
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol. , 28 June 2019
Sec. Stroke
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00693
This article is part of the Research Topic Cerebral Small Vessel Diseases: From Vessel Alterations to Cortical Parenchymal Injury View all 21 articles
 Masahiro Uemura1
Masahiro Uemura1 Hiroaki Nozaki2
Hiroaki Nozaki2 Akihide Koyama1,3Naoko Sakai1
Akihide Koyama1,3Naoko Sakai1 Shoichiro Ando1
Shoichiro Ando1 Masato Kanazawa1Taisuke Kato4
Masato Kanazawa1Taisuke Kato4 Osamu Onodera1*
Osamu Onodera1*Background: Mutations in the high-temperature requirement A serine peptidase 1 (HTRA1) cause cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). Most carriers for HTRA1 mutations are asymptomatic, but more than 10 mutations have been reported in symptomatic carriers. The molecular differences between the mutations identified in symptomatic carriers and mutations identified only in CARASIL patients are unclear. HTRA1 is a serine protease that forms homotrimers, with each HTRA1 subunit activating the adjacent HTRA1 via the sensor domain of loop 3 (L3) and the activation domain of loop D (LD). Previously, we analyzed four HTRA1 mutant proteins identified in symptomatic carriers and found that they were unable to form trimers or had mutations in the LD or L3 domain. The mutant HTRA1s with these properties are presumed to inhibit trimer-dependent activation cascade. Indeed, these mutant HTRA1s inhibited wild-type (WT) protease activity. In this study, we further analyzed 15 missense HTRA1s to clarify the molecular character of mutant HTRA1s identified in symptomatic carriers.
Methods: We analyzed 12 missense HTRA1s identified in symptomatic carriers (hetero-HTRA1) and three missense HTRA1s found only in CARASIL (CARASIL-HTRA1). The protease activity of the purified recombinant mutant HTRA1s was measured using fluorescein isothiocyanate-labeled casein as substrate. Oligomeric structure was evaluated by size-exclusion chromatography. The protease activities of mixtures of WT with each mutant HTRA1 were also measured.
Results: Five hetero-HTRA1s had normal protease activity and were excluded from further analysis. Four of the seven hetero-HTRA1s and one of the three CARASIL-HTRA1s were unable to form trimers. The other three hetero-HTRA1s had mutations in the LD domain. Together with our previous work, 10 of 11 hetero-HTRA1s and two of six CARASIL-HTRA1s were either defective in trimerization or had mutations in the LD or L3 domain (P = 0.006). By contrast, eight of 11 hetero-HTRA1s and two of six CARASIL-HTRA1 inhibited WT protease activity (P = 0.162).
Conclusions: HTRA1 mutations identified in symptomatic carriers have the property of interfering the trimer-dependent activation cascade of HTRA1.
Loss-of-function mutations in the high-temperature requirement A serine peptidase 1 (HTRA1) gene cause cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) (1, 2). HTRA1 is a serine protease that forms a homotrimer. The sensor domain of loop 3 (L3) and the activation domain of loop D (LD) play essential roles in trimer-mediated activation of the neighboring HTRA1 (3, 4).
Recently, symptomatic carriers have been reported for several HTRA1 mutant alleles (5–9). We analyzed four HTRA1 alleles identified in symptomatic carriers and found that the proteins inhibited WT protease activity (6). In contrast, two of three missense HTRA1s observed only in CARASIL patients did not inhibit WT protease activity. Therefore, we proposed that the missense HTRA1s identified in symptomatic carriers have unique molecular characteristics (6). However, these characteristics have not been evaluated in other missense HTRA1s identified in symptomatic carriers or in homozygous CARASIL patients. The prediction of the pathogenicity of HTRA1 mutation in carriers is important to relatives of homozygous CARASIL patients, and carriers for variant HTRA1 alleles. In this study, we have evaluated an additional 15 missense HTRA1 mutants identified in symptomatic carriers and CARASIL patients.
From a literature search performed up to December 2017, we retrieved 12 missense HTRA1 alleles identified in symptomatic carriers: S121R, A123S, R133G, R166C, R166L, A173P, S284G, S284R, P285Q, F286V, G295R, and D450H; and 3 missense HTRA1 alleles reported only in CARASIL patients: A173T, A321T, and L364P (5, 8–12). Clinical characteristics of the symptomatic carriers are summarized in Supplemental Table 1. This study was approved by the Niigata University institutional review board.
