 Pratik S. Chougule
Pratik S. Chougule Raymond P. Najjar
Raymond P. Najjar Maxwell T. Finkelstein
Maxwell T. Finkelstein Nagaendran Kandiah3,4
Nagaendran Kandiah3,4 Dan Milea
Dan Milea
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 12 April 2019
Sec. Neuro-Ophthalmology
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00360
This article is part of the Research Topic The Pupil: Behavior, Anatomy, Physiology and Clinical Biomarkers View all 25 articles
The impact of Alzheimer's disease (AD) on the pupillary light response (PLR) is controversial, being dependent on the stage of the disease and on the experimental pupillometric protocols. The main hypothesis driving pupillometry research in AD is based on the concept that the AD-related neurodegeneration affects both the parasympathetic and the sympathetic arms of the PLR (cholinergic and noradrenergic theory), combined with additional alterations of the afferent limb, involving the melanopsin expressing retinal ganglion cells (mRGCs), subserving the PLR. Only a few studies have evaluated the value of pupillometry as a potential biomarker in AD, providing various results compatible with parasympathetic dysfunction, displaying increased latency of pupillary constriction to light, decreased constriction amplitude, faster redilation after light offset, decreased maximum velocity of constriction (MCV) and maximum constriction acceleration (MCA) compared to controls. Decreased MCV and MCA appeared to be the most accurate of all PLR parameters allowing differentiation between AD and healthy controls while increased post-illumination pupillary response was the most consistent feature, however, these results could not be replicated by more recent studies, focusing on early and pre-clinical stages of the disease. Whether static or dynamic pupillometry yields useful biomarkers for AD screening or diagnosis remains unclear. In this review, we synopsize the current knowledge on pupillometric features in AD and other neurodegenerative diseases, and discuss potential roles of pupillometry in AD detection, diagnosis and monitoring, alone or in combination with additional biomarkers.
Dementia is a global epidemic and has become a public health priority. Alzheimer's disease (AD) is the most common cause for dementia worldwide (1), accounting for 50–70% of dementia cases. The two major neuropathological landmarks of AD are deposition of insoluble amyloid-β (Aβ) plaques and formation of neurofibrillary tangles, composed of hyperphosphorylated tau proteins. These pathologic abnormalities are found in the central nervous system, as well as in the retina (2–4). The pathophysiology of AD is poorly understood, but a common hypothesis postulates that aggregation of Aβ is a pre-requisite for tau accumulation, neurodegeneration, and ultimately, to clinical manifestations. Clinical features of AD include progressive cognitive decline, affecting memory, learning, language, visuospatial abilities, and executive functions, but also deterioration of sleep and normal circadian rhythms (5). Most often extensive and largely irreversible neuronal histopathological changes precede clinical features of AD (6), which may explain the current failure of all disease modifying agents in this condition. For these reasons, it is believed that early diagnosis of AD is crucial for early and effective therapeutic interventions, improving AD outcomes.
Several in vivo biomarkers have been proposed for early identification of AD pathology, including brain imaging biomarkers (positron emission tomography after Aβ labeling) (7), as well as fluid biomarkers (within the cerebrospinal fluid and, possibly, in the blood) (8). These, and other novel genetic, biological, deep-learning based, or behavioral biomarkers aim to surpass the current performance of classical clinical evaluations in AD, which are subjective, time-consuming and deliver variable results. The eye, which is embryologically, anatomically, and physiologically an extension of the brain, has been an early explored target for identification of neurodegeneration biomarkers in AD (9, 10). Functionally, various ocular biomarkers have been tested for detection and evaluation of AD, such as eye movement recordings and pupillary responses to light or to cognitive load (9–11). Several studies have suggested that AD may be associated with altered pupillary light responses (PLR), as a consequence of abnormalities in the retina and/or the efferent pupillary system. Several arguments support the possible pupillary involvement in AD, including pathological changes in the retina, as well as existence of parasympathetic (cholinergic) and sympathetic (adrenergic) dysfunctions in the disease. Indeed, neurodegeneration commonly affects the locus coeruleus (LC), located in pons and involved in the sympathetic control of pupil size and PLR (2, 12), as well as the Edinger Westphal nucleus (EWN) (4, 13), involved in the parasympathetic control of the pupil. Pupillometry is an easy, non-invasive and affordable tool, allowing the evaluation of the PLR in AD and other ocular and neurological diseases. Whether static or dynamic pupillometry yields useful biomarkers for AD screening or diagnosis remains unclear. In this review, we synopsize the current knowledge on pupillometric features in AD and other neurodegenerative diseases, and discuss potential roles of pupillometry in AD detection, diagnosis and monitoring, alone or in combination with additional biomarkers.
