 Fuxiang Chen
Fuxiang Chen Wenzhong Mei1†
Wenzhong Mei1† Dezhi Kang
Dezhi Kang- 1Department of Neurosurgery, The First Affiliated Hospital of Fujian Medical University, Fuzhou, China
- 2Department of Disease Prevention and Healthcare, Fujian Provincial Hospital, Fuzhou, China
Atypical teratoid/rhabdoid tumor (AT/RT) is a highly malignant central nervous system neoplasm predominantly found in children under the age of 3 years, and is extremely rare in adults. There is no specific clinical presentations or radiological features in reported cases of AT/RT. Diagnosis of brain AT/RT is mainly dependent on the classical pathological characteristics. We report a rare case of AT/RT arising from the trigeminal nerve and leading to progressively multiple cranial nerve palsies in a 25-year-old male patient. Microsurgical resection of the tumor has been performed and confirmed the diagnosis by postoperative pathology. To our knowledge, this is the second case of adult-onset AT/RT originating from the trigeminal nerve.
Introduction
Atypical teratoid/rhabdoid tumor (AT/RT) is a highly malignant tumor of the central nervous system (CNS), that is mostly discovered in infancy and young children, and is extremely rare in adults (1). It was firstly named by Rorke et al. (2) and was defined to be a grade IV tumor according to the 2016 World Health Organization classification of CNS tumors (3). Combination treatment of surgical resection, chemotherapy, and radiation therapy is currently widely used. However, the overall prognosis of AT/RT is still very unsatisfactory.
AT/RT distinctively presents with the deactivation of integrase interactor 1 (INI-1) tumor suppressor gene, also known as hSNF5/SMARCB1, which is located on chromosome 22q11.2. It has been widely accepted that mutation of INI-1gene or lack of protein expression plays a decisive role in the diagnosis of AT/RT (4, 5). The clinical manifestations of AT/RT mainly depend on the tumor location. Patients with AT/RT deriving from the cranial nerves have been described in few literature to date (6, 7), but these cases are still rarely reported in adults (8, 9). Here, we report a case of adult-onset AT/RT suspected to be derived from the trigeminal nerve followed by the involvement of multiple cranial nerves in a short period of time. In addition, a review of adult-onset AT/RT arising from the cranial nerves is provided.
Case Description
A 25-year-old male with an unremarkable family history presented with a 2-week history of right facial pain, numbness, and double vision. On clinical examination, the young adult was found to be experiencing facial hypesthesia and was unable to abduct the right eye. Cerebrospinal fluid obtained by lumbar puncture was basically normal. Initial magnetic resonance imaging (MRI) of the brain demonstrated a 3.0 × 2.0 cm solid mass originated from the right trigeminal nerve, with heterogeneous contrast enhancement (Figures 1A–C), which was believed to be a trigeminal schwannoma despite less typical imaging features. This patient refused surgical removal of the tumor over concerns about postoperative complications.
