 Njoud Al-Naama
Njoud Al-Naama Rafah Mackeh
Rafah Mackeh Tomoshige Kino
Tomoshige Kino
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 14 February 2020
Sec. Pediatric Neurology
Volume 11 - 2020 | https://doi.org/10.3389/fneur.2020.00032
This article is part of the Research Topic Pediatric Neurology Editor's Pick 2021 View all 10 articles
Neurodevelopmental disorders (NDDs) are multifaceted pathologic conditions manifested with intellectual disability, autistic features, psychiatric problems, motor dysfunction, and/or genetic/chromosomal abnormalities. They are associated with skewed neurogenesis and brain development, in part through dysfunction of the neural stem cells (NSCs) where abnormal transcriptional regulation on key genes play significant roles. Recent accumulated evidence highlights C2H2-type zinc finger proteins (C2H2-ZNFs), the largest transcription factor family in humans, as important targets for the pathologic processes associated with NDDs. In this review, we identified their significant accumulation (74 C2H2-ZNFs: ~10% of all human member proteins) in brain physiology and pathology. Specifically, we discuss their physiologic contribution to brain development, particularly focusing on their actions in NSCs. We then explain their pathologic implications in various forms of NDDs, such as morphological brain abnormalities, intellectual disabilities, and psychiatric disorders. We found an important tendency that poly-ZNFs and KRAB-ZNFs tend to be involved in the diseases that compromise gross brain structure and human-specific higher-order functions, respectively. This may be consistent with their characteristic appearance in the course of species evolution and corresponding contribution to these brain activities.
Neurodevelopmental disorders (NDDs) are multifaceted pathologic conditions caused by skewed development of the central nervous system (CNS) and subsequent morphological and/or functional abnormalities (1). Manifestations associated with NDDs include, but are not limited to, neuropsychiatric problems, cognitive impairment, motor dysfunctions, language/speech abnormalities, and affective deficits (2). Intellectual disability (ID), autism spectrum disorders (ASDs), motor diseases including developmental coordination disorder, communication, speech and language disorders, attention-deficit/hyperactivity disorder (ADHD), and various genetic disorders, such as Down syndrome and fragile-X syndrome, all fall into the NDD entity (1). Neuropsychiatric disorders like schizophrenia, major depressive disorder (MDD), and bipolar affective disorder (BAD) are also considered as part of the NDDs (1). There are significantly overlapping clinical symptoms between different types of NDDs (3), suggesting the presence of commonalities shared among them. The pathologic mechanisms developing NDDs emerge during the early stage of brain development organized in utero and in childhood, and this is largely due to significant involvement of genome deficits in various key genes required for normal brain development (2). Thus, identifying causative mutations/genetic abnormalities greatly facilitates our understanding of the overall pathogenesis and neuropathological processes of NDDs.
One of the major requirements for normal brain development is the precise coordination of neural stem cell (NSC) activity throughout the embryonic period to early childhood (4). NSCs are self-renewing multipotent cells that give rise to three distinct types of CNS cells: neurons, astrocytes, and oligodendrocytes (4). Differentiated neurons are critical for virtually all brain activities including the coordination of sensory and motor systems, cognitive functions, and mood maintenance (5). On the other hand, astrocytes and oligodendrocytes, also known as glial cells, support proper functioning of the differentiated neurons (5). During the early stage of embryonic brain development, NSCs originate from the neuroepithelial stem cells of the embryonic neural tube (6). NSCs undergo three major stages: (1) proliferation and renewal of the lineage, (2) migration to appropriate brain areas, and (3) differentiation into neurons, astrocytes, or oligodendrocytes; precise transitioning between these stages is critical for normal brain development (7). For example, transitioning from proliferation to differentiation and subsequent induction of the programmed cell death are crucial for the formation of normal anatomical structure of the developing brain by maintaining appropriate cell numbers (8, 9). Importantly, many of these NSCs activities are orchestrated and driven by the spatio-temporal expression of the groups of genes responsible for fine-tuning of transcriptional activity (10). Thus, dysregulation in any processes supported by these key genes impacts proper NSC activities, resulting in the development of NDDs (11, 12).
Transcription factors (TFs) are a family of protein molecules that drive gene transcription by binding directly/indirectly to the upstream genome regulatory elements of protein-coding genes (13). Accumulated evidence indicates that TFs are pivotal for brain development by influencing the ability of NSCs to differentiate into different neural cell lineages and the subsequent formation of various brain areas and substructures (14). Some TFs are also key for the precise neural cell migration to their final brain destinations (14, 15). Among such TFs, the C2H2-type zinc finger proteins (C2H2-ZNFs) are highlighted to play significant roles in the regulation of NSCs activities and subsequent brain development (16–19). Many of their family members also participate in the pathogenesis and pathophysiology of NDDs (20). In this article, we will discuss the biological activities of C2H2-ZNFs in brain development and their pathologic contribution to NDDs.
C2H2-ZNFs form the largest TF family in the animal kingdom with significant expansion of their members through species evolution (20). The family consists of ~800 members in humans (20, 21). In addition to C2H2-type zinc fingers (ZFs), these proteins contain other functional domains, such as BTB (BR-C, ttk, and bab)/POZ (Pox virus and Zinc finger), KRAB (Krüppel-associated box), and/or SCAN (SRE-ZBP, CTfin51, AW-1, and Number 18 cDNA), and are classified into four subtypes depending on the possession of these domains: (1) poly-ZNFs without any other domains, (2) BTB/POZ-ZNFs, (3) KRAB-ZNFs, and (4) SCAN-ZNFs (20, 21) (Figure 1).
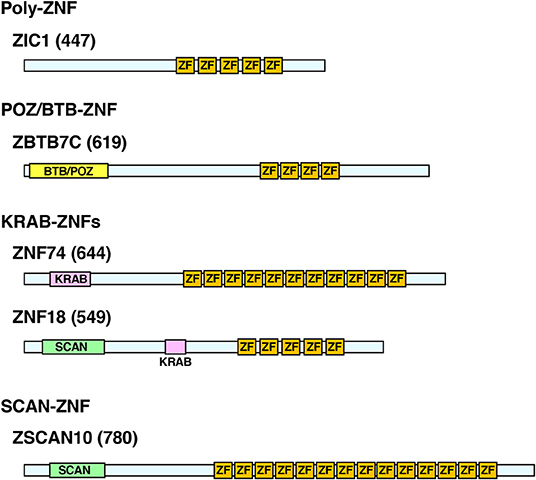
Figure 1. Protein structure of the representative human C2H2-ZNFs. Protein structure of ZIC1 (Poly-ZF), ZBTB7C (POZ/BTB-ZNF), ZNF74, ZNF18 (KRAB-ZNFs), and ZSCAN10 (SCAN-ZNF) are shown as representatives of respective subtypes. Some C2H2-ZNFs have multiples of the same or different domains as indicated in ZNF18. Location and length of the respective domains are based on the data from the UniProt (https://www.uniprot.org/) and/or the National Center of Biotechnology Information (NCBI) Conserved Domain (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Numbers in brackets indicate amino acid numbers. ZF, C2H2-type zinc finger; KRAB, KRAB domain; POZ/BTB, POZ/BTB domain; SCAN, SCAN domain.
ZFs are small peptide domains forming a secondary structure supported by a zinc ion, which makes ionic bonds to the cysteine and/or histidine residues of the finger (22). The C2H2-type ZF is composed of up to 30 amino acids with the consensus sequence CX2−4CX12HX2−8H (X refers to any amino acid), which forms one α-helix and two β-sheets, respectively, in the carboxyl- and amino-terminal portions (23–25). These secondary structures of the C2H2-type ZF fold into a stable three-dimensional assembly through hydrophobic interactions and enclosure of a zinc ion (26, 27). In C2H2-ZNFs, multiple ZFs are usually present in tandem and are connected by linkers with conserved amino acid sequences (28). C2H2-type ZNFs bind genome DNA at their cognate recognition sequences located in the regulatory region of their target protein-coding genes by forming various modes of contacts to target DNA double helices with their ZFs. DNA-bound C2H2-type ZNFs then recruit cofactors, chromatin remodeling proteins and the RNA polymerase II together with other TFs, and modulate the transcription rates of downstream coding sequences (29). In addition to the primary role as DNA-binding factors, some C2H2-type ZNFs use their ZFs for interacting with other proteins or double-stranded RNAs, which may be important for their communication with other proteins/RNAs also attracted to the multi-molecule transcriptional complex formed on DNA (28, 30, 31).
Among the functional domains of C2H2-type ZNFs, BTB/POZ, and KRAB domains have transcriptional regulatory activity (mainly repressive but sometimes enhancing) by attracting various repressive cofactor molecules, such as the histone deacetylases, corepressor complex, heterochromatin protein 1 (HP1), and/or the KRAB-associated protein-1 (KAP1). In contrast, the SCAN domain does not have such activities (21, 32). About seven percent (%) of the human C2H2-type ZNFs have a BTB/POZ domain, while 43% harbor a KRAB domain and 7% contain a SCAN domain (33). Sixty-seven percent of them have only ZFs without any of these domains (33). Some C2H2-type ZNFs have multiples of the same or of different domains (29) (Figure 1).