An expression plasmid for each variant HTRA1 cDNA was generated using the GENEART® Site-Directed Mutagenesis System (Invitrogen, Carlsbad, CA). The WT-HTRA1 cDNA tagged with a C-terminal myc-His6 was subcloned into the pcDNA 3.1 vector (Invitrogen) was the substrate for mutagenesis. The concentration of each plasmid was measured using the Quant-iTTM PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). The HTRA1 cDNA plasmid vectors were transfected into FreeStyle 293 cells (Thermo Fisher Scientific) and incubated for 72 h. After incubation, secreted HTRA1 protein was purified from the culture medium using a HisTrap FF crude column (GE Healthcare, Cleveland, OH, USA). The concentration of recombinant HTRA1 proteins was measured using a Bicinchoninic Acid (BCA) Protein Assay Kit (Wako, Osaka, Japan). After pre-incubating 1 μg of the recombinant HTRA1 protein, protease activities were evaluated at 37°C using fluorescein isothiocyanate (FITC)–labeled casein as a substrate (Fluorescent Protease Assay Kit; Pierce, Rockford, IL, USA) (2, 6). WT was used as the positive control and S328A, which is deficient in protease activity, was used as the negative control. Fluorescence was measured using a FilterMax F5 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). Protease activities were calculated from the slope of the linear portion of the normalized fluorescence vs. time plots at 30, 60, and 90 min using MATLAB® R2017b (9.3.0.713579). The amount of each HTRA1 protein was analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), stained by SYPRO® Ruby Protein Gel Stain (Thermo Fisher Scientific). Five mutations, S121R, A123S, R133G, S284G, and D450H, were excluded from further analysis because the protease activities of these mutations were normal (Figure 1B); two of these five mutations were appeared to be benign (A123S and R133G) (5).
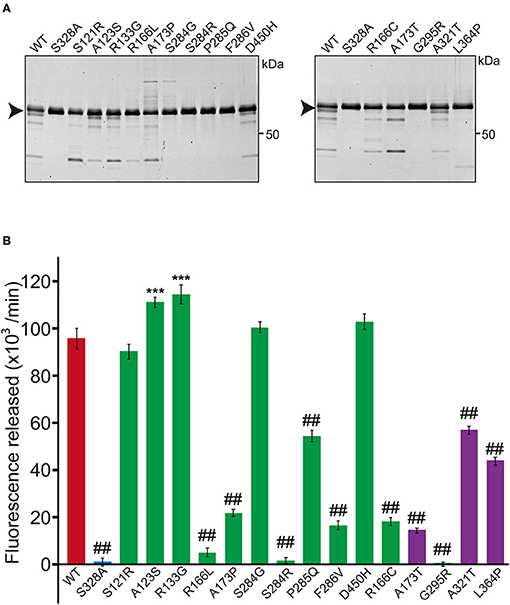
Figure 1. Protease activity of missense HTRA1s identified in symptomatic carriers. (A) SDS-PAGE of WT and missense mutant HTRA1 proteins used in the protease assay. Black arrows indicate the full-length band of HTRA1 tagged with myc-His6. (B) Protease activities of missense HTRA1s identified in symptomatic carriers and CARASIL patients. Activities were calculated from the slope of the linear portion of the normalized fluorescence vs. time (30, 60, 90 min) plots. Mean values from 3 independent experiments are shown. Red and blue bars indicate protease activities of WT and S328A, the positive and negative controls, respectively. Green bars indicate protease activities of missense HTRA1s identified in symptomatic carriers. Purple bars indicate protease activities of missense HTRA1s identified only in CARASIL patients. I-bars indicate standard errors (SE). Statistical comparisons of protease activities between WT and each missense HTRA1 protein were performed with one-way analysis of variance followed by the Dunnett's post hoc test. ***P < 0.0001 for protease activities of each HTRA1 relative to WT. ##P < 0.0001 for protease activities of HTRA1 relative to WT.