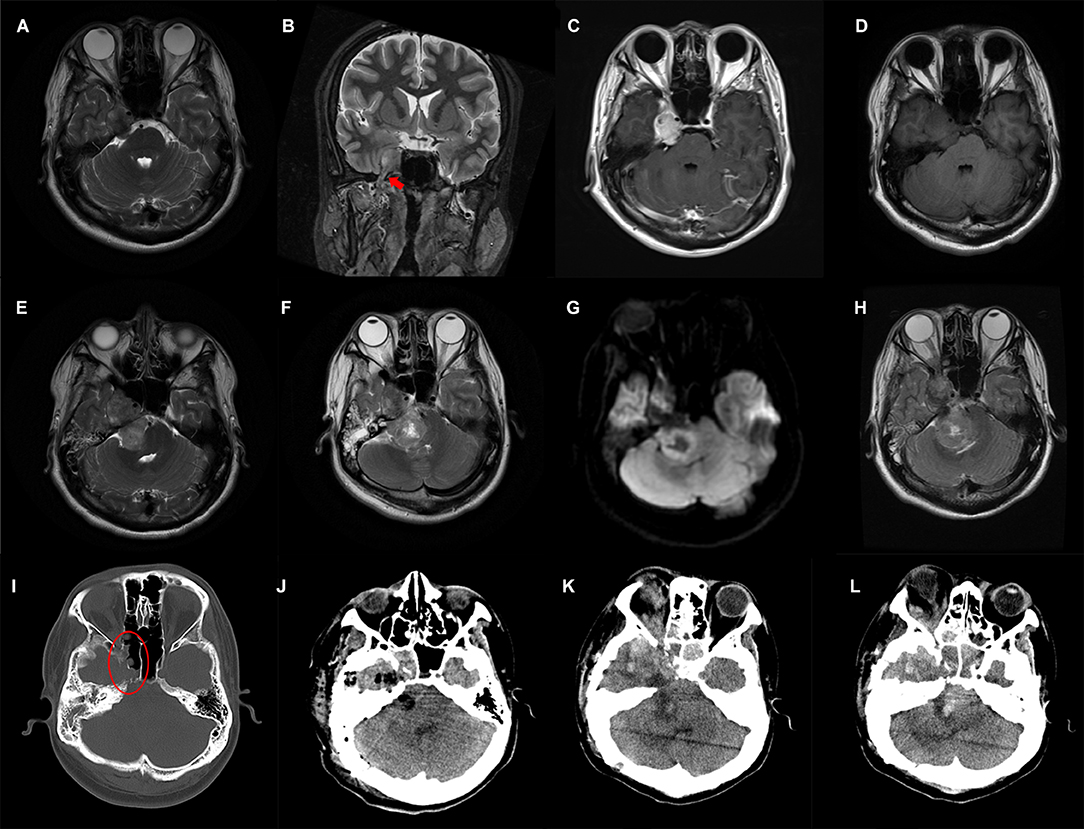
Figure 1. Preoperative and postoperative imaging features of the patient. T2-weighted MRI axial (A) and coronal (B) images show a solid tumor arising from the trigeminal nerve (red arrow) with heterogeneous contrast-enhanced (C); (D–H) Axial MRI images reveal gradual enlargement of the tumor; (I) Destruction of the adjacent skull base bone is shown in the oval; (J) Postoperative CT shows subtotal removal of the tumor; (K,L) Follow-up CT scanned 3 weeks after surgery shows tumor recurrence.
A brain MRI a month later showed slightly enlargement of the tumor (Figure 1D), ~4.2 × 2.3 cm in size. The young adult presented with unstable walking, a constant headache, a shallower right nasolabial groove, and contralateral mouth angle droop over the next 2 weeks, which made him readmit to the previous hospital. Subsequently, this patient received a 3-day CyberKnife radiosurgery (DT1800cGy/3f) in short intervals because of his continued rejection of the surgical suggestion. The young man was conscious of the partial alleviation of the above-mentioned symptoms after radiotherapy, while the rescanned MRI (Figure 1E) showed further enlargement of tumor size apart from radiation-induced edema. Emergences of new clinical symptoms, such as right-side hearing loss and ptosis, developed in this male patient in the following 2 months, with tumor extension toward the right cerebellopontine angle (6.3 × 3.2 cm), pushing the brainstem and cerebellum, as well as destruction of the adjacent bones (Figures 1F–I).
The patient was then referred to our institution and surgical removal of the dumbbell-shaped mass was performed on December 2019 via a combination of the suboccipital retrosigmoid approach and the subtemporal approach. The tumor was found to be hard with abundant blood supply and tightly adhered to the surrounding cranial nerves during the operation, resulting in subtotal resection of the mass. There was no significant hemorrhage in the operative field according to the postoperative computed tomography (CT) carried out the next day (Figure 1J). The patient's neurological status remained basically unchanged during the early postoperative period, and he felt relief of his headache and facial pain.
Histologically, the tumor was composed of sheets of cells with eosinophilic cytoplasm, prominent nucleolus, and invasive growth. Parts of the tumor cell appeared with a high nucleocytoplasmic ratio and presented with a blue small cell-like appearance (Figure 2A). Immunohistochemical (IHC) staining revealed partial immunoreactivity for epithelial membrane antigen (EMA), vimentin (VIM), CD99, synaptophysin, smooth-muscle actin (SMA), and brahma-related gene 1 (BRG1, also known as SMARCA4), while desmin, S100, and glial fibrillary acidic protein (GFAP) were negatively expressed in the tumor specimen (Figures 2B–G). In addition, the proliferative index (Ki-67) was 90%. Unexpectedly, loss of expression of the INI-1 protein was discovered in tumor cells (Figure 2H). All these pathological features, especially the lack of INI-1 protein expression, facilitated the diagnosis of AT/RT in this case.
