 Ru Liu1,2,3,4
Ru Liu1,2,3,4 Xiaofeng Yang
Xiaofeng Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 17 January 2020
Sec. Epilepsy
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.01407
This article is part of the Research Topic Demarcating the Epileptogenic Zone - From Bench to Bedside View all 6 articles
As a main inhibitory neurotransmitter in the central nervous system, γ-aminobutyric acid (GABA) activates chloride-permeable GABAa receptors (GABAa Rs) and induces chloride ion (Cl−) flow, which relies on the intracellular chloride concentration ([Cl−]i) of the postsynaptic neuron. The Na-K-2Cl cotransporter isoform 1 (NKCC1) and the K-Cl cotransporter isoform 2 (KCC2) are two main cation-chloride cotransporters (CCCs) that have been implicated in human epilepsy. NKCC1 and KCC2 reset [Cl−]i by accumulating and extruding Cl−, respectively. Previous studies have shown that the profile of NKCC1 and KCC2 in neonatal neurons may reappear in mature neurons under some pathophysiological conditions, such as epilepsy. Although increasing studies focusing on the expression of NKCC1 and KCC2 have suggested that impaired chloride plasticity may be closely related to epilepsy, additional neuroelectrophysiological research aimed at studying the functions of NKCC1 and KCC2 are needed to understand the exact mechanism by which they induce epileptogenesis. In this review, we aim to briefly summarize the current researches surrounding the expression and function of NKCC1 and KCC2 in epileptogenesis and its implications on the treatment of epilepsy. We will also explore the potential for NKCC1 and KCC2 to be therapeutic targets for the development of novel antiepileptic drugs.
Many factors have been shown to play important roles in facilitating the development of epilepsy in humans and animal disease models, including genetic mutations, environmental factors during development, and brain injury (1–3). Although the exact mechanism of epileptogenesis is not yet fully understood, epilepsy is widely believed to be associated with an excitatory-inhibitory imbalance (4, 5). There is increasing evidence in recent literature to suggest that epileptogenesis results primarily from a deficit of GABA inhibition (6, 7).
Seventy to eighty percent of neocortical neurons are excitatory glutamatergic pyramidal neurons while the remaining are mostly GABAergic inhibitory interneurons with diverse characteristics (8, 9). Based on experimental studies involving seizure models and epilepsy patients, we know that the down-regulation of GABA receptor subunits, loss of GABAergic interneurons, and decreased frequency and amplitude of spontaneous synaptic GABA currents from pyramidal neurons contribute to decreased GABAergic inhibition (7, 10–12). In neurons, NKCC1 and KCC2 serve as two main regulators of GABA receptors' function by maintaining [Cl−]i homeostasis (13). A disruption in the balance between NKCC1 and KCC2 induced by unknown causes may result in ineffective GABA inhibition (14, 15). This suggests a more complex scope of functions for GABAergic signaling than just global inhibition.
Approximately 30% of patients with epilepsy fail to respond to current therapies, so there is increasing demand for novel antiepileptic drugs (AEDs) and other therapeutic strategies (16, 17). Current AEDs enhance GABAergic function (18), either by modulating the post-synaptic GABAa Rs (barbiturates and benzodiazepines) or by acting on synaptic GABA availability (vigabatrin). The challenge faced by physicians is that conventional AEDs approved for use in adults have poor effects in neonates and may actually potentiate seizures in this patient population (19–22). In this review, we introduce the main chloride transporters associated with GABA signaling and discuss their regulatory dynamics on GABA functions at various levels ranging from genotypes to functional alterations and phenotypes. We will also discuss the possible antiepileptic effect of the NKCC1 blocker, bumetanide, as a promising AED.
Intracellular chloride concentration is regulated by cation-chloride cotransporters in neural cells and it changes during development. The function of GABA may vary between different developmental stages (23).
There are nine kinds of glycoproteins in the cation-chloride cotransporter (CCC) family. They each have apparent molecular weights of 120–200 kDa and are encoded by Slc12a1-9 genes (24). Seven of the nine are plasmalemmal ion transporters, which are divided into three categories: two NKCCs (NKCC1 and NKCC2), one Na-Cl cotransporter (NCC), and four KCCs (KCC1-4) (24, 25). All CCCs except for NKCC2 and NCC are found in the brain and each is expressed in specific cell types, brain regions, or developmental stages (26, 27). NKCC1 and KCC2 are two main secondary active transporters which work by accumulating and extruding Cl−, respectively.
KCC2 is encoded by Slc12a5 and is exclusively expressed in the plasma membrane of somata and dendrites on pyramidal neurons and interneurons from the hippocampus and neocortex. It pumps Cl− across the plasma membrane out of the cell. In contrast, NKCC1, which is widely expressed in central and peripheral neurons as well as glial cells, is encoded by SLC12a2 and facilitates uptake of Cl− into cells (13, 28). KCC2 is composed of two splice variants: KCC2a and KCC2b. KCC2b is responsible for establishing hyperpolarizing GABAa receptor-mediated transmission (29). NKCC1a and NKCC1b are two highly homologous NKCC1 isoforms. The NKCC1a isoform exhibits mRNA expression primarily in the brain (30). NKCC1 and KCC2 are comprised of 12 membrane-spanning segments, 6 extracellular loops, and intracellular N- and C-terminals. They differ in the position of regulatory sequences, phosphorylation sites, and long extracellular loops (25, 31, 32). In immature neurons, an age-specific upregulation of NKCC1 and a relative deficiency in KCC2 loads more Cl− into the cell, resulting in a net Cl− outflow and subsequent depolarization when GABA activates GABAa Rs. Conversely, higher KCC2 and less NKCC1 expression results in a net Cl− influx in adult neurons (33–35). The developmental shift in expression and/or function of NKCC1 and KCC2 has sparked a large number of studies to understand the physiological and pathological mechanisms of this inreverse function in GABA.
There are a number of studies stating that downregulated NKCC1 and upregulated KCC2 lead to a switch in equilibrium potential of chloride () from depolarizing to hyperpolarizing during the neonatal period in “altricial” species (23, 36–38). Dzhala found that [Cl−]i was maintained at low levels in adult neurons with high KCC2 and low NKCC1 expression at postnatal day 21 (P21) in rodents. In contrast, immature neurons accumulated Cl−, leading to membrane depolarization at postnatal day 5 (P5) (34, 39). However, quite different from what is observed in rodents, KCC2 mRNA is already present in abundant amounts at embryonic day 42 (E42) in guinea pigs (40). Notably, similar results was got in the perinatal human hippocampus and neocortex (34, 41). Recent research has shown that the expression of NKCC1 and KCC2 may occur outside the aforementioned developmental windows (42). There are compelling differences in epilepsy subtypes between the sexes (43), and the developmental expression of KCC2 and NKCC1 also exhibit sexual dimorphism (44–46). The expression profiles of NKCC1 in male rats have previously been reported by Dzhala (34, 46). There is a peak in the relative expression of NKCC1 observed in males but not in females at P9 and P11 separately in the hippocampus and entorhinal cortex. The expression of KCC2 was significantly higher in females than in males, particularly at P1 and P9-15 both in the hippocampus and entorhinal cortex (46). This is also consistent with more depolarizing GABAergic signaling previously reported in P4-14 male rats compared to female rats (45). In addition, in terms of CCCs, levels of transcription and protein expression may not be consistent with expected [Cl−]i. However, it should not be ignored that inward and outward transport rates and changes of post-transcription related modification, such as protein phosphorylation, participate in chloride homeostasis (47, 48).
GABA, working as a major inhibitory transmitter of the mature central nervous system, is important in controlling neuron excitability and maturation and forming emerging networks. It binds to GABAa receptors, which are ligand-gated chloride channels, and triggers subsequent conformational changes (49). Then, the complex facilitates the passive flow of Cl− depending on the neuron's Ecl− (50). In immature neurons, Ecl−, which is affected significantly by minor changes in [Cl−]i, becomes positive relative to the resting membrane potential (Vm) when [Cl−]i is relatively high. However, is more negative than Vm when [Cl−]i is low and the activation of GABAa Rs triggers Cl− influx (Figure 1A).
