and decreased Bacteroides and Lactobacillus in multiple sclerosis patients compared to healthy matched controls (62–67). Additionally, disease-modifying therapies for multiple sclerosis have been shown to alter gut microbial composition, though the functional consequences of these drug-induced changes are not known (65, 68). Gnotobiotic mice colonized with multiple sclerosis-associated fecal microbiota exhibit more severe EAE compared to gnotobiotic mice colonized with fecal microbiota from matched controls, suggesting that the gut microbiota can influence multiple sclerosis (66, 67). Indeed, specific bacteria including Bacteroides, Lactobacillus, and Prevotella have been shown to protect against EAE by modulation of the immune response, including induction of regulatory T cells and suppression of proinflammatory T-helper (Th) type 1 and Th17 cells (69–73). Importantly, pilot studies of probiotic administration or dietary invention with associated downstream effects on the gut microbiota have been shown to benefit multiple sclerosis patients (74–76).
Parkinson's disease is a neurodegenerative disease associated with pathological aggregation of α-synuclein in the CNS (77). Degeneration of the dopaminergic neurons of the substantia nigra pars compacta leads to the characteristic symptoms of Parkinson's including tremor, muscle rigidity, and impaired balance (77). Interestingly, patients with Parkinson's disease exhibit gastrointestinal inflammation and increased intestinal permeability years prior to neurological deficits and α-synuclein can be found early in the enteric nervous system and the glossopharyngeal and vagal nerves (78, 79). Indeed, patients with Parkinson's disease exhibit alterations in certain gut commensal taxa including decreased Prevotellaceae and increased Verrucomicrobiaceae (80–83). Utilizing a murine model of synucleinopathy, colonization of gnotobiotic mice with the fecal microbiota of Parkinson's disease patients accelerated the accumulation of α-synuclein in the CNS compared to mice colonized with microbiota from matched controls (84). Short-chain fatty acids, which are derived from the gut microbiota, was also shown to be sufficient to accelerate accumulation of α-synuclein in the CNS, though the mechanism is unclear (84). Interestingly, the human commensal bacterium, Enterococcus faecalis, has the ability to metabolize levodopa, a dopaminergic agent used to treat Parkinson's disease, and decrease its bioavailability in an animal model (85). Together, these data suggest a functional role of the gut microbiota in Parkinson's disease pathogenesis and treatment.
The above examples demonstrate the wide-reaching influence the gut microbiota has on neurological disorders of varying etiologies. The ability of the gut microbiota to modulate the immune system, produce neuroactive compounds, affect gut barrier function, among other functions, adds a layer of complexity to neuroinflammatory diseases. The mechanisms of communication between the gut microbiota and the CNS along the gut-brain axis remain to be fully elucidated. Still, the positive results from pilot trials using probiotics, antibiotics, and fecal microbiota transfer in certain neurological conditions suggests that microbiota-based therapies may be on the horizon for specific contexts.
Neurological Manifestations of Gastrointestinal Disease
In the opposite direction, certain immune-mediated gastrointestinal diseases, including inflammatory bowel disease (IBD) and celiac disease, have been shown to have neurological involvement. While the exact mechanisms linking gastrointestinal inflammation to neurological dysfunction remain to be elucidated, future investigation will be critical in addressing these manifestations.
IBD, which includes ulcerative colitis and Crohn's disease, is characterized by chronic inflammation of the gastrointestinal tract. Imaging studies have demonstrated increased white matter lesions (86–88), decreased gray matter volume (88, 89), and increased axonal damage (88) in patients with IBD compared to healthy matched controls. While other studies have failed to identify these IBD-associated neurological changes (90, 91), this may be due to differences in the severity of IBD at the time of imaging. Indeed, gray matter atrophy has been reported to correlate with IBD duration (89). The etiology of these structural brain lesions is not completely understood, though several mechanisms have been proposed. These include thromboembolism, vasculitis, neurotoxic inflammation, chronic pain-induced excito-toxicity, demyelination, iatrogenic progressive multifocal leukoencephalopathy, and opportunistic infections (92, 93). IBD-associated brain lesions can be asymptomatic, however the association of IBD with peripheral neuropathy, myopathy, myelopathy, and cerebrovascular disease suggest that IBD can lead to true dysfunction of the nervous system (94).
Unlike IBD, where the cause of gastrointestinal inflammation is often not clear, patients with celiac disease exhibit gastrointestinal inflammatory disease triggered by dietary gluten (95). Celiac disease is frequently associated with neurological symptoms ranging from ataxia, neuropathy, headache, cognitive impairment, and psychiatric disorders, though celiac disease is not likely the primary cause of these conditions (96). Neurological involvement has been attributed to nutritional deficiencies, including deficiencies in vitamin B12, vitamin E, folate, zinc, or copper (96), due to due to loss of brush border proteins and enzymes needed for the absorption of these nutrients, and also inflammation of the small intestine (97). Other deficiencies are also common but not often directly related to neurologic symptoms, including reduced levels of iron, folate, vitamin D, and magnesium (97). Overlapping nervous system autoimmunity may also contribute to neurological symptoms as anti-transglutaminase antibodies, a biomarker of celiac disease, have been reported in the CNS of celiac disease patients (98) Indeed, intraventricular injection of anti-transglutaminase antibodies into mice results in ataxia (99). Auto-antibodies to gangliosides have also been reported (100); the significance remains unclear but the generation of anti-ganglioside antibodies is speculated to be linked to the intestinal immune response to ingested gluten. Similar to ganglioside generation in acute immune-mediated neuropathies, where molecular mimicry between gangliosides and bacterial or viral oligosaccharides is proposed as a mechanism (101) in celiac disease as well (100). Following a gluten-free diet, which alleviates gastrointestinal symptoms of celiac disease, has mixed success in relieving the neurological manifestations of the disease (102–105). This observation reinforces the possibility that celiac disease may be an association of some neurologic symptoms but not the primary cause. For example, in some hereditary ataxia syndromes, CD antibodies are more commonly found than the normal population (106) Whether primary, secondary, or otherwise genetically associated, recognition and treatment of underlying celiac disease is worthy of investigation and treatment.
In animal models, gastrointestinal inflammation is often accompanied by systemic inflammation, chronic pain and stress, and nutritional deficiencies that can impede neurological function (107). While unethical to model chronic stress in humans, adequate intervention to disrupt systemic inflammation (and chronic stress as able) by early detection and mechanistic investigation of neurological dysfunction in the context of immune-mediated gastrointestinal diseases will be key.
Functional Gastrointestinal Disorders
Among the most common gastroenterology diagnoses are functional gastrointestinal disorders (108). According to the most recent ROME IV criteria on functional gastrointestinal disorders, these are classified by symptoms related to any combination of gastrointestinal motility disturbance, visceral hypersensitivity, altered mucosal and immune function, altered gut microbiome, or altered CNS processing (109). While it is acknowledged that functional gastrointestinal disorders may be associated with processes such as motility disturbances or altered immune function, the recommendation by the ROME committee is to conduct a careful physical examination and cost-efficient investigation, with the diagnosis of a functional disorder relying largely on symptom description and duration, rather than objective findings. The ROME committee also reminds the clinician to acknowledge the worsening of certain symptoms with stress, without themselves acknowledging that the fundamental role of the autonomic nervous system in responding to both physical and emotional stress. Treatment recommendations most commonly include education, reassurance, and symptom management. This approach leaves the vast majority of patients undiagnosed and improperly treated, neglecting potential mechanism-directed therapeutic options. In contrast, it is our experience that a true etiology can be identified in many individuals with a diagnosis of a functional gastrointestinal disorder, if an expanded investigation is undertaken.
Irritable bowel syndrome (IBS) serves as the prototypical functional gastrointestinal disorder, as it is the most prevalent of the functional gastrointestinal disorders. IBS is estimated to affect ~11% of the world's population and is defined by abdominal pain and a change in bowel function (110). The diagnostic criteria for IBS include recurrent abdominal pain, on average at least 1 day per week in the last 3 months, associated with two of the following three conditions: (1) pain with defecation, (2) change in frequency of stool, or (3) change in the appearance of stool. An astute clinician can quickly realize that this simply categorizes individuals based on symptoms, and does not provide insight into the pathophysiologic determinants of the symptoms. Despite the recognition that genetic, environmental, and immune factors can increase the risk of functional gastrointestinal disorders, very few patients will undergo a thorough clinical evaluation. Within this paradigm, patients may be diagnosed with functional gastrointestinal disorders, with the true diagnosis never identified, leading to sub-optimal care. It is within this clinical space that the Gastroenterologist and Neurologist working together can provide true diagnoses and improve clinical care.
The inflammatory component of IBS has been recognized since early histological observations of increased inflammatory cells, including mast cells and T cells, in the intestinal mucosa of IBS patients (111, 112). These leukocytes are located in close proximity to nerves of the enteric nervous system that can respond to immune products such as cytokines and chemokines (113, 114). The role of inflammation is also substantiated by genome-wide association studies of IBS patients, identifying major risk alleles in immune-related genes such as TNFSF15 (115), which encodes for the cytokine tumor necrosis factor-like ligand 1A, and TLR9 (116), which encodes for a Toll-like receptor that senses microbe-derived DNA. IBS has also been linked to autoimmune conditions, including rheumatoid arthritis and psoriasis, though neural autoantibodies are only rarely found in IBS patients (117, 118). Still, stress commonly exacerbates IBS symptoms, which may be due to catecholamine-mediated induction of inflammation (119). Thus, the immune system emerges as a critical effector in IBS that can integrate signals from the nervous system and the gut microbial community to give rise to disease.
Gastroparesis
Similar to IBS, gastroparesis is defined as a syndrome of delayed emptying of solid gastric contents without mechanical obstruction. The prevalence of gastroparesis is estimated to be 9.6 per 100,000 for men and 37.8 per 100,000 for women (120). Patients with gastroparesis exhibit symptoms including nausea, early satiety, postprandial fullness, gastroesophageal reflux, abdominal pain, and often vomiting. While diabetes and surgical trauma have been identified as major etiologies for gastroparesis, idiopathic gastroparesis is by far the most common (121). Gastroparesis is reported in 20% of individuals with a ROME IV diagnosis of functional dyspepsia (122), highlighting the difficultly in distinguishing gastroparesis from functional gastrointestinal disorders. Within idiopathic gastroparesis, immunologic, neurologic, neuroimmunologic, neuromuscular, connective tissue, genetic, and metabolic dysfunction may underlie the gastroparesis, further highlighting the missed opportunities for identifying the true diagnosis. Thus, by an error of attribution, a diagnosis of a functional gastrointestinal disorder limits our ability to appropriately investigate and treat these patients.
Among the underappreciated mechanisms of gastroparesis, neuroimmunological etiologies such as infectious and autoimmune should be considered. Interestingly, some patients develop gastroparesis after acute viral infections, including Epstein-Barr, Herpes simplex, and Varicella Zoster virus (123). Viral triggering of neuroimmune pathology (124) and/or autoimmunity (125) is a well-studied phenomenon that can lead to gastroparesis due to immune-mediated damage of the enteric nervous system. In addition, gastroparesis is associated with a number of autoimmune conditions including autoimmune gastritis, Sjogren's syndrome, and scleroderma (126–128). Indeed, inflammatory cytokines and reactive species, including interleukin-6, interleukin-1β, tumor necrosis factor-α, and inducible nitric oxide synthase, have been associated with impaired intestinal motility (129). Although most cases of gastroparesis are categorized as idiopathic, neuroimmunological etiologies explain at least some of these cases, and could provide opportunity for therapeutic intervention.
Autoimmune Gastrointestinal Dysmotility
Autoimmune gastrointestinal dysmotility (AGID) is an autoimmune dysautonomia that remains underappreciated within the realm of gastroenterology. AGID presents with findings of abnormal gastrointestinal motility along with symptoms of nausea, vomiting, abdominal pain, constipation, and diarrhea. Greater than 70% of these individuals will have serologic evidence of detectable autoantibodies and others may have evidence of myenteric inflammation on full thickness biopsies from the gastrointestinal tract (130). The incidence and prevalence of AGID is unknown, as it is not routinely screened for in clinical practice, however the disease appears to be more prevalent in females (131). Patients with AGID may exhibit autonomic nervous system abnormalities limited to the gastrointestinal tract or systemic dysautonomia (130, 131). Some AGID patients also have overlapping autoimmune conditions such as Graves' disease, Celiac disease, lupus, and type I diabetes (130, 131). AGID can be idiopathic, meaning that the cause of autoantibody elevations is not yet understood, however, some cases may have a paraneoplastic etiology related to an underlying malignant process. Increased detection of AGID—and subsequent immune-directed treatment—has the potential to significantly improve outcomes and quality of life for patients with an otherwise refractory gastrointestinal motility disorder.