The pupil size is under the control of a closed autonomic loop. The pupil constrictor and dilator muscles receive antagonistic impulses from the parasympathetic (cholinergic) and sympathetic (adrenergic) autonomic nervous systems, respectively (Figure 1). The PLR is also dependent on the integrity of the retina, and in particular on the integrity of the intrinsically photosensitive melanopsin expressing retinal ganglion cells (mRGCs) (15). Although the mRGCs are activated by rods and cones, they are also intrinsically photosensitive through the melanopsin photopigment, subserving the PLR via central projections to the olivary pretectal nucleus (OPN) (16), which projects to the EWN, as demonstrated in non-human primates. The parasympathetic EWN is a cholinergic nucleus in the oculomotor complex, at the origin of preganglionic efferent neurons which synapse in the ciliary ganglion, subsequently innervating the constrictor pupillae (14, 17). The mRGCs also project to the central circadian clock located in the hypothalamic suprachiasmatic nucleus (SCN), which governs various bodily circadian rhythms and projects to the sympathetic locus coeruleus (LC), located in pons (Figure 1) (18, 19) [for review see (20)]. The sympathetic efferent system, including LC and SCN, regulates the resting pupil size at different levels of background illumination (17), by controlling the tone of the dilator muscle. Beyond the intervention of these motor structures, the pupillary size and function can be modulated by supranuclear neuronal influences. Other possible confounding factors, affecting the size of the pupils include the respiration rate (21), emotional status (22), vigilance (23), and age (24).
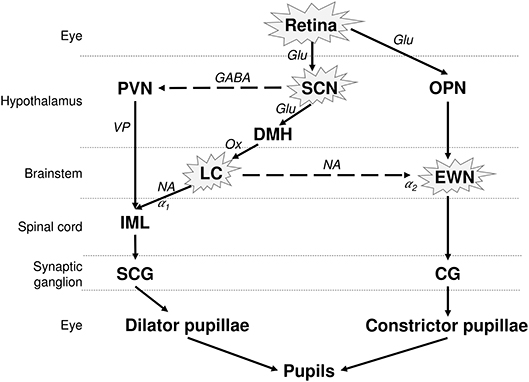
Figure 1. Neuronal control of the PLR and affected structures in AD. The pupil size depends on the interplay between antagonistic parasympathetic and sympathetic impulses. The photoreceptors in retina, including the melanopsin expressing retinal ganglion cells (mRGCs), stimulate the OPN, connected to the parasympathetic EWN. mRGCs also project to the SCN, which is connected to the sympathetic LC. EWN is the major cholinergic center subserving the pupillary constriction to light. Light has an inhibitory effect on sympathetic activity and PVN via SCN. This inhibitory effect is attenuated by sympatho-excitation, which is mediated via the SCN and LC (14). The encircled structures are affected in AD. Solid lines, excitatory connections; dashed lines, inhibitory connections; Hypothalamic nuclei: SCN, suprachiasmatic nucleus; PVN, paraventricular nucleus; DMH, dorsomedial hypothalamus. Autonomic premotor nuclei: OPN, olivary pretectal nucleus; LC, locus coeruleus. Parasympathetic nucleus/ganglion: EWN, Edinger Westphal nucleus; CG, Cilliary ganglion. Sympathetic nucleus/ganglion: IML, intermedio-lateral column of spinal cord; SCG, superior cervical ganglia. Neurotransmitters: Glu, glutamate; GABA, γ-amino-butyric acid; VP, vasopressin; Ox, orexin; Ach, acetylcholine; NA, noradrenaline. Adrenoceptors: α1, excitatory; α2, inhibitory.
Recorded using an infra-red pupilometer, the PLR is governed by rods, cones and mRGCs. The intrinsically photosensitive mRGCs, located in the inner retina, produce a sustained constriction of the pupil in response to bright blue light which persists even after light offset; in addition to integrating signals from rods and cones (25). Different light wavelengths, at different intensities stimulate specifically different retinal photoreceptors (26–29). Thus, chromatic pupillometry, using different wavelengths and light intensities has been used for the evaluation of inner and outer retina integrity, in various conditions (26–29).
Loss of cholinergic neurons is a common event in AD, possibly leading to alteration of cognitive processes. Specific loss of cholinergic neurons, mainly located in the medial septum and in the para-hippocampal area, is associated with memory impairment (30), but also with other cognitive deficits seen in AD (31, 32). The hypothesis of cholinergic deficit fails however, to explain other impairments in AD, i.e., disruption of circadian rhythms, sleep, and executive functions.
AD affects the cholinergic EWN, which is the central brainstem sub-nucleus of the oculomotor complex, involved in the control of the pupil constriction. Pathologic studies have shown that the EWN is affected even at early stages of AD, displaying deposition of Aβ amyloid plaques and neurofibrillary tangles (4, 13, 33). AD is associated with increased glutaminyl cyclase activity, resulting in formation of highly neurotoxic Aβ amyloid precursors (pyroglutamate Aβ peptide), identified in the EWN and in the preganglionic cholinergic PLR-governing neurons, as well as in other cholinergic neurons in the nucleus basalis of Meynert (34). In AD, the EWN neurons, display a decrease in their total dendritic length per neuron as well as a severe loss of distal dendritic branches and dendritic spines, leading to severe decrease in synaptic contacts (35). It has been suggested that these pathological changes of the EWN may be an early and specific feature of AD and they may result in decreased cholinergic control of pupillary responses (35). Unfortunately, little is known about the involvement of other parasympathetic structures involved in the PLR, i.e., the olivary pretectal nucleus and ciliary ganglia.