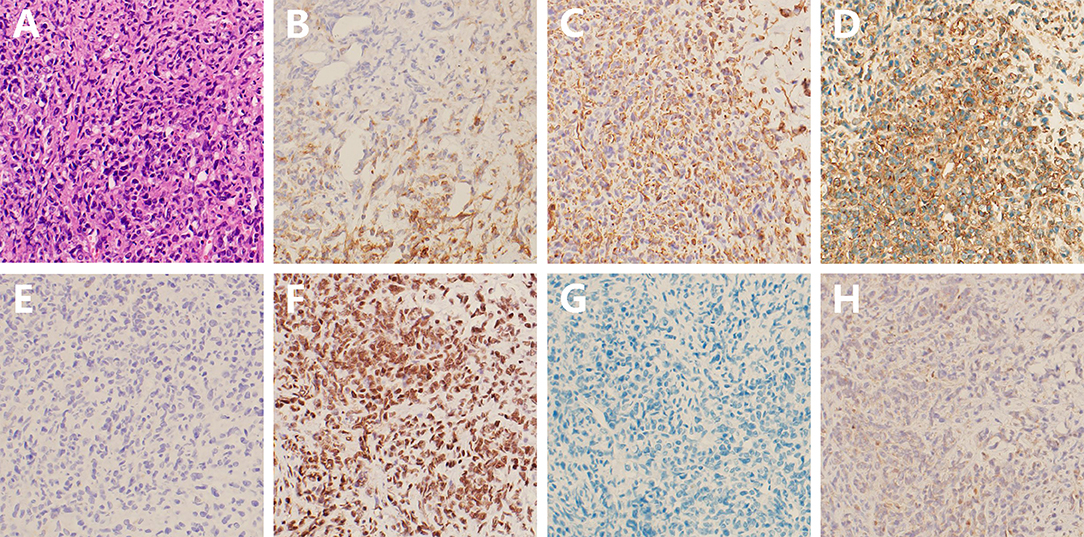
Figure 2. Hematoxylin-eosin and immunohistochemical staining of the tumor tissue. (A) Histological staining shows a high cell density with eosinophilic cytoplasm and prominent nucleolus; (B–F) Positive expression of EMA, VIM, CD99, synaptophysin, and BRG1, respectively; (G,H) Negative expression of desmin and INI-1 (at 200 magnification).
The patient was transferred to the oncology department for his first-stage chemotherapy after surgery recovery, but he suddenly developed respiratory dysfunction on the same day. Instead, treatment for pulmonary infection and respiratory support were performed during those days. Unexpectedly, a brain CT scan 3 weeks after surgery showed an obvious increase in tumor size (Figures 1K–L). He then gave up further treatment and died several days after, out of hospital.
Discussion
AT/RT has a similar incidence rate in young pediatric patients as medulloblastoma and primitive neuroectodermal tumor (PNET), at approximately 2%, but it is very rare in adults (9, 10). AT/RT occurs more frequently in men than in women, with a ratio of ~2: 1 (8). The patients suffer a poor clinical outcome regardless of their age and gender (11). To date, a total of nearly 55 adult AT/RTs have been reported in the literature (12). Only one of them originated from the trigeminal nerve (9), and this young adult reported here belongs to another one. In the present report, multiple brain nerves were involved successively due to the expansion of the tumor, which then manifested as third to eighth cranial nerve dysfunctions.
Adult-onset AT/RT can occur anywhere in the central nervous system, such as the cerebellar hemispheres, sellar region, spinal cord, pineal region, and cranial nerves (9, 13–16), even secondarily developing from other primary tumors (17, 18). In addition, the clinical manifestations and neuroimaging characteristics of patients often lacked specificity (19). Although mixed intensity in T2-weighted image, heterogeneous contrast enhancement, mild peritumoral edema, high density, and bone destruction on computed tomography have been reported frequently in adult-onset AT/RT (9, 20), there are still great challenges in accurate diagnosis of AT/RT preoperatively. In clinical practice, AT/RT is easily misdiagnosed as other types of intracranial tumor before pathological confirmation. For example, previous reports misdiagnosed cases of AT/RT arising from the optic nerve as the optic pathway glioma because of their strikingly similar characteristics on MRI (7, 21). Two other cases of adult-onset AT/RT spreading along the cranial nerve, together with ours, were also misdiagnosed as schwannomas preoperatively, and even received radiation therapy (Table 1).