C2H2-ZNFs are highly expressed in the developing brain, and control early patterning of the CNS (16). They significantly contribute to the regulation of brain morphogenesis, influencing the proliferation, migration, and cell fate of NSCs or one-step committed neural progenitor cells (NPCs), and their differentiation into neuronal cells (see below). Their implications to brain disorders still remain elusive, but recent clinical studies have identified various mutations in the coding sequences of many C2H2-ZNF genes in patients with NDDs (20). Hence, we will first discuss the physiologic roles of C2H2-ZNFs in normal brain development by focusing on their involvement in the actions of NSCs or NPCs (Table 1). We will then describe their involvement in the formation of some structural components of the CNS by introducing experimental and clinical evidence. Further, we will discuss their pathologic contribution to particular forms of NDDs.

Table 1. The C2H2-ZNFs involved in brain development, NDDs, and/or other neuropsychiatric disorders.
Embryonic brain development or morphogenesis begins with neurulation, the invagination of the neural plate to form the neural tube (4). Upon closure of the neural tube, the neuroepithelial cells residing in the ventricular zone shift from proliferative to neurogenic, and are committed into the radial glial progenitor cells (RGCs), which serve as the primary NPCs for generating neurons and glial cells (8). Neocortical development relies on different NPCs depending on their localization, such as apical progenitors (APs) and basal progenitors (BPs: also known as intermediate progenitors: IPs), which are, respectively localized in the apical surface and the basal side of the ventricular zone (117). Neurogenesis starts at E9-E13 in the mouse embryo in which RGCs go into two modes of cell division: “symmetric” to produce two daughter cells that retain the properties of RGCs, and “asymmetric” dividing into one daughter cell with the property of RGCs and one differentiated neural cell (8). The transition from symmetric to asymmetric division of RGCs is extremely critical for determining the numbers of residing neurons and subsequent brain size, whereas intrinsically coordinated cell cycle progression in these cells plays a role in balancing their proliferating vs. differentiating properties (9). Disruption of these processes thus leads to abnormal brain development (9).
Various C2H2-ZNFs are significantly involved in the above indicated process of neurogenesis organized by RGCs. The birth of cortical neurons is severely reduced or lost in Gli3-mutated mice (9). Gli3 is a poly-ZNF functioning in the sonic hedgehog (Shh) signaling and controls the cell cycle of RGCs by changing the length of the G1 phase (9, 42). Inactivation of Gli3 shortens the length of their entire cell cycle and causes delays in the formation of cortical neurons and the process of cortical lamination (9, 42). Several mutations in the GLI3 gene are reported in patients with Greig cephalopolysyndactyly syndrome, Acrocallosal syndrome, and Pallister-Hall syndrome, which develop various morphological abnormalities in CNS and polydactyly (43).
The Zeb family of C2H2-ZNFs (Zeb1 and Zeb2), which are poly-ZNFs with one atypical homeodomain, is essential for normal brain development. Among them, Zeb1 is required for neocortical development (81). Its peak expression reaches during the period of neocortical development, persists at high levels throughout the embryonic neurogenesis and then decreases postnatally (81). Zeb1 acts as a transcriptional repressor and regulates proliferation, migration, and differentiation of RGCs by affecting the division mode of these cells (81). It promotes and accelerates maturation of the generated neurons and their ability to develop electrophysiological properties (81). Interestingly, inactivation of Zeb1 significantly decreases trans-differentiation from mouse embryonic fibroblasts into functional neurons in an in vitro system (81). Zeb2 (also known as Smadip1, Aip1, and Zfhxib) is essential for the transition of RGCs to Bergmann glia cells and astrocytes in mouse cerebellum (118). In humans, the ZEB2 mutation is associated with Mowat-Wilson syndrome, a genetic disorder characterized by ID, epilepsy, and motor defects (119–121).
The Zic-type poly-ZNFs (Zic1, Zic2, Zic3, Zic4, and Zic5) are expressed in the specific regions of neuroectoderm during the early embryonic phase in mice, and they have essential roles in CNS development (18, 51, 54). Specifically, Zics are pivotal for regulating the proliferation and the differentiation of NPCs in the medial forebrain and cerebellum (122), and are involved in the neurulation process and neural tissue formation (63). They are essential for the neural tube formation, particularly the neural plate closure (122). Zics expressed in the neural tube seem to play a role in the formation of the neural crests as well (122). They also contribute significantly in forebrain development, as mutations in Zic1, Zic2, and Zic3 result in an inadequate division of forebrain, which fails to develop into two hemispheres (123). Indeed, Zic1, Zic2, and Zic3 are expressed in the NPCs residing in the septum and cortical hem, the sites of generation of the Cajal-Retzius (CR) cells. Mice defective in these Zics demonstrate a reduction in the number of CR cells in the rostral cortex and develop altered localization of the CR cells and cortical lamination defects that resemble the changes noted in type II (cobblestone) lissencephaly (124). Zic1 and Zic4 are also involved in cerebellar morphogenesis (123). Simultaneous deletion of the ZIC1 and ZIC4 genes due to their close proximity in chromosome 3 results in a congenital brain anomaly called Dany-Walker malformation (DWM) in humans, which is characterized by hypoplasia of the cerebellar vermis and other brain abnormalities, and develops delayed motor development and cognitive problems in the affected individuals (18).
Several C2H2-ZNFs are implicated in the etiology of microcephaly as well. Microcephaly refers to a reduction in brain circumference and diminution in brain volume (125). The majority of cases with microcephaly are congenital forms in which the processes of neuronal proliferation, migration and/or death are affected (125). De novo deletions in the 19q13.11 region encompassing four KRAB-ZNFs (ZNF30, ZNF81, ZNF302, and ZNF599) are identified in two unrelated cases of microcephaly (86). Both patients demonstrated mild to severe ID and speech disturbances (86) and, in mice, microcephaly developed when the Znf568 gene was knocked out (104). Znf568 is the KRAB-ZNF essential for NSC maintenance and brain size regulation (104). Znf568 is expressed in NSCs of fetal mouse brain (104). It is also expressed in the adult NSCs residing in two neurogenic niches, the subgranular zone (SGZ) and the subventricular zone (SVZ) of the hippocampal dentate gyrus (104). Mice defective in Znf568 develop a significantly smaller brain compared to wild type mice (104). Reduction of the brain size in these mice is mainly due to defective neuronal migration and subsequent abnormal cortical layering (104). Further, particular single nucleotide variants in the ZNF568 gene are associated with the head size in humans (104).
ZNF519 is a poly-ZNF highly expressed in brain and is involved in the etiology of microcephaly and lissencephaly (126). The latter is a developmental malformation of the brain cortex (smooth brain without normal convolutions) caused by improper neuronal migration (126). Investigation on a four-generation Pakistani consanguineous family exhibiting congenital microcephaly (Jawad syndrome) and remarkable learning deficits mapped the causative gene(s) to the chromosome 18p11.22-q11.2, which harbors six candidate genes including ZNF519 (97). The potential contribution of ZNF519 to the development of lissencephaly is also supported by the evidence that its expression is downregulated in mice with Lis1, Dcx, or Ywhae knockouts, whose gene mutations are causative for lissencephaly in humans (126). Zfp462, a poly-ZNF involved in the pluripotency and differentiation of embryonic stem cells by regulating the expression of Sox2, Oct4, and Nanog TFs in mice (127), modulates the expression of the genes specific to neuronal differentiation (59). It is predominantly expressed in the embryonic cerebral cortex particularly in the ventricular zone and hippocampus (59). Homozygotic Zfp462 knockout is lethal in mice, whereas the heterozygotic mice exhibit developmental delay with low brain weight and anxiety-like behavior with excessive self-grooming (59). ZNF148 is a poly-ZNF associated with congenital brain structural defects in humans. ZNF148 is highly expressed in the developing fetal brain in humans and is crucial for the development of the corpus callosum (19). Four patients harboring de novo truncating mutations in the ZNF148 gene shared core syndromic features including abnormal development of corpus callosum, microcephaly, ID, short stature, and facial dimorphisms (19).
ZNF521 is the KRAB-ZNF acting as one of the intrinsic factors for driving commitment of NSCs to NPCs (61). It also promotes proliferation of these cells and delays their differentiation (61). ZNF24/ZNF191 is a KRAB-ZNF also harboring one SCAN domain (128). ZNF24/ZNF191 is expressed in NPCs, and is required for the maintenance of their proliferation potency by promoting cell cycle progression (115). Accordingly, ZNF24/ZNF191 expression is pronounced during early brain development and its expression decreases after all differentiation occurs (115).
ID is one type of the generalized NDDs characterized by significant impairment of intellectual (such as learning and reasoning) and adaptive functioning (activities for daily living, such as communication and independent living) (129). It is a heterogeneous disorder with regard to its clinical and genetic characteristics (130). Some C2H2-ZNFs are involved in the development of ID, particularly the form called X-linked intellectual disability (XLID) (131). This genetic disease is inherited in an X-linked repressive fashion, and thus, affected boys demonstrate more obvious phenotypes than girls (131). Several KRAB-ZNF genes, such as ZNF41, ZNF81, ZNF148, ZNF673, and ZNF674, residing on chromosome X are reported as novel causative genes for XLID with strong association to particular phenotypes among the other ~200 candidate genes (109, 132). Several unrelated ID patients displaying similar manifestations and developmental delays shared the same mutations in the ZNF674 and ZNF673 genes (108, 109). Four patients harboring de novo truncation mutations in the ZNF148 gene demonstrated overlapping clinical manifestations including ID, microcephaly, and mal-development of the corpus callosum (19). Two mutations in the ZNF711 gene, which is also located on chromosome X and encodes a poly-ZNF protein, were identified in 11 XLID patients from two families, some of whom additionally demonstrated autistic features (64). ZNF711 has a role in brain development by binding to the PHD finger protein 8 (PHF8) that is the histone demethylase highly expressed in neurons, and failure of ZNF711 to bind to PHF8 affects normal neuronal migration (64, 65). Mutations in the PHF8 gene, which is also located on chromosome X, cause Siderius-type XLID, characterized by facial dysmorphism, cleft lip/palate, and occasionally microcephaly and ID (133, 134).