The LC modulates pupil size in 2 possible ways; by direct stimulation of preganglionic sympathetic neurons, as well as by inhibitory regulation of the EWN (Figure 1) (17, 36). Various factors stimulating the LC may modulate the PLR. For example, anxiety, associated with excitation of the LC (37, 38) or drugs increasing noradrenergic output to the EWN like noradrenaline re-uptake inhibitors (14, 39), lead to an increased sympathetic response on PLR like prolonged latency, reduced amplitude of constriction and faster redilation. Conversely, drugs inhibiting LC activity like clonidine (α2-adrenoceptor agonist) cause pupillary miosis and reduce the sympathetic effect on PLR (40–42). In monkeys, an electrophysiologically detectable activity in LC has been associated with mydriasis at rest (43).
Patients with AD and mild cognitive impairment (MCI), considered as the pre-clinical stage for AD, undergo significant loss of noradrenergic neurons in the LC (55 and 30%, respectively), compared to healthy controls, a finding which may impact the PLR (44). Neuronal loss in the LC of patients with AD may lead to decreased sympathetic supply to the iris and reduce the baseline pupil size (45).
Aging is associated with optical (46, 47) photoreceptoral and retinal neuronal changes (48–50). Optically, in spite of decreased lens transmittance for short wavelength blue light in aging and cataract, the mRGCs induced pupillary response by blue light are well-preserved (51–53), and the pupillary responses are reduced irrespective of the wavelength of light. Although, aging has been associated with axonal and retinal ganglion cell loss (54), AD has been associated with greater thinning of retinal nerve fiber layer compared to age matched healthy controls (55–57), suggesting an accelerated loss of RGCs in AD patients. Pathological studies have shown presence of Aβ amyloid plaques in the retina of AD patients (58, 59) including in the inner layers of retina (3). These changes were often associated with blood vessel abnormalities and areas of cellular degeneration, similar to what is seen histologically in the brain of patients with AD (60, 61).
Moreover, retinas of patients with advanced AD display not only histological evidence of mRGC loss, but also selective deposition of Aβ amyloid plaques within these cells, which subserve the PLR (3). Less is known about the early selective loss of mRGC and possible mRGC Aβ deposition, occurring in AD. Occurrence of such a phenomenon should allow discrimination between normal aging patients and AD, using chromatic pupillometry. Chromatic pupillometry has been used in other conditions as a marker of mRGC integrity (62). In primary open angle glaucoma, a condition associated with histological mRGC loss (63), abnormal melatonin secretion profile (64) and sleep and circadian rhythm dysfunction (65), various pupillometric studies have shown abnormal PLR responses (28, 66–70). Conversely, in mitochondrial hereditary optic neuropathies, mRGCs are resistant to neurodegeneration, explaining the relatively preserved chromatic pupillometry parameters (71–73) and melatonin profiles (74). It is possible that mRGC loss, alone or combined with neuronal loss occurring in the suprachiasmatic nuclei, may be associated with circadian rhythm dysfunctions which can occur even at early stages of AD (3, 5).
The consequence of AD on the pupillary diameter at rest has been controversial in various studies, probably due to methodological differences, i.e., measurement conditions and sample sizes. A few studies with small sample size (range 9 to 23 AD patients) have reported reduced baseline pupillary diameters in AD compared to those of healthy subjects (45, 75–77). Other studies did not find any difference in baseline pupil diameters between AD, MCI and controls (78, 79), however, the groups were not age-matched in the study with largest sample size (n = 66 AD, 42 MCI) (78), while AD patients in the other study were using cholinergic and anti-depressant medications which may alter the baseline pupil size (n = 15 AD) (79). An increased baseline pupillary diameter has also been reported in AD (n = 20 AD), but no details of age of the two groups were mentioned (80) (Table 1).