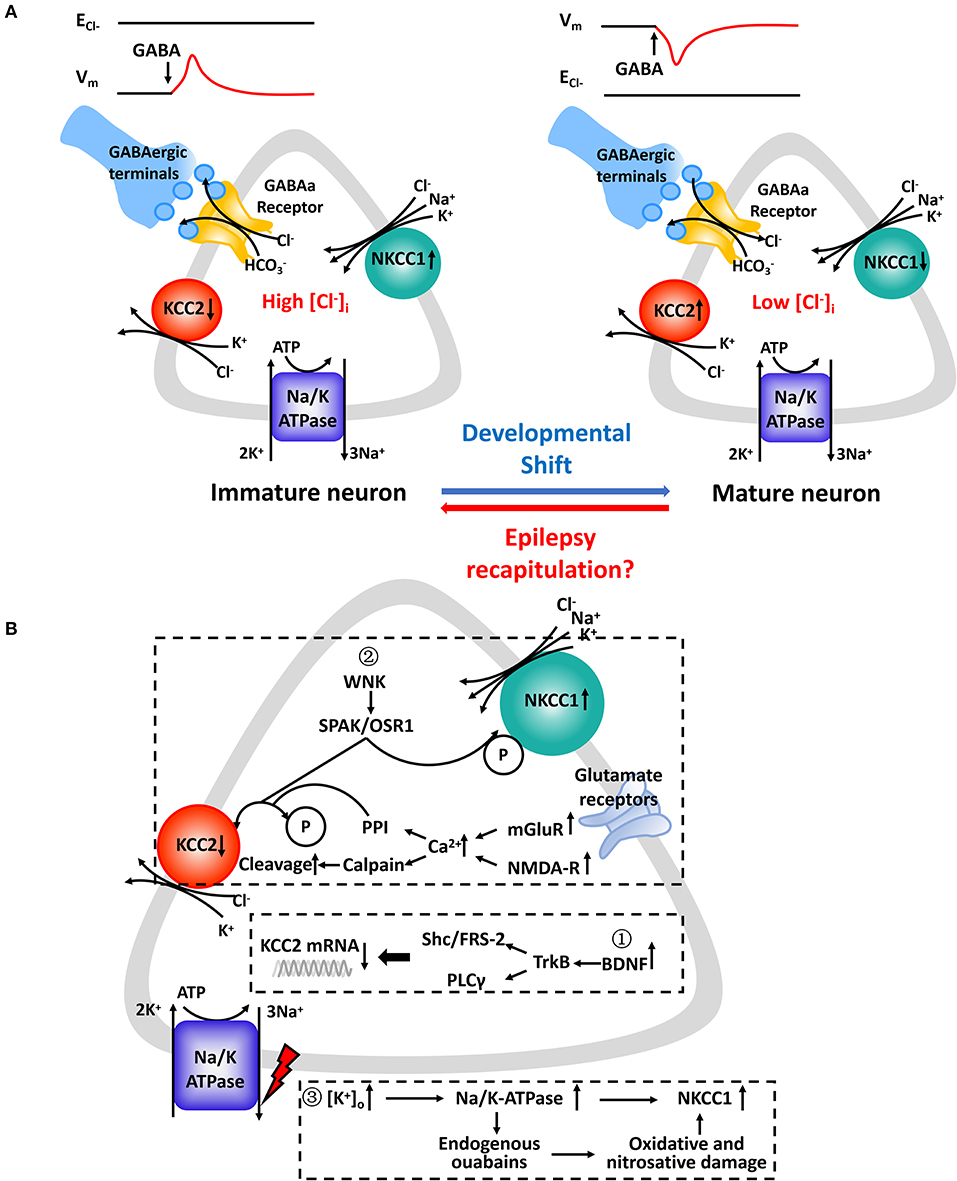
Figure 1. (A) Chloride concentration regulatory mechanisms underlying GABAa receptor-mediated responses in immature and mature CNS neurons. (Left): Upregulated NKCC1 is the main regulator which mediates Cl− uptake in immature CNS neurons in parallel with downregulated KCC2 or in its absence. (Right): KCC2 is the principal K-Cl cotransporter in parallel with subsequent downregulated NKCC1 in mature CNS neurons. Cl− transported by NKCC1 and KCC2 is facilitated by an electrochemical gradient of Na+ and K+ created by the Na+/K+-ATPase. The relative activity of NKCC1 and KCC2 and their opposing effects on [Cl−]i determines the value of ECl− relative to the membrane potential (Vm). GABA binds to corresponding GABAa receptors and opens ligand-gated Cl− channels which are also permeable to HCO3−. Expression profiles of NKCC1 and KCC2 varies at different developmental stages. In several pathophysiologic conditions, such as epilepsy, neurons experience “recapitulation” and dedifferentiation to some key and special stages of early neuronal development. (B) Molecular cascades leading to expressive and functional abnormalities in NKCC1 and KCC2. Expression levels of NKCC1 and KCC2 mRNA ①, protein levels ②, and designated regulatory pathways ③ have been shown to be altered. Summarized findings are from animal models as well as human studies.
It is a major misconception that depolarizing GABA actions mean excitation and that a more negative equilibrium potential for GABA (EGABA) is a prerequisite for inhibitory GABAergic function. First, because of intrinsic outward rectification of GABAa currents, inactivation of Na+ channels and activation of K+ channels caused by depolarizing GABA action may lead to more hyperlarizing GABAergic action (24). Second, depolarized GABA affects targeted cells via its shunting action. Postsynaptic activation of GABAa Rs leads to decreasing membrane resistance, increasing background conductance, and reduced efficacy of excitatory signals in space and in time. In other words, it may lead to a more effective inhibitory action by reducing the action potential threshold (51–53). Third, when GABA binds to GABAa Rs, bicarbonate (HCO3−) flows through GABAa Rs to a lesser extent compared to Cl− (54). A net efflux of HCO3− depolarizes EGABA and may even control the polarity of GABAergic responses (55). It has been reported that HCO3− generated by the catalytic activity of carbonic anhydrase not only paricipates in the regulation of neuronal volume and pH but also contributes to chloride homeostasis by affecting the Na+-driven Cl−/bicarbonate exchanger and the Cl−/HCO3− exchanger AE3 (56, 57). However, due to the ~5-fold increase in permeability of Cl− over HCO3−, EGABA lies closer to the Ecl− than EHCO3− in most neurons. Thus, [Cl−]i is still a critical determinant in fast-synaptic inhibition (12, 53). It had been reported that depolarization by GABA controls early network activity in the developing hippocampus and neocortex (58, 59). During development, the activation of GABAa Rs leads to neuron depolarization and activation of Ca2+ signals in immature cells (60). GABAa receptor-mediated depolarization appears to play a decisive role in early network activities (58), circuit formation (61, 62), neuronal migration (63), and synapse maturation (64).
Clinical studies have shown that children are more likely to have epileptic seizures than adults, and the neonatal period in particular has the greatest incidence of seizures (1.8–3.5%) (65, 66). There is evidence that GABA has a depolarizing effect in the healthy newborn rat cortex (67). During the postnatal period, an increased expression of KCC2 shifts the polarity of GABAergic responses from depolarizing to hyperpolarizing in the mature neuron (34, 40). Researchers observed that elevated [Cl−]i caused pathologic neuronal and network hyperexcitability in animal models, including knockout mice carrying a genetic loss of NKCC1 or KCC2 (1, 34, 68, 69). Wang et al. described the expression of NKCC1 and KCC2 based on electrophysiological studies of patients with epilepsy (12). Bumetanide, a specific inhibitor of NKCC1, may be a powerful tool to test whether NKCC1 is responsible for the accumulation [Cl−]i (34).
In different animal models of chronic epilepsy, a significant upregulation of NKCC1 and downregulation of KCC2, both in mRNA and protein levels, have been reported (12). Depolarized EGABA and accumulation of [Cl−]i were consistent with the corresponding increased expression of NKCC1 and decreased expression of KCC2. However, studies on EGABA shifts have reported inconsistent findings in animal models of epilepsy and are currently controversial (Table 1).