Diagnostic Considerations
Scintigraphy based tests are considered the gold-standard method of measuring gastrointestinal transit (132), however these tests are limited in that they can only provide information related to transit. Furthermore, only scintigraphy-based gastric emptying studies are available at most centers, with small intestinal and colonic scintigraphy limited to only very specialized centers. This translates to underdiagnosis of the vast majority of small intestinal and colonic dysmotility. Manometric based testing and wireless motility capsule (WMC) analysis can both provide information regarding intraluminal pressure measurements and coordination of muscular contractions. Until very recently, antroduodenal-jejunal and colonic manometry were considered the gold-standard for diagnosing complicated gastrointestinal motility disorders, as it was thought this technology could differentiate between a myopathic and neuropathic pathology. However, recent systematic comparison of full thickness small intestinal histopathology to antroduodenal-jejunal manometry could not reliably differentiate myopathic and neuropathic gastrointestinal disorders of motility (133). Therefore, in some individuals the identification of a gastrointestinal motility disorder, whether made by manometry-based studies or WMC, should prompt the consideration for full thickness biopsy of the gastrointestinal tract to aid in diagnosis. The WMC provides important measures of regional and global gastrointestinal transit times, intraluminal pressure measurements, pH, and temperature measurements (134, 135) and has been incredibly useful in helping to diagnose many cases of AGID and characterizing the gastrointestinal motility disturbances secondary to underling neurologic disorders. An additional advantage of the WMC, over small intestinal and colonic scintigraphy, is that it can be performed outside of highly specialized centers.
Serology is a useful tool to identify autoimmune processes. While there is a vast array of neuronal-specific autoantibodies (136), certain autoantibodies are especially useful in the context of neuroimmune-mediated gastrointestinal disease (Table 1). Classically, these anti-neuronal antibodies are thought of as paraneoplastic processes that arise against onconeural antigens expressed by the tumor environment (170), however these autoantibodies can arise even in the absence of a tumor (171). Antibodies associated with AGID include: type 1 anti-neuronal nuclear autoantibody, ganglionic nicotinic acetylcholine receptor antibody, voltage-gated potassium channel-complex antibody, N-type calcium channel antibody, P/Q-type calcium channel antibody, muscle acetylcholine receptor antibody, striational antibody, collapsing response-mediator protein-5 antibody, and glutamic acid decarboxylase 65-kilodalton isoform antibody (131). Other autoantibodies associated with gastrointestinal dysmotility include: peripherin antibody, Purkinje cell cytoplasmic antibody type 1, and smooth muscle L-type calcium channel antibody. Antibodies that target cell surface epitopes, such as receptors and channels, commonly play a direct role in the pathogenesis of neurological dysfunction (172). For example, transfer of purified ganglionic nicotinic acetylcholine receptor antibody to rodents can recapitulate dysautonomia (144). Conversely, antibodies that target intracellular epitopes are largely thought to be a marker of disease generated from the release of autoantigens during neurodegeneration (172). Serology is limited to probing the humoral arm of the immune response and does not necessarily predict response to immunomodulation. Still, serology for neuronal antibodies may have utility in the setting of significant gastrointestinal symptoms unresponsive to conventional therapy.
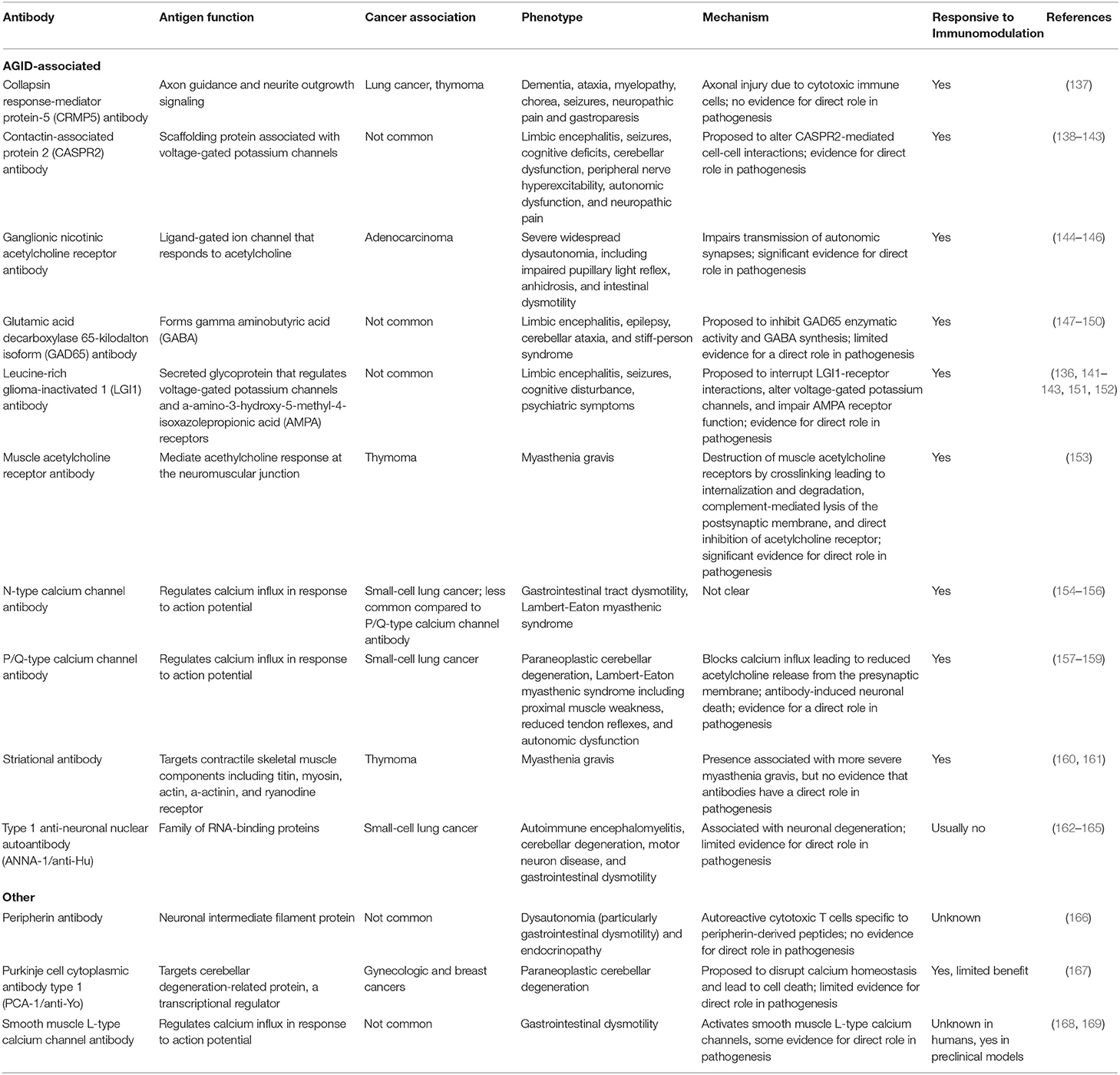
Table 1. Neural autoantibodies associated with gastrointestinal dysmotility.
Various imaging modalities can be used to capture signs of inflammation and gastrointestinal dysfunction. Abdominal CT is also commonly used to visualize the bowels and adjacent structures to uncover obstructions and other anatomical problems affecting the gastrointestinal tract (173). Similarly, FDG-PET/CT is an excellent method for the non-invasive quantification of bowel inflammation, and has been studied extensively in patients with IBD (174). These imaging modalities are critical to localize the inflammatory insult, however imaging must be combined with other diagnostics to determine the underlying cause. Specific to inflammation of the nervous system, inflammation can be more challenging to detect unless profound, but MRI of the brain and spinal cord (with contrast) can be a useful test to detect neuroinflammatory lesions on T2-weighted and fluid attenuated inversion recovery (FLAIR) sequences (175). Likewise, FDG-PET/CT can be useful, and specific to autoimmune encephalitis, FDG-PET/CT has been found to be more often abnormal than initial EEG, MRI, and CSF studies in neurology inpatients with autoimmune encephalitis, with brain region hypometabolism the most frequently observed pattern (176). Obtaining FDG-PET/CT scanning, either of the brain or body, can be quite challenging, as it is often not covered by insurance for investigation of inflammatory or immune-mediated conditions in the United States (177) or internationally.
Autonomic testing is a critical diagnostic to assess for dysautonomia. The sensitivity and quantitative nature of autonomic testing allows for the localization and severity of autonomic dysfunction to be measured. Broadly, autonomic testing evaluates adrenergic, cardiovagal, and sudomotor functions (178). Adrenergic function is assayed by monitoring blood pressure recovery during Valsalva, monitoring blood pressure and heart rate in response to head-up tilt, measuring plasma norepinephrine in response to standing, and measuring the uptake of labeled norepinephrine analogs (179). Cardiovagal function is assayed by monitoring heart rate in response to deep breathing and Valsalva (180). Sudomotor function is assayed by quantitative sudomotor axon reflex test and thermoregulatory sweat test (180). In addition to these standard autonomic tests, other tests including pupillometry, testing lacrimal and salivary gland production, and urodynamic studies can provide additional read outs of autonomic function (181–183). Combined, these tests can help delineate whether gastrointestinal dysmotility is derived from lesions of the autonomic nervous system and can also monitor patient response to treatment.
Proper workup of overlapping neurological and gastrointestinal symptoms requires the use of various diagnostic modalities (Table 2). Combined, the goals of these modalities are to localize the neurological insult, identify an immune etiology, and quantify gastrointestinal dysfunction.
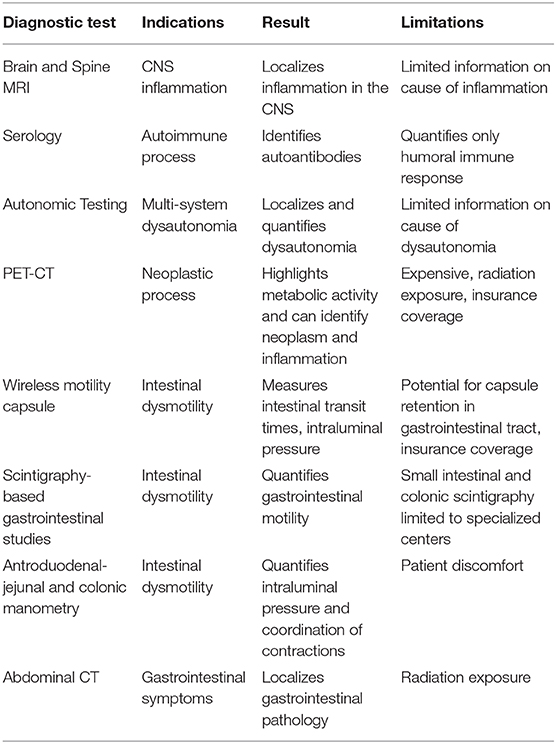
Table 2. Diagnostic modalities useful in neuroimmunogastroenterological conditions.
Therapeutic Considerations
Mild cases of IBS are commonly managed with diet and lifestyle modifications by avoidance of trigger foods and management of stress, though more severe manifestations of IBS may require drugs such as prokinetics, anticholinergics, opioid antagonists, antidiarrheals, or antibiotics (184). Significantly, mast cell-targeted drugs have shown to be beneficial in IBS. Disodium cromoglycate is an example of such an agent, which reduces the release of the major mast cell effector tryptase in jejunal biopsies, and provided symptomatic benefit to patients with diarrhea-predominant IBS (185, 186). Ebastin, a histamine 1-receptor antagonist, targets the signaling of another major mast cell product, histamine, and was found to relieve abdominal pain associated with IBS (187).
While gastroparesis is treated symptomatically with antiemetic and prokinetic medications or with gastric electrical stimulation devices (188), a subset of gastroparesis patients fail to respond to these interventions. In a retrospective study of drug and device resistant gastroparesis patients with evidence of neuroinflammation, as determined by the presence of anti-glutamic acid decarboxylase antibodies and inflammation on full-thickness gastric biopsy, intravenous immunoglobulin improved nausea, vomiting, and abdominal pain (189). This was followed up by a prospective study in a small cohort of refractory gastroparesis patients with evidence of immune dysfunction and similarly found symptomatic benefit of IVIG (190).