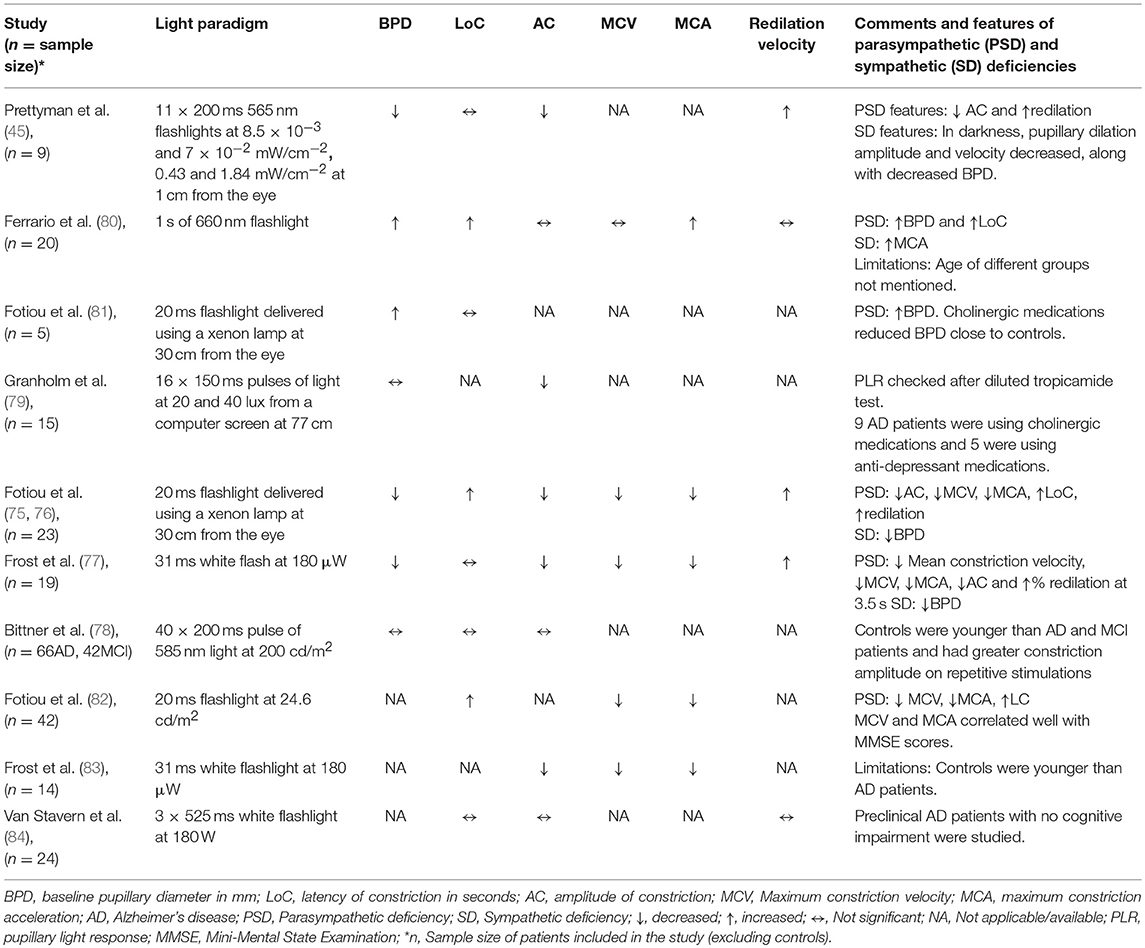
Table 1. Summary of studies on PLR in AD.
Most pupillometric studies in patients with AD have reported results compatible with parasympathetic deficiency, translating to increased latency of pupillary constriction to light, decreased constriction amplitude, reduced mean constriction velocity and faster redilation after light offset (45, 75, 76, 83, 85) (Table 1). Pupil constriction velocity is obtained as the first derivative of change in pupil size with respect to time and acceleration as the second derivative (change in constriction velocity with respect to time) (Figure 2). Patients with AD typically display decreased maximum velocity of constriction (MCV) and maximum constriction acceleration (MCA) compared to controls, suggesting a parasympathetic deficiency. Amongst all pupillometric features, MCA and MCV have been reported as the most accurate parameters to differentiate AD patients from healthy controls (75, 76, 83). Nonetheless, other studies have failed to find such differences between AD patients and healthy individuals (78, 80). These differences may be the result of the different illumination protocols used, since studies using white light typically are associated with larger constriction amplitudes and shorter latencies (24), compared to studies using red light at 660 nm (80) or 585 nm (78). Considering that different studies have used different intensity and wavelength stimuli, this effect cannot be completely attributed to the wavelength alone. However, other studies have demonstrated that when photon density is kept constant, shorter wavelength light produced greater constriction amplitude than longer wavelength (86, 87).