Oligomerization was evaluated by size-exclusion chromatography using a Superdex 200 10/300 GL column (GE Healthcare, Chicago, IL, USA) on an AKTA FPLC workstation equilibrated with Tris-buffered saline (100 mM Tris-HCl, pH 8.0 and 150 mM NaCl). After pre-incubation at 37°C for 30 min, each HTRA1 protein sample was injected at 500 ng/μl. Y169E/F171E HTRA1, an artificial, known monomeric HTRA1, was used as the reference monomer and S328A HTRA1 was used as a reference trimer (Figure 2C) (4, 6). SDS-PAGE was used to evaluate fractions from size-exclusion chromatography (6).
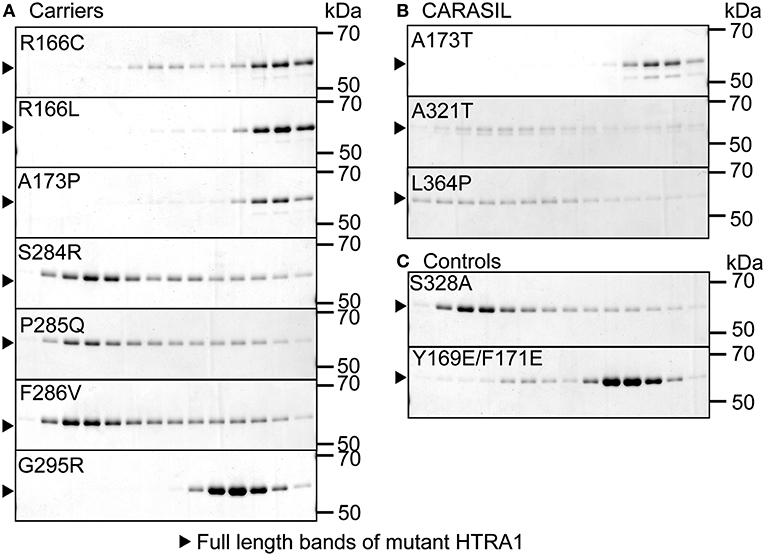
Figure 2. Oligomerization of missense HTRA1s. Size-exclusion chromatography of mutant and WT HTRA1 protein preparations. Fractions were separated by SDS-PAGE and visualized by Coomassie Brilliant Blue staining. (A) Oligomeric state analysis of each missense HTRA1 identified in symptomatic carriers. (B) Oligomeric state analysis of each missense HTRA1 found only in CARASIL. (C) Oligomeric state analysis of S328A and Y169E/F171E HTRA1.
Each HTRA1 variant vector was cotransfected with an equal amount of WT vector into FreeStyle 293 cells. Purification of the mixed HTRA1 proteins and evaluation of their protease activity were performed as described above. We compared the protease activities of the mixtures of WT with each variant HTRA1 to that of a mixture of WT and S328A, an artificially inactive HTRA1 that forms a trimer with WT and does not have a dominant-negative effect; this was used as a reference to adjust the protein concentration in the reaction mixture (6). The mixtures of G283E with WT and A252T with WT were used as positive and negative controls for dominant-negative effects, respectively (6).
Statistical analyses were performed using R 3.2.2. Groups were compared using one-way analysis of variance (ANOVA) for independent samples, followed by Dunnett's multiple-comparison test when overall P was < 0.05. Fisher's exact test was used to compare the frequencies of mutant HTRA1s with dominant-negative effect between symptomatic carriers and CARASIL.
Five missense HTRA1s (S121R, A123S, R133G, S284G, and D450H) showed normal protease activity, thus we excluded these HTRA1s from further analysis (Figures 1A,B). Protease activities of other HTRA1s were significantly decreased relative to WT activity. We assayed trimerization of the other HTRA1s and found that four hetero-HTRA1s (R166C, R166L, A173P, and G295R) and one CARASIL-HTRA1 (A173T) were unable to form trimers (Figures 2A,B). Among mutant HTRA1s form trimers, all hetero-HTRA1s had mutations in the LD domain (residues 284–290) or L3 loop domain, (residues 301–314) while two CARASIL-HTRA1s had mutations in the protease domain (residues 204–356) (Table 1).