Patients with AGID are often identified after failing symptomatic treatment of their gastrointestinal dysmotility. In the setting of autoimmune autonomic disorders, plasma exchange has been shown to be efficacious in relieving gastrointestinal dysmotility due to dysautonomia (191). IVIG and IV methylprednisolone have also been used as diagnostic trials in patients with suspected AGID with some success in addressing gastrointestinal symptoms (130). As an example of a targeted approach, an AGID patient positive for ganglionic neuronal acetylcholine receptor was treated with pyridostigmine, an acetylcholinesterase inhibitor, resulting in alleviated gastrointestinal symptoms (192). Due to the low prevalence of AGID, studies of AGID therapy have been limited to a non-randomized, retrospective design. Still, the promising reports of immunomodulation in this population should encourage future randomized, controlled trials.
With appropriate recognition and characterization, inflammatory and autoimmune neurological disorders afford the treating physician an opportunity to improve the quality of life of patients with concomitant neurologic and gastrointestinal dysfunction.
Illustrative Case Series
We now present three clinical vignettes to illustrate our interdisciplinary approach to the diagnosis and treatment of neuroimmunogastrointestinal disorders.
Case 1
A 21-year-old female with known systemic autoimmunity including autoimmune interstitial lung disease (long-standing positive cyclic citrullinated peptide (CCP) antibody and rheumatoid factor (RF); phenotypically meeting Sjögren's syndrome criteria) and a history of neurofibromatosis type I, presented to Neurogastroenterology Clinic with atypical, episodic spells of intractable nausea and vomiting accompanied by profound dizziness. A brain MRI with and without contrast was obtained during the course of evaluation, and revealed abnormal T2/FLAIR hyperintense signal within the bilateral hippocampi suggestive of autoimmune limbic encephalitis (Figure 2). Since childhood, she had reported heartburn symptoms, frequent nausea with vomiting, motion sickness, and chronic constipation. However, these episodic spells represented a dramatic change from her baseline symptoms. At the time of her initial evaluation in Neurogastroenterology Clinic, she already had been given a diagnosis of a functional nausea and vomiting disorder, and her interstitial lung disease had been stabilized with long-standing mycophenolate mofetil. Given the description of her episodes of profound nausea and vomiting, she was referred for an MRI of the brain (results discussed above) and EEG, which was normal, along with an evaluation of gastrointestinal motility. She completed SmartPill WMC (Medtronic, Dublin, Ireland) testing with small intestinal, colonic, and global gastrointestinal motility delays noted, with preservation of appropriate colonic responses to sleep and waking. She also underwent anorectal manometry, given her reports of significant straining and sensation of incomplete evacuation with defecation, which revealed high anal sphincter pressures (maximum resting pressure 157 vs. 40–100 mm Hg; maximum squeezing pressure 298 vs. 100–180 mm Hg), visceral hypersensitivity, and evidence of an evacuation disorder. She also underwent standardized autonomic reflex testing including 10- min 70° head-up tilt (HUT), heart rate variability with deep breathing (HRDB), Valsalva maneuver, quantitative pupilometry, and sudomotor testing (Q-SWEAT). While blood pressure and heart rate responses to HUT and pupillometry were normal, Q-SWEAT responses (right forearm sweat output 0 mL/cm2 vs. 0.08 mL/cm2, right proximal leg sweat output 0.09 mL/cm2 vs. 0.19 mL/cm2, right distal leg 0.1 mL/cm2 vs. 0.14 mL/cm2), HRDB (11.86 beats per minute (BPM) vs. >14 BPM) and Valsalva ratio (1.42 vs. > 1.46) were reduced, suggesting an underlying autonomic neuropathy. Given her clinical symptoms, abnormalities noted on brain imaging, and the finding of an underlying autonomic neuropathy, her immunotherapy was changed to rituximab, resulting in a dramatic improvement, but not complete resolution, of her gastrointestinal symptoms. Addition of linaclotide 290 mcg once daily improved her complaints of chronic constipation associated with straining and sensation of incomplete evacuation with defecation, which then nearly resolved the remainder of her gastrointestinal symptoms.
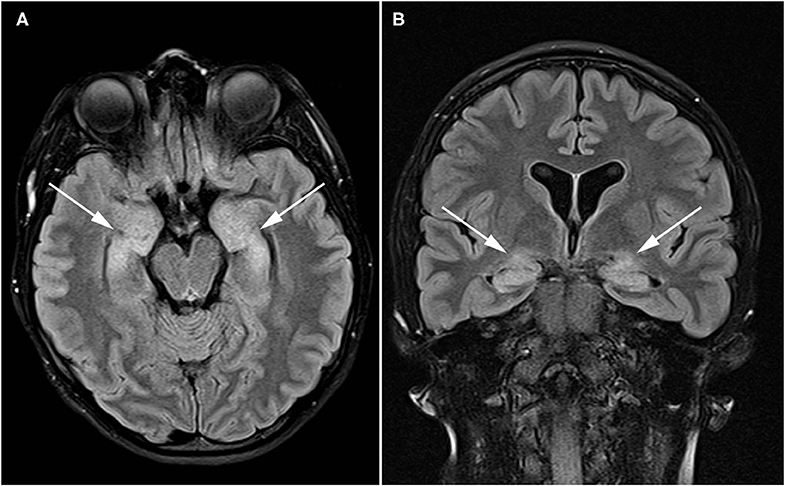
Figure 2. Patient with autoimmune enteric nervous system dysfunction and limbic encephalitis. Axial (A) and coronal (B) FLAIR images demonstrate high signal throughout the bilateral hippocampi and amygdala (arrows) compatible with limbic encephalitis.
This case highlights the danger of categorizing patients exhibiting chronic gastrointestinal symptoms with a functional disorder, or similarly, misattributing a significant change in symptomology to evolution of pre-existing gastrointestinal symptom. It is important to consider the possibility of an underlying neurological disorder when faced with atypical gastrointestinal complaints (193). This case also confirms that insults to the temporal lobe can mimic true gastrointestinal symptoms (194–196). Utilization of manometry and autonomic testing allowed for the acquisition of objective data regarding autonomic and enteric nervous system function. Imaging was also instrumental in supporting a central nervous system inflammatory process, justifying a diagnostic trial of rituximab, which alleviated many of her gastrointestinal symptoms and severe episodes of profound nausea and vomiting with dizziness.
Case 2
A 43-year-old male with a remote history of Burkitt's lymphoma treated with R-CHOP (rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone), along with multiple courses of intrathecal methotrexate and history of small bowel resection, presented to Neuroimmunology Clinic with worsening fatigue and paresthesias over the course of 5 years. A lumbar puncture performed to rule out recurrence of CNS cancer cells revealed elevated protein levels (52 mg/dL vs. 14–45 mg/dL). Prior to presentation, he had experienced a 15-pound unintentional weight loss associated with complaints of early satiety, post-prandial fullness, abdominal bloating, constipation, and anorexia. Serological testing revealed positive titers of anti-nuclear antibodies (ANA) (1:160 vs. <1:40; speckled pattern) and borderline levels of myositis specific antibodies (anti-SAE1, SUMO activating enzyme, and anti-MDA5, CADM-140), however he did not clearly fall into a specific myositis profile, as he had a normal creatine kinase and electromyography (EMG). As part of his clinical evaluation, he underwent cross-sectional imaging of his abdomen that demonstrated dilation of the stomach suggestive of profound gastroparesis. Given his symptoms, and the concern for recurrence of malignancy, a whole-body PET-CT was obtained, which demonstrated diffuse, non-specific muscle activity, along with marked gastric distention and retention of gastric contents despite being NPO for greater than 12 h prior to the imaging study (Figure 3). This finding was again suggestive of severe gastroparesis. Therefore, a SmartPill WMC study was performed, and demonstrated prolonged gastrointestinal transit times in all regions (stomach >16 h, normal <5 h; small intestine 7 h, normal 2.5–6 h; and colon >71 h, normal <59 h) and globally (>94 h, normal <73 h) (Figure 4). Given concerns for an autoimmune/autoinflammatory myositis as contributing to symptoms, abnormalities noted on PET-CT, and gastrointestinal dysmotility, he was started on infusions of intravenous methylprednisolone (eventually transitioning to oral prednisone) and rituximab. This resulted in a dramatic improvement in fatigue and paresthesias, along with gastrointestinal symptoms including early satiety, post-prandial fullness, abdominal bloating, constipation, and anorexia. Follow up SmartPill analysis three months post-treatment was again abnormal, however there was demonstrated improvements in transit times which correlated with significant improvements in gastrointestinal symptoms (Gastric emptying time, 1 hour 31 minutes; small intestine transit time, 2 h 12 minutes; colonic transit time, 25 h, 27 minutes; and global gastrointestinal transit time, 29 h 12 minutes). He was slowly tapered off 7.5 mg prednisone daily over the course of 12 months, with continuation of rituximab monotherapy and ongoing improvement of symptoms.
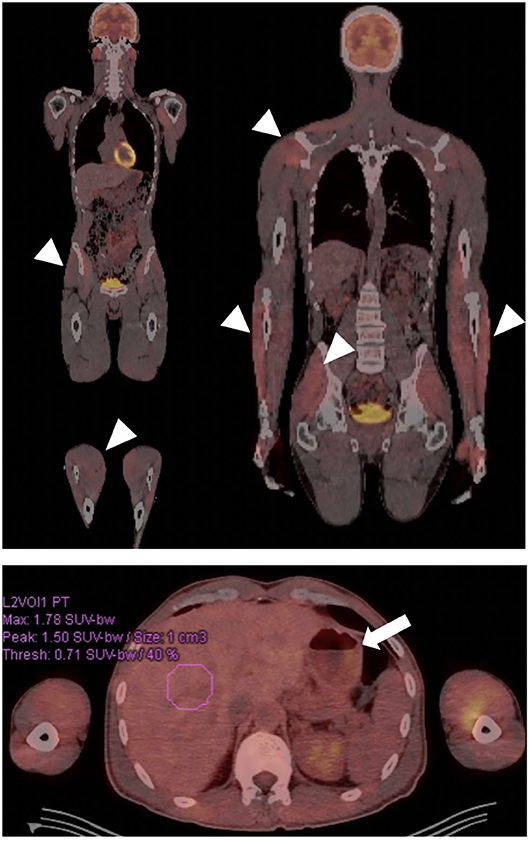
Figure 3. Patient with autoimmune gastroparesis and diffuse myositis. Body FDG-PET revealed diffuse uptake throughout the musculature compatible with the patient's known myositis (arrow heads). Gastroparesis was also incidentally demonstrated, given presence of a large amount of stomach contents despite fasting for >12 h (arrow).
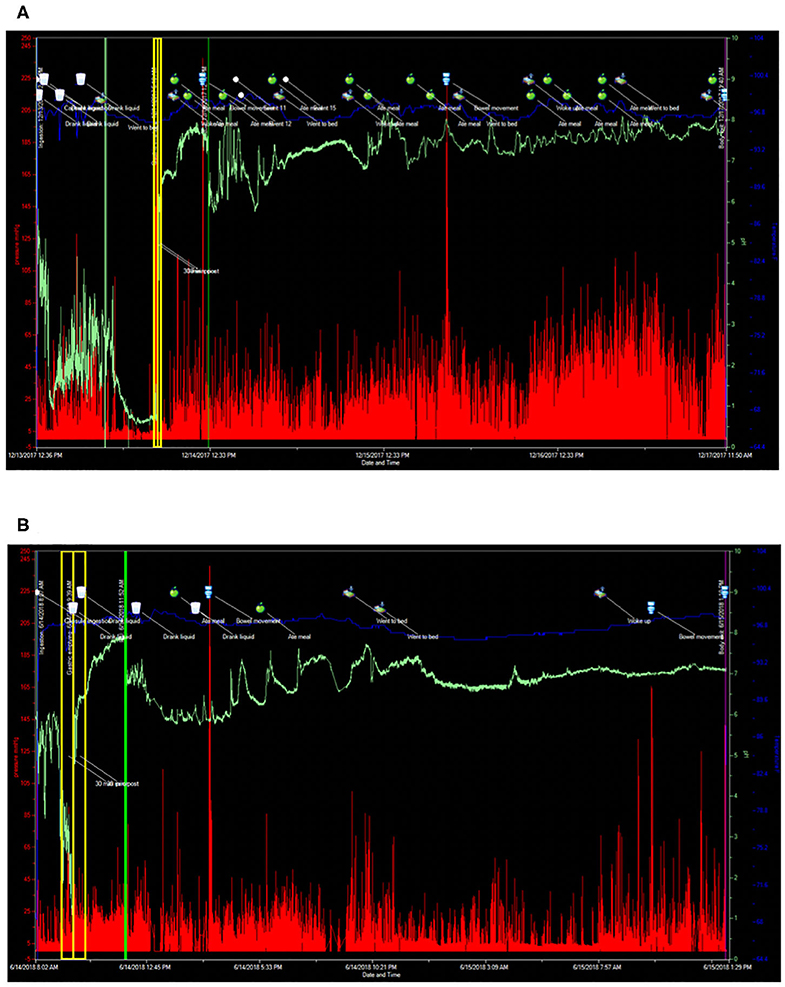
Figure 4. SmartPill wireless motility capsule studies before and after immunomodulatory therapy in a patient with autoimmune myositis. (A) Prior to commencement of Rituximab. Gastric emptying time 16 h, 38 min (normal <4 h), small intestinal transit time 7 h (normal <6 h), colonic transit time 71 h, 20 min (normal <59 h), and global gastrointestinal transit time 94 h, 58 min (normal <73 h). (B) SmartPill wireless motility capsule after commencement of steroids and Rituximab. Gastric emptying time 1 h, 31 min (normal <4 h), small intestinal transit time 2 h, 12 min (normal <6 h), colonic transit time 25 h, 27 min (normal <59 h), and global gastrointestinal transit time 29 h, 12 min (normal <73 h). For each SmartPill study below, the blue tracing represents temperature measurements, the green tracing is pH, the vertical red lines represent intraluminal pressure measurements with the height of the red line corresponding to the intensity of the pressure measurement. The vertical yellow lines correspond to the 30 min before and 30 min after gastric emptying. The vertical green line corresponds to passage of the capsule from the small intestine into the colon. The cup symbol marks fluid ingestion, the apple symbol marks solid food ingestion, the toilet symbol marks bowel movements, the bed symbol marks periods of sleep and waking.