ASDs are a group of pervasive NDDs demonstrating heterogeneous manifestations mainly characterized by deficits in social cognition, communication, and restricted behavior with repetitive phenotypes (135). They can range from mild social cognitive impairment to debilitating cognitive abilities (135). Accumulating evidence indicates the significant contribution of C2H2-ZNFs in the pathogenesis and pathophysiology of ASDs and autistic features. These include BCLLA, FEZF1, FEZF2, GLIS1, POGZ, TSHZ3, ZBTB16, ZBTB20, ZNF8, ZNF18, ZNF81, ZNF182, ZNF292, ZNF385B, ZNF407, ZNF462, ZNF517, ZNF548, ZNF559, ZNF626, ZNF713, ZNF774, ZNF778, ZNF804A, ZNF827, and many of them are KRAB-ZNFs (Table 1). Below, we explain the contributions of some of these C2H2-ZNFs in the development of ASDs and autistic features.
A 335.4 Kb-size microduplication located in the Xp11.2p11.3 segment of chromosome X, which includes KRAB-ZNF-expressing ZNF81 and ZNF182, was identified in a patient demonstrating developmental retardation, autistic features, and delayed growth and speech (91). Elevated ZNF182 expression was identified in another ASD case displaying hyperactivity, learning and visual-spatial difficulties, and microcephaly (136). The latter patient harbored a 1.3 Mb-size micro-duplication in Xp11.23p11.3 that includes ZNF182 (136). These two cases suggest that elevated expression of ZNF182 with the dosage nature of its encoding gene contributes to the development of their ASD phenotypes. ZNF292 is also a potential target gene for ASDs (55, 56). One study employing a large cohort of the ASD probands obtained from the Autism Clinical and Genetic Resourced in China (ACGC) indicated ZNF292 as a novel autism risk gene, as the patients harboring various mutations in this gene demonstrated ID and severe language impairment (55). Another study using a large ASD cohort collected from several countries identified four unrelated individuals who had deletions of the ZNF292 gene (56). Homozygotic and compound heterozygotic mutations in the ZNF18 gene were identified in the Autism Genetic Research Exchange (AGRE) cohort consisting of ~1,000 multiplex ASD families (85). ZNF18 is a KRAB-ZNF with one SCAN domain, and is upregulated upon depolarization in mouse neuronal cells, suggesting its potential activity-dependent roles (85). The ZBTB20 gene is also involved in the development of ASDs in addition to other types of NDDs including 3q13.31 microdeletion and microduplication syndrome, Primrose syndrome and ID (72, 137–139). ZBTB20 is a BTB/POZ-ZNF mainly expressed in the developing forebrain neocortex and is involved in cortical neurogenesis, hippocampal neuronal differentiation and connectivity, and promotes astrocytogenesis (140). Four unrelated individuals with de novo inactivating mutations in the Krüppel-like factor 7 (KLF7) gene exhibited autistic features along with ID (141). KLF7 is a poly-ZNF, and is essential for neurogenesis and is involved in neuronal differentiation and morphogenesis (141, 142). Klf7-knockout mice showed impaired axon projection in several brain regions including the cerebral cortex and hippocampus, and exhibited reduced dendritic branching in hippocampus (142). The pogo transposable element with ZNF domain (POGZ) gene is also a plausible candidate for ASDs, as de novo missense or nonsense mutations in this gene were identified in at least eight independent ASD patients (143–145). POGZ, a unique poly-ZNF harboring the transposase domain at its C-terminus in addition to nine ZFs (145), is highly expressed in the human fetal brain and is involved in mitosis and regulation of neural proliferation (145). POGZ is also implicated in the development of NDDs and microcephaly, as several de novo loss-of-function mutations in this gene were identified in seven patients showing these manifestations (146).
Several C2H2-ZNFs have etiologic roles in the manifestations associated with Down syndrome. Down syndrome is a common chromosomal disease caused by the chromosome 21 trisomy or its various rearrangements, and develops ID and constellations of morphological abnormalities (147). Some patients also demonstrate the manifestations reminiscent of ASDs (148). The Tc1 mouse model of Down syndrome shows elevated expression of Znf295 (also known as Zbtb21) in the brain cortex, and its human ortholog is located on chromosome 21, thus dosage abnormality in this BTB/POZ-ZNF may contribute to the development of some neurological manifestations associated with Down syndrome (149). Since ZNF298 is located on chromosome 21q22.3 and duplication of this segment is strongly associated with the development of Down syndrome (150, 151), dosage abnormality in ZNF298 appears to contribute to the development of some manifestations of this disease (152). ZNF298 is a poly-ZNF and has a SET [Su(var)3-9, Enhancer-of-zeste, Trithorax] domain in its N-terminal portion (152).
Schizophrenia is a complex NDD characterized by psychotic symptoms, such as hallucinations and delusions, accompanied by variable degrees of loss of insight (153). Interplay between genetic, biological, environmental, and psychological factors are supposed to play roles in the development of these manifestations (89, 153). ZNF74 was identified as a candidate gene for modifying the development of schizophrenia in particular patient groups (89). ZNF74 encodes a KRAB-ZNF, is highly expressed in the developing brain and is located on chromosome 22q11, a gene segment previously identified as a positional candidate locus for the susceptibility to schizophrenia as part of the 22q11 deletion syndrome (154). ZNF74 is highly expressed in the developing fetal brain in humans (19). Several polymorphisms identified in ZNF74 are significantly associated with age-at-onset of schizophrenia, although no statistical difference was detected for their frequencies between the patients and control subjects (89). Systematic meta-analysis on the psychotic diseases including schizophrenia, BAD, and ADHD identified several gene variants in ZNF804A, the zinc finger DHHC-type-containing 8 (ZDHHC8) and the zinc finger with KRAB and SCAN domain 4 (ZKSCAN4) genes (67). The ZNF804A variants, particularly rs1344706, located in the intronic sequence of this gene are highly associated with the development of schizophrenia and its various manifestations (155). ZNF804A expresses a poly-ZNF harboring just one ZF, and its reduced expression is likely important for the development of schizophrenia in part by changing the expression of the genes involved in neural cell adhesion, neurite outgrowth, and synapse formation (155). Although ZDHHC8 is located on chromosome 22q11 and was initially identified as a potential candidate gene for schizophrenia, it turned out not to be involved in this disease in later studies (156, 157). The ZKSCAN4 gene, also known as ZNF307 or ZNF427, expresses a KRAB-ZNF that harbors a SCAN domain in its amino-terminus (158). This gene is located on chromosome 6p21p22.1, which was previously identified as one of the schizophrenia-associated gene loci (159). Several ZKSCAN4 polymorphisms were strongly associated with schizophrenia in the Chinese Han population (160), although underlying molecular mechanisms are not known.
Mood disorders, such as MDD and BAD, are among the most common brain disorders caused by various abnormalities in the brain (e.g., imbalance of neurotransmitters), and particular genetic backgrounds precipitate these diseases (161). Several C2H2-ZNFs are involved in their pathogenesis. A novel point mutation in the ZNF34 gene that replaces proline at the amino acid position 17 to arginine (P17R) was identified in a multi-generationally affected family with early-onset MDD (87). The mutation P17R is located in the KRAB-A domain of ZNF34, which is required for the repressive transcriptional activity of this protein, suggesting defective transcriptional regulation by the mutant protein appears to contribute to the development of MDD. ZNF34 is also associated with BAD; ZNF34 mRNA was differentially expressed in the postmortem brain samples obtained from patients with BAD (162). ZNF34 also contains common variants precipitated in this disease (163). Further, ZNF34 is located on chromosome 8p24.3, which is included in the region shown to be associated with BAD (164, 165). One ZNF536 polymorphism (rs77554113) is correlated with remission rates of MDD patients who are under anti-depressant treatment, indicating its potential roles in MDD-related pathophysiologic processes (166). ZNF536 is a poly-ZNF highly expressed in neuronal cells and known to suppress neuronal differentiation (21, 63).
Brain development is organized by the sophisticated coordination of the proliferation, differentiation, migration, and cell death of its component neural cells (4). This is accomplished, mainly, by the intrinsic program of the self-renewing cell lineages, NSCs, and NPCs, through coordinated regulation of their transcriptional network by numerous TFs and transcriptional regulatory molecules (4). Importantly, these processes are under the influence of the individual's genetic background as well as the vulnerability to extrinsic factors, such as infectious agents, toxic substances, and various maternal conditions including immunity (2). Skewing any part of this regulatory network causes NDDs, leading to the development of various degrees of social, emotional, cognitive, and motor deficits (2).