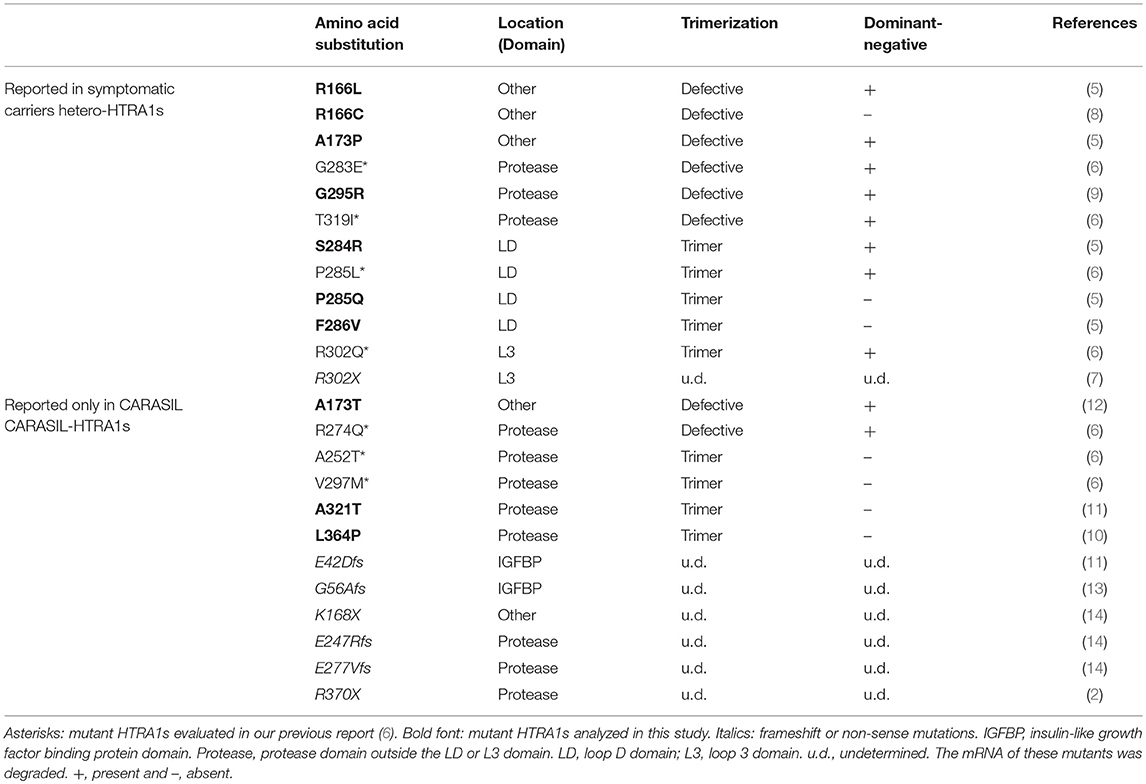
Table 1. Summary information of previously-reported HTRA1 mutations identified in symptomatic carriers and CARASIL patients.
Next we investigated whether these missense HTRA1s have dominant-negative. To serve as a control for this assay, the half dose of WT was not suitable because protein and substrate concentrations in the reaction mixture differ from those expressing WT and each mutant HTRA1 (6). Therefore, we used a mixture of WT and S328A, an artificial inactive HTRA1 that trimerizes, as a control (3, 4) and compared the protease activities of the mixtures of WT and each mutant HTRA1 with that of the control. dominant-negative was then defined as a protease activity less than that of the control. Four of six hetero-HTRA1s and one of four CARASIL-HTRA1 showed dominant-negative (Figure 3 and Supplemental Figure 1).