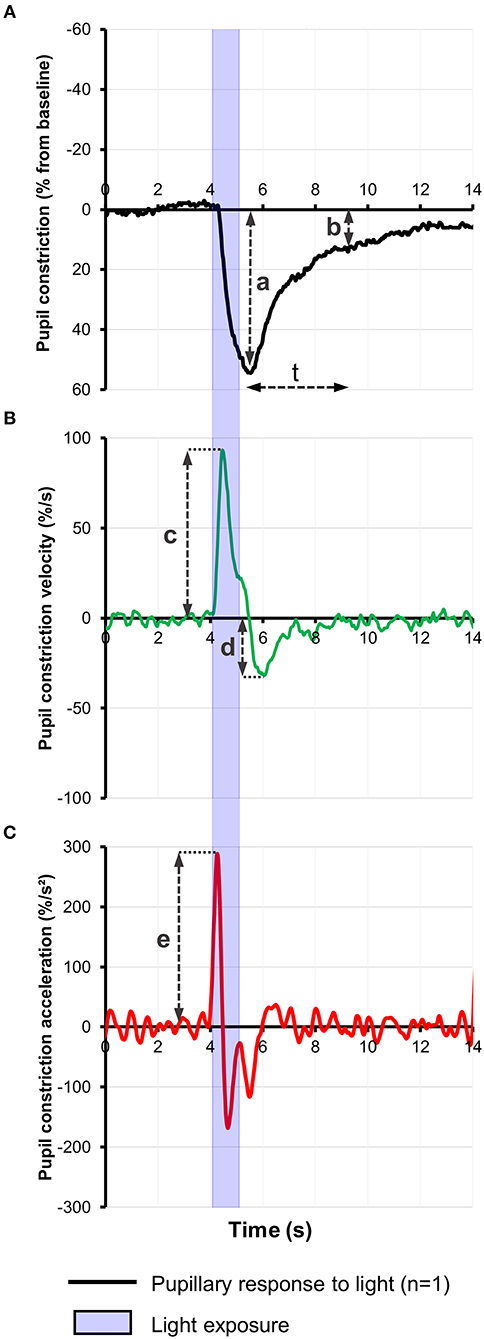
Figure 2. (A) Representative baseline-adjusted pupillary response to a 1-s bright blue light (480 nm) stimulus of 13.0 Log photons/cm2/s in a healthy individual. Pupil constriction velocity (B) and acceleration (C) curves were computed, respectively, from the trace in (A) as the first and second derivatives of change in pupil constriction with respect to time. Pupillometric features extracted include: a, amplitude of pupillary constriction; b, post-illumination pupillary response after (t) seconds from light offset; c, Maximum constriction velocity (MCV); d, maximum redilation velocity; e, Maximum constriction acceleration (MCA).
In a recent study in cognitively normal pre-clinical AD subjects, diagnosed on the basis of high cortical binding potential of Aβ amyloid on PET imaging and/or low CSF Aβ amyloid levels, there was no significant difference in any of the pupillary parameters in response to light emitting diodes (LEDs) producing a white flash of 180 W for 525 ms, compared to healthy controls (84). Only a marginal difference was observed in PLR in pre-clinical stages, suggesting that the effect of AD on PLR in pre-clinical stages is very small and may not be detectable. This suggests that PLR using achromatic light stimulus may be a valid biomarker for established Alzheimer's disease but it may have limited clinical utility in screening for AD in pre-clinical stages, perhaps due to the very small disease effect on PLR (84).
A pupillometric study, using non-Maxwellian retinal stimulation with repetitive, brief, long wavelength light (585 nm), aimed to compare patients with MCI, AD and normal controls, but failed to find inter-group differences (evaluating baseline diameters, constriction amplitudes, and constriction latencies), after adjustment for age (78). However, the use of specific repetitive light stimuli induced stronger pupillary responses in controls in terms of relative constriction amplitude (difference in the PLR measurements of the first and last stimuli), compared to AD and MCI patients, suggesting that repetitive pupillary stimulation might be a more appropriate stimulus to discriminate patients with AD and MCI from controls (78). It was proposed that repetitive stimuli caused a fatigue of the sympathetic inhibitory system to the parasympathetic pathway revealing the true effect of parasympathetic system alone on PLR. Smaller relative miosis and amplitude of constriction in AD patients suggests a decreased parasympathetic innervation to the pupillary system which may not be detected in the presence of inhibitory sympathetic system. Although relative amplitude was independent of age, the difference between MCI and control groups was not statistically significant while between AD and controls may fail to reach statistical significance if Bonferroni correction is applied. Moreover, its definition and calculation is not clearly explained in the study. Constriction phase due to different wavelength light stimuli in AD patients has not been studied yet.