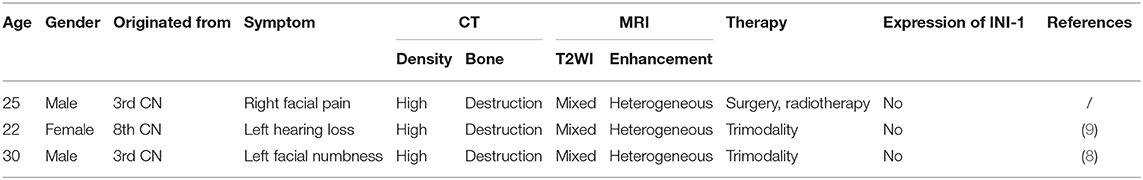
Table 1. Summary of the adult-onset AT/RT arising from cranial nerves.
The majority of AT/RTs predominantly contain characteristic rhabdoid cells, with additional neuroepithelial, epithelial, and mesenchymal constituents (22). But aforementioned histopathological features are limited to diagnosis of AT/RT due to these findings, and can also be seen in PNET and medulloblastoma (23). AT/RTs have been reported to exist with 70% mutation or deletion of INI-1 tumor suppressor gene on 22q11.2 (8). However, almost all cases of AT/RT show a loss of expression of INI-1 protein, which has been regarded as a sensitive and specific diagnostic marker (23, 24). As described in Table 1, these three cases of adult-onset AT/RT arising from the cranial nerves were also negative for expression of INI-1 protein. Consistent with previous reports (8, 12), the tumor was also positive for SMA, VIM, CD99, and EMA expression, and negative for desmin, S100, and GFAP expression. Classical immunohistochemical profiles have a high predictive value for differential diagnosis of other brain tumors (8). Notably, the proliferative index was up to 90% in our case, suggesting a high risk of poor clinical outcome.
Multiple therapeutic strategies have been taken for the treatment of pediatric AT/RT (25, 26). Due to the low incidence of AT/RT in adults, the treatment regimen was basically referenced to that of children. Trimodality therapy consisting of maximal tumor resection followed by chemotherapy and radiation therapy is currently the widely accepted treatment (26). Unfortunately, the prognosis of AT/RT is still very poor despite aggressive therapy. Therefore, there is an urgent need for more effective treatments, and exploring pathological mechanisms of adult-onset AT/RT has been one of the key factors. Recent studies have revealed that LIN28B is highly expressed in AT/RT tumors, as well as knockdown of LIN28B inhibited cell proliferation and tumorigenicity. Further studies showed that LIN28B might be regulated through INI-1, suggesting that LIN28B in AT/RT cells might be utilized as a potential therapeutic target (27, 28). In another in vitro study, the author found that pretreatment with histone deacetylase inhibitors decreased the proliferation of AT/RT cells and augmented effects of radiation therapy (29). Therefore, further studies toward the pathogenic mechanism of AT/RT may be promising to improve clinical outcomes.
Conclusion
Adult-onset AT/RT arising from the trigeminal nerve is extremely rare. Initial clinical symptoms depend on the affected cranial nerve. The early diagnosis of AT/RT before surgical resection is very difficult due to its nonspecific imaging features. Patients with AT/RT are still suffering from poor prognosis despite various therapeutic strategies. Understanding of the pathogenesis may provide a new direction for the treatment of adult-onset AT/RT in the future.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
Written informed consent was obtained from the patient's mother for the publication of any potentially identifiable images or data included in this article.