This case emphasizes the benefits of coordinated efforts between a neuroimmunologist and a neurogastroenterologist, and the utility of PET-CT and the SmartPill wireless motility capsule, to provide objective data in an otherwise difficult to diagnose immune-mediated myositis. The history of Burkitt's lymphoma initially raised the concern for an underlying paraneoplastic process. PET-CT was helpful in ruling out recurrence of malignancy and in providing objective evidence of myositis without the need for muscle biopsy (Figure 3); an unanticipated finding was the evidence of gastroparesis, which guided further testing and led to confirmation of this previously unappreciated aspect of his disease burden. Using wireless motility capsule measurements, we demonstrated diffuse gastrointestinal dysmotility that dramatically improved after commencement of immune-modulating therapy. The objective improvements in gastrointestinal motility correlated with the dramatic improvement in the patient's gastrointestinal symptoms.
Case 3
A 44-year-old woman with clinically diagnosed hypermobile Ehlers-Danlos syndrome (EDS), confirmed by a geneticist with expertise in connective tissue disorders and based on physical examination findings along with negative genetic testing, presented to Neurogastroenterology Clinic with complaints of nausea, oral tingling, rhinorrhea, tinnitus, and urgent diarrhea with certain foods, abdominal bloating, distention, pain, and constipation. She also reported frequent dizziness and lightheadedness with standing, though without any episodes of true syncope. She was found to have positive titers of N-type calcium channel autoantibody (0.15 nmol/L vs. <0.03 nmol/L), elevated ANA (1:160) and a lip biopsy (focal lymphocytic sialadenitis) consistent with a diagnosis of seronegative Sjögren's Syndrome. For evaluation of her abdominal pain, she underwent a series of studies to look for neurogenic median arcuate ligament syndrome (nMALS). Her duplex ultrasound revealed stenosis of the celiac artery, a CTA of her abdomen demonstrated a lower lying median arcuate ligament that was closely approximating the origin of the celiac artery during both inspiration and expiration (Figure 5). There was also evidence of collateral artery formation noted on CTA. An MRI protocolized to identify the celiac ganglia was not approved by insurance. However, she had a dramatic improvement in her abdominal pain immediately following a CT guided celiac plexus block, confirming the diagnosis of nMALS. An MRI brain revealed bifrontal white matter signal abnormalities consistent with neurologic involvement of her seronegative Sjögren's Syndrome.
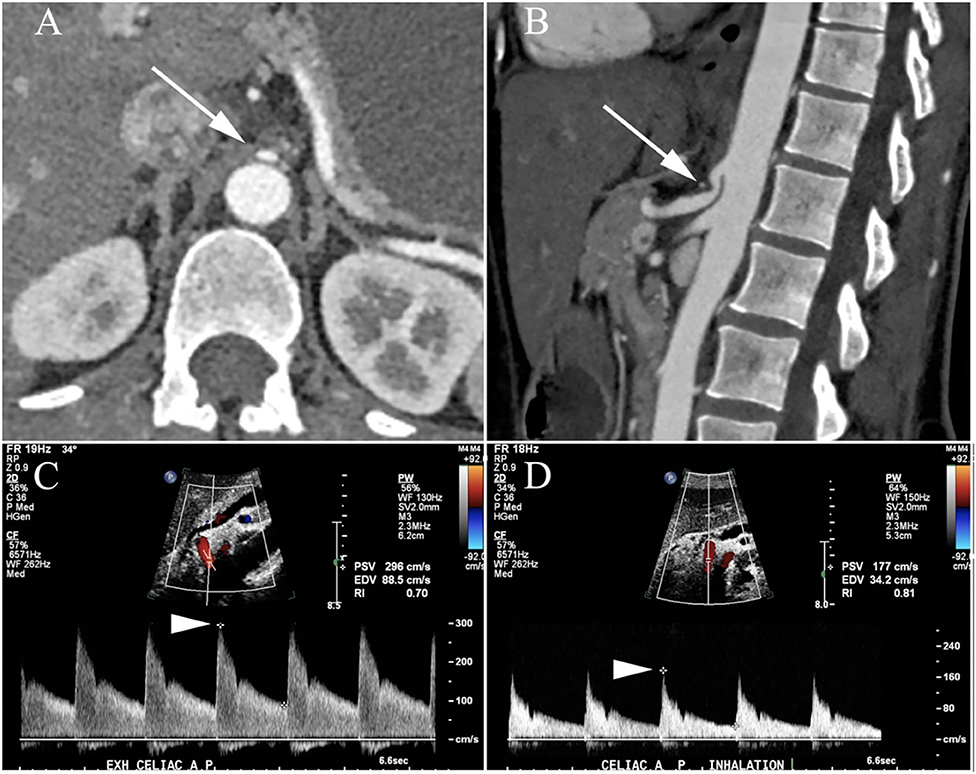
Figure 5. Patient with neurogenic median arcuate ligament syndrome (nMALS). Axial (A) and sagittal (B) CTA images demonstrate severe narrowing of the celiac artery origin (arrow) related to extrinsic compression by the median arcuate ligament. Duplex ultrasound demonstrated normal arterial flow during expiratory ventilation with a peak systolic velocity (PSV) of 286 cm/s (arrowhead, C) but marked flow impairment during inspiration with PSV of 177 cm/s (arrowhead, D) equivalent to >70% stenosis and hemodynamic compromise.
Because she also reported symptoms of oral tingling, tinnitus, and urgent diarrhea, she underwent upper endoscopy with mucosal biopsy sampling. Gastrointestinal mucosal biopsy revealed elevated mast cell counts (via CD-117 staining) in the stomach (gastric antrum; 20.4 mast cells per 10 high-powered fields (HPFs); gastric body, 20 mast cells per 10 HPFs) and duodenum (proximal duodenum, 31 mast cells per 10 HPFs; distal duodenum, 21.1 mast cells per 10 HPFs). Her laboratory testing also revealed an elevated plasma histamine. These results were consistent with a diagnosis of mast cell activation syndrome (MCAS).
Autonomic reflex testing, including 70° HUT, Q-SWEAT, and quantitative pupillometry, did not reveal an overt autonomic neuropathy, although it did demonstrate blunted late phase II blood pressure response to Valsalva and a COMPASS-31 score of 44.85 (domain scores: Orthostatic intolerance 7, Vasomotor 0, Secretomotor 0, Gastrointestinal 17, Bladder 0, and Pupillomotor 5). Current standardized autonomic reflex testing primarily focuses on the cardiovascular complications of autonomic nervous system disorders. Her testing did reveal blunted late phase II blood pressure response to Valsalva, which may be related to underlying seronegative Sjögren's Syndrome, MCAS, or EDS (Figure 6). To address her symptoms of orthostatic dizziness, she was started on NormaLyte oral rehydration salts with significant reduction in her orthostatic symptoms. To address her MCAS, she was started on ranitidine, Montelukast, cetirizine, ketotifen, and cromolyn sodium with dramatic improvement in MCAS related symptoms, including many of her neurologic complaints.
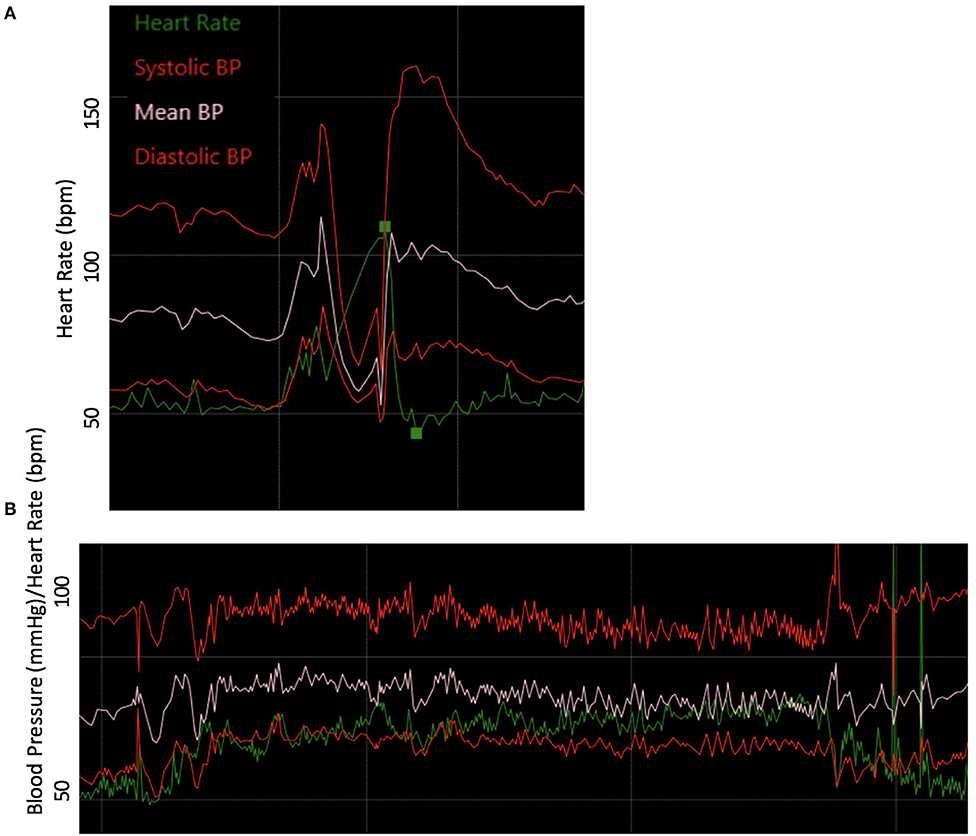
Figure 6. Abnormal autonomic testing reveals poor vasomotor tone. (A) Blood pressure responses to the Valsalva maneuver were notable for blunted late phase II. (B) The blood pressure and heart rate responses to 10- min head-up tilt accompanied by modest drop in blood pressure and heart rate increment.
Given that she had near complete, but temporary, resolution of her chronic abdominal pain with a CT guided celiac plexus block she was diagnosed with nMALS and referred for surgical decompression of the celiac ganglia and plexus. Surgical decompression completely resolved her chronic abdominal pain.
Interestingly, patients with genetic hypermobile connective tissue disorders have increased co-morbid gastrointestinal symptoms, gastrointestinal motility disorders (197), neurologic disorders, immune mediated disorders, and neuroimmunologic disorders. The mechanism by which genetic connective tissue disorders lead to the development of gastrointestinal symptoms are not completely understood. Gastrointestinal dysmotility, nMALS, and MCAS are very common comorbid illnesses within this patient population and this comorbid illness burden further complicate their care.
In this case, we were able to diagnose seronegative Sjögren's syndrome and detect N-type calcium channel autoantibodies, raising concern for neurological autoimmune disease and recommended immunomodulatory therapy, which she has declined to date, given the above overall improvement in her symptom burden. Utilizing multi-modal imaging techniques and a diagnostic celiac plexus block, we were able to support a diagnosis of nMALS (Figure 5) for which she has successfully undergone surgical decompression with complete resolution of abdominal pain, bloating, distention, and constipation. MCAS is a common occurrence in EDS patients and very commonly presents with numerous gastrointestinal and systemic symptoms (198). MCAS is defined by aberrant activation of mast cells resulting in excessive secretion of mast cell mediators including histamine, leukotrienes, and prostaglandins. The pathophysiologic mechanism that leads to higher rates of MCAS within the EDS population is not yet known. Importantly, treatment with widely available, over the counter agents that suppress mast cell effector molecules, provide significant relief from symptoms in many patients.