Our literature-based analysis on the brain development and NDDs revealed that numerous C2H2-ZNF proteins (74, ~10% of all human member proteins) are essential or involved in these conditions (Table 1). Indeed, many of them play critical roles in the proper functioning of NSCs, such as their potencies of proliferation and commitment into differentiated neural cell lineages. We found that different C2H2-ZNFs act on specific functions of these self-renewing cells at the particular developmental stages and their residing brain areas, and defective actions of C2H2-ZNFs develop characteristic morphological and/or functional abnormalities depending on their actions, expressed timing and residing cells.
Although there are substantial numbers of exceptions, defective poly-ZNFs (e.g., BLI3, ZEBs, and ZICs) tend to be associated with the NDDs with gross abnormality in brain morphology and/or structure, whereas dysfunction of the C2H2-ZNFs harboring additional domains, such as KRAB and SCAN (e.g., ZNF18, ZNF34, ZNF81, ZNF427, ZNF673, ZNF804A, and ZBTB20) are linked to the development of NDDs with abnormality in higher-order brain functions, such as cognitive deficit, memory loss, and emotional changes, represented by ID, ASDs, schizophrenia, MDD, and/or BAD. C2H2-ZNFs are found throughout the organisms from yeasts to humans, whereas their numbers have exponentially expanded following the species evolution, particularly in vertebrates including humans (33). Poly-ZNFs tend to present from lower to higher organisms and mediate the fundamental functions shared by most of them, such as embryonic/fetal development, organogenesis, and limb formation (21). On the other hand, KRAB-ZNFs and SCAN-ZNFs, which appeared in the animal kingdom from vertebrates and mammals, respectively, show their numbers have significantly expanded in higher organisms, with the former demonstrating this trend more obviously (33). It is likely that the addition of these domains to C2H2-ZNFs, particularly the KRAB domain, appears to be required for supporting the functions specific to higher organisms, for example, sophisticated cognitive functions unique to humans (21). These pieces of evolutionary evidence on C2H2-ZNFs may explain our successful identification of specific C2H2-ZNF subtypes in particular forms of NDDs. For example, we found high accumulation of KRAB-ZNFs in ID, ASDs, and psychotic diseases that are associated with dysfunctions in higher-order brain functions, whereas defective poly-ZNFs appears to be linked to gross morphological brain abnormalities, including microcephaly, lissencephaly, and local hypoplasia/anomaly. This is also consistent with the previous finding that the characteristic expression of KRAB-ZNFs in the human brain compared to other primates including chimpanzees appears to be required for driving human-specific brain functions (16).
About two thirds of the KRAB-ZNF proteins are reported to bind retrotransposon sequences incorporated in the genome DNA, and act as protecting agents against reactivation and subsequent genome migration of these mobile elements (167). Retrotransposons cause various genetic diseases with their property of genome mutagenesis and chromosomal rearrangement (168). On the other hand, they are major driving forces for species evolution, participating in the development of a sophisticated gene regulatory network characteristic found in higher organisms by providing new regulatory elements and/or TF-binding sites through insertion of their long terminal repeat promoters (169). Thus, dense involvement of KRAB-ZNFs in neurobiology and neurogenesis might have been established in part through insertion of the regulatory elements originated from retrotransposons that harbor binding sites for KRAB-ZNFs into relevant key genes. Because retrotransposons facilitate the development of non-inherited gene regulatory diversity in brain neurons through their genome migration and subsequent mutagenic property in these non-dividing cells (170, 171), it is possible that dysfunction of the KRAB-ZNFs might influence this unique process mediated by retrotransposons by impacting their reactivation and further increase phenotypic variation of the NDD patients.
In conclusion, we performed the literature-based analysis on the roles of C2H2-ZNFs in brain development and pathogenesis of NDDs. We found that numerous C2H2-ZNFs play important roles in these physiologic and pathologic processes. We hope that this literature assessment will encourage the researchers' focus on C2H2-ZNFs in helping us extend our understanding of brain physiology and pathophysiology.
NA-N wrote the manuscript draft. RM created the table and edited the text. TK edited and created the final manuscript.
This literature work was supported by the internal grant of the Sidra Medicine, part of the Qatar Foundation, to TK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Mr. E. K. Zachman for critically reading this manuscript and the lab members for supporting this literature work.
1. Mullin AP, Gokhale A, Moreno-De-Luca A, Sanyal S, Waddington JL, Faundez V. Neurodevelopmental disorders: mechanisms and boundary definitions from genomes, interactomes and proteomes. Transl Psychiatry. (2013) 3:e329. doi: 10.1038/tp.2013.108
2. Homberg JR, Kyzar EJ, Scattoni ML, Norton WH, Pittman J, Gaikwad S, et al. Genetic and environmental modulation of neurodevelopmental disorders: translational insights from labs to beds. Brain Res Bull. (2016) 125:79–91. doi: 10.1016/j.brainresbull.2016.04.015
3. Thapar A, Cooper M, Rutter M. Neurodevelopmental disorders. Lancet Psychiatry. (2017) 4:339–46. doi: 10.1016/S2215-0366(16)30376-5
4. Bergstrom T, Forsberg-Nilsson K. Neural stem cells: brain building blocks and beyond. Ups J Med Sci. (2012) 117:132–42. doi: 10.3109/03009734.2012.665096
5. Androutsellis-Theotokis A, Rueger MA, Mkhikian H, Korb E, McKay RD. Signaling pathways controlling neural stem cells slow progressive brain disease. Cold Spring Harb Symp Quant Biol. (2008) 73:403–10. doi: 10.1101/sqb.2008.73.018
6. Jin X. The role of neurogenesis during development and in the adult brain. Eur J Neurosci. (2016) 44:2291–9. doi: 10.1111/ejn.13251
7. Bond AM, Ming GL, Song H. Adult mammalian neural stem cells and neurogenesis: five decades later. Cell Stem Cell. (2015) 17:385–95. doi: 10.1016/j.stem.2015.09.003
8. Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. (2005) 6:777–88. doi: 10.1038/nrm1739
9. Hasenpusch-Theil K, West S, Kelman A, Kozic Z, Horrocks S, McMahon AP, et al. Gli3 controls the onset of cortical neurogenesis by regulating the radial glial cell cycle through Cdk6 expression. Development. (2018) 145:dev163147. doi: 10.1242/dev.163147
10. Albert M, Huttner WB. Epigenetic and transcriptional pre-patterning -an emerging theme in cortical neurogenesis. Front Neurosci. (2018) 12:359. doi: 10.3389/fnins.2018.00359
11. Sacco R, Cacci E, Novarino G. Neural stem cells in neuropsychiatric disorders. Curr Opin Neurobiol. (2018) 48:131–8. doi: 10.1016/j.conb.2017.12.005
12. Ardhanareeswaran K, Mariani J, Coppola G, Abyzov A, Vaccarino FM. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat Rev Neurol. (2017) 13:265–78. doi: 10.1038/nrneurol.2017.45
13. Santiago C, Bashaw GJ. Transcription factors and effectors that regulate neuronal morphology. Development. (2014) 141:4667–80. doi: 10.1242/dev.110817
14. Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron. (2016) 89:248–68. doi: 10.1016/j.neuron.2015.12.008
15. Lein ES, Belgard TG, Hawrylycz M, Molnar Z. Transcriptomic perspectives on neocortical structure, development, evolution, and disease. Annu Rev Neurosci. (2017) 40:629–52. doi: 10.1146/annurev-neuro-070815-013858
16. Nowick K, Gernat T, Almaas E, Stubbs L. Differences in human and chimpanzee gene expression patterns define an evolving network of transcription factors in brain. Proc Natl Acad Sci USA. (2009) 106:22358–63. doi: 10.1073/pnas.0911376106
17. Dias C, Estruch SB, Graham SA, McRae J, Sawiak SJ, Hurst JA, et al. BCL11A haploinsufficiency causes an intellectual disability syndrome and dysregulates transcription. Am J Hum Genet. (2016) 99:253–74. doi: 10.1016/j.ajhg.2016.05.030
18. Grinberg I, Northrup H, Ardinger H, Prasad C, Dobyns WB, Millen KJ. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat Genet. (2004) 36:1053–5. doi: 10.1038/ng1420