The pupillary redilation phase has been explored in several studies, but these results are sometimes difficult to compare, given the variable definitions of this parameter such as (i) percentage of pupillary redilation after 3.5 s of white flash light offset (75–77), (ii) 75% redilation time (45, 77), and (iii) average dilation velocity (77, 84). The majority of these studies have reported a faster pupillary redilation phase [analogous to decreased Post-illumination pupil response (PIPR)] in AD patients compared to controls, making it the most consistent PLR feature in AD patients (45, 75–77). In a study which extensively studied the redilation phase, the percentage recovery at 3.5 s and the 75% redilation time was significantly greater in AD patients compared to aging controls. Although the mean dilation velocity (mm/sec) was slower in AD patients, it can be ignored since the pupil size was not calculated as a percentage of baseline pupil diameter which was significantly smaller in AD (77). A recent study in preclinical AD alone did not find a significant increase in dilation velocity compared to controls following an achromatic light stimulus, however, it is not clear whether the measurements were adjusted to baseline pupil diameter (84). The faster pupillary redilation after light offset has been attributed to the diminished parasympathetic tonus associated with the cholinergic deficit, translating into failure to maintain a tonic pupillary constriction. An alternative explanation, which was not yet considered in previous studies, is that the accelerated pupillary redilation may be the result of selective mRGC loss in AD, causing an abnormal, faster PIPR as seen in other conditions affecting the mRGC, such as glaucoma (66). This hypothesis is in line with the findings of another study, which did not find any difference of the redilation phase between a group of AD patients and controls, after exposure to red light (660 nm) (80). Indeed, red light at medium high intensities is less prone to stimulate the mRGC and might have failed to explore their dysfunction. Although, redilation was not found to be different in preclinical AD patients in a recent study using a 180 W achromatic stimulus for 525 ms (84), chromatic pupillometry studying different parameters like constriction amplitude or PIPR to blue light, can still be a viable option in such cases due to selective mRGCs loss as reported in AD patients (3). No study till date has investigated pupillometric signs of mRGC dysfunction or impaired PIPR to blue light in AD patients and needs to be explored in the future. However, these interpretations are speculative, it is well-possible that the faster redilation in AD patients may be the result of an interplay between the two factors, i.e., the mRGC loss and the cholinergic deficit. It is noticeable that mRGC loss in AD should result in selective faster redilation after offset of blue light (460 nm), which specifically stimulates the mRGCs. On the opposite, faster redilation due to cholinergic deficiency should be independent of wavelength of stimulus used, occurring after offset of both blue and red light. Future studies taking into account these factors, should be able to disentangle the respective role of the afferent vs. efferent system in the pupillary redilation phase.
The gold standard for AD diagnosis is still based on pathology findings. However, in clinical practice, screening for AD or cognitive impairment uses various questionnaires and interactive tests, including the Mini Mental Scoring Examination (MMSE) or the Montreal cognitive assessment test (MoCA). These tests have numerous limitations, including language barriers, geographical adaptability, subjectivity, long implementation time, and the necessity for constant supervision by trained and skilled personnel. Several objective tests have been developed for early AD detection and diagnosis including cerebro-spinal fluid (CSF) analysis to measure beta amyloid (Aβ), total tau proteins and phosphorylated tau peptides quantification (8, 88), and magnetic resonance imaging (MRI) imaging of the brain and positron emission tomography (PET) scan measuring the brain Aβ plaque burden (6, 7, 89). These modalities are expensive, invasive and potentially dangerous (90, 91). In addition, they detect only structural and not functional changes in AD.
From the published studies, pupillometric evaluations may not suffice for early AD detection. However, they may constitute an adjunctive method in a, multimodal approach, combining (i) novel, cognitive, visuospatial, and memory tests involving portable virtual reality devices, (ii) retinal imaging for detection of neuronal loss and/or amyloid deposition, and (iii) objective functional outcomes provided by targeted color pupillometry.
Apolipoprotein E is a fat-binding protein involved in the metabolism of fat, produced by APOE gene found on chromosome 19, being the only genetic factor associated with the common late onset AD. APOE mutation is not a causative mutation, but is rather considered as a risk factor for AD (92, 93). Although the PLR is not directly influenced by the APOE ε4 carrier status (78), their combination may increase the area under the curve for the combined test performance (83).
A pupillometric study has evaluated participants from a single family harboring an Amyloid-Beta Precursor Protein genetic mutation (APPGlu693Gln) (6 carriers with no cognitive impairment and 6 non-carriers) (94). This mutation results in a rare form of autosomal dominant Alzheimer's disease with phenotypical penetration approaching 100% and which is responsible for an early onset of AD. The pupillometric assessment yielded a slower pupil 75% recovery time in mutation carriers compared to non-mutation carriers. Globally, pupillometric changes were detected in pre-symptomatic carriers of the mutations, but were not statistically significant.
MMSE is routinely used to screen elderly subjects for dementia and has a AUROC of 0.89 (95). PLR in patients with AD having higher MMSE and Wechsler Memory Scale (better cognition) scores correlated moderately with MCV, MCA and latency of constriction (82). On repetitive pupillary stimulation, higher MMSE correlated with larger increase in amplitude and relative amplitude and greater decrease in the latency (p < 0.05) of constriction (78). These outcomes suggest that the pupillary light response may depend on the severity of the disease and can be used for monitoring the disease progression. However, combined efficacy of MMSE and PLR has not been explored as both tools are practical, easy, non-invasive, and affordable and may yield better accuracy if combined together, compared to individual outcomes.
Decrease in pupillary constriction amplitude with repetitive stimulation in AD patients correlated with lower Aβ42 protein levels (p = 0.01) and a trend with higher tau levels in CSF (p = 0.08) (78). This suggests a possible association between cholinergic deficit, decreased Aβ42 protein levels and a trend with higher tau levels in CSF which supports a causative role of Aβ amyloid plaques in central cholinergic deficit (92). To date, the efficacy of a combined PLR-CSF screening method remains unknown.