Conclusion
Patients with inflammatory and autoimmune neurological disorders often have comorbid gastrointestinal symptoms that are overlooked. Providers in both Neurology and Gastroenterology should no longer be satisfied with labeling patients with functional disorders, especially when these patients exhibit multi-systemic symptoms. In such cases, both neurologists and gastroenterologists may individually lack the training or capacity to address these cross-disciplinary disorders, and therefore should work in close collaboration. Case 1 demonstrated the dangers of missing a diagnosis of autoimmune limbic encephalitis by labeling the patient with a functional nausea and vomiting disorder. Case 2 highlighted the quality of care a patient can receive when clinicians take the time to integrate all the information they receive from clinical testing, as the PET-CT revealed not only myositis, but also profound gastroparesis. Furthermore, this patient met criteria for idiopathic gastroparesis, which he clearly did not have as his gastrointestinal dysmotility great improved with therapies targeted to treat his myositis. Case 3 highlighted the underappreciated neuroimmunological and gastrointestinal co-morbidities of patients with connective tissue disorders, and the utility of surgical and mast cell-based interventions. Importantly, we show examples of patients with immune mediated neurogastrointestinal disorders who were responsive to a combination of symptomatic management and immune-modulating therapy, which markedly alleviated symptoms and improved their quality of life. Finally, following expanded diagnostic investigation, therapies targeted at the underlying pathophysiology of the disease process may have the ability to prevent further disease progression along with reducing systemic complications.
Challenges and Future Directions
Neuroimmune etiologies for gastrointestinal dysfunction are underappreciated and patients with such conditions are often misdiagnosed as having functional disorders. Unfortunately, these patients may be subject to a diagnostic odyssey before identifying their underlying condition. The extensive workup required to diagnose these patients is a significant clinical challenge (Table 2). In the setting of AGID, specific autoantibodies (listed in Table 1) are highly associated with the disease and should increase clinical suspicion of this entity. However, in other neuroimmune-mediated conditions affecting the gastrointestinal tract, serological markers may be absent or may be not be clearly defined. The above cases highlight the importance of broad testing using a variety of modalities to evidence neuroinflammatory processes, though further investigation is required to define more specific diagnostic markers.
Given the lack of clinical detection of neuroimmune-mediated gastrointestinal dysfunction, investigation into treatment of such conditions has been limited to case reports and retrospective studies. The use of immunomodulatory agents, including IVIG and corticosteroids, carry risks of opportunistic infections among other side effects. Thus, such treatment should be reserved for patients that exhibit significant gastrointestinal symptoms that are not appropriately managed with symptomatic treatments. Randomized, placebo-controlled, double-blind clinical trials are needed for the variety of neuroinflammatory gastrointestinal dysmotility conditions.
Ethics Statement
We would like to confirm that written informed consent was obtained from the individual(s) AND/OR minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JS was responsible for literature review and manuscript preparation. JM was responsible for interpretation of data and critical revision of manuscript for intellectual content. MC was responsible for study concept and design, interpretation of data, and critical revision of manuscript for intellectual content. JH was responsible for critical revision of manuscript for intellectual content. LP and SC were responsible for study concept and design, interpretation of data, literature review, and critical revision of manuscript for intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH 5T32AI055434 (to JS), American Heart Association Scientist Development Grant 17SDG33460420 (to JM), NIH NIMHD LRP, American Academy of Neurology Research Training Scholarship (to MC), Office of Research on Women's Health of the NIH K12HD085852, and Accelerating Research and Innovation in Autism Through Precision Medicine (ARIA) (to LP).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge Barbara Gural Steinmetz and the Transverse Myelitis Association for their support. Artwork from the Servier Medical Art collection was used under the Creative Commons Attribution 3.0 Unported License to create Figure 1.
References
1. Furness JB, Callaghan BP, Rivera LR, Cho H-J. The enteric nervous system and gastrointestinal innervation: integrated local and central control. Adv Exp Med Biol. (2014) 817:39–71. doi: 10.1007/978–1-4939–0897-4_3
2. Krowicki ZK, Hornby PJ. Substance P and serotonin independently affect intragastric pressure when microinjected into the nucleus raphe obscurus of the rat. J Auton Nerv Syst. (1995) 51:175–9. doi: 10.1016/0165–1838(94)00133–5
3. Taché Y, Yang H, Kaneko H. Caudal raphe-dorsal vagal complex peptidergic projections: role in gastric vagal control. Peptides. (1995) 16:431–5. doi: 10.1016/0196–9781(94)00212-O
4. Miller AD, Nonaka S, Jakus J. Brain areas essential or non-essential for emesis. Brain Res. (1994) 647:255–64. doi: 10.1016/0006–8993(94)91325–0
5. Henke PG. Hippocampal pathway to the amygdala and stress ulcer development. Brain Res Bull. (1990) 25:691–5. doi: 10.1016/0361–9230(90)90044-Z
6. Hennessey A, Robertson NP, Swingler R, Compston DA. Urinary, faecal and sexual dysfunction in patients with multiple sclerosis. J Neurol. (1999) 246:1027–32. doi: 10.1007/s004150050508
7. Popescu BFG, Lennon VA, Parisi JE, Howe CL, Weigand SD, Cabrera-Gómez JA, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology. (2011) 76:1229–37. doi: 10.1212/WNL.0b013e318214332c
8. Grover M, Gibbons SJ, Nair AA, Bernard CE, Zubair AS, Eisenman ST, et al. Transcriptomic signatures reveal immune dysregulation in human diabetic and idiopathic gastroparesis. BMC Med Genomics. (2018) 11:62. doi: 10.1186/s12920–018-0379–1
9. Kraichely RE, Farrugia G, Pittock SJ, Castell DO, Lennon VA. Neural autoantibody profile of primary achalasia. Dig Dis Sci. (2010) 55:307–11. doi: 10.1007/s10620–009-0838–9
10. Furuzawa-Carballeda J, Aguilar-León D, Gamboa-Domínguez A, Valdovinos MA, Nuñez-Álvarez C, Martín-del-Campo LA, et al. Achalasia–an autoimmune inflammatory disease: a cross-sectional study. J Immunol Res. (2015) 2015:729217–18. doi: 10.1155/2015/729217
12. Browning KN, Travagli RA. Central Nervous System Control of Gastrointestinal Motility and Secretion and Modulation of Gastrointestinal Functions. Hoboken, NJ: American Cancer Society (2011).
13. Mayer EA. Gut feelings: the emerging biology of gut-brain communication. Nat Rev Neurosci. (2011) 12:453–66. doi: 10.1038/nrn3071
14. Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The Central nervous system and the gut microbiome. Cell. (2016) 167:915–32. doi: 10.1016/j.cell.2016.10.027
15. Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell. (2016) 164:378–91. doi: 10.1016/j.cell.2015.12.023
16. Grisanti LA, Woster AP, Dahlman J, Sauter ER, Combs CK, Porter JE. α1-adrenergic receptors positively regulate Toll-like receptor cytokine production from human monocytes and macrophages. J Pharmacol Exp Ther. (2011) 338:648–57. doi: 10.1124/jpet.110.178012
17. Chow AK, Gulbransen BD. Potential roles of enteric glia in bridging neuroimmune communication in the gut. Am J Physiol Gastrointest Liver Physiol. (2017) 312:G145–52. doi: 10.1152/ajpgi.00384.2016
18. Kabata H, Artis D. Neuro-immune crosstalk and allergic inflammation. J Clin Invest. (2019) 130:1475–82. doi: 10.1172/JCI124609
19. Yamasaki Y, Shimamura O, Kizu A, Nakagawa M, Ijichi H. Modulation by alpha 2-adrenergic stimulation of IgE-mediated 14C-serotonin release from rat mast cells. Agents Actions. (1983) 13:310–7. doi: 10.1007/BF01971482
20. Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med. (2003) 9:125–34. doi: 10.1007/BF03402177
21. Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. (2011) 334:98–101. doi: 10.1126/science.1209985
22. Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. (2007) 132:397–414. doi: 10.1053/j.gastro.2006.11.002
23. de las Casas-Engel M, Domínguez-Soto A, Sierra-Filardi E, Bragado R, Nieto C, Puig-Kroger A, et al. Serotonin skews human macrophage polarization through HTR2B and HTR7. J Immunol. (2013) 190:2301–10. doi: 10.4049/jimmunol.1201133
24. Csaba G, Kapa E, Cserhalmi M. Hormone receptor studies on frog macrophage cells by means of histamine, serotonin and indoleacetic acid. Endokrinologie. (1975) 65:219–23.
25. Song J, Zhang L, Bai T, Qian W, Li R, Hou X. Mast cell-dependent mesenteric afferent activation by mucosal supernatant from different bowel segments of guinea pigs with post-infectious irritable bowel syndrome. J Neurogastroenterol Motil. (2015) 21:236–46. doi: 10.5056/jnm14095
26. Starodub AM, Wood JD. Histamine suppresses A-type potassium current in myenteric neurons from guinea pig small intestine. J Pharmacol Exp Ther. (2000) 294:555–61.
27. Lotz M, Vaughan JH, Carson DA. Effect of neuropeptides on production of inflammatory cytokines by human monocytes. Science. (1988) 241:1218–21. doi: 10.1126/science.2457950
28. de la Fuente M, Bernaez I, Del Rio M, Hernanz A. Stimulation of murine peritoneal macrophage functions by neuropeptide Y and peptide YY. Involvement of protein kinase C. Immunology. (1993) 80:259–65.
29. Chorny A, Gonzalez-Rey E, Fernandez-Martin A, Pozo D, Ganea D, Delgado M. Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc Natl Acad Sci USA. (2005) 102:13562–7. doi: 10.1073/pnas.0504484102
30. Delgado M, Gonzalez-Rey E, Ganea D. The neuropeptide vasoactive intestinal peptide generates tolerogenic dendritic cells. J Immunol. (2005) 175:7311–24. doi: 10.4049/jimmunol.175.11.7311
31. Sun W, Tadmori I, Yang L, Delgado M, Ganea D. Vasoactive intestinal peptide (VIP) inhibits TGF-β1 production in murine macrophages. J Neuroimmunol. (2000) 107:88–99. doi: 10.1016/S0165–5728(00)00245–9
32. Delgado M, Leceta J, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide stimulate the induction of Th2 responses by up-regulating B7.2 expression. J Immunol. (1999) 163:3629–35.