19. Stevens SJ, van Essen AJ, van Ravenswaaij CM, Elias AF, Haven JA, Lelieveld SH, et al. Truncating de novo mutations in the Krüppel-type zinc-finger gene ZNF148 in patients with corpus callosum defects, developmental delay, short stature, and dysmorphisms. Genome Med. (2016) 8:131. doi: 10.1186/s13073-016-0386-9
20. Nowick K, Hamilton AT, Zhang H, Stubbs L. Rapid sequence and expression divergence suggest selection for novel function in primate-specific KRAB-ZNF genes. Mol Biol Evol. (2010) 27:2606–17. doi: 10.1093/molbev/msq157
21. MacKeh R, Marr AK, Fadda A, Kino T. C2H2-type zinc finger proteins: Evolutionarily old and new partners of the nuclear hormone receptors. Nucl Recept Signal. (2018) 15:1550762918801071. doi: 10.1177/1550762918801071
22. Gamsjaeger R, Liew CK, Loughlin FE, Crossley M, MacKay JP. Sticky fingers: zinc-fingers as protein-recognition motifs. Trends Biochem Sci. (2007) 32:63–70. doi: 10.1016/j.tibs.2006.12.007
23. Berg JM. Proposed structure for the zinc-binding domains from transcription factor IIIA and related proteins. Proc Natl Acad Sci USA. (1988) 85:99–102. doi: 10.1073/pnas.85.1.99
24. Lee MS, Gippert GP, Soman KV, Case DA, Wright PE. Three-dimensional solution structure of a single zinc finger DNA-binding domain. Science. (1989) 245:635–7. doi: 10.1126/science.2503871
25. Neuhaus D, Nakaseko Y, Schwabe JW, Klug A. Solution structures of two zinc-finger domains from SWI5 obtained using two-dimensional 1H nuclear magnetic resonance spectroscopy. A zinc-finger structure with a third strand of β-sheet. J Mol Biol. (1992) 228:637–51. doi: 10.1016/0022-2836(92)90846-C
26. Miller J, McLachlan AD, Klug A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. (1985) 4:1609–14. doi: 10.1002/j.1460-2075.1985.tb03825.x
27. Pavletich NP, Pabo CO. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. (1991) 252:809–17. doi: 10.1126/science.2028256
28. Klug A. The discovery of zinc fingers and their development for practical applications in gene regulation and genome manipulation. Q Rev Biophys. (2010) 43:1–21. doi: 10.1017/S0033583510000089
29. Urrutia R. KRAB-containing zinc-finger repressor proteins. Genome Biol. (2003) 4:231. doi: 10.1186/gb-2003-4-10-231
30. Frietze S, O'Geen H, Littlepage LE, Simion C, Sweeney CA, Farnham PJ, et al. Global analysis of ZNF217 chromatin occupancy in the breast cancer cell genome reveals an association with ERα. BMC Genomics. (2014) 15:520. doi: 10.1186/1471-2164-15-520
31. Brown RS. Zinc finger proteins: getting a grip on RNA. Curr Opin Struct Biol. (2005) 15:94–8. doi: 10.1016/j.sbi.2005.01.006
32. Collins T, Stone JR, Williams AJ. All in the family: the BTB/POZ, KRAB, and SCAN domains. Mol Cell Biol. (2001) 21:3609–15. doi: 10.1128/MCB.21.11.3609-3615.2001
33. Emerson RO, Thomas JH. Adaptive evolution in zinc finger transcription factors. PLoS Genet. (2009) 5:e1000325. doi: 10.1371/journal.pgen.1000325
34. Malishkevich A, Leyk J, Goldbaum O, Richter-Landsberg C, Gozes I. ADNP/ADNP2 expression in oligodendrocytes: implication for myelin-related neurodevelopment. J Mol Neurosci. (2015) 57:304–13. doi: 10.1007/s12031-015-0640-4
35. Dresner E, Agam G, Gozes I. Activity-dependent neuroprotective protein (ADNP) expression level is correlated with the expression of the sister protein ADNP2: deregulation in schizophrenia. Eur Neuropsychopharmacol. (2011) 21:355–61. doi: 10.1016/j.euroneuro.2010.06.004
36. Shimbo H, Yokoi T, Aida N, Mizuno S, Suzumura H, Nagai J, et al. Haploinsufficiency of BCL11A associated with cerebellar abnormalities in 2p15p16.1 deletion syndrome. Mol Genet Genomic Med. (2017) 5:429–37. doi: 10.1002/mgg3.289
37. Yoshida M, Nakashima M, Okanishi T, Kanai S, Fujimoto A, Itomi K, et al. Identification of novel BCL11A variants in patients with epileptic encephalopathy: Expanding the phenotypic spectrum. Clin Genet. (2018) 93:368–73. doi: 10.1111/cge.13067
38. Tang B, Di Lena P, Schaffer L, Head SR, Baldi P, Thomas EA. Genome-wide identification of Bcl11b gene targets reveals role in brain-derived neurotrophic factor signaling. PLoS ONE. (2011) 6:e23691. doi: 10.1371/journal.pone.0023691
39. Shimizu T, Nakazawa M, Kani S, Bae YK, Shimizu T, Kageyama R, et al. Zinc finger genes Fezf1 and Fezf2 control neuronal differentiation by repressing Hes5 expression in the forebrain. Development. (2010) 137:1875–85. doi: 10.1242/dev.047167
40. Liu X, Su P, Lu L, Feng Z, Wang H, Zhou J. Function of FEZF1 during early neural differentiation of human embryonic stem cells. Sci China Life Sci. (2018) 61:35–45. doi: 10.1007/s11427-017-9155-4
41. Chen B, Wang SS, Hattox AM, Rayburn H, Nelson SB, McConnell SK. The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc Natl Acad Sci USA. (2008) 105:11382–7. doi: 10.1073/pnas.0804918105
42. Theil T. Gli3 is required for the specification and differentiation of preplate neurons. Dev Biol. (2005) 286:559–71. doi: 10.1016/j.ydbio.2005.08.033
43. Biesecker LG. What you can learn from one gene: GLI3. J Med Genet. (2006) 43:465–9. doi: 10.1136/jmg.2004.029181
44. Takata A, Miyake N, Tsurusaki Y, Fukai R, Miyatake S, Koshimizu E, et al. Integrative analyses of de novo mutations provide deeper biological insights into autism spectrum disorder. Cell Rep. (2018) 22:734–47. doi: 10.1016/j.celrep.2017.12.074
45. Maekawa M, Yamaguchi K, Nakamura T, Shibukawa R, Kodanaka I, Ichisaka T, et al. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature. (2011) 474:225–9. doi: 10.1038/nature10106
46. Zhang F, Nakanishi G, Kurebayashi S, Yoshino K, Perantoni A, Kim YS, et al. Characterization of Glis2, a novel gene encoding a Gli-related, Krüppel-like transcription factor with transactivation and repressor functions. Roles in kidney development and neurogenesis. J Biol Chem. (2002) 277:10139–49. doi: 10.1074/jbc.M108062200
47. Caubit X, Gubellini P, Andrieux J, Roubertoux PL, Metwaly M, Jacq B, et al. TSHZ3 deletion causes an autism syndrome and defects in cortical projection neurons. Nat Genet. (2016) 48:1359–69. doi: 10.1038/ng.3681
48. Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, et al. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. (1998) 20:180–3. doi: 10.1038/2484
49. Solomon BD, Lacbawan F, Mercier S, Clegg NJ, Delgado MR, Rosenbaum K, et al. Mutations in ZIC2 in human holoprosencephaly: description of a novel ZIC2 specific phenotype and comprehensive analysis of 157 individuals. J Med Genet. (2010) 47:513–24. doi: 10.1136/jmg.2009.073049
50. Brown L, Paraso M, Arkell R, Brown S. In vitro analysis of partial loss-of-function ZIC2 mutations in holoprosencephaly: alanine tract expansion modulates DNA binding and transactivation. Hum Mol Genet. (2005) 14:411–20. doi: 10.1093/hmg/ddi037
51. Nakata K, Nagai T, Aruga J, Mikoshiba K. Xenopus Zic3, a primary regulator both in neural and neural crest development. Proc Natl Acad Sci USA. (1997) 94:11980–5. doi: 10.1073/pnas.94.22.11980
52. Li S, Liu S, Chen W, Yuan Y, Gu R, Song Y, et al. A novel ZIC3 gene mutation identified in patients with heterotaxy and congenital heart disease. Sci Rep. (2018) 8:12386. doi: 10.1038/s41598-018-30204-3
53. Fujimi TJ, Hatayama M, Aruga J. Xenopus Zic3 controls notochord and organizer development through suppression of the Wnt/β-catenin signaling pathway. Dev Biol. (2012) 361:220–31. doi: 10.1016/j.ydbio.2011.10.026
54. Inoue T, Hatayama M, Tohmonda T, Itohara S, Aruga J, Mikoshiba K. Mouse Zic5 deficiency results in neural tube defects and hypoplasia of cephalic neural crest derivatives. Dev Biol. (2004) 270:146–62. doi: 10.1016/j.ydbio.2004.02.017
55. Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. (2016) 7:13316. doi: 10.1038/ncomms13316
56. Engwerda A, Frentz B, den Ouden AL, Flapper BCT, Swertz MA, Gerkes EH, et al. The phenotypic spectrum of proximal 6q deletions based on a large cohort derived from social media and literature reports. Eur J Hum Genet. (2018) 26:1478–89. doi: 10.1038/s41431-018-0172-9
57. Liang S, Wang XL, Zou MY, Wang H, Zhou X, Sun CH, et al. Family-based association study of ZNF533, DOCK4 and IMMP2L gene polymorphisms linked to autism in a northeastern Chinese Han population. J Zhejiang Univ Sci B. (2014) 15:264–71. doi: 10.1631/jzus.B1300133
58. Kambouris M, Maroun RC, Ben-Omran T, Al-Sarraj Y, Errafii K, Ali R, et al. Mutations in zinc finger 407 [ZNF407] cause a unique autosomal recessive cognitive impairment syndrome. Orphanet J Rare Dis. (2014) 9:80. doi: 10.1186/1750-1172-9-80