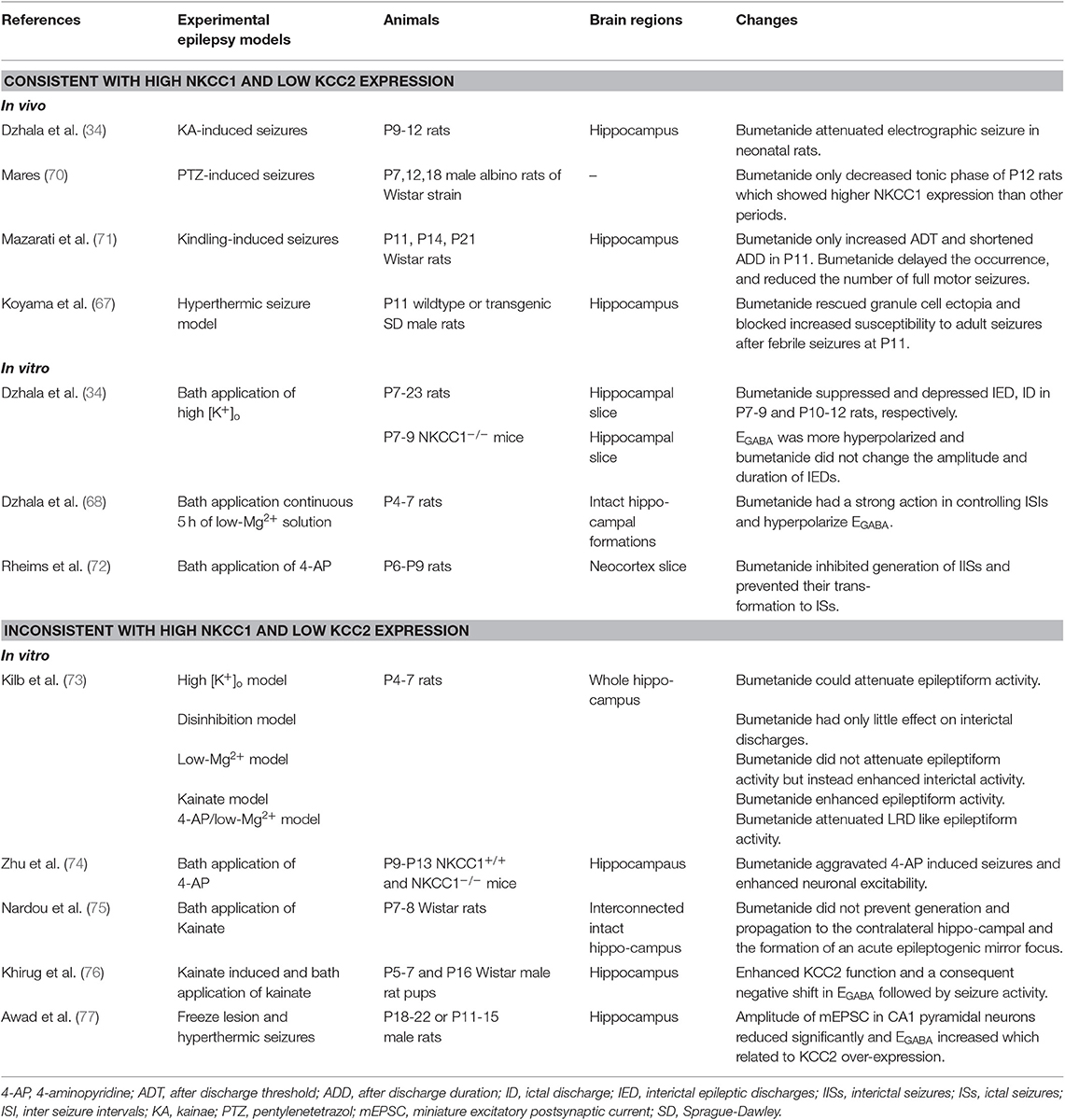
Table 1. Changes in function in experimental neonatal epilepsy models.
Dzhala found that bumetanide contributed to more hyperpolarized EGABA. He depressed synchronous bursts of action potentials by combining local field potential and whole-cell recordings of epileptiform activity induced by 8.5 mM [K+]o and illustrated that NKCC1 facilitates seizures in the developing brain (34). Genetic loss of KCC2 in diverse model organisms including worms, flies, fish, and mice resulted in less Cl− extrusion, more increase in [Cl−]i, and pathologic neuronal and network hyperexcitability (78). Dysregulation of NKCC1 and KCC2 has been shown to occur in various conditions, such as hyperthermic seizure, hypoxic-ischemic encephalopathy, and sevoflurane- induced seizures (67, 79, 80). However, there are still differing opinions on the effects of altered NKCC1 and KCC2 on epileptic activity. For example, a different point proposed by Zhu et al. illustrated that NKCC1 acts as an anticonvulsant by inhibiting the generation of seizures instead of facilitating seizure activity (74). Another view is that increased KCC2 protein levels are accompanied by a depolarized EGABA in cortical dysplasia and hyperthermia-induced seizures, suggesting that KCC2 overexpression may increase febrile seizure susceptibility (77). In contrast to downregulation of KCC2 in seizure models, several scholars have shown that a single seizure episode induced by kainate injection of P5-7 in rats can increase the Cl− extrusion capacity of rat hippocampal CA1 neurons and increase the activity-dependent regulation of KCC2 function (76). In addition to understand the regulation of NKCC1 and KCC2 in various neonatal seizure and epilepsy models, it is worth exploring changes in NKCC1 and KCC2 in mature neurons (Table 2).
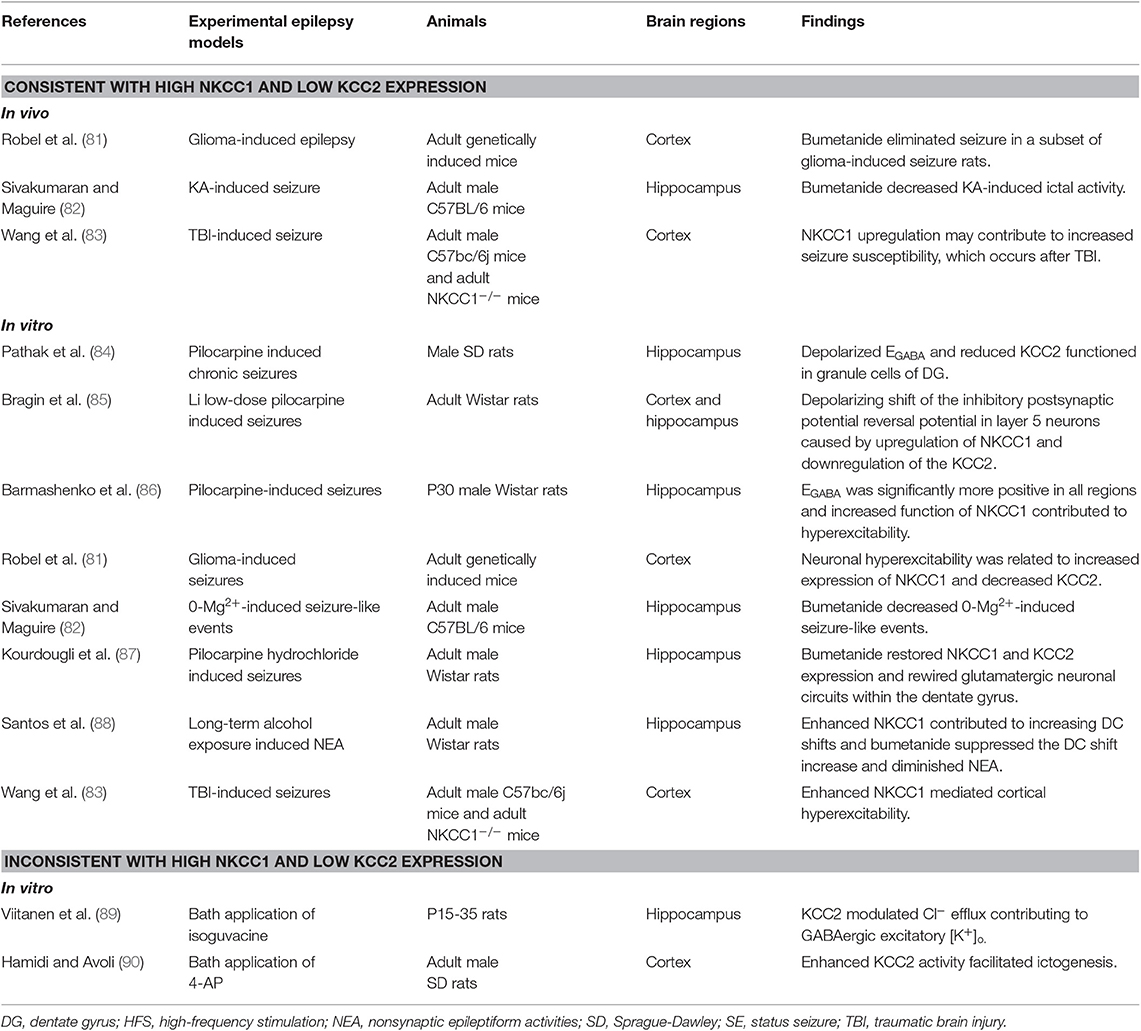
Table 2. Changes in function of NKCC1 in experimental adult epilepsy models.
Several studies have shown that enhanced NKCC1 and decreased KCC2 function is consistent with increased NKCC1 and decreased KCC2 mRNA and protein levels in different adult animal seizure models. The phenomenon is named-“ontogenetic recapitulation,” which implies that neurons in the adult models had dedifferentiated to some key and special stage of early neuronal development. Bumetanide significantly decreases kainate-induced ictal activity in vivo and 0-Mg2+-induced seizure-like events in vitro, which implies that impaired chloride plasticity may contribute to in seizure progression (82). It is hypothesized that an abnormal depolarizing GABAergic function caused by an imbalance in NKCC1 and KCC2, may cause rewiring of excitatory glutamatergic neuronal circuits and play a role in the epileptogenic processes (87). NKCC1 not only plays a key role in facilitating synaptic epileptiform activity, but also in non-synaptic epileptiform activity in the dentate gyrus (91). Increased seizure susceptibility and depolarized EGABA caused by chronic stress may imply an imbalance in chloride homeostasis and can be reversed with bumetanide (92). KCC2 drives Cl− out of the neuron against its electrochemical driving force through the “Cl− pump” mechanism and is postulated to be the force for mediating Cl− outflow in mammalian cortical neurons (93). Moreover, significantly decreased KCC2 protein and mRNA levels with an impaired chloride clearance causes a positive shift in EGABA, resulting in decreased inhibition in the dentate gyrus. This functions as a filter impeding excessive or aberrant activity from propagating further into the circuit (94). The aforementioned studies in adult animal seizure models are controversial, but it is generally accepted that recurrent seizures and other traumatic insults may lead to downregulation of KCC2 and remodeling of NKCC1-dependent depolarizing GABAergic signaling (35). Hamidi detected high-frequency oscillatory events from field potential recordings and suggested that enhancing KCC2 activity may facilitate ictogenesis by increasing extracellular K+ (90). KCC2 expression returned to normal level and was even upregulated during the chronic phase, suggesting that a certain level of reorganization on seizure activity might be necessary for this increase (84).