Author Contributions
FC and WL drafted the manuscript. WM participated in literature collection. TZ and DK analyzed the MRI features. XW and HY reviewed and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the Startup Fund for scientific research, Fujian Medical University (Nos. 2017XQ1084, 2018QH1154, and 2018QH1060), and the Joint Funds for the innovation of science and Technology, Fujian province (No. 2018Y9085).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Meel MH, Guillen Navarro M, de Gooijer MC, Metselaar DS, Waranecki P, Breur M, et al. MEK/MELK inhibition and blood-brain barrier deficiencies in atypical teratoid/rhabdoid tumors. Neuro Oncol. (2000) 22:58–69. doi: 10.1093/neuonc/noz151
2. Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. (1996) 85:56–65. doi: 10.3171/jns.1996.85.1.0056
3. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
4. Erkek S, Johann PD, Finetti MA, Drosos Y, Chou HC, Zapatka M, et al. Comprehensive analysis of chromatin states in atypical teratoid/rhabdoid tumor identifies diverging roles for SWI/SNF and polycomb in gene regulation. Cancer Cell. (2019) 35:95–110.e118. doi: 10.1016/j.ccell.2018.11.014
5. Pezuk JA, Salomao KB, Baroni M, Pereira CA, Geron L, Brassesco MS. Aberrantly expressed microRNAs and their implications in childhood central nervous system tumors. Cancer Metastasis Rev. (2019) 38:813–28. doi: 10.1007/s10555-019-09820-6
6. Wykoff CC, Lam BL, Brathwaite CD, Biegel JA, McKeown CA, Rosenblum MK, et al. Atypical teratoid/rhabdoid tumor arising from the third cranial nerve. J Neuroophthalmol. (2008) 28:207–11. doi: 10.1097/WNO.0b013e318183c770
7. Mahdi Y, Kharmoum J, Alouan A, Elouarradi H, Elkhiyat I, Maher M, et al. Primary atypical teratoid/rhabdoid tumor of the optic nerve: a rare entity in an exceptional location. Diagn Pathol. (2015) 10:47. doi: 10.1186/s13000-015-0284-2
8. Wang X, Liu X, Lin Z, Chen Y, Wang P, Zhang S. Atypical teratoid/rhabdoid tumor. (AT/RT) arising from the acoustic nerve in a young adult: a case report and a review of literature. Medicine. (2015) 94:e439. doi: 10.1097/MD.0000000000000439
9. Yu F, Chiang F, Bazan C. Atypical teratoid/rhabdoid tumor arising from the trigeminal nerve in an adult. Neuroradiol J. (2016) 29:447–9. doi: 10.1177/1971400916654319
10. Oh CC, Orr BA, Bernardi B, Garre ML, Rossi A, Figa-Talamanca L, et al. Atypical teratoid/rhabdoid tumor. (ATRT) arising from the 3rd cranial nerve in infants: a clinical-radiological entity? J Neurooncol. (2015) 124:175–83. doi: 10.1007/s11060-015-1787-0
11. Quinn TJ, Almahariq MF, Siddiqui ZA, Thompson AB, Hamstra DA, Kabolizadeh P, et al. Trimodality therapy for atypical teratoid/rhabdoid tumor is associated with improved overall survival: a surveillance, epidemiology, and end results analysis. Pediatr Blood Cancer. (2019) 66:e27969. doi: 10.1002/pbc.27969
12. Monteiro J, Santiago B, Manilha R, Viegas C, Oliveira A, Cunha ESM. Adult atypical teratoid/rhabdoid tumor in the pineal region: case report and literature review. World Neurosurg. (2019) 134:428–33. doi: 10.1016/j.wneu.2019.11.075
13. Roy S, Mallik C, Maiti S, Chaudhuri T. Temporal lobe atypical teratoid/ rhabdoid tumor in a 24-year old adult female. South Asian J Cancer. (2013) 2:210. doi: 10.4103/2278-330X.119909
14. Wang J, Liu Z, Fang J, Du J, Cui Y, Xu L, et al. Atypical teratoid/rhabdoid tumors with multilayered rosettes in the pineal region. Brain Tumor Pathol. (2016) 33:261–6. doi: 10.1007/s10014-016-0267-3
15. Paolini MA, Kipp BR, Sukov WR, Jenkins SM, Barr Fritcher EG, Aranda D, et al. Sellar region atypical teratoid/rhabdoid tumors in adults: clinicopathological characterization of five cases and review of the literature. J Neuropathol Exp Neurol. (2018) 77:1115–21. doi: 10.1093/jnen/nly091
16. Benesch M, Nemes K. Spinal cord atypical teratoid/rhabdoid tumors in children: clinical, genetic, and outcome characteristics in a representative European cohort. Pediatric Blood Cancer. (2000) 67:e28022. doi: 10.1002/pbc.28022