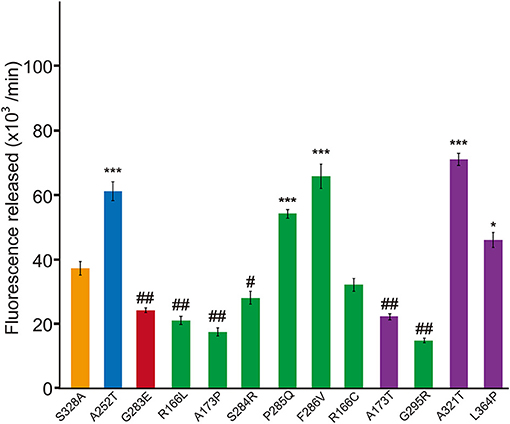
Figure 3. Dominant-negative effects of missense HTRA1s identified in symptomatic carriers. Protease activities of mixtures of each missense HTRA1 with WT calculated from the slope of the linear portion of normalized fluorescence vs. time (30, 60, and 90 min) plots. Orange, S328A/WT, a positive control for a dominant-negative effect. Blue and red bars indicate protease activities of A252T/WT and G283E/WT, respectively, negative and positive controls for dominant-negative effect, respectively. Green bars, missense HTRA1s identified in symptomatic carriers. Purple bars, missense HTRA1s found only in CARASIL patients. I-bars indicate standard errors (SE). Statistical comparisons of protease activities between each mutant HTRA1/WT and S328A/WT were performed with one-way analysis of variance followed by Dunnett's post hoc test. ***P < 0.0001; *P < 0.05 for increases in protease activities for each HTRA1/WT relative to S328A/WT. ##P < 0.0001; #p < 0.05 for differences for HTRA1/WT mixtures relative to S328A/WT.
The results showed the possibility that the HTRA1 mutant proteins identified in the symptomatic carriers cannot form trimers or have mutations in the LD or L3 domain. Therefore, we examined the frequency of mutant HTRA1s with these characteristics between missense mutations identified in symptomatic carriers and only in CARASIL. Combined with our previous results, all missense HTRA1s identified in symptomatic carriers were defective in trimerization or had mutations in the LD or L3 domain, but only two of six missense HTRA1s found only in CARASIL patients were defective in trimerization and none had mutations in the LD or L3 domain (P = 0.006; Table 1). In contrast, the frequency of mutant HTRA1s with dominant-negative effects in symptomatic carriers was not significantly higher than that in CARASIL patients (72.7 vs. 33.3%, P = 0.162; Table 1).
In this study, we found that missense HTRA1 mutations identified in symptomatic carriers display two characteristics: defective trimerization or mutations in the LD or L3 domain. We newly identified that four HTRA1 missense mutants identified in symptomatic carriers were defective trimerization (R166C, R166L, A173P, and G295R). We also demonstrated that monomeric HTRA1 inhibits WT protease activity (6). Thus, these mutants have a more deleterious effect on carriers relative to mutants that did not inhibit WT protease activity, resulting in a symptomatic carrier. Although A173T and R274Q were also defective trimerization, no symptomatic carriers with A173T or and R274Q have been identified to date (12, 15), carriers with these mutations should be carefully evaluated for cerebral small vessel disease (CSVD).
Among the other missense HTRA1s observed in the symptomatic carriers, four of the five missense HTRA1s had a mutation in the LD domain. The simulation analysis revealed that these mutations induced an instability in the helical structure of the LD loop (16). For the L3 domain, R302 is the only portion observed in the symptomatic carrier. R302 is essential for inter-monomer communication and substrate binding (16). Thus, a mutation in the LD or L3 domain that interferes with signal transduction between the monomers result in inhibition of the WT activity.
Regarding the molecular pathogenesis in symptomatic carriers, we hypothesized that these mutations inhibit the WT protease activity by interfering with the trimer-dependent activation cascade, resulting in <50% of protease activity in symptomatic carriers (6). However, the data from this study show that not all mutations identified in symptomatic carriers showed <50% of protease activity. Moreover, symptomatic carriers with non-sense or frameshift mutations have also been reported (7, 17). Thus, we consider that the residual HTRA1 protease activity might correlate with the risk for developing CSVD. To elucidate the precise correlation between HTRA1 protease activity and risk of CSVD, we and others must identify and evaluate additional carriers for HTRA1 mutations.
Our study has several limitations. First, protease activity was measured by using casein, a non-physiological substrate. Several substrates, including fibronectin and transforming growth factor-beta binding protein 1 have been reported (18, 19). Thus, results of protease assays may be different if other substrates are used. Second, the pathogenicity of mutations outside the protease domain remains unknown. Several studies have investigated the role of the Kazal-like and PDZ domains in regulating the protease activity of HTRA1s (20–22). The Kazal-like domain has been implicated in autolysis (21), and mutations in this domain could influence its expression. Thus, the possibility that a mutation may decrease the amount of protein in vivo by decreasing stability or secretion to the extracellular matrix cannot be excluded.