The molecular cascades that lead to imbalanced CCCs and the possibility of progression from “activity-dependent” chloride plasticity to “pathologic” chloride plasticity remain largely unknown (12). As studies stand presently, there are some factors that lead to an upregulation of NKCC1 and downregulation of KCC2 in neurons in epilepsy (Figure 1B). First, the brain-derived neurotrophic factor (BDNF) and tyrosine kinase receptor B (TrkB) signaling pathways endogenously release more BDNF, which binds to TrkB receptors and activate src homology domains that contain transforming protein/FGF receptor substrate 2 (Shc/FRS-2) and phospholipase Cγ (PLC-γ). This has been shown to reduce the expression of KCC2 mRNA in various animal models of epilepsy (79, 95, 96). Second, different phosphorylation sites play different roles in the surface stability and activity of KCC2. The phosphorylation of residue serine-940 (S940) within the KCC2 cytoplasmic domain leads to increased KCC2 activity as well as its increased accumulation on the plasma membrane (97). Ca2+-dependent mechanisms initiated by the accumulation Ca2+ from metabotropic glutamate receptors (mGluRs) and glutamatergic NMDA receptors lead to downregulation of KCC2. It also coincides with protein phosphatase 1 dependent dephosphorylation of S940 and activation of calcium-activated protease calpain, which is involved in the cleavage of KCC2 (98, 99). Moreover, phosphorylation of Tyr 903/1087 of the KCC2 protein in pilocarpine-induced adult seizure mouse models leads to increased lysosomal degradation of KCC2 (100). Activation of with-no-lysine kinase (WNK), SPS1-related proline/alanine-rich kinases (SPAK), or SPAK homolog oxidative stress-responsive kinase1 (OSR1) pathways causes phosphorylation of threonin residues T906 and T1007, causing decreased KCC2 activity (47). The WNK-SPAK/OSR1 pathway also phosphorylates NKCC1 and promotes NKCC1 activity (101). Third, the role that Na+/K+-ATPase plays in epileptogenesis is not yet fully understood. The downregulation of ion transporters, such as plasmalemmal Na+/K+-ATPase, occurs via translocation of the protein and functional loss of transport activity and may reflect an adaptive response to an “energy crisis” (102, 103). Extracellular potassium uptake in neurons occurs via the Na+/K+-ATPase (104). Ion gradients created by the catalytic effect of Na+/K+-ATPase drives NKCC1 function by activating production of endogenous ouabains that cause oxidative and nitrosative damage (105). Reduced GABAergic potentials and enhanced coupling between excitatory postsynaptic potentials and spike firing are the primary mechanisms underlying the hyperexcitability associated with impaired Na+/K+-ATPase activity (106). Furthermore, molecular mechanisms related to the regulation of neuronal NKCC1 in epilepsy needs to be further explored.
Increasingly more studies have taken an interest in animal models that artificially and specifically lower the expression of ortholog KCC2. Genetic ablation of KCC2 resulted in increased seizure susceptibility (107), and may work through not only impaired Cl− homeostasis but also dendritic spine dysfunction due to inability of KCC2 to bind to the cytoskeleton (32, 108). Transgenic technology made it possible to analyze the gene sequence for human SLC12a5, a mutant human gene in patients with seizure disorders (1, 109–112). This provided a potential target for electrophysiological studies aimed at exploring the pathogenesis of epileptic attacks. Unfortunately, while epileptiform discharges can be induced in animal seizure models, whether scientists can reproduce all aspects of human pathology in epilepsy remains uncertain (113).
Although there have been many acute and chronic animal models studies on altered NKCC1 and KCC2, there is still a need to perform studies on human epileptic brain tissue (114). Following are several studies investigating the changes of NKCC1 and KCC2 in epilepsy patients.
Dzhala investigated changes in NKCC1 and KCC2 levels in the human cortex and showed that NKCC1 level peaked from post-conceptional week (PCW) 31–41. KCC2 expression was ~2–25% of adult levels at PCW 31–41 and increased over the first year of life (34). Described as a specific malformation of cortical development, focal cortical dysplasia (FCD) is categorized into 3 main types. FCD type II is divided into two subtypes, FCD IIa and FCD IIb whose only difference histopathologically is the appearance of balloon cells (115). Lesions in FCD IIb have higher NKCC1: KCC2 ratios compared to lesions in FCD IIa. In other words, decreased GABAergic inhibition may represent a critical mechanism of epileptogenesis in FCD IIb compared to FCD IIa (11). In a recent study on pediatric FCD patients, interictal discharges sustained by GABAergic signaling were suppressed by blocking NKCC1. Meanwhile, perturbed Cl− homeostasis related to decreased expression and changes in KCC2 have been observed in the same patients (7). Sharing similar pathological characteristics with FCD, dysplastic neurons in tuberous sclerosis complex (TSC) exhibit depolarizing GABAa Rs mediated spontaneous post-synaptic currents (PSCs), which are controlled by bumetanide (11). In general, a shift toward the excitatory effect of GABA may contribute to FCD and TSC epileptogenicity.
Based on results from several animal models of chronic epilepsy using kainate or pilocarpine, downregulation of KCC2 may play a critical role in the pathophysiology of temporal lobe epilepsy (TLE). The hippocampal subiculum serves as an origin for synchronous firing to spread to other regions of the temporal lobe during seizures. This region shows significant upregulation of NKCC1 mRNA and downregulation of KCC2 mRNA compared with the hippocampus proper or neocortex in drug-resistant mesial temporal lobe epilepsy (MTLE) (116, 117). Electrophysiology studies have shown that aberrant GABA excitation caused by altered NKCC1 and KCC2 contributes to the precipitation of epileptic seizures in TLE (117, 118).
What roles do different types of neurons play in the generation of epileptic activities? Depolarizing GABAa receptor-mediated PSCs in a minority of subiculum pyramidal cells have been shown to impair Cl− homeostasis. Remodeling Cl− homeostasis using bumetanide produced a hyperpolarizing shift in EGABA and suppressed interictal activity. Among depolarized cells, most were immunonegative for KCC2, and all hyperpolarized cells in interictal events were immune-positive for KCC2 (119). In TLE, high-affinity glycine receptors (haGlyR), which are homologous to GABAa receptors, are increased in the hippocampus and this is inversely associated with a decrease in KCC2 expression (120). Contrary to what was mentioned previously, Karlocai et al. investigated differences in KCC2 expression in the hippocampus of TLE patients and control brain samples using immunocytochemistry and noted an overall increase in KCC2 expression in epilepsy patients (121). It has been disputed that the differences in KCC2 expression may result from differences in epileptic tissues, epileptic phases (i.e., acute, latent, chronic) (84), and brain regions studied (85, 119, 122, 123).
Analogous to FCD and TLE, brain tumors also exhibit a shift in GABAergic signaling from inhibitory to excitatory, which likely contribute to their epileptogenicity (124). Conti et al. studied peritumoral cortical tissues from epileptic patients afflicted by gliomas and found alterations in NKCC1 and KCC2 activity and a reduction in the hyperpolarizing effects of GABA, which likely contribute to epileptogenesis (124). Pallud et al. showed that in patients with gliomas, GABA depolarized 65% of pyramidal cells, which exhibited a perturbation of chloride homeostasis with a 144% increase in NKCC1 expression and a 42% decrease in KCC2 expression (125).
Human hypothalamic hamartomas associated with gelastic seizures is difficult to diagnose early in life and notoriously refractory to medical therapy (126, 127). Kim et al. expounded that altered expression of NKCC1 and KCC2 via GABAa receptor-mediated excitation contributes to epileptogenesis in hypothalamic hamartomas (128). In a recent study, a shift of EGABA toward more depolarizing values and a parallel increase in the ratio of NKCC1 to KCC2 expression were found in Xenopus oocyte cell membranes obtained from brain tissues of Dravet syndrome patients (129). Differences in gene silencing also play a role as significantly lower NKCC1 DNA methylation and higher KCC2 DNA methylation has been shown in juvenile myoclonic epilepsy (130). However, we need to await further experiments based on human tissues to verify these altered patterns in brain disorders associated with epilepsy. Bumetanide, which rescues the accumulation of [Cl−]i by suppressing NKCC1 specifically, reestablishes the hyperpolarizing GABA response and decreases epileptiform activity. Thus, it is a potential novel therapeutic avenue for the treatment of severe and intractable epilepsy.