33. Olofsson PS, Levine YA, Caravaca A, Chavan SS, Pavlov VA, Faltys M, et al. Single-pulse and unidirectional electrical activation of the cervical vagus nerve reduces tumor necrosis factor in endotoxemia. Bioelectronic Med. (2015) 2:37–42. doi: 10.15424/bioelectronmed.2015.00006
34. Nakai A, Hayano Y, Furuta F, Noda M, Suzuki K. Control of lymphocyte egress from lymph nodes through β2-adrenergic receptors. J Exp Med. (2014) 211:2583–98. doi: 10.1084/jem.20141132
35. Swanson MA, Lee WT, Sanders VM. IFN-gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J Immunol. (2001) 166:232–40. doi: 10.4049/jimmunol.166.1.232
36. Slota C, Shi A, Chen G, Bevans M, Weng N-P. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain Behav Immun. (2015) 46:168–79. doi: 10.1016/j.bbi.2015.01.015
37. Besser MJ, Ganor Y, Levite M. Dopamine by itself activates either D2, D3 or D1/D5 dopaminergic receptors in normal human T-cells and triggers the selective secretion of either IL-10, TNFalpha or both. J Neuroimmunol. (2005) 169:161–71. doi: 10.1016/j.jneuroim.2005.07.013
38. Levite M. Neuropeptides, by direct interaction with T cells, induce cytokine secretion and break the commitment to a distinct T helper phenotype. Proc Natl Acad Sci USA. (1998) 95:12544–9. doi: 10.1073/pnas.95.21.12544
39. Wheway J, Mackay CR, Newton RA, Sainsbury A, Boey D, Herzog H, et al. A fundamental bimodal role for neuropeptide Y1 receptor in the immune system. J Exp Med. (2005) 202:1527–38. doi: 10.1084/jem.20051971
40. Talbot S, Abdulnour R-EE, Burkett PR, Lee S, Cronin SJF, Pascal MA, et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron. (2015) 87:341–54. doi: 10.1016/j.neuron.2015.06.007
41. Cryan JF, O'Riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, et al. The microbiota-gut-brain axis. Physiol Rev. (2019) 99:1877–2013. doi: 10.1152/physrev.00018.2018
42. Ma Q, Xing C, Long W, Wang HY, Liu Q, Wang R-F. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J Neuroinflammation. (2019) 16:53–4. doi: 10.1186/s12974–019-1434–3
43. König J, Wells J, Cani PD, García-Ródenas CL, MacDonald T, Mercenier A, et al. Human intestinal barrier function in health and disease. Clin Transl Gastroenterol. (2016) 7:e196. doi: 10.1038/ctg.2016.54
44. Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. (2010) 16:228–31. doi: 10.1038/nm.2087
45. Tremlett H, Bauer KC, Appel-Cresswell S, Finlay BB, Waubant E. The gut microbiome in human neurological disease: a review. Ann Neurol. (2017) 81:369–82. doi: 10.1002/ana.24901
46. Bron PA, Kleerebezem M, Brummer R-J, Cani PD, Mercenier A, MacDonald TT, et al. Can probiotics modulate human disease by impacting intestinal barrier function? Br J Nutr. (2017) 117:93–107. doi: 10.1017/S0007114516004037
47. Lord C, Brugha TS, Charman T, Cusack J, Dumas G, Frazier T, et al. Autism spectrum disorder. Nat Rev Dis Primers. (2020) 6:5–23. doi: 10.1038/s41572–019-0138–4
48. Fattorusso A, Di Genova L, Dell'Isola GB, Mencaroni E, Esposito S. Autism spectrum disorders and the gut microbiota. Nutrients. (2019) 11:521. doi: 10.3390/nu11030521
49. Xu M, Xu X, Li J, Li F. Association between gut microbiota and autism spectrum disorder: a systematic review and meta-analysis. Front Psychiatry. (2019) 10:473. doi: 10.3389/fpsyt.2019.00473
50. Sharon G, Cruz NJ, Kang D-W, Gandal MJ, Wang B, Kim Y-M, et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell. (2019) 177:1600–18.e17. doi: 10.1016/j.cell.2019.05.004
51. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. (2013) 155:1451–63. doi: 10.1016/j.cell.2013.11.024
52. Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, Costa-Mattioli M. Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell. (2016) 165:1762–75. doi: 10.1016/j.cell.2016.06.001
53. Sgritta M, Dooling SW, Buffington SA, Momin EN, Francis MB, Britton RA, et al. Mechanisms underlying microbial-mediated changes in social behavior in mouse models of autism spectrum disorder. Neuron. (2019) 101:246–59.e6. doi: 10.1016/j.neuron.2018.11.018
54. Kang D-W, Adams JB, Gregory AC, Borody T, Chittick L, Fasano A, et al. Microbiota transfer therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome. (2017) 5:10–6. doi: 10.1186/s40168–016-0225–7
55. Sandler RH, Finegold SM, Bolte ER, Buchanan CP, Maxwell AP, Väisänen ML, et al. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J Child Neurol. (2000) 15:429–35. doi: 10.1177/088307380001500701
56. Grimaldi R, Gibson GR, Vulevic J, Giallourou N, Castro-Mejía JL, Hansen LH, et al. A prebiotic intervention study in children with autism spectrum disorders (ASDs). Microbiome. (2018) 6:133. doi: 10.1186/s40168–018-0523–3
57. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
58. Preziosi G, Raptis DA, Raeburn A, Thiruppathy K, Panicker J, Emmanuel A. Gut dysfunction in patients with multiple sclerosis and the role of spinal cord involvement in the disease. Eur J Gastroenterol Hepatol. (2013) 25:1044–50. doi: 10.1097/MEG.0b013e328361eaf8
59. Spear ET, Holt EA, Joyce EJ, Haag MM, Mawe SM, Hennig GW, et al. Altered gastrointestinal motility involving autoantibodies in the experimental autoimmune encephalomyelitis model of multiple sclerosis. Neurogastroenterol Motil. (2018) 30:e13349. doi: 10.1111/nmo.13349
60. Yacyshyn B, Meddings J, Sadowski D, Bowen-Yacyshyn MB. Multiple sclerosis patients have peripheral blood CD45RO+ B cells and increased intestinal permeability. Dig Dis Sci. (1996) 41:2493–8. doi: 10.1007/BF02100148
61. Nouri M, Bredberg A, Weström B, Lavasani S. Intestinal barrier dysfunction develops at the onset of experimental autoimmune encephalomyelitis, and can be induced by adoptive transfer of auto-reactive T cells. PLoS ONE. (2014) 9:e106335. doi: 10.1371/journal.pone.0106335
62. Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, et al. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS ONE. (2015) 10:e0137429. doi: 10.1371/journal.pone.0137429
63. Tremlett H, Fadrosh DW, Faruqi AA, Zhu F, Hart J, Roalstad S, et al. Gut microbiota in early pediatric multiple sclerosis: a case-control study. Eur J Neurol. (2016) 23:1308–21. doi: 10.1111/ene.13026
64. Chen J, Chia N, Kalari KR, Yao JZ, Novotna M, Soldan MMP, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep. (2016) 6:28484. doi: 10.1038/srep28484
65. Jangi S, Gandhi R, Cox LM, Li N, Glehn von F, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. (2016) 7:12015. doi: 10.1038/ncomms12015
66. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA. (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
67. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA. (2017) 114:10719–24. doi: 10.1073/pnas.1711233114
68. Katz Sand I, Zhu Y, Ntranos A, Clemente JC, Cekanaviciute E, Brandstadter R, et al. Disease-modifying therapies alter gut microbial composition in MS. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e517. doi: 10.1212/NXI.0000000000000517
69. Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Begum-Haque S, Dasgupta S, et al. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J Immunol. (2010) 185:4101–8. doi: 10.4049/jimmunol.1001443
70. Lavasani S, Dzhambazov B, Nouri M, Fåk F, Buske S, Molin G, et al. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS ONE. (2010) 5:e9009. doi: 10.1371/journal.pone.0009009
71. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. (2017) 551:585–9. doi: 10.1038/nature24628
72. Libbey JE, Sanchez JM, Doty DJ, Sim JT, Cusick MF, Cox JE, et al. Variations in diet cause alterations in microbiota and metabolites that follow changes in disease severity in a multiple sclerosis model. Benef Microbes. (2018) 9:495–513. doi: 10.3920/BM2017.0116
73. Mangalam A, Shahi SK, Luckey D, Karau M, Marietta E, Luo N, et al. Human gut-derived commensal bacteria suppress CNS inflammatory and demyelinating disease. Cell Rep. (2017) 20:1269–77. doi: 10.1016/j.celrep.2017.07.031
74. Tankou SK, Regev K, Healy BC, Cox LM, Tjon E, Kivisäkk P, et al. Investigation of probiotics in multiple sclerosis. Mult Scler. (2018) 24:58–63. doi: 10.1177/1352458517737390
75. Choi IY, Piccio L, Childress P, Bollman B, Ghosh A, Brandhorst S, et al. A diet mimicking fasting promotes regeneration and reduces autoimmunity and multiple sclerosis symptoms. Cell Rep. (2016) 15:2136–46. doi: 10.1016/j.celrep.2016.05.009
76. Cignarella F, Cantoni C, Ghezzi L, Salter A, Dorsett Y, Chen L, et al. Intermittent fasting confers protection in CNS autoimmunity by altering the gut microbiota. Cell Metab. (2018) 27:1222–35.e6. doi: 10.1016/j.cmet.2018.05.006
77. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. (2017) 3:17013–21. doi: 10.1038/nrdp.2017.13
78. Santos SF, de Oliveira HL, Yamada ES, Neves BC, Pereira A. The gut and parkinson's disease-a bidirectional pathway. Front Neurol. (2019) 10:574. doi: 10.3389/fneur.2019.00574
79. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson's disease. PLoS ONE. (2011) 6:e28032. doi: 10.1371/journal.pone.0028032
80. Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, et al. Colonic bacterial composition in Parkinson's disease. Mov Disord. (2015) 30:1351–60. doi: 10.1002/mds.26307
81. Scheperjans F, Aho V, Pereira PAB, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov Disord. (2015) 30:350–8. doi: 10.1002/mds.26069
82. Hasegawa S, Goto S, Tsuji H, Okuno T, Asahara T, Nomoto K, et al. Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson's disease. PLoS ONE. (2015) 10:e0142164. doi: 10.1371/journal.pone.0142164
83. Li C, Cui L, Yang Y, Miao J, Zhao X, Zhang J, et al. Gut microbiota differs between parkinson's disease patients and healthy controls in Northeast China. Front Mol Neurosci. (2019) 12:171. doi: 10.3389/fnmol.2019.00171
84. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell. (2016) 167:1469–80.e12. doi: 10.1016/j.cell.2016.11.018
85. Maini Rekdal V, Bess EN, Bisanz JE, Turnbaugh PJ, Balskus EP. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science. (2019) 364:eaau6323. doi: 10.1126/science.aau6323
86. Geissler A, Andus T, Roth M, Kullmann F, Caesar I, Held P, et al. Focal white-matter lesions in brain of patients with inflammatory bowel disease. Lancet. (1995) 345:897–8. doi: 10.1016/S0140–6736(95)90013–6
87. Chen M, Lee G, Kwong LN, Lamont S, Chaves C. Cerebral white matter lesions in patients with Crohn's disease. J Neuroimaging. (2012) 22:38–41. doi: 10.1111/j.1552–6569.2010.00538.x
88. Zikou AK, Kosmidou M, Astrakas LG, Tzarouchi LC, Tsianos E, Argyropoulou MI. Brain involvement in patients with inflammatory bowel disease: a voxel-based morphometry and diffusion tensor imaging study. Eur Radiol. (2014) 24:2499–506. doi: 10.1007/s00330–014-3242–6
89. Agostini A, Benuzzi F, Filippini N, Bertani A, Scarcelli A, Farinelli V, et al. New insights into the brain involvement in patients with Crohn's disease: a voxel-based morphometry study. Neurogastroenterol Motil. (2013) 25:147–82. doi: 10.1111/nmo.12017
90. Hart PE, Gould SR, MacSweeney JE, Clifton A, Schon F. Brain white-matter lesions in inflammatory bowel disease. Lancet. (1998) 351:1558. doi: 10.1016/S0140–6736(05)61123–3
91. Agostini A, Campieri M, Bertani A, Scarcelli A, Ballotta D, Calabrese C, et al. Absence of change in the gray matter volume of patients with ulcerative colitis in remission: a voxel based morphometry study. Biopsychosoc Med. (2015) 9:1. doi: 10.1186/s13030–014-0028–7