59. Wang B, Zheng Y, Shi H, Du X, Zhang Y, Wei B, et al. Zfp462 deficiency causes anxiety-like behaviors with excessive self-grooming in mice. Genes Brain Behav. (2017) 16:296–307. doi: 10.1111/gbb.12339
60. Curtis D. Practical experience of the application of a weighted burden test to whole exome sequence data for obesity and schizophrenia. Ann Hum Genet. (2016) 80:38–49. doi: 10.1111/ahg.12135
61. Shen S, Pu J, Lang B, McCaig CD. A zinc finger protein Zfp521 directs neural differentiation and beyond. Stem Cell Res Ther. (2011) 2:20. doi: 10.1186/scrt61
62. Kamiya D, Banno S, Sasai N, Ohgushi M, Inomata H, Watanabe K, et al. Intrinsic transition of embryonic stem-cell differentiation into neural progenitors. Nature. (2011) 470:503–9. doi: 10.1038/nature09726
63. Qin Z, Ren F, Xu X, Ren Y, Li H, Wang Y, et al. ZNF536, a novel zinc finger protein specifically expressed in the brain, negatively regulates neuron differentiation by repressing retinoic acid-induced gene transcription. Mol Cell Biol. (2009) 29:3633–43. doi: 10.1128/MCB.00362-09
64. van der Werf IM, Van Dijck A, Reyniers E, Helsmoortel C, Kumar AA, Kalscheuer VM, et al. Mutations in two large pedigrees highlight the role of ZNF711 in X-linked intellectual disability. Gene. (2017) 605:92–8. doi: 10.1016/j.gene.2016.12.013
65. Kleine-Kohlbrecher D, Christensen J, Vandamme J, Abarrategui I, Bak M, Tommerup N, et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol Cell. (2010) 38:165–78. doi: 10.1016/j.molcel.2010.03.002
66. Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. (2015) 21:185–91. doi: 10.1038/nm.3792
67. Sun Y, Hu D, Liang J, Bao YP, Meng SQ, Lu L, et al. Association between variants of zinc finger genes and psychiatric disorders: systematic review and meta-analysis. Schizophr Res. (2015) 162:124–37. doi: 10.1016/j.schres.2015.01.036
68. Walters JT, Corvin A, Owen MJ, Williams H, Dragovic M, Quinn EM, et al. Psychosis susceptibility gene ZNF804A and cognitive performance in schizophrenia. Arch Gen Psychiatry. (2010) 67:692–700. doi: 10.1001/archgenpsychiatry.2010.81
69. Wang Y, Koh K, Ichinose Y, Yasumura M, Ohtsuka T, Takiyama Y. A de novo mutation in the NALCN gene in an adult patient with cerebellar ataxia associated with intellectual disability and arthrogryposis. Clin Genet. (2016) 90:556–7. doi: 10.1111/cge.12851
70. Gilling M, Lind-Thomsen A, Mang Y, Bak M, Moller M, Ullmann R, et al. Biparental inheritance of chromosomal abnormalities in male twins with non-syndromic mental retardation. Eur J Med Genet. (2011) 54:e383–388. doi: 10.1016/j.ejmg.2011.03.008
71. Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum Mol Genet. (2012) 21:4781–92. doi: 10.1093/hmg/dds301
72. Jones KA, Luo Y, Dukes-Rimsky L, Srivastava DP, Koul-Tewari R, Russell TA, et al. Neurodevelopmental disorder-associated ZBTB20 gene variants affect dendritic and synaptic structure. PLoS ONE. (2018) 13:e0203760. doi: 10.1371/journal.pone.0203760
73. Guedj F, Pennings JL, Massingham LJ, Wick HC, Siegel AE, Tantravahi U, et al. An integrated human/murine transcriptome and pathway approach to identify prenatal treatments for Down syndrome. Sci Rep. (2016) 6:32353. doi: 10.1038/srep32353
74. Song GG, Kim JH, Lee YH. Genome-wide pathway analysis in major depressive disorder. J Mol Neurosci. (2013) 51:428–36. doi: 10.1007/s12031-013-0047-z
75. Sodersten E, Lilja T, Hermanson O. The novel BTB/POZ and zinc finger factor Zbtb45 is essential for proper glial differentiation of neural and oligodendrocyte progenitor cells. Cell Cycle. (2010) 9:4866–75. doi: 10.4161/cc.9.24.14154
76. Wang Z, Mo XT, Zhang X, Xu MY, Zhao J, Wang L. Isolation and functional characterization of a stress-responsive transcription factor ZmC2H2-1 in Zea mays. Yi Chuan. (2018) 40:767–78.
77. Gardiner K, Slavov D, Bechtel L, Davisson M. Annotation of human chromosome 21 for relevance to Down syndrome: gene structure and expression analysis. Genomics. (2002) 79:833–43. doi: 10.1006/geno.2002.6782
78. Xia YY, Ding YB, Liu XQ, Chen XM, Cheng SQ, Li LB, et al. Allelic methylation status of CpG islands on chromosome 21q in patients with Trisomy 21. Mol Med Rep. (2014) 9:1681–8. doi: 10.3892/mmr.2014.1985
79. Fattahi Z, Sheikh TI, Musante L, Rasheed M, Taskiran II, Harripaul R, et al. Biallelic missense variants in ZBTB11 can cause intellectual disability in humans. Hum Mol Genet. (2018) 27:3177–88. doi: 10.1093/hmg/ddy220
80. Depienne C, Nava C, Keren B, Heide S, Rastetter A, Passemard S, et al. Genetic and phenotypic dissection of 1q43q44 microdeletion syndrome and neurodevelopmental phenotypes associated with mutations in ZBTB18 and HNRNPU. Hum Genet. (2017) 136:463–79. doi: 10.1007/s00439-017-1772-0
81. Yan L, Li Y, Shi Z, Lu X, Ma J, Hu B, et al. The zinc finger E-box-binding homeobox 1 (Zeb1) promotes the conversion of mouse fibroblasts into functional neurons. J Biol Chem. (2017) 292:12959–70. doi: 10.1074/jbc.M116.771493
82. Leduc-Galindo D, Qvist P, Toth AE, Fryland T, Nielsen MS, Borglum AD, et al. The effect of hypoxia on ZEB1 expression in a mimetic system of the blood-brain barrier. Microvasc Res. (2019) 122:131–5. doi: 10.1016/j.mvr.2018.08.004
83. Saunders CJ, Zhao W, Ardinger HH. Comprehensive ZEB2 gene analysis for Mowat-Wilson syndrome in a North American cohort: a suggested approach to molecular diagnostics. Am J Med Genet A. (2009) 149a:2527–31. doi: 10.1002/ajmg.a.33067
84. Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. (2008) 321:218–23. doi: 10.1126/science.1157657
85. Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS, et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. (2012) 8:e1002635. doi: 10.1371/journal.pgen.1002635
86. Gana S, Veggiotti P, Sciacca G, Fedeli C, Bersano A, Micieli G, et al. 19q13.11 cryptic deletion: description of two new cases and indication for a role of WTIP haploinsufficiency in hypospadias. Eur J Hum Genet. (2012) 20:852–6. doi: 10.1038/ejhg.2012.19
87. Subaran RL, Odgerel Z, Swaminathan R, Glatt CE, Weissman MM. Novel variants in ZNF34 and other brain-expressed transcription factors are shared among early-onset MDD relatives. Am J Med Genet B Neuropsychiatr Genet. (2016) 171b:333–41. doi: 10.1002/ajmg.b.32408
88. Shoichet SA, Hoffmann K, Menzel C, Trautmann U, Moser B, Hoeltzenbein M, et al. Mutations in the ZNF41 gene are associated with cognitive deficits: identification of a new candidate for X-linked mental retardation. Am J Hum Genet. (2003) 73:1341–54. doi: 10.1086/380309
89. Takase K, Ohtsuki T, Migita O, Toru M, Inada T, Yamakawa-Kobayashi K, et al. Association of ZNF74 gene genotypes with age-at-onset of schizophrenia. Schizophr Res. (2001) 52:161–5. doi: 10.1016/S0920-9964(00)00191-2
90. Berto S, Perdomo-Sabogal A, Gerighausen D, Qin J, Nowick K. A consensus network of gene regulatory factors in the human frontal lobe. Front Genet. (2016) 7:31. doi: 10.3389/fgene.2016.00031
91. Alesi V, Bertoli M, Barrano G, Torres B, Pusceddu S, Pastorino M, et al. 335.4 kb microduplication in chromosome band Xp11.2p11.3 associated with developmental delay, growth retardation, autistic disorder and dysmorphic features. Gene. (2012) 505:384–7. doi: 10.1016/j.gene.2012.05.031
92. Fullard JF, Giambartolomei C, Hauberg ME, Xu K, Voloudakis G, Shao Z, et al. Open chromatin profiling of human postmortem brain infers functional roles for non-coding schizophrenia loci. Hum Mol Genet. (2017) 26:1942–51. doi: 10.1093/hmg/ddx103
93. Matsunami K, Nishida N, Kaneko N, Ikeo K, Toyo-Oka L, Takeuchi H, et al. Genome-wide association study identifies ZNF354C variants associated with depression from interferon-based therapy for chronic hepatitis C. PLoS ONE. (2016) 11:e0164418. doi: 10.1371/journal.pone.0164418
94. Andrieux J, Cuvellier JC, Duban-Bedu B, Joriot-Chekaf S, Dieux-Coeslier A, Manouvrier-Hanu S, et al. A 6.9 Mb 1qter deletion/4.4 Mb 18pter duplication in a boy with extreme microcephaly with simplified gyral pattern, vermis hypoplasia and corpus callosum agenesis. Eur J Med Genet. (2008) 51:87–91. doi: 10.1016/j.ejmg.2007.10.004
95. Vengoechea J, Parikh AS, Zhang S, Tassone F. De novo microduplication of the FMR1 gene in a patient with developmental delay, epilepsy and hyperactivity. Eur J Hum Genet. (2012) 20:1197–200. doi: 10.1038/ejhg.2012.78
96. Prasad A, Merico D, Thiruvahindrapuram B, Wei J, Lionel AC, Sato D, et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3. (2012) 2:1665–85. doi: 10.1534/g3.112.004689