Depolarizing responses mediated by GABAa Rs were first seen in hippocampal tissues obtained from human epileptic tissue (131). Although many adult epilepsy patients respond well to conventional AEDs—particularly those that potentiate GABAa Rs—these drugs are largely ineffective in controling neonatal epilepsy (68, 132). Consequently, it is critical that we pursue novel AEDs for epilepsy treatment.
Two widely used diuretic drugs have been implicated in the regulation of NKCC1 and KCC2: bumetanide which blocks NKCC1 and furosemide which targets several types of ion-transporters (133, 134). Furosemide, acts on multiple protein targets, such as KCC2, AE3, some subtypes of GABAa Rs, as well as carbonic anhydrase (135, 136). It exerts its anticonvulsant effect through a variety of complex mechanisms involving the regulation of [K+]o and [Cl−]i homeostasis, cell volume, and pH. Uwera et al. investigated the effects of furosemide on Cs-induced epileptiform activity and found that it worked by blocking neuronal KCC2 and AE3 transporters (134).
Kharod summarized the results of pre-clinical and clinical studies regarding model-specific efficacies of bumetanide (137). Here, we revisit these clinical studies briefly. Kahle et al. explored the clinical antiepileptic effects of bumetanide and found that bumetanide did not show any antiepileptic effects in the human neonate (138). Disappointingly, the first clinical phase I/II trial with bumetanide in 14 newborn infants with seizures showed that bumetanide was not effective enough as an adjunct to phenobarbital and increased the risk of hearing loss (139). Eftekhari et al. were the first to demonstrate the efficacy of bumetanide on reduction of seizure frequency in three adult patients with temporal lobe epilepsy (140). It was enlightening and inspiring to see decreased seizure frequency in all three patients and decreased epileptiform discharges in two of the three patients (140). A recent study of 30 patients with drug-resistant TLE showed that bumetanide can be an effective and relatively tolerable drug for patients with recalcitrant disease (141, 142). Of course, there are limitations and adverse side effects of bumetanide, which limit its application in clinical practice (35, 143). Therefore, it remains to be determined whether bumetanide will become a novel AED.
NKCC1 and KCC2, two main members of the cell-type-specific and subcellular expression patterns of functional CCCs, regulate the effects of GABAergic signals by mediating steady-state [Cl−]i gradients. Temporal shifts in GABAergic signaling have been identified during development. In early stages of embryonic development, depolarizing GABA activates synaptic receptors and regulates neuronal migration. Consistent with the excitatory effects of GABA, NKCC1, and KCC2 undergo similar changes in ontogenesis. However, in several pathophysiologic conditions, such as epilepsy, neurons revert to an immature state and demonstrate downregulation of KCC2 and upregulation of NKCC1.
We have summarized electrophysiology studies investigating the role of NKCC1 and KCC2 in animal epileptic models and in vitro studies using tissue from epilepsy patients. The most common conclusion of the role of these two cotransporters in epileptogenesis is that increased [Cl−]i, mediated by upregulation of NKCC1 and downregulation of KCC2, diminishes the efficacy of GABAergic inhibition or even reverts it to excitation. Changes in NKCC1 and KCC2 likely interact with multiple complex mechanisms and contribute to the pathogenesis of epilepsy. The diuretic bumetanide is widely used to inhibit neuronal NKCC1, particularly when its expression is abnormally increased in patients with epilepsy. However, there is still no convincing data confirming that bumetanide could be used in the clinic as an effective AED.
Several studies have shown that, at the cellular level, plasmalemmal channels and ion transporters are often multifunctional (144). Future experimental work on polymorphisms may provide additional insights into functional and dysfunctional CCCs. Further studies in human subjects are necessary to uncover the exact mechanism by which CCCs contribute to epileptogenesis.
RL, JW, SL, and GZ contributed to the conceptualization, designed the manuscript, and conducted literature searches. RL wrote the manuscript and made tables. XY and GZ revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the National Natural Science Foundation of China (81971202, 81671367, 81790653, and 81471391).
1. Stodberg T, McTague A, Ruiz AJ, Hirata H, Zhen J, Long P, et al. Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat Commun. (2015) 6:8038. doi: 10.1038/ncomms9038
2. Morganti-Kossmann MC, Semple BD, Hellewell SC, Bye N, Ziebell JM. The complexity of neuroinflammation consequent to traumatic brain injury: from research evidence to potential treatments. Acta Neuropathol. (2019) 137:731–55. doi: 10.1007/s00401-018-1944-6
3. Thijs RD, Surges R, O'Brien TJ, Sander JW. Epilepsy in adults. Lancet. (2019) 393:689–701. doi: 10.1016/S0140-6736(18)32596-0
4. Kardos J, Heja L, Jemnitz K, Kovacs R, Palkovits M. The nature of early astroglial protection-Fast activation and signaling. Prog Neurobiol. (2017) 153:86–99. doi: 10.1016/j.pneurobio.2017.03.005
5. Palma E, Ruffolo G, Cifelli P, Roseti C, Vliet EAV, Aronica E. Modulation of GABAA receptors in the treatment of epilepsy. Curr Pharm Des. (2017) 23:5563–8. doi: 10.2174/1381612823666170809100230
6. Kato M, Dobyns WB. X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”. J Child Neurol. (2005) 20:392–7. doi: 10.1177/08830738050200042001
7. Blauwblomme T, Dossi E, Pellegrino C, Goubert E, Iglesias BG, Sainte-Rose C, et al. Gamma-aminobutyric acidergic transmission underlies interictal epileptogenicity in pediatric focal cortical dysplasia. Ann Neurol. (2019) 85:204–17. doi: 10.1002/ana.25403
8. Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. (2004) 5:793–807. doi: 10.1038/nrn1519
9. Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. (2014) 505:318–26. doi: 10.1038/nature12983
10. Zhu WJ, Roper SN. Reduced inhibition in an animal model of cortical dysplasia. J Neurosci. (2000) 20:8925–31. doi: 10.1523/JNEUROSCI.20-23-08925.2000
11. Talos DM, Sun H, Kosaras B, Joseph A, Folkerth RD, Poduri A, et al. Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann Neurol. (2012) 71:539–51. doi: 10.1002/ana.22696
12. Wang Y, Wang Y, Chen Z. Double-edged GABAergic synaptic transmission in seizures: the importance of chloride plasticity. Brain Res. (2018) 1701:126–36. doi: 10.1016/j.brainres.2018.09.008
13. Rahmati N, Hoebeek FE, Peter S, De Zeeuw CI. Chloride homeostasis in neurons with special emphasis on the olivocerebellar system: differential roles for transporters and channels. Front Cell Neurosci. (2018) 12:101. doi: 10.3389/fncel.2018.00101
14. Moore YE, Kelley MR, Brandon NJ, Deeb TZ, Moss SJ. Seizing control of KCC2: a new therapeutic target for epilepsy. Trends Neurosci. (2017) 40:555–71. doi: 10.1016/j.tins.2017.06.008
15. Hsu YT, Chang YG, Chern Y. Insights into GABAAergic system alteration in Huntington's disease. Open Biol. (2018) 8:180165. doi: 10.1098/rsob.180165
16. Kwan P, Brodie MJ. Definition of refractory epilepsy: defining the indefinable? Lancet Neurol. (2010) 9:27–9. doi: 10.1016/S1474-4422(09)70304-7
17. Krauss GL, Sperling MR. Treating patients with medically resistant epilepsy. Neurol Clin Pract. (2011) 1:14–23. doi: 10.1212/CPJ.0b013e31823d07d1
18. Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat Med. (2004) 10:685–92. doi: 10.1038/nm1074
19. Rennie J, Boylan G. Treatment of neonatal seizures. Arch Dis Child Fetal Neonatal Ed. (2007) 92:F148–50. doi: 10.1136/adc.2004.068551
20. Bonifacio SL, Glass HC, Peloquin S, Ferriero DM. A new neurological focus in neonatal intensive care. Nat Rev Neurol. (2011) 7:485–94. doi: 10.1038/nrneurol.2011.119
21. Low E, Stevenson NJ, Mathieson SR, Livingstone V, Ryan AC, Rennie JM, et al. Short-term effects of phenobarbitone on electrographic seizures in neonates. Neonatology. (2016) 110:40–6. doi: 10.1159/000443782