In a highly controversial study, Scinto et al. reported that patients with AD exhibit larger pupil dilation compared to age-matched controls after instillation of diluted anticholinergic eyedrops (Tropicamide 0.01%) (96) Several studies have contradicted this finding (97–99) which could be due to ethnicity, age, ocular penetration of drug, properties of the solution and background luminance (79). A combination of topical weak anticholinesterase and PLR showed significant reduction in constriction amplitude for AD and Parkinson's patients compared to controls, but no significant difference between Alzheimer's and Parkinson's patients was found, while latency of constriction was similar within the 3 groups (79). However, others did not find any such significant difference in PLR pre or post weak anticholinergic eyedrops use, between AD patients and controls (80). Hence, the use of weak anticholinergic eyedrops may not improve the efficacy of PLR in detecting AD, since it may not give consistent results and decrease in amplitude of constriction is noted in AD even without using topical anticholinesterase.
Most of the previously published pupillometric studies in AD have various methodologic limitations. The intensity, light wavelength and duration of light exposure were variable in all the above mentioned studies. Yet, these parameters can affect, independently, or in combination, the PLR outcomes (24, 100). Therefore, there is a high need for standardization of experimental conditions in AD studies, similar to what has been described in studies using light therapy (101) and in other animal studies (102). Most current pupillometric studies agree of the need for standardized analysis of baseline pupil diameters (103, 104). Interestingly, most of the previous PLR studies in AD have not normalized the baseline diameter in their subjects, making any comparison very difficult. In a few studies evaluating pupillometric results in AD, there was no age-matching between the groups of patients and controls (78, 79, 83). Indeed, the decreased pupillary diameter with age (105, 106) may constitute a confounding factor. Last, but not least, the severity of AD was rarely taken into account in the evaluation of the PLR.
Only a few, small sample studies have reported the effect on the PLR of cholinergic drugs, commonly used in AD (79, 81). Thus, AD patients without cholinergic medications displayed larger baseline pupillary diameter, reduced pupillary miosis and higher number of oscillations at rest, compared to AD patients on cholinergic treatments and to healthy controls. Patients on medications had a greater latency of onset of constriction compared to both the controls and the medication free AD patients (81). However, other study in AD patients, have not found an effect of cholinergic medications on pupillary miosis but supported the increase in latency of constriction (79). PLR in AD patients on cholinergic medications behaved more like controls with no significant difference in constriction amplitude and baseline pupil diameter than their medication free counterparts (81). Taken together, these findings suggest that cholinergic medications might improve the pupillary responses in AD patients. Due to the very small sample size of these studies, it is difficult to conclude regarding a possible effect of cholinergic medications on the PLR in these patients. Additional studies are needed to understand the effect of cholinergic medications on PLR.
The most common ocular condition associated with aging is cataract which can attenuate the PLR response to both red and blue light. But the senescence of the lens does not selectively reduce the mRGCs responses to intense blue light and is well-preserved, in spite of its decreased lens transmittance in aging and cataract (51). Different retinal and optic nerve conditions can affect PLR and using chromatic pupillometry it is possible to localize the loss of photoreceptoral function i.e., inner or outer retina (107). Primary open angle glaucoma is associated with decreased PLR in response to exposure to both red and blue light with decreased PIPR for blue light (28, 70), while retinal dystrophies affecting rods and cones lead to decreased PLR responses to red and low intensity blue light with an increased PIPR to bright blue light stimulus (108, 109). Diabetic retinopathy and age-related macular degeneration can also affect the PLR (110, 111), but there is little indication to what extent these PLR alterations might be disease-specific, or whether they may confound co-existence of AD in the aging population.
Autonomic nervous system dysfunction has been described in Parkinson's disease (PD) (112), including cholinergic deficit (113), Various PLR abnormalities have been described in PD, including reduced amplitude of constriction, increased latency and decreased velocity and acceleration of constriction, while the baseline pupil diameter may be increased or not significantly different compared to healthy controls (Table 2) (76, 114). Pupillary unrest has also been increased in PD patients which were not on medications compared to healthy individuals or in treated patients (115), Pupillary redilation has not been significantly different in PD studies using white flash light stimuli (114, 115). However, a recent chromatic pupillometry study in PD patients has suggested that the PIPR following a short wavelength blue light elicits a faster redilation compared to healthy controls. This finding is consistent with loss of mRGCs in PD (116), possibly related to deposition of α-synuclein in the retinal ganglion cells in the inner plexiform layer of the retina (117, 118). An alternative explanation might be related to reduction in the dopamine expression in the amacrine cells which relay information from rods and cones to mRGCS.