92. Zois CD, Katsanos KH, Kosmidou M, Tsianos EV. Neurologic manifestations in inflammatory bowel diseases: current knowledge and novel insights. J Crohns Colitis. (2010) 4:115–24. doi: 10.1016/j.crohns.2009.10.005
93. Casella G, Tontini GE, Bassotti G, Pastorelli L, Villanacci V, Spina L, et al. Neurological disorders and inflammatory bowel diseases. World J Gastroenterol. (2014) 20:8764–82. doi: 10.3748/wjg.v20.i27.8764
94. Lossos A, River Y, Eliakim A, Steiner I. Neurologic aspects of inflammatory bowel disease. Neurology. (1995) 45:416–21. doi: 10.1212/WNL.45.3.416
95. Leonard MM, Sapone A, Catassi C, Fasano A. Celiac disease and nonceliac gluten sensitivity: a review. JAMA. (2017) 318:647–56. doi: 10.1001/jama.2017.9730
96. McKeon A, Lennon VA, Pittock SJ, Kryzer TJ, Murray J. The neurologic significance of celiac disease biomarkers. Neurology. (2014) 83:1789–96. doi: 10.1212/WNL.0000000000000970
97. Caruso R, Pallone F, Stasi E, Romeo S, Monteleone G. Appropriate nutrient supplementation in celiac disease. Ann Med. (2013) 45:522–31. doi: 10.3109/07853890.2013.849383
98. Schrödl D, Kahlenberg F, Peter-Zimmer K, Hermann W, Kühn H-J, Mothes T. Intrathecal synthesis of autoantibodies against tissue transglutaminase. J Autoimmun. (2004) 22:335–40. doi: 10.1016/j.jaut.2004.02.001
99. Boscolo S, Lorenzon A, Sblattero D, Florian F, Stebel M, Marzari R, et al. Anti transglutaminase antibodies cause ataxia in mice. PLoS ONE. (2010) 5:e9698. doi: 10.1371/journal.pone.0009698
100. Yu XB, Uhde M, Green PH, Alaedini A. Autoantibodies in the Extraintestinal manifestations of celiac disease. Nutrients. (2018) 10:1123. doi: 10.3390/nu10081123
101. Yuki N, Susuki K, Koga M, Nishimoto Y, Odaka M, Hirata K, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barre syndrome. Proc Natl Acad Sci USA. (2004) 101:11404–9. doi: 10.1073/pnas.0402391101
102. Kaplan JG, Pack D, Horoupian D, deSouza T, Brin M, Schaumburg H. Distal axonopathy associated with chronic gluten enteropathy: a treatable disorder. Neurology. (1988) 38:642–5. doi: 10.1212/WNL.38.4.642
103. Luostarinen L, Himanen S-L, Luostarinen M, Collin P, Pirttilä T. Neuromuscular and sensory disturbances in patients with well treated coeliac disease. J Neurol Neurosurg Psychiatry. (2003) 74:490–4. doi: 10.1136/jnnp.74.4.490
104. Canales P, Mery VP, Larrondo F-J, Bravo FL, Godoy J. Epilepsy and celiac disease: favorable outcome with a gluten-free diet in a patient refractory to antiepileptic drugs. Neurologist. (2006) 12:318–21. doi: 10.1097/01.nrl.0000250950.35887.6c
105. Hadjivassiliou M, Grünewald RA, Lawden M, Davies-Jones GA, Powell T, Smith CM. Headache and CNS white matter abnormalities associated with gluten sensitivity. Neurology. (2001) 56:385–8. doi: 10.1212/WNL.56.3.385
106. Bushara KO, Goebel SU, Shill H, Goldfarb LG, Hallett M. Gluten sensitivity in sporadic and hereditary cerebellar ataxia. Ann Neurol. (2001) 49:540–3. doi: 10.1002/ana.108
107. Collins SM. Stress and the gastrointestinal tract IV. Modulation of intestinal inflammation by stress: basic mechanisms and clinical relevance. Am J Physiol Gastrointest Liver Physiol. (2001) 280:G315–8. doi: 10.1152/ajpgi.2001.280.3.G315
108. Peery AF, Dellon ES, Lund J, Crockett SD, McGowan CE, Bulsiewicz WJ, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. (2012) 143:1179–87.e3. doi: 10.1053/j.gastro.2012.08.002
109. Drossman DA, Chang L, Chey WD, Kellow J, Tack J. Rome IV: functional GI disorders: disorders of gut-brain interaction. Gastroenterology. (2016) 150:1257–61. doi: 10.24890/pc
110. Mearin F, Lacy BE, Chang L, Chey WD, Lembo AJ, Simren M, et al. Bowel disorders. Gastroenterology. (2016) 150:1393–407.e5. doi: 10.1053/j.gastro.2016.02.031
111. O'Sullivan M, Clayton N, Breslin NP, Harman I, Bountra C, McLaren A, et al. Increased mast cells in the irritable bowel syndrome. Neurogastroenterol Motil. (2000) 12:449–57. doi: 10.1046/j.1365–2982.2000.00221.x
112. Spiller RC, Jenkins D, Thornley JP, Hebden JM, Wright T, Skinner M, et al. Increased rectal mucosal enteroendocrine cells, T lymphocytes, and increased gut permeability following acute Campylobacter enteritis and in post-dysenteric irritable bowel syndrome. Gut. (2000) 47:804–11. doi: 10.1136/gut.47.6.804
113. Taché Y, Perdue MH. Role of peripheral CRF signalling pathways in stress-related alterations of gut motility and mucosal function. Neurogastroenterol Motil. (2004) 16(Suppl. 1):137–42. doi: 10.1111/j.1743–3150.2004.00490.x
114. Bischoff SC, Mailer R, Pabst O, Weier G, Sedlik W, Li Z, et al. Role of serotonin in intestinal inflammation: knockout of serotonin reuptake transporter exacerbates 2,4,6-trinitrobenzene sulfonic acid colitis in mice. Am J Physiol Gastrointest Liver Physiol. (2009) 296:G685–95. doi: 10.1152/ajpgi.90685.2008
115. Zucchelli M, Camilleri M, Andreasson AN, Bresso F, Dlugosz A, Halfvarson J, et al. Association of TNFSF15 polymorphism with irritable bowel syndrome. Gut. (2011) 60:1671–77. doi: 10.1136/gut.2011.241877
116. Villani A-C, Lemire M, Thabane M, Belisle A, Geneau G, Garg AX, et al. Genetic risk factors for post-infectious irritable bowel syndrome following a waterborne outbreak of gastroenteritis. Gastroenterology. (2010) 138:1502–13. doi: 10.1053/j.gastro.2009.12.049
117. Koloski N, Jones M, Walker MM, Veysey M, Zala A, Keely S, et al. Population based study: atopy and autoimmune diseases are associated with functional dyspepsia and irritable bowel syndrome, independent of psychological distress. Aliment Pharmacol Ther. (2019) 49:546–55. doi: 10.1111/apt.15120
118. Pittock SJ, Lennon VA, Dege CL, Talley NJ, Locke GR. Neural autoantibody evaluation in functional gastrointestinal disorders: a population-based case-control study. Dig Dis Sci. (2011) 56:1452–9. doi: 10.1007/s10620–010-1514–9
119. Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, et al. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. (2005) 135:1295–307. doi: 10.1016/j.neuroscience.2005.06.090
120. Jung H-K, Choung RS, Locke GR, Schleck CD, Zinsmeister AR, Szarka LA, et al. The incidence, prevalence, and outcomes of patients with gastroparesis in Olmsted County, Minnesota, from 1996 to 2006. Gastroenterology. (2009) 136:1225–33. doi: 10.1053/j.gastro.2008.12.047
121. Camilleri M, Chedid V, Ford AC, Haruma K, Horowitz M, Jones KL, et al. Gastroparesis. Nat Rev Dis Primers. (2018) 4:41. doi: 10.1038/s41572–018-0038-z
122. Talley NJ. Functional dyspepsia: advances in diagnosis and therapy. Gut Liver. (2017) 11:349–57. doi: 10.5009/gnl16055
123. Vijayvargiya P, Camilleri M. Gastroparesis. In: Lacy BE, DiBaise JK, Pimentel M, Ford AC, editors. Essential Medical Disorders of the Stomach and Small Intestine. A Clinical Casebook. Cham: Springer (2019). p. 23–50. doi: 10.1007/978–3-030–01117-8_2
124. Tenembaum S, Chitnis T, Ness J, Hahn JS, Group FTIPMS. Acute disseminated encephalomyelitis. Neurology. (2007) 68:S23–36. doi: 10.1212/01.wnl.0000259404.51352.7f
125. Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. (2006) 19:80–94. doi: 10.1128/CMR.19.1.80–94.2006
126. Kalkan Ç, Soykan I, Soydal Ç, Özkan E, Kalkan E. Assessment of gastric emptying in patients with autoimmune gastritis. Dig Dis Sci. (2016) 61:1597–602. doi: 10.1007/s10620–015-4021–1
127. Hammar O, Ohlsson B, Wollmer P, Mandl T. Impaired gastric emptying in primary Sjogren's syndrome. J Rheumatol. (2010) 37:2313–8. doi: 10.3899/jrheum.100280
128. Miller JB, Gandhi N, Clarke J, McMahan Z. Gastrointestinal involvement in systemic sclerosis: an update. J Clin Rheumatol. (2018) 24:328–37. doi: 10.1097/RHU.0000000000000626
129. Verheijden S, Boeckxstaens GE. Neuroimmune interaction and the regulation of intestinal immune homeostasis. Am J Physiol Gastrointest Liver Physiol. (2018) 314:G75–80. doi: 10.1152/ajpgi.00425.2016
130. Flanagan EP, Saito YA, Lennon VA, McKeon A, Fealey RD, Szarka LA, et al. Immunotherapy trial as diagnostic test in evaluating patients with presumed autoimmune gastrointestinal dysmotility. Neurogastroenterol Motil. (2014) 26:1285–97. doi: 10.1111/nmo.12391
131. Dhamija R, Tan KM, Pittock SJ, Foxx-Orenstein A, Benarroch E, Lennon VA. Serologic profiles aiding the diagnosis of autoimmune gastrointestinal dysmotility. Clin Gastroenterol Hepatol. (2008) 6:988–92. doi: 10.1016/j.cgh.2008.04.009
132. Keller J, Bassotti G, Clarke J, Dinning P, Fox M, Grover M, et al. Expert consensus document: advances in the diagnosis and classification of gastric and intestinal motility disorders. Nat Rev Gastroenterol Hepatol. (2018) 15:291–308. doi: 10.1038/nrgastro.2018.7
133. Malagelada C, Karunaratne TB, Accarino A, Cogliandro RF, Landolfi S, Gori A, et al. Comparison between small bowel manometric patterns and full-thickness biopsy histopathology in severe intestinal dysmotility. Neurogastroenterol Motil. (2018) 30:e13219. doi: 10.1111/nmo.13219
134. Tran K, Brun R, Kuo B. Evaluation of regional and whole gut motility using the wireless motility capsule: relevance in clinical practice. Therap Adv Gastroenterol. (2012) 5:249–260. doi: 10.1177/1756283X12437874
135. Farmer AD, Scott SM, Hobson AR. Gastrointestinal motility revisited: the wireless motility capsule. United Eur Gastroenterol J. (2013) 1:413–21. doi: 10.1177/2050640613510161
136. Lancaster E, Dalmau J. Neuronal autoantigens–pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. (2012) 8:380–90. doi: 10.1038/nrneurol.2012.99
137. Dubey D, Lennon VA, Gadoth A, Pittock SJ, Flanagan EP, Schmeling JE, et al. Autoimmune CRMP5 neuropathy phenotype and outcome defined from 105 cases. Neurology. (2018) 90:e103–10. doi: 10.1212/WNL.0000000000004803
138. Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. (2003) 162:1149–60. doi: 10.1083/jcb.200305018
139. Joubert B, Saint-Martin M, Noraz N, Picard G, Rogemond V, Ducray F, et al. Characterization of a subtype of autoimmune encephalitis with anti-contactin-associated protein-like 2 antibodies in the cerebrospinal fluid, prominent limbic symptoms, and seizures. JAMA Neurol. (2016) 73:1115–24. doi: 10.1001/jamaneurol.2016.1585
140. van Sonderen A, Ariño H, Petit-Pedrol M, Leypoldt F, Körtvélyessy P, Wandinger K-P, et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology. (2016) 87:521–8. doi: 10.1212/WNL.0000000000002917
141. Plantone D, Renna R, Koudriavtseva T. Neurological diseases associated with autoantibodies targeting the voltage-gated potassium channel complex: immunobiology and clinical characteristics. Neuroimmunol Neuroinflammation. (2015) 3:69–78. doi: 10.4103/2347–8659.169883
142. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain. (2012) 135:1622–38. doi: 10.1093/brain/aws082
143. Petit-Pedrol M, Sell J, Planagumà J, Mannara F, Radosevic M, Haselmann H, et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain. (2018) 141:3144–59. doi: 10.1093/brain/awy253
144. Lennon VA, Ermilov LG, Szurszewski JH, Vernino S. Immunization with neuronal nicotinic acetylcholine receptor induces neurological autoimmune disease. J Clin Invest. (2003) 111:907–13. doi: 10.1172/JCI17429
145. Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. (2000) 343:847–55. doi: 10.1056/NEJM200009213431204