22. Verrotti A, Moavero R, Panzarino G, Di Paolantonio C, Rizzo R, Curatolo P. The Challenge of pharmacotherapy in children and adolescents with epilepsy-ADHD comorbidity. Clin Drug Investig. (2018) 38:1–8. doi: 10.1007/s40261-017-0585-1
23. Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. (2007) 87:1215–84. doi: 10.1152/physrev.00017.2006
24. Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. (2009) 61:820–38. doi: 10.1016/j.neuron.2009.03.003
25. Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. (2005) 85:423–93. doi: 10.1152/physrev.00011.2004
26. Watanabe M, Fukuda A. Development and regulation of chloride homeostasis in the central nervous system. Front Cell Neurosci. (2015) 9:371. doi: 10.3389/fncel.2015.00371
27. Schulte JT, Wierenga CJ, Bruining H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci Biobehav Rev. (2018) 90:260–71. doi: 10.1016/j.neubiorev.2018.05.001
28. Benarroch EE. Cation-chloride cotransporters in the nervous system: general features and clinical correlations. Neurology. (2013) 80:756–63. doi: 10.1212/WNL.0b013e318283bb1c
29. Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. (2005) 93:1557–68. doi: 10.1152/jn.00616.2004
30. Cutler CP, Cramb G. Two isoforms of the Na+/K+/2Cl- cotransporter are expressed in the European eel (Anguilla anguilla). Biochim Biophys Acta. (2002) 1566:92–103. doi: 10.1016/S0005-2736(02)00596-5
31. Acton BA, Mahadevan V, Mercado A, Uvarov P, Ding Y, Pressey J, et al. Hyperpolarizing GABAergic transmission requires the KCC2 C-terminal ISO domain. J Neurosci. (2012) 32:8746–51. doi: 10.1523/JNEUROSCI.6089-11.2012
32. Hartmann AM, Nothwang HG. Molecular and evolutionary insights into the structural organization of cation chloride cotransporters. Front Cell Neurosci. (2014) 8:470. doi: 10.3389/fncel.2014.00470
33. Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol. (1997) 33:781–95. doi: 10.1002/(SICI)1097-4695(19971120)33:6<781::AID-NEU6>3.0.CO;2-5
34. Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. (2005) 11:1205–13. doi: 10.1038/nm1301
35. Loscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. (2013) 69:62–74. doi: 10.1016/j.neuropharm.2012.05.045
36. Hubner CA, Lorke DE, Hermans-Borgmeyer I. Expression of the Na-K-2Cl-cotransporter NKCC1 during mouse development. Mech Dev. (2001) 102:267–9. doi: 10.1016/S0925-4773(01)00309-4
37. Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hubner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. (2004) 468:57–64. doi: 10.1002/cne.10983
38. Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. (2004) 557(Pt 3):829–41. doi: 10.1113/jphysiol.2004.062471
39. Jansen LA, Peugh LD, Roden WH, Ojemann JG. Impaired maturation of cortical GABA(A) receptor expression in pediatric epilepsy. Epilepsia. (2010) 51:1456–67. doi: 10.1111/j.1528-1167.2009.02491.x
40. Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, et al. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. (1999) 397:251–5. doi: 10.1038/16697
41. Vanhatalo S, Palva JM, Andersson S, Rivera C, Voipio J, Kaila K. Slow endogenous activity transients and developmental expression of K+-Cl- cotransporter 2 in the immature human cortex. Eur J Neurosci. (2005) 22:2799–804. doi: 10.1111/j.1460-9568.2005.04459.x
42. Sedmak G, Jovanov-Milosevic N, Puskarjov M, Ulamec M, Kruslin B, Kaila K, et al. Developmental expression patterns of KCC2 and functionally associated molecules in the human brain. Cereb Cortex. (2016) 26:4574–89. doi: 10.1093/cercor/bhv218
43. Giorgi FS, Galanopoulou AS, Moshe SL. Sex dimorphism in seizure-controlling networks. Neurobiol Dis. (2014) 72(Pt B):144–52. doi: 10.1016/j.nbd.2014.05.010
44. Nunez JL, McCarthy MM. Evidence for an extended duration of GABA-mediated excitation in the developing male versus female hippocampus. Dev Neurobiol. (2007) 67:1879–90. doi: 10.1002/dneu.20567
45. Galanopoulou AS. Dissociated gender-specific effects of recurrent seizures on GABA signaling in CA1 pyramidal neurons: role of GABA(A) receptors. J Neurosci. (2008) 28:1557–67. doi: 10.1523/JNEUROSCI.5180-07.2008
46. Murguia-Castillo J, Beas-Zarate C, Rivera-Cervantes MC, Feria-Velasco AI, Urena-Guerrero ME. NKCC1 and KCC2 protein expression is sexually dimorphic in the hippocampus and entorhinal cortex of neonatal rats. Neurosci Lett. (2013) 552:52–7. doi: 10.1016/j.neulet.2013.07.038
47. Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. (2009) 138:525–36. doi: 10.1016/j.cell.2009.05.031
48. Friedel P, Kahle KT, Zhang J, Hertz N, Pisella LI, Buhler E, et al. WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci Signal. (2015) 8:ra65. doi: 10.1126/scisignal.aaa0354
49. Zhu S, Noviello CM, Teng J, Walsh RMJr, Kim JJ, Hibbs RE. Structure of a human synaptic GABAA receptor. Nature. (2018) 559:67–72. doi: 10.1038/s41586-018-0255-3
50. Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist. (2012) 18:467–86. doi: 10.1177/1073858412438697
51. Gulledge AT, Stuart GJ. Excitatory actions of GABA in the cortex. Neuron. (2003) 37:299–309. doi: 10.1016/S0896-6273(02)01146-7
52. Branchereau P, Cattaert D, Delpy A, Allain AE, Martin E, Meyrand P. Depolarizing GABA/glycine synaptic events switch from excitation to inhibition during frequency increases. Sci Rep. (2016) 6:21753. doi: 10.1038/srep21753
53. Doyon N, Vinay L, Prescott SA, De Koninck Y. Chloride regulation: a dynamic equilibrium crucial for synaptic inhibition. Neuron. (2016) 89:1157–72. doi: 10.1016/j.neuron.2016.02.030
54. Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol. (1987) 385:243–86. doi: 10.1113/jphysiol.1987.sp016493
55. Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. (1994) 42:489–537. doi: 10.1016/0301-0082(94)90049-3
56. Hentschke M, Wiemann M, Hentschke S, Kurth I, Hermans-Borgmeyer I, Seidenbecher T, et al. Mice with a targeted disruption of the Cl-/HCO3- exchanger AE3 display a reduced seizure threshold. Mol Cell Biol. (2006) 26:182–91. doi: 10.1128/MCB.26.1.182-191.2006
57. Chen LM, Kelly ML, Parker MD, Bouyer P, Gill HS, Felie JM, et al. Expression and localization of Na-driven Cl-HCO(3)(-) exchanger (SLC4A8) in rodent CNS. Neuroscience. (2008) 153:162–74. doi: 10.1016/j.neuroscience.2008.02.018
58. Cherubini E, Griguoli M, Safiulina V, Lagostena L. The depolarizing action of GABA controls early network activity in the developing hippocampus. Mol Neurobiol. (2011) 43:97–106. doi: 10.1007/s12035-010-8147-z
59. Kirmse K, Hubner CA, Isbrandt D, Witte OW, Holthoff K. GABAergic transmission during brain development: multiple effects at multiple stages. Neuroscientist. (2018) 24:36–53. doi: 10.1177/1073858417701382
60. Fritschy JM. Significance of GABA(A) receptor heterogeneity: clues from developing neurons. Adv Pharmacol. (2015) 73:13–39. doi: 10.1016/bs.apha.2014.11.006
61. Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. (2002) 3:728–39. doi: 10.1038/nrn920
62. Akerman CJ, Cline HT. Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J Neurosci. (2006) 26:5117–30. doi: 10.1523/JNEUROSCI.0319-06.2006
63. Bortone D, Polleux F. KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron. (2009) 62:53–71. doi: 10.1016/j.neuron.2009.01.034
64. Aguado F, Carmona MA, Pozas E, Aguilo A, Martinez-Guijarro FJ, Alcantara S, et al. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl- co-transporter KCC2. Development. (2003) 130:1267–80. doi: 10.1242/dev.00351
65. Cowan LD. The epidemiology of the epilepsies in children. Ment Retard Dev Disabil Res Rev. (2002) 8:171–81. doi: 10.1002/mrdd.10035
66. Wirrell EC. Neonatal seizures: to treat or not to treat? Semin Pediatr Neurol. (2005) 12:97–105. doi: 10.1016/j.spen.2005.03.004
67. Koyama R, Tao K, Sasaki T, Ichikawa J, Miyamoto D, Muramatsu R, et al. GABAergic excitation after febrile seizures induces ectopic granule cells and adult epilepsy. Nat Med. (2012) 18:1271–8. doi: 10.1038/nm.2850
68. Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. (2008) 63:222–35. doi: 10.1002/ana.21229
69. Gagnon KB, Delpire E. Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am J Physiol Cell Physiol. (2013) 304:C693–714. doi: 10.1152/ajpcell.00350.2012