We found that a either a deficiency in trimerization or location of the amino-acid mutation in the LD or L3 domain was highly observed in individual missense HTRA1 alleles in symptomatic carriers. Our findings will have significant utility for improving genetic counseling both for the relatives of CARASIL patients and for carriers with HTRA1 variants with sporadic CSVDs.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
MU: draft of manuscript, study concept and design, acquisition of data and analysis. HN: revision of manuscript, interpretation of data, and study supervision. NS, SA, and MK: revision of manuscript and interpretation of data. AK and TK: acquisition of data and interpretation of data. OO: revision of the manuscript, study concept and design, and study supervision.
This study was funded by a grant-in-aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control; 26117006) from MEXT; a grant-in-aid for Practical Research Project for Rare/Intractable Diseases (17928469) from AMED; a grant-in-aid for Scientific Research (A) (16804840); a grant-in-aid for Medical Research from the Takeda Science Foundation; and a grant-in-aid for Research on Intractable Diseases from the Japanese Ministry of Health, Labor, and Welfare.
OO has received speaking honoraria from Kyowa Hakko Kirin Co., Ltd., Bristol-Myers Squibb, Ono Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharm, Takeda, Daiichi-Sankyo, FUJIFILM, SANOFI, and FP-pharm.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00693/full#supplementary-material
Supplemental Figure 1. Mixtures of missense HTRA1 proteins with WT protein. Mixture of each missense HTRA1 and WT protein evaluated by SDS-PAGE stained with SYPRO® Ruby. Broken arrows indicate the full-length band of missense HTRA1s tagged with myc-His6. Arrowheads indicate the full-length band of WT HTRA1 tagged with His6.
Supplemental Table 1. Summary of the clinical features of symptomatic heterozygotes with HTRA1 mutations.
1. Fukutake T, Hirayama K. Familial young-adult-onset arteriosclerotic leukoencephalopathy with alopecia and lumbago without arterial hypertension. Eur Neurol. (1995) 35:69–79. doi: 10.1159/000117096
2. Hara K, Shiga A, Fukutake T, Nozaki H, Miyashita A, Yokoseki A, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. (2009) 360:1729–39. doi: 10.1056/NEJMoa0801560
3. Clausen T, Kaiser M, Huber R, Ehrmann M. HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol. (2011) 12:152–62. doi: 10.1038/nrm3065
4. Truebestein L, Tennstaedt A, Monig T, Krojer T, Canellas F, Kaiser M, et al. Substrate-induced remodeling of the active site regulates human HTRA1 activity. Nat Struct Mol Biol. (2011) 18:386–8. doi: 10.1038/nsmb.2013
5. Verdura E, Herve D, Scharrer E, Amador Mdel M, Guyant-Marechal L, Philippi A, et al. Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain. (2015) 138:2347–58. doi: 10.1093/brain/awv155
6. Nozaki H, Kato T, Nihonmatsu M, Saito Y, Mizuta I, Noda T, et al. Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology. (2016) 86:1964–74. doi: 10.1212/WNL.0000000000002694
7. Tateoka T, Onda H, Hirota K, Kasuya H, Shinohara T, Kinouchi H, et al. Unusual case of cerebral small vessel disease with a heterozygous nonsense mutation in HTRA1. J Neurol Sci. (2016) 362:144–6. doi: 10.1016/j.jns.2016.01.037
8. Bougea A, Velonakis G, Spantideas N, Anagnostou E, Paraskevas G, Kapaki E, et al. The first Greek case of heterozygous cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy: an atypical clinico-radiological presentation. Neuroradiol J. (2017) 30:583–5. doi: 10.1177/1971400917700168