97. Hassan MJ, Chishti MS, Jamal SM, Tariq M, Ahmad W. A syndromic form of autosomal recessive congenital microcephaly (Jawad syndrome) maps to chromosome 18p11.22-q11.2. Hum Genet. (2008) 123:77–82. doi: 10.1007/s00439-007-0452-x
98. Schuurs-Hoeijmakers JH, Vulto-van Silfhout AT, Vissers LE, van de Vondervoort II, van Bon BW, de Ligt J, et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J Med Genet. (2013) 50:802–11. doi: 10.1136/jmedgenet-2013-101644
99. Spreiz A, Haberlandt E, Baumann M, Baumgartner Sigl S, Fauth C, Gautsch K, et al. Chromosomal microaberrations in patients with epilepsy, intellectual disability, and congenital anomalies. Clin Genet. (2014) 86:361–6. doi: 10.1111/cge.12288
100. Castillo A, Kramer N, Schwartz CE, Miles JH, DuPont BR, Rosenfeld JA, et al. 19q13.32 microdeletion syndrome: three new cases. Eur J Med Genet. (2014) 57:654–658. doi: 10.1016/j.ejmg.2014.08.009
101. Ahmed I, Rafiq MA, Vincent JB, Bhatti A, Ayub M, John P. Homozygosity mapping of autosomal recessive intellectual disability loci in 11 consanguineous Pakistani families. Acta Neuropsychiatr. (2015) 27:38–47. doi: 10.1017/neu.2014.37
102. Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. (2015) 87:1215–33. doi: 10.1016/j.neuron.2015.09.016
103. De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. (2014) 515:209–15. doi: 10.1038/nature13772
104. Chien HC, Wang HY, Su YN, Lai KY, Lu LC, Chen PC, et al. Targeted disruption in mice of a neural stem cell-maintaining, KRAB-Zn finger-encoding gene that has rapidly evolved in the human lineage. PLoS ONE. (2012) 7:e47481. doi: 10.1371/journal.pone.0047481
105. Venturini L, Stadler M, Manukjan G, Scherr M, Schlegelberger B, Steinemann D, et al. The stem cell zinc finger 1 (SZF1)/ZNF589 protein has a human-specific evolutionary nucleotide DNA change and acts as a regulator of cell viability in the hematopoietic system. Exp Hematol. (2016) 44:257–68. doi: 10.1016/j.exphem.2015.12.005
106. Agha Z, Iqbal Z, Azam M, Ayub H, Vissers LE, Gilissen C, et al. Exome sequencing identifies three novel candidate genes implicated in intellectual disability. PLoS ONE. (2014) 9:e112687. doi: 10.1371/journal.pone.0112687
107. Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, et al. A de novo paradigm for mental retardation. Nat Genet. (2010) 42:1109–12. doi: 10.1038/ng.712
108. Ramaswamy V, Castillo M, Bolduc FV. Developmental disability: duplication of zinc finger transcription factors 673 and 674. Pediatr Neurol. (2010) 43:209–12. doi: 10.1016/j.pediatrneurol.2010.04.016
109. Lugtenberg D, Yntema HG, Banning MJ, Oudakker AR, Firth HV, Willatt L, et al. ZNF674: a new krüppel-associated box-containing zinc-finger gene involved in nonsyndromic X-linked mental retardation. Am J Hum Genet. (2006) 78:265–78. doi: 10.1086/500306
110. Metsu S, Rainger JK, Debacker K, Bernhard B, Rooms L, Grafodatskaya D, et al. A CGG-repeat expansion mutation in ZNF713 causes FRA7A: association with autistic spectrum disorder in two families. Hum Mutat. (2014) 35:1295–300. doi: 10.1002/humu.22683
111. Lei H, Yan Z, Sun X, Zhang Y, Wang J, Ma C, et al. Axon guidance pathways served as common targets for human speech/language evolution and related disorders. Brain Lang. (2017) 174:1–8. doi: 10.1016/j.bandl.2017.06.007
112. Willemsen MH, Fernandez BA, Bacino CA, Gerkes E, de Brouwer AP, Pfundt R, et al. Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. Eur J Hum Genet. (2010) 18:429–35. doi: 10.1038/ejhg.2009.192
113. Li Z, Chen J, Yu H, He L, Xu Y, Zhang D, et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat Genet. (2017) 49:1576–83. doi: 10.1038/ng.3973
114. Ruiz-Botero F, Pachajoa H. Deletion 21q22.3 and duplication 7q35q36.3 in a Colombian girl: a case report. J Med Case Rep. (2016) 10:204. doi: 10.1186/s13256-016-0988-2
115. Khalfallah O, Ravassard P, Lagache CS, Fligny C, Serre A, Bayard E, et al. Zinc finger protein 191 (ZNF191/Zfp191) is necessary to maintain neural cells as cycling progenitors. Stem Cells. (2009) 27:1643–53. doi: 10.1002/stem.88
116. Luo XJ, Mattheisen M, Li M, Huang L, Rietschel M, Borglum AD, et al. Systematic integration of brain eQTL and GWAS identifies ZNF323 as a novel schizophrenia risk gene and suggests recent positive selection based on compensatory advantage on pulmonary function. Schizophr Bull. (2015) 41:1294–308. doi: 10.1093/schbul/sbv017
117. Attardo A, Calegari F, Haubensak W, Wilsch-Brauninger M, Huttner WB. Live imaging at the onset of cortical neurogenesis reveals differential appearance of the neuronal phenotype in apical versus basal progenitor progeny. PLoS ONE. (2008) 3:e2388. doi: 10.1371/journal.pone.0002388
118. He L, Yu K, Lu F, Wang J, Wu LN, Zhao C, et al. Transcriptional regulator ZEB2 is essential for Bergmann glia development. J Neurosci. (2018) 38:1575–87. doi: 10.1523/JNEUROSCI.2674-17.2018
119. Mowat DR, Wilson MJ, Goossens M. Mowat-Wilson syndrome. J Med Genet. (2003) 40:305–10. doi: 10.1136/jmg.40.5.305
120. Cacheux V, Dastot-Le Moal F, Kaariainen H, Bondurand N, Rintala R, Boissier B, et al. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum Mol Genet. (2001) 10:1503–10. doi: 10.1093/hmg/10.14.1503
121. Verschueren K, Remacle JE, Collart C, Kraft H, Baker BS, Tylzanowski P, et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5'-CACCT sequences in candidate target genes. J Biol Chem. (1999) 274:20489–98. doi: 10.1074/jbc.274.29.20489
122. Aruga J. The role of Zic genes in neural development. Mol Cell Neurosci. (2004) 26:205–21. doi: 10.1016/j.mcn.2004.01.004
123. Merzdorf CS. Emerging roles for zic genes in early development. Dev Dyn. (2007) 236:922–40. doi: 10.1002/dvdy.21098
124. Inoue T, Ogawa M, Mikoshiba K, Aruga J. Zic deficiency in the cortical marginal zone and meninges results in cortical lamination defects resembling those in type II lissencephaly. J Neurosci. (2008) 28:4712–25. doi: 10.1523/JNEUROSCI.5735-07.2008
125. Passemard S, Kaindl AM, Verloes A. Microcephaly. Handb Clin Neurol. (2013) 111:129–41. doi: 10.1016/B978-0-444-52891-9.00013-0
126. Pramparo T, Libiger O, Jain S, Li H, Youn YH, Hirotsune S, et al. Global developmental gene expression and pathway analysis of normal brain development and mouse models of human neuronal migration defects. PLoS Genet. (2011) 7:e1001331. doi: 10.1371/journal.pgen.1001331
127. Masse J, Piquet-Pellorce C, Viet J, Guerrier D, Pellerin I, Deschamps S. ZFPIP/Zfp462 is involved in P19 cell pluripotency and in their neuronal fate. Exp Cell Res. (2011) 317:1922–34. doi: 10.1016/j.yexcr.2011.04.015
128. Han ZG, Zhang QH, Ye M, Kan LX, Gu BW, He KL, et al. Molecular cloning of six novel Krüppel-like zinc finger genes from hematopoietic cells and identification of a novel transregulatory domain KRNB. J Biol Chem. (1999) 274:35741–8. doi: 10.1074/jbc.274.50.35741
129. Vasudevan P, Suri M. A clinical approach to developmental delay and intellectual disability. Clin Med. (2017) 17:558–61. doi: 10.7861/clinmedicine.17-6-558
130. Srour M, Shevell M. Genetics and the investigation of developmental delay/intellectual disability. Arch Dis Child. (2014) 99:386–9. doi: 10.1136/archdischild-2013-304063
131. Mandel JL, Chelly J. Monogenic X-linked mental retardation: is it as frequent as currently estimated? The paradox of the ARX (Aristaless X) mutations. Eur J Hum Genet. (2004) 12:689–93. doi: 10.1038/sj.ejhg.5201247
132. Raymond FL, Tarpey P. The genetics of mental retardation. Hum Mol Genet. (2006) 15:R110–6. doi: 10.1093/hmg/ddl189
133. Laumonnier F, Holbert S, Ronce N, Faravelli F, Lenzner S, Schwartz CE, et al. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J Med Genet. (2005) 42:780–6. doi: 10.1136/jmg.2004.029439
134. Abidi F, Miano M, Murray J, Schwartz C. A novel mutation in the PHF8 gene is associated with X-linked mental retardation with cleft lip/cleft palate. Clin Genet. (2007) 72:19–22. doi: 10.1111/j.1399-0004.2007.00817.x
135. Sztainberg Y, Zoghbi HY. Lessons learned from studying syndromic autism spectrum disorders. Nat Neurosci. (2016) 19:1408–17. doi: 10.1038/nn.4420
136. El-Hattab AW, Bournat J, Eng PA, Wu JB, Walker BA, Stankiewicz P, et al. Microduplication of Xp11.23p11.3 with effects on cognition, behavior, and craniofacial development. Clin Genet. (2011) 79:531–8. doi: 10.1111/j.1399-0004.2010.01496.x
137. Koul R, Dukes-Rimsky L, Luo Y, Jones KA, Lemyre E, Sowell SM, et al. “American society of human genetics,” in Annual Meeting (Boston, MA) (2013).