70. Mares P. Age- and dose-specific anticonvulsant action of bumetanide in immature rats. Physiol Res. (2009) 58:927–30.
71. Mazarati A, Shin D, Sankar R. Bumetanide inhibits rapid kindling in neonatal rats. Epilepsia. (2009) 50:2117–22. doi: 10.1111/j.1528-1167.2009.02048.x
72. Rheims S, Represa A, Ben-Ari Y, Zilberter Y. Layer-specific generation and propagation of seizures in slices of developing neocortex: role of excitatory GABAergic synapses. J Neurophysiol. (2008) 100:620–8. doi: 10.1152/jn.90403.2008
73. Kilb W, Sinning A, Luhmann HJ. Model-specific effects of bumetanide on epileptiform activity in the in-vitro intact hippocampus of the newborn mouse. Neuropharmacology. (2007) 53:524–33. doi: 10.1016/j.neuropharm.2007.06.015
74. Zhu L, Polley N, Mathews GC, Delpire E. NKCC1 and KCC2 prevent hyperexcitability in the mouse hippocampus. Epilepsy Res. (2008) 79:201–12. doi: 10.1016/j.eplepsyres.2008.02.005
75. Nardou R, Ben-Ari Y, Khalilov I. Bumetanide, an NKCC1 antagonist, does not prevent formation of epileptogenic focus but blocks epileptic focus seizures in immature rat hippocampus. J Neurophysiol. (2009) 101:2878–88. doi: 10.1152/jn.90761.2008
76. Khirug S, Ahmad F, Puskarjov M, Afzalov R, Kaila K, Blaesse P. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci. (2010) 30:12028–35. doi: 10.1523/JNEUROSCI.3154-10.2010
77. Awad PN, Sanon NT, Chattopadhyaya B, Carrico JN, Ouardouz M, Gagne J, et al. Reducing premature KCC2 expression rescues seizure susceptibility and spine morphology in atypical febrile seizures. Neurobiol Dis. (2016) 91:10–20. doi: 10.1016/j.nbd.2016.02.014
78. Kahle KT, Khanna AR, Duan J, Staley KJ, Delpire E, Poduri A. The KCC2 cotransporter and human epilepsy: getting excited about inhibition. Neuroscientist. (2016) 22:555–62. doi: 10.1177/1073858416645087
79. Kang SK, Johnston MV, Kadam SD. Acute TrkB inhibition rescues phenobarbital-resistant seizures in a mouse model of neonatal ischemia. Eur J Neurosci. (2015) 42:2792–804. doi: 10.1111/ejn.13094
80. Cabrera OH, Tesic V, Tat QL, Chastain S, Quillinan N, Jevtovic-Todorovic V. Sevoflurane-induced dysregulation of cation-chloride cotransporters NKCC1 and KCC2 in neonatal mouse brain. Mol Neurobiol. (2019). doi: 10.1007/s12035-019-01751-1. [Epub ahead of print].
81. Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, et al. Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci. (2015) 35:3330–45. doi: 10.1523/JNEUROSCI.1574-14.2015
82. Sivakumaran S, Maguire J. Bumetanide reduces seizure progression and the development of pharmacoresistant status epilepticus. Epilepsia. (2016) 57:222–32. doi: 10.1111/epi.13270
83. Wang F, Wang X, Shapiro LA, Cotrina ML, Liu W, Wang EW, et al. NKCC1 up-regulation contributes to early post-traumatic seizures and increased post-traumatic seizure susceptibility. Brain Struct Funct. (2017) 222:1543–56. doi: 10.1007/s00429-016-1292-z
84. Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, et al. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. (2007) 27:14012–22. doi: 10.1523/JNEUROSCI.4390-07.2007
85. Bragin DE, Sanderson JL, Peterson S, Connor JA, Muller WS. Development of epileptiform excitability in the deep entorhinal cortex after status epilepticus. Eur J Neurosci. (2009) 30:611–24. doi: 10.1111/j.1460-9568.2009.06863.x
86. Barmashenko G, Hefft S, Aertsen A, Kirschstein T, Kohling R. Positive shifts of the GABAA receptor reversal potential due to altered chloride homeostasis is widespread after status epilepticus. Epilepsia. (2011) 52:1570–8. doi: 10.1111/j.1528-1167.2011.03247.x
87. Kourdougli N, Pellegrino C, Renko JM, Khirug S, Chazal G, Kukko-Lukjanov TK, et al. Depolarizing gamma-aminobutyric acid contributes to glutamatergic network rewiring in epilepsy. Ann Neurol. (2017) 81:251–65. doi: 10.1002/ana.24870
88. Santos LEC, Rodrigues AM, Lopes MR, Costa VDC, Scorza CA, Scorza FA, et al. Long-term alcohol exposure elicits hippocampal nonsynaptic epileptiform activity changes associated with expression and functional changes in NKCC1, KCC2 co-transporters and Na+/K+-ATPase. Neuroscience. (2017) 340:530–41. doi: 10.1016/j.neuroscience.2016.11.015
89. Viitanen T, Ruusuvuori E, Kaila K, Voipio J. The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol. (2010) 588(Pt 9):1527–40. doi: 10.1113/jphysiol.2009.181826
90. Hamidi S, Avoli M. KCC2 function modulates in vitro ictogenesis. Neurobiol Dis. (2015) 79:51–8. doi: 10.1016/j.nbd.2015.04.006
91. Nogueira GS, Santos LE, Rodrigues AM, Scorza CA, Scorza FA, Cavalheiro EA, et al. Enhanced nonsynaptic epileptiform activity in the dentate gyrus after kainate-induced status epilepticus. Neuroscience. (2015) 303:59–72. doi: 10.1016/j.neuroscience.2015.06.057
92. MacKenzie G, Maguire J. Chronic stress shifts the GABA reversal potential in the hippocampus and increases seizure susceptibility. Epilepsy Res. (2015) 109:13–27. doi: 10.1016/j.eplepsyres.2014.10.003
93. Thompson SM, Deisz RA, Prince DA. Outward chloride/cation co-transport in mammalian cortical neurons. Neurosci Lett. (1988) 89:49–54. doi: 10.1016/0304-3940(88)90479-X
94. Bonislawski DP, Schwarzbach EP, Cohen AS. Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol Dis. (2007) 25:163–9. doi: 10.1016/j.nbd.2006.09.002
95. Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, et al. BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol. (2002) 159:747–52. doi: 10.1083/jcb.200209011
96. Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipila S, et al. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. (2004) 24:4683–91. doi: 10.1523/JNEUROSCI.5265-03.2004
97. Lee HH, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem. (2007) 282:29777–84. doi: 10.1074/jbc.M705053200
98. Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci. (2011) 14:736–43. doi: 10.1038/nn.2806
99. Puskarjov M, Ahmad F, Kaila K, Blaesse P. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci. (2012) 32:11356–64. doi: 10.1523/JNEUROSCI.6265-11.2012
100. Lee HH, Jurd R, Moss SJ. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol Cell Neurosci. (2010) 45:173–9. doi: 10.1016/j.mcn.2010.06.008
101. Begum G, Yuan H, Kahle KT, Li L, Wang S, Shi Y, et al. Inhibition of WNK3 kinase signaling reduces brain damage and accelerates neurological recovery after stroke. Stroke. (2015) 46:1956–65. doi: 10.1161/STROKEAHA.115.008939
102. Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev. (1985) 65:101–48. doi: 10.1152/physrev.1985.65.1.101
103. Pylova SI, Majkowska J, Hilgier W, Kapuscinski A, Albrecht J. Rapid decrease of high affinity ouabain binding sites in hippocampal CA1 region following short-term global cerebral ischemia in rat. Brain Res. (1989) 490:170–3. doi: 10.1016/0006-8993(89)90446-0
104. Xiong ZQ, Stringer JL. Sodium pump activity, not glial spatial buffering, clears potassium after epileptiform activity induced in the dentate gyrus. J Neurophysiol. (2000) 83:1443–51. doi: 10.1152/jn.2000.83.3.1443
105. Hertz L, Song D, Peng L, Chen Y. Multifactorial effects on different types of brain cells contribute to ammonia toxicity. Neurochem Res. (2017) 42:721–36. doi: 10.1007/s11064-016-1966-1
106. Vaillend C, Mason SE, Cuttle MF, Alger BE. Mechanisms of neuronal hyperexcitability caused by partial inhibition of Na+-K+-ATPases in the rat CA1 hippocampal region. J Neurophysiol. (2002) 88:2963–78. doi: 10.1152/jn.00244.2002
107. Chamma I, Chevy Q, Poncer JC, Levi S. Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front Cell Neurosci. (2012) 6:5. doi: 10.3389/fncel.2012.00005
108. Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. (2014) 15:637–54. doi: 10.1038/nrn3819
109. Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. (2009) 87:41–57. doi: 10.1016/j.pneurobio.2008.09.016