9. Di Donato I, Bianchi S, Gallus GN, Cerase A, Taglia I, Pescini F, et al. Heterozygous mutations of HTRA1 gene in patients with familial cerebral small vessel disease. CNS Neurosci Ther. (2017) 23:759–65. doi: 10.1111/cns.12722
10. Wang XL, Li CF, Guo HW, Cao BZ. A novel mutation in the HTRA1 gene identified in Chinese CARASIL pedigree. CNS Neurosci Ther. (2012) 18:867–9. doi: 10.1111/j.1755-5949.2012.00373.x
11. Bianchi S, Di Palma C, Gallus GN, Taglia I, Poggiani A, Rosini F, et al. Two novel HTRA1 mutations in a European CARASIL patient. Neurology. (2014) 82:898–900. doi: 10.1212/WNL.0000000000000202
12. Khaleeli Z, Jaunmuktane Z, Beaufort N, Houlden H, Haffner C, Brandner S, et al. A novel HTRA1 exon 2 mutation causes loss of protease activity in a Pakistani CARASIL patient. J Neurol. (2015) 262:1369–72. doi: 10.1007/s00415-015-7769-5
13. Cai B, Zeng J, Lin Y, Lin Y, Lin W, Lin W, et al. A frameshift mutation in HTRA1 expands CARASIL syndrome and peripheral small arterial disease to the Chinese population. Neurol Sci. (2015) 36:1387–91. doi: 10.1007/s10072-015-2121-5
14. Preethish-Kumar V, Nozaki H, Tiwari S, Vengalil S, Bhat M, Prasad C, et al. CARASIL families from India with 3 novel null mutations in the HTRA1 gene. Neurology. (2017) 89:2392–4. doi: 10.1212/WNL.0000000000004710
15. Nishimoto Y, Shibata M, Nihonmatsu M, Nozaki H, Shiga A, Shirata A, et al. A novel mutation in the HTRA1 gene causes CARASIL without alopecia. Neurology. (2011) 76:1353–5. doi: 10.1212/WNL.0b013e318215281d
16. Cabrera AC, Melo E, Roth D, Topp A, Delobel F, Stucki C, et al. HtrA1 activation is driven by an allosteric mechanism of inter-monomer communication. Sci Rep. (2017) 7:14804. doi: 10.1038/s41598-017-14208-z
17. Lee YC, Chung CP, Chao NC, Fuh JL, Chang FC, Soong BW, et al. Characterization of heterozygous HTRA1 mutations in Taiwanese patients with cerebral small vessel disease. Stroke. (2018) 49:1593–601. doi: 10.1161/STROKEAHA.118.021283
18. Tiaden AN, Richards PJ. The emerging roles of HTRA1 in musculoskeletal disease. Am J Pathol. (2013) 182:1482–8. doi: 10.1016/j.ajpath.2013.02.003
19. Beaufort N, Scharrer E, Kremmer E, Lux V, Ehrmann M, Huber R, et al. Cerebral small vessel disease-related protease HtrA1 processes latent TGF-beta binding protein 1 and facilitates TGF-beta signaling. Proc Natl Acad Sci USA. (2014) 111:16496–501. doi: 10.1073/pnas.1418087111
20. Eigenbrot C, Ultsch M, Lipari MT, Moran P, Lin SJ, Ganesan R, et al. Structural and functional analysis of HtrA1 and its subdomains. Structure. (2012) 20:1040–50. doi: 10.1016/j.str.2012.03.021
21. Risor MW, Poulsen ET, Thomsen LR, Dyrlund TF, Nielsen TA, Nielsen NC, et al. The autolysis of human HtrA1 is governed by the redox state of its N-terminal domain. Biochemistry. (2014) 53:3851–7. doi: 10.1021/bi401633w
Keywords: heritability, vascular dementia, mutations, HTRA1, carriers, CARASIL
Citation: Uemura M, Nozaki H, Koyama A, Sakai N, Ando S, Kanazawa M, Kato T and Onodera O (2019) HTRA1 Mutations Identified in Symptomatic Carriers Have the Property of Interfering the Trimer-Dependent Activation Cascade. Front. Neurol. 10:693. doi: 10.3389/fneur.2019.00693
Received: 21 March 2019; Accepted: 13 June 2019;
Published: 28 June 2019.
Edited by:
Andreas Charidimou, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Alessandra Rufa, University of Siena, ItalyCopyright © 2019 Uemura, Nozaki, Koyama, Sakai, Ando, Kanazawa, Kato and Onodera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Osamu Onodera, b25vZGVyYUBicmkubmlpZ2F0YS11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.