146. McKeon A, Lennon VA, Lachance DH, Fealey RD, Pittock SJ. Ganglionic acetylcholine receptor autoantibody: oncological, neurological, and serological accompaniments. Arch Neurol. (2009) 66:735–41. doi: 10.1001/archneurol.2009.78
147. McKeon A, Tracy JA. GAD65 neurological autoimmunity. Muscle Nerve. (2017) 56:15–27. doi: 10.1002/mus.25565
148. McKeon A, Robinson MT, McEvoy KM, Matsumoto JY, Lennon VA, Ahlskog JE, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. (2012) 69:230–8. doi: 10.1001/archneurol.2011.991
149. Nemni R, Caniatti LM, Gironi M, Bazzigaluppi E, de Grandis D. Stiff person syndrome does not always occur with maternal passive transfer of GAD65 antibodies. Neurology. (2004) 62:2101–2. doi: 10.1212/01.WNL.0000127446.61806.2F
150. Hansen N, Grünewald B, Weishaupt A, Colaço MN, Toyka KV, Sommer C, et al. Human Stiff person syndrome IgG-containing high-titer anti-GAD65 autoantibodies induce motor dysfunction in rats. Exp Neurol. (2013) 239:202–9. doi: 10.1016/j.expneurol.2012.10.013
151. Mayasi Y, Takhtani D, Garg N. Leucine-rich glioma-inactivated protein 1 antibody encephalitis: a case report. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e51. doi: 10.1212/NXI.0000000000000051
152. Lai M, Huijbers MGM, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. (2010) 9:776–85. doi: 10.1016/S1474–4422(10)70137-X
153. Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K, et al. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J. (2007) 274:3799–845. doi: 10.1111/j.1742–4658.2007.05935.x
154. Finkel L, Koh S. N-type calcium channel antibody-mediated autoimmune encephalitis: an unlikely cause of a common presentation. Epilepsy Behav Case Rep. (2013) 1:92–6. doi: 10.1016/j.ebcr.2013.06.001
155. Morgan TT, Armitage A, Stone B, Benge J. Non paraneoplastic immune-mediated calcium channel chorea. Proc (Bayl Univ Med Cent). (2019) 32:281–2. doi: 10.1080/08998280.2019.1581318
156. Bekircan-Kurt CE, Derle Çiftçi E, Kurne AT, Anlar B. Voltage gated calcium channel antibody-related neurological diseases. World J Clin Cases. (2015) 3:293–300. doi: 10.12998/wjcc.v3.i3.293
157. Martín-García E, Mannara F, Gutiérrez-Cuesta J, Sabater L, Dalmau J, Maldonado R, et al. Intrathecal injection of P/Q type voltage-gated calcium channel antibodies from paraneoplastic cerebellar degeneration cause ataxia in mice. J Neuroimmunol. (2013) 261:53–9. doi: 10.1016/j.jneuroim.2013.05.003
158. McKasson M, Clardy SL, Clawson SA, Hill KE, Wood B, Carlson N, et al. Voltage-gated calcium channel autoimmune cerebellar degeneration: Case and study of cytotoxicity. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e222. doi: 10.1212/NXI.0000000000000222
159. Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. (2011) 10:1098–107. doi: 10.1016/S1474–4422(11)70245–9
160. Romi F, Skeie GO, Gilhus NE, Aarli JA. Striational antibodies in myasthenia gravis: reactivity and possible clinical significance. Arch Neurol. (2005) 62:442–6. doi: 10.1001/archneur.62.3.442
161. Suzuki S, Utsugisawa K, Nagane Y, Suzuki N. Three types of striational antibodies in myasthenia gravis. Autoimmune Dis. (2011) 2011:740583–7. doi: 10.4061/2011/740583
162. Szabo A, Dalmau J, Manley G, Rosenfeld M, Wong E, Henson J, et al. HuD, a paraneoplastic encephalomyelitis antigen, contains RNA-binding domains and is homologous to Elav and Sex-lethal. Cell. (1991) 67:325–33. doi: 10.1016/0092–8674(91)90184-Z
163. Greenlee JE, Clawson SA, Hill KE, Wood B, Clardy SL, Tsunoda I, et al. Neuronal uptake of anti-Hu antibody, but not anti-Ri antibody, leads to cell death in brain slice cultures. J Neuroinflammation. (2014) 11:160. doi: 10.1186/s12974–014-0160–0
164. Greenlee JE. Treatment of paraneoplastic neurologic disorders. Curr Treat Options Neurol. (2010) 12:212–30. doi: 10.1007/s11940–010-0066–9
165. Chu G, Wilson PC, Carter CD, Lennon VA, Roberts-Thomson IC. Intestinal pseudo-obstruction, type 1 anti-neuronal nuclear antibodies and small-cell carcinoma of the lung. J Gastroenterol Hepatol. (1993) 8:604–6. doi: 10.1111/j.1440–1746.1993.tb01659.x
166. Chamberlain JL, Pittock SJ, Oprescu A-M, Dege C, Apiwattanakul M, Kryzer TJ, et al. Peripherin-IgG association with neurologic and endocrine autoimmunity. J Autoimmun. (2010) 34:469–77. doi: 10.1016/j.jaut.2009.12.004
167. Venkatraman A, Opal P. Paraneoplastic cerebellar degeneration with anti-Yo antibodies - a review. Ann Clin Transl Neurol. (2016) 3:655–63. doi: 10.1002/acn3.328
168. Jackson MW, Gordon TP, Waterman SA. Disruption of intestinal motility by a calcium channel-stimulating autoantibody in type 1 diabetes. Gastroenterology. (2004) 126:819–28. doi: 10.1053/j.gastro.2003.12.015
169. Wan E-C, Gordon TP, Jackson MW. Autoantibodies to calcium channels in type 1 diabetes mediate autonomic dysfunction by different mechanisms in colon and bladder and are neutralized by antiidiotypic antibodies. J Autoimmun. (2008) 31:66–72. doi: 10.1016/j.jaut.2008.03.004
170. Kashyap P, Farrugia G. Enteric autoantibodies and gut motility disorders. Gastroenterol Clin North Am. (2008) 37:397–410, vi–vii. doi: 10.1016/j.gtc.2008.02.005
171. Knowles CH, Lang B, Clover L, Scott SM, Gotti C, Vincent A, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol. (2002) 37:166–70. doi: 10.1080/003655202753416821
172. Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol. (2010) 257:509–17. doi: 10.1007/s00415–009-5431–9
173. Frøkjaer JB, Drewes AM, Gregersen H. Imaging of the gastrointestinal tract-novel technologies. World J Gastroenterol. (2009) 15:160–8. doi: 10.3748/wjg.15.160
174. Perlman SB, Hall BS, Reichelderfer M. PET/CT imaging of inflammatory bowel disease. Semin Nucl Med. (2013) 43:420–6. doi: 10.1053/j.semnuclmed.2013.06.006
175. Quarantelli M. MRI/MRS in neuroinflammation: methodology and applications. Clin Transl Imaging. (2015) 3:475–89. doi: 10.1007/s40336–015-0142-y
176. Probasco JC, Solnes L, Nalluri A, Cohen J, Jones KM, Zan E, et al. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e352. doi: 10.1212/NXI.0000000000000352
177. National Coverage Determination Abnormal brain metabolism on FDG-PET/CT is a common early finding in autoimmune encephalitis. (NCD) for PET Scans (220.6). National Coverage Determination (NCD) for PET Scans (220.6) (2020). Available online at: https://www.cms.gov/medicare-coverage-database/details/ncd-details.aspx?NCDId=211&ncdver=3&bc=AgAAgAAACAAA& (accessed July 17, 2020).
178. Low PA, Tomalia VA, Park K-J. Autonomic function tests: some clinical applications. J Clin Neurol. (2013) 9:1–8. doi: 10.3988/jcn.2013.9.1.1
179. Illigens BMW, Gibbons CH. Autonomic testing, methods and techniques. Handb Clin Neurol. (2019) 160:419–33. doi: 10.1016/B978–0-444–64032-1.00028-X
180. Ziemssen T, Siepmann T. The investigation of the cardiovascular and Sudomotor autonomic nervous system-a review. Front Neurol. (2019) 10:53. doi: 10.3389/fneur.2019.00053
181. Muppidi S, Adams-Huet B, Tajzoy E, Scribner M, Blazek P, Spaeth EB, et al. Dynamic pupillometry as an autonomic testing tool. Clin Auton Res. (2013) 23:297–303. doi: 10.1007/s10286–013-0209–7
182. Kohler PF, Winter ME. A quantitative test for xerostomia. The Saxon test, an oral equivalent of the Schirmer test. Arthritis Rheum. (1985) 28:1128–32. doi: 10.1002/art.1780281008
183. Senchyna M, Wax MB. Quantitative assessment of tear production: a review of methods and utility in dry eye drug discovery. J Ocul Biol Dis Infor. (2008) 1:1–6. doi: 10.1007/s12177–008-9006–2
184. Camilleri M, Boeckxstaens G. Dietary and pharmacological treatment of abdominal pain in IBS. Gut. (2017) 66:966–74. doi: 10.1136/gutjnl-2016–313425
185. Lobo B, Vicario M, Martinez C, Ramos L, Alonso C, Guilarte M, et al. 156 clinical benefit in IBS after disodium cromoglycate involves mast cell-mediated recovery of healthy-like innate immunity genes expression profile in the Jejunal Mucosa. Gastroenterology. (2009) 136:A−30. doi: 10.1016/S0016–5085(09)60139–6
186. Lobo B, Vicario M, Martinez C, Gonzalez-Castro AM, Ramos L, Alonso C, et al. Clinical improvement in IBS after disodium cromoglycate involves mast cell-mediated toll-like receptor signaling downregulation. Gastroenterology. (2011) 140:499–500. doi: 10.1016/S0016–5085(11)62067–2
187. van Wanrooij S, Wouters MM, van Oudenhove L, Vermeire S, Rutgeerts PJ, Boeckxstaens GE. 901 Effect of the H1-receptor antagonist ebastin on visceral perception clinical symptoms in IBS. Gastroenterology. (2013) 144:160. doi: 10.1016/S0016–5085(13)60576–4
188. Cutts TF, Luo J, Starkebaum W, Rashed H, Abell TL. Is gastric electrical stimulation superior to standard pharmacologic therapy in improving GI symptoms, healthcare resources, and long-term health care benefits? Neurogastroenterol Motil. (2005) 17:35–43. doi: 10.1111/j.1365–2982.2004.00609.x
189. Soota K, Kedar A, Nikitina Y, Arendale E, Vedanarayanan V, Abell TL. Immunomodulation for treatment of drug and device refractory gastroparesis. Results Immunol. (2016) 6:11–4. doi: 10.1016/j.rinim.2016.02.001
190. Ashat M, Lewis A, Liaquat H, Stocker A, McElmurray L, Vedanarayanan V, et al. Intravenous immunoglobulin in drug and device refractory patients with the symptoms of gastroparesis-an open-label study. Neurogastroenterol Motil. (2018) 30:e13256. doi: 10.1111/nmo.13256
191. Schroeder C, Vernino S, Birkenfeld AL, Tank J, Heusser K, Lipp A, et al. Plasma exchange for primary autoimmune autonomic failure. N Engl J Med. (2005) 353:1585–90. doi: 10.1056/NEJMoa051719
192. Pasha SF, Lunsford TN, Lennon VA. Autoimmune gastrointestinal dysmotility treated successfully with pyridostigmine. Gastroenterology. (2006) 131:1592–6. doi: 10.1053/j.gastro.2006.06.018
193. Keane JR. Neurologic symptoms mistaken for gastrointestinal disease. Neurology. (1998) 50:1189–90. doi: 10.1212/WNL.50.4.1189
194. Murai T, Tohyama T, Kinoshita M. Recurrent diarrhea as a manifestation of temporal lobe epilepsy. Epilepsy Behav Case Rep. (2014) 2:57–9. doi: 10.1016/j.ebcr.2014.03.007
195. Thompson C, Ahmad M, Ouyang A. A case report of abdominal epilepsy in an adult with recurrent emesis. Am J Gastroenterol. (2000) 95:2610. doi: 10.1111/j.1572–0241.2000.03047.x
196. Mertz H. Role of the brain and sensory pathways in gastrointestinal sensory disorders in humans. Gut. (2002) 51(Suppl. 1):i29–33. doi: 10.1136/gut.51.suppl_1.i29
197. Zarate N, Farmer AD, Grahame R, Mohammed SD, Knowles CH, Scott SM, et al. Unexplained gastrointestinal symptoms and joint hypermobility: is connective tissue the missing link? Neurogastroenterol Motil. (2010) 22:252–78. doi: 10.1111/j.1365–2982.2009.01421.x
Keywords: autonomic disease, neuroimmunology, gastroenterology, autoimmune disease, motility disorders
Citation: Sanchez JMS, McNally JS, Cortez MM, Hemp J, Pace LA and Clardy SL (2020) Neuroimmunogastroenterology: At the Interface of Neuroimmunology and Gastroenterology. Front. Neurol. 11:787. doi: 10.3389/fneur.2020.00787
Received: 30 December 2019; Accepted: 25 June 2020;
Published: 31 July 2020.
Edited by:
Valentina Tomassini, University of Studies G. d'Annunzio Chieti and Pescara, ItalyReviewed by:
Silvia Romano, Sapienza University of Rome, ItalyCarlos Rodrigo Camara-Lemarroy, University of Calgary, Canada
Copyright © 2020 Sanchez, McNally, Cortez, Hemp, Pace and Clardy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stacey L. Clardy, c3RhY2V5LmNsYXJkeUBoc2MudXRhaC5lZHU=