17. Kleinschmidt-DeMasters BK, Birks DK, Aisner DL, Hankinson TC, Rosenblum MK. Atypical teratoid/rhabdoid tumor arising in a ganglioglioma: genetic characterization. Am J Surg Pathol. (2011) 35:1894–901. doi: 10.1097/PAS.0b013e3182382a3f
18. Nobusawa S, Hirato J, Sugai T, Okura N, Yamazaki T, Yamada S, et al. Atypical teratoid/rhabdoid tumor. (AT/RT) arising from ependymoma: a type of AT/RT secondarily developing from other primary central nervous system tumors. J Neuropathol Exp Neurol. (2016) 75:167–74. doi: 10.1093/jnen/nlv017
19. Nowak J, Nemes K, Hohm A, Vandergrift LA, Hasselblatt M, Johann PD, et al. Magnetic resonance imaging surrogates of molecular subgroups in atypical teratoid/rhabdoid tumor. Neuro Oncol. (2018) 20:1672–9. doi: 10.1093/neuonc/noy111
20. Kanoto M, Toyoguchi Y, Hosoya T, Kuchiki M, Sugai Y. Radiological image features of the atypical teratoid/rhabdoid tumor in adults: a systematic review. Clin Neuroradiol. (2015) 25:55–60. doi: 10.1007/s00062-013-0282-2
21. Verma A, Morriss C. Atypical teratoid/rhabdoid tumor of the optic nerve. Pediatr Radiol. (2008) 38:1117–21. doi: 10.1007/s00247-008-0923-9
22. Dufour C, Beaugrand A, Le Deley MC, Bourdeaut F, Andre N, Leblond P, et al. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer. (2012) 118:3812–21. doi: 10.1002/cncr.26684
23. Varlet P, Beaugrand A, Lacroix L, Lequin D, Pierron G, Puget S, et al. Atypical teratoid rhabdoid tumor mimicking beta-catenin-positive nodular medulloblastoma. Acta Neuropathol. (2011) 121:429–30. doi: 10.1007/s00401-010-0796-5
24. Ng JM, Martinez D, Marsh ED, Zhang Z, Rappaport E, Santi M, et al. Generation of a mouse model of atypical teratoid/rhabdoid tumor of the central nervous system through combined deletion of Snf5 and p53. Cancer Res. (2015) 75:4629–39. doi: 10.1158/0008-5472.CAN-15-0874
25. McGovern SL, Okcu MF, Munsell MF, Kumbalasseriyil N, Grosshans DR, McAleer MF, et al. Outcomes and acute toxicities of proton therapy for pediatric atypical teratoid/rhabdoid tumor of the central nervous system. Int J Radiat Oncol Biol Phys. (2014) 90:1143–52. doi: 10.1016/j.ijrobp.2014.08.354
26. Ren YM, Wu X, You C, Zhang YK, Li Q, Ju Y. Multimodal treatments combined with gamma knife surgery for primary atypical teratoid/rhabdoid tumor of the central nervous system: a single-institute experience of 18 patients. Childs Nerv Syst. (2018) 34:627–38. doi: 10.1007/s00381-017-3688-3
27. Choi SA, Kim SK, Lee JY, Wang KC, Lee C, Phi JH. LIN28B is highly expressed in atypical teratoid/rhabdoid tumor. (AT/RT) and suppressed through the restoration of SMARCB1. Cancer Cell Int. (2016) 16:32. doi: 10.1186/s12935-016-0307-4
28. Rubens JA, Wang SZ, Price A, Weingart MF, Allen SJ, Orr BA, et al. The TORC1/2 inhibitor TAK228 sensitizes atypical teratoid rhabdoid tumors to cisplatin-induced cytotoxicity. Neuro Oncol. (2017) 19:1361–71. doi: 10.1093/neuonc/nox067
Keywords: atypical teratoid/rhabdoid tumor, adult, trigeminal nerve, cranial nerve, surgery
Citation: Chen F, Mei W, Lu W, Zeng T, Kang D, Wu X and You H (2020) Atypical Teratoid/Rhabdoid Tumor Originated From the Trigeminal Nerve in a Young Male Adult: Case Report and Review of the Literature. Front. Neurol. 11:265. doi: 10.3389/fneur.2020.00265
Received: 16 January 2020; Accepted: 20 March 2020;
Published: 21 April 2020.
Edited by:
Theodore Nicolaides, New York University, United StatesReviewed by:
John Crawford, University of California, San Diego, United StatesAlireza Mansouri, Pennsylvania State University (PSU), United States
Copyright © 2020 Chen, Mei, Lu, Zeng, Kang, Wu and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiyue Wu, MTM3MTg2MDg2OSYjeDAwMDQwO3FxLmNvbQ==; Honghai You, Mjk3MTgzMDY3JiN4MDAwNDA7cXEuY29t
†These authors have contributed equally to this work