110. Dibbens LM, Reid CA, Hodgson B, Thomas EA, Phillips AM, Gazina E, et al. Augmented currents of an HCN2 variant in patients with febrile seizure syndromes. Ann Neurol. (2010) 67:542–6. doi: 10.1002/ana.21909
111. Mulley JC, Hodgson B, McMahon JM, Iona X, Bellows S, Mullen SA, et al. Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia. (2013) 54:e122–6. doi: 10.1111/epi.12323
112. Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, et al. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep. (2014) 15:723–9. doi: 10.1002/embr.201438749
113. Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. (2002) 298:1418–21. doi: 10.1126/science.1076510
114. Thevathasan A, Naylor J, Churilov L, Mitchell PJ, Dowling RJ, Yan B, et al. Association between hemorrhagic transformation after endovascular therapy and poststroke seizures. Epilepsia. (2018) 59:403–9. doi: 10.1111/epi.13982
115. Blumcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. (2011) 52:158–74. doi: 10.1111/j.1528-1167.2010.02777.x
116. Cohen I, Navarro V, Le Duigou C, Miles R. Mesial temporal lobe epilepsy: a pathological replay of developmental mechanisms? Biol Cell. (2003) 95:329–33. doi: 10.1016/S0248-4900(03)00081-9
117. Palma E, Amici M, Sobrero F, Spinelli G, Di Angelantonio S, Ragozzino D, et al. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci USA. (2006) 103:8465–8. doi: 10.1073/pnas.0602979103
118. Palma E, Spinelli G, Torchia G, Martinez-Torres A, Ragozzino D, Miledi R, et al. Abnormal GABAA receptors from the human epileptic hippocampal subiculum microtransplanted to Xenopus oocytes. Proc Natl Acad Sci USA. (2005) 102:2514–8. doi: 10.1073/pnas.0409687102
119. Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, et al. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. (2007) 27:9866–73. doi: 10.1523/JNEUROSCI.2761-07.2007
120. Eichler SA, Kirischuk S, Juttner R, Schaefermeier PK, Legendre P, Lehmann TN, et al. Glycinergic tonic inhibition of hippocampal neurons with depolarizing GABAergic transmission elicits histopathological signs of temporal lobe epilepsy. J Cell Mol Med. (2008) 12:2848–66. doi: 10.1111/j.1582-4934.2008.00357.x
121. Karlocai MR, Wittner L, Toth K, Magloczky Z, Katarova Z, Rasonyi G, et al. Enhanced expression of potassium-chloride cotransporter KCC2 in human temporal lobe epilepsy. Brain Struct Funct. (2016) 221:3601–15. doi: 10.1007/s00429-015-1122-8
122. Aronica E, Boer K, Redeker S, Spliet WG, van Rijen PC, Troost D, et al. Differential expression patterns of chloride transporters, Na+-K+-2Cl–cotransporter and K+-Cl–cotransporter, in epilepsy-associated malformations of cortical development. Neuroscience. (2007) 145:185–96. doi: 10.1016/j.neuroscience.2006.11.041
123. Munoz A, Mendez P, DeFelipe J, Alvarez-Leefmans FJ. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia. (2007) 48:663–73. doi: 10.1111/j.1528-1167.2007.00986.x
124. Conti L, Palma E, Roseti C, Lauro C, Cipriani R, de Groot M, et al. Anomalous levels of Cl- transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia. (2011) 52:1635–44. doi: 10.1111/j.1528-1167.2011.03111.x
125. Pallud J, Le Van Quyen M, Bielle F, Pellegrino C, Varlet P, Cresto N, et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci Transl Med. (2014) 6:244ra289. doi: 10.1126/scitranslmed.3008065
126. Berkovic SF, Andermann F, Melanson D, Ethier RE, Feindel W, Gloor P. Hypothalamic hamartomas and ictal laughter: evolution of a characteristic epileptic syndrome and diagnostic value of magnetic resonance imaging. Ann Neurol. (1988) 23:429–39. doi: 10.1002/ana.410230502
127. Iannetti P, Chessa L, Raucci U, Basile LA, Fantozzi LM, Bozzao L. Gelastic epilepsy. A clinical contribution. Clin Pediatr. (1992) 31:467–70. doi: 10.1177/000992289203100804
128. Kim DY, Fenoglio KA, Simeone TA, Coons SW, Wu J, Chang Y, et al. GABAA receptor-mediated activation of L-type calcium channels induces neuronal excitation in surgically resected human hypothalamic hamartomas. Epilepsia. (2008) 49:861–71. doi: 10.1111/j.1528-1167.2007.01455.x
129. Ruffolo G, Cifelli P, Roseti C, Thom M, van Vliet EA, Limatola C, et al. A novel GABAergic dysfunction in human Dravet syndrome. Epilepsia. (2018) 59:2106–17. doi: 10.1111/epi.14574
130. Genc F, Kara M, Unal Y, Uygur Kucukseymen E, Bicer Gomceli Y, Kaynar T, et al. Methylation of cation-chloride cotransporters NKCC1 and KCC2 in patients with juvenile myoclonic epilepsy. Neurol Sci. (2019) 40:1007–13. doi: 10.1007/s10072-019-03743-4
131. Sepkuty JP, Cohen AS, Eccles C, Rafiq A, Behar K, Ganel R, et al. A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J Neurosci. (2002) 22:6372–9. doi: 10.1523/JNEUROSCI.22-15-06372.2002
132. Cleary RT, Sun H, Huynh T, Manning SM, Li Y, Rotenberg A, et al. Bumetanide enhances phenobarbital efficacy in a rat model of hypoxic neonatal seizures. PLoS ONE. (2013) 8:e57148. doi: 10.1371/annotation/48a011e6-e4d0-4706-9a28-857eba8cfb31
133. Vanhatalo S, Hellstrom-Westas L, De Vries LS. Bumetanide for neonatal seizures: based on evidence or enthusiasm? Epilepsia. (2009) 50:1292–3. doi: 10.1111/j.1528-1167.2008.01894.x
134. Uwera J, Nedergaard S, Andreasen M. A novel mechanism for the anticonvulsant effect of furosemide in rat hippocampus in vitro. Brain Res. (2015) 1625:1–8. doi: 10.1016/j.brainres.2015.08.014
135. Staley KJ. Diuretics as antiepileptic drugs: should we go with the flow? Epilepsy Curr. (2002) 2:35–8. doi: 10.1046/j.1535-7597.2002.00018.x
136. Maa EH, Kahle KT, Walcott BP, Spitz MC, Staley KJ. Diuretics and epilepsy: will the past and present meet? Epilepsia. (2011) 52:1559–69. doi: 10.1111/j.1528-1167.2011.03203.x
137. Kharod SC, Kang SK, Kadam SD. Off-label use of bumetanide for brain disorders: an overview. Front Neurosci. (2019) 13:310. doi: 10.3389/fnins.2019.00310
138. Kahle KT, Barnett SM, Sassower KC, Staley KJ. Decreased seizure activity in a human neonate treated with bumetanide, an inhibitor of the Na(+)-K(+)-2Cl(-) cotransporter NKCC1. J Child Neurol. (2009) 24:572–6. doi: 10.1177/0883073809333526
139. Pressler RM, Boylan GB, Marlow N, Blennow M, Chiron C, Cross JH, et al. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): an open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. (2015) 14:469–77. doi: 10.1016/S1474-4422(14)70303-5
140. Eftekhari S, Mehvari Habibabadi J, Najafi Ziarani M, Hashemi Fesharaki SS, Gharakhani M, Mostafavi H, et al. Bumetanide reduces seizure frequency in patients with temporal lobe epilepsy. Epilepsia. (2013) 54:e9–12. doi: 10.1111/j.1528-1167.2012.03654.x
141. Rahmanzadeh R, Mehrabi S, Barati M, Ahmadi M, Golab F, Kazmi S, et al. Effect of co-administration of bumetanide and phenobarbital on seizure attacks in temporal lobe epilepsy. Basic Clin Neurosci. (2018) 9:408–16. doi: 10.32598/bcn.9.6.408
142. Gharaylou Z, Tafakhori A, Agah E, Aghamollaii V, Kebriaeezadeh A, Hadjighassem M. A preliminary study evaluating the safety and efficacy of bumetanide, an NKCC1 inhibitor, in patients with drug-resistant epilepsy. CNS Drugs. (2019) 33:283–91. doi: 10.1007/s40263-019-00607-5
143. Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. (2014) 55:806–18. doi: 10.1111/epi.12620
Keywords: epilepsy, NKCC1, KCC2, electrophysiology, bumetanide
Citation: Liu R, Wang J, Liang S, Zhang G and Yang X (2020) Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front. Neurol. 10:1407. doi: 10.3389/fneur.2019.01407
Received: 11 April 2019; Accepted: 23 December 2019;
Published: 17 January 2020.
Edited by:
Liang Chen, Huashan Hospital, Fudan University, ChinaReviewed by:
Damir Janigro, Case Western Reserve University, United StatesCopyright © 2020 Liu, Wang, Liang, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Yang, eGlhb2Zlbmd5YW5nQHlhaG9vLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.