138. Rasmussen MB, Nielsen JV, Lourenco CM, Melo JB, Halgren C, Geraldi CV, et al. Neurodevelopmental disorders associated with dosage imbalance of ZBTB20 correlate with the morbidity spectrum of ZBTB20 candidate target genes. J Med Genet. (2014) 51:605–13. doi: 10.1136/jmedgenet-2014-102535
139. Mattioli F, Piton A, Gerard B, Superti-Furga A, Mandel JL, Unger S. Novel de novo mutations in ZBTB20 in Primrose syndrome with congenital hypothyroidism. Am J Med Genet A. (2016) 170:1626–9. doi: 10.1002/ajmg.a.37645
140. Nagao M, Ogata T, Sawada Y, Gotoh Y. Zbtb20 promotes astrocytogenesis during neocortical development. Nat Commun. (2016) 7:11102. doi: 10.1038/ncomms11102
141. Powis Z, Petrik I, Cohen JS, Escolar D, Burton J, van Ravenswaaij-Arts CMA, et al. De novo variants in KLF7 are a potential novel cause of developmental delay/intellectual disability, neuromuscular and psychiatric symptoms. Clin Genet. (2018) 93:1030–8. doi: 10.1111/cge.13198
142. Laub F, Lei L, Sumiyoshi H, Kajimura D, Dragomir C, Smaldone S, et al. Transcription factor KLF7 is important for neuronal morphogenesis in selected regions of the nervous system. Mol Cell Biol. (2005) 25:5699–711. doi: 10.1128/MCB.25.13.5699-5711.2005
143. Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. (2012) 485:242–5. doi: 10.1038/nature11011
144. Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. (2014) 515:216–21. doi: 10.1038/nature13908
145. Fukai R, Hiraki Y, Yofune H, Tsurusaki Y, Nakashima M, Saitsu H, et al. A case of autism spectrum disorder arising from a de novo missense mutation in POGZ. J Hum Genet. (2015) 60:277–9. doi: 10.1038/jhg.2015.13
146. Ye Y, Cho MT, Retterer K, Alexander N, Ben-Omran T, Al-Mureikhi M, et al. De novo POGZ mutations are associated with neurodevelopmental disorders and microcephaly. Cold Spring Harb Mol Case Stud. (2015) 1:a000455. doi: 10.1101/mcs.a000455
148. Rasmussen P, Borjesson O, Wentz E, Gillberg C. Autistic disorders in Down syndrome: background factors and clinical correlates. Dev Med Child Neurol. (2001) 43:750–4. doi: 10.1017/S0012162201001372
149. Spellman C, Ahmed MM, Dubach D, Gardiner KJ. Expression of trisomic proteins in Down syndrome model systems. Gene. (2013) 512:219–25. doi: 10.1016/j.gene.2012.10.051
150. Capkova P, Misovicova N, Vrbicka D. Partial trisomy and tetrasomy of chromosome 21 without Down Syndrome phenotype and short overview of genotype-phenotype correlation. A case report. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. (2014) 158:321–5. doi: 10.5507/bp.2013.077
151. Mekkawy MK, Mazen IM, Kamel AK, Vater I, Zaki MS. Genotype/phenotype correlation in a female patient with 21q22.3 and 12p13.33 duplications. Am J Med Genet A. (2016) 170a:1050–8. doi: 10.1002/ajmg.a.37523
152. Shibuya K, Kudoh J, Okui M, Shimizu N. Identification of a novel zinc finger protein gene (ZNF298) in the GAP2 of human chromosome 21q. Biochem Biophys Res Commun. (2005) 332:557–68. doi: 10.1016/j.bbrc.2005.04.159
154. Bassett AS, Chow EW. 22q11 deletion syndrome: a genetic subtype of schizophrenia. Biol Psychiatry. (1999) 46:882–91. doi: 10.1016/S0006-3223(99)00114-6
155. Chang H, Xiao X, Li M. The schizophrenia risk gene ZNF804A: clinical associations, biological mechanisms and neuronal functions. Mol Psychiatry. (2017) 22:944–53. doi: 10.1038/mp.2017.19
156. Xu M, St. Clair D, He L. Testing for genetic association between the ZDHHC8 gene locus and susceptibility to schizophrenia: an integrated analysis of multiple datasets. Am J Med Genet B Neuropsychiatr Genet. (2010) 153b:1266–75. doi: 10.1002/ajmg.b.31096
157. Ota VK, Gadelha A, Assuncao IB, Santoro ML, Christofolini DM, Bellucco FT, et al. ZDHHC8 gene may play a role in cortical volumes of patients with schizophrenia. Schizophr Res. (2013) 145:33–5. doi: 10.1016/j.schres.2013.01.011
158. Ecker K, Lorenz A, Wolf F, Ploner C, Bock G, Duncan T, et al. A RAS recruitment screen identifies ZKSCAN4 as a glucocorticoid receptor-interacting protein. J Mol Endocrinol. (2009) 42:105–17. doi: 10.1677/JME-08-0087
159. Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, et al. Common variants conferring risk of schizophrenia. Nature. (2009) 460:744–7. doi: 10.1038/nature08186
160. Zhang Y, Lu T, Yan H, Ruan Y, Wang L, Zhang D, et al. Replication of association between schizophrenia and chromosome 6p21-6p22.1 polymorphisms in Chinese Han population. PLoS ONE. (2013) 8:e56732. doi: 10.1371/journal.pone.0056732
161. Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. (2003) 301:386–9. doi: 10.1126/science.1083968
162. Zhao Z, Xu J, Chen J, Kim S, Reimers M, Bacanu SA, et al. Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol Psychiatry. (2015) 20:563–72. doi: 10.1038/mp.2014.82
163. Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. (2013) 45:984–94. doi: 10.1038/ng.2711
164. Cichon S, Schumacher J, Muller DJ, Hurter M, Windemuth C, Strauch K, et al. A genome screen for genes predisposing to bipolar affective disorder detects a new susceptibility locus on 8q. Hum Mol Genet. (2001) 10:2933–44. doi: 10.1093/hmg/10.25.2933
165. McInnis MG, Lan TH, Willour VL, McMahon FJ, Simpson SG, Addington AM, et al. Genome-wide scan of bipolar disorder in 65 pedigrees: supportive evidence for linkage at 8q24, 18q22, 4q32, 2p12, and 13q12. Mol Psychiatry. (2003) 8:288–98. doi: 10.1038/sj.mp.4001277
166. Lin E, Kuo PH, Liu YL, Yu YW, Yang AC, Tsai SJ. A deep learning approach for predicting antidepressant response in major depression using clinical and genetic niomarkers. Front Psychiatry. (2018) 9:290. doi: 10.3389/fpsyt.2018.00290
167. Yang P, Wang Y, Macfarlan TS. The role of KRAB-ZFPs in transposable element repression and mammalian evolution. Trends Genet. (2017) 33:871–81. doi: 10.1016/j.tig.2017.08.006
168. Reilly MT, Faulkner GJ, Dubnau J, Ponomarev I, Gage FH. The role of transposable elements in health and diseases of the central nervous system. J Neurosci. (2013) 33:17577–86. doi: 10.1523/JNEUROSCI.3369-13.2013
169. Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. (2008) 9:397–405. doi: 10.1038/nrg2337
170. Erwin JA, Marchetto MC, Gage FH. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci. (2014) 15:497–506. doi: 10.1038/nrn3730
Keywords: brain development, structural abnormality, KRAB domain, mutation, neural stem cells, transcriptional regulation
Citation: Al-Naama N, Mackeh R and Kino T (2020) C2H2-Type Zinc Finger Proteins in Brain Development, Neurodevelopmental, and Other Neuropsychiatric Disorders: Systematic Literature-Based Analysis. Front. Neurol. 11:32. doi: 10.3389/fneur.2020.00032
Received: 14 November 2019; Accepted: 10 January 2020;
Published: 14 February 2020.
Edited by:
Piero Pavone, University of Catania, ItalyReviewed by:
Diego Iacono, Biomedical Research Institute of New Jersey, United StatesCopyright © 2020 Al-Naama, Mackeh and Kino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoshige Kino, dGtpbm9Ac2lkcmEub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.