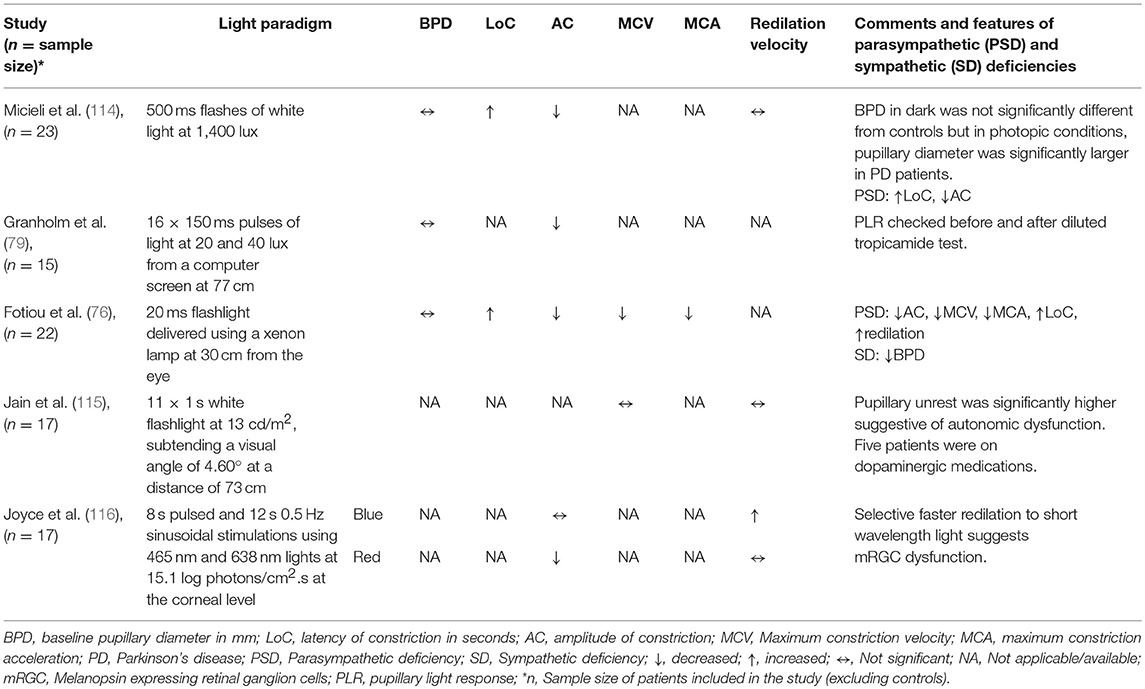
Table 2. Summary of studies on PLR in PD.
Autonomic nervous system dysfunction has also been described in dementia with Lewy bodies and to a lesser extent in fronto-temporal dementia, which can be associated with retinal abnormalities (119, 120). However, the specific effects of autonomic dysfunctions and retinal changes on pupillary light reflexes have not yet been studied in these disorders.
In summary, MCV and MCA appear to be the most accurate PLR features, but also the least studied, while redilation velocity/rate (corresponding to PIPR) appears to be the most consistently altered PLR feature in AD. In conjunction with other features (baseline pupillary diameter, amplitude and latency of constriction), these parameters predominantly suggest parasympathetic deficiency, associated with mRGCs dysfunction. Longitudinal and adequately designed studies are necessary to validate the use of pupillometry in the early detection and follow-up of AD. Further studies are needed to establish the respective contribution of retinal (afferent) vs. efferent pupillary pathways in the alteration of the pupillary responses, for which chromatic pupillometry can potentially be used and translated into clinical application. Studies may also be designed to investigate the effect of cholinergic medication on PLR in AD patients and the potential use of artificial intelligence on pupillometric traces and video recordings. Using low-cost hardware, pupillometry can now easily be implemented in both remote tele-ophthalmology settings (121), as well as in continuous home monitoring (122). Combined with cognitive game-based investigations and wearables (123, 124), pupillometry may allow a more accurate screening, follow-up, and management of patients with AD.
PC conducted the review of literature. PC, DM, and RN wrote the manuscript. All authors (PC, RN, DM, MF, and NK) reviewed and approved the manuscript.
This work was supported by the National Medical Research Council, Singapore (NMRC/CIRG/1401/2014) to DM.
DM has a patent application based on a pupillometry protocol (PCT/SG2015/050494): A method and system for monitoring and/or assessing pupillary responses. DM and RN have a patent application based on a hand held device for ophthalmic and neurological screening (PCT/SG2018/050204): Hand held ophthalmic and neurological screening device.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Prince M, Jackson MJ, Ferri DCP, Sousa R, Albanese DE, Ribeiro MWS, et al. World Alzheimer Report 2009. Alzheimer's Disease International (2009).
2. Bondareff W, Mountjoy CQ, Roth M, Rossor MN, Iversen LL, Reynolds GP, et al. Neuronal degeneration in locus ceruleus and cortical correlates of Alzheimer disease. Alzheimer Dis Assoc Disord. (1987) 1:256–62. doi: 10.1097/00002093-198701040-00005
3. La Morgia C, Ross-Cisneros FN, Koronyo Y, Hannibal J, Gallassi R, Cantalupo G, et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann Neurol. (2016) 79:90–109. doi: 10.1002/ana.24548
4. Scinto LF, Wu CK, Firla KM, Daffner KR, Saroff D, Geula C. Focal pathology in the Edinger-Westphal nucleus explains pupillary hypersensitivity in Alzheimer's disease. Acta Neuropathol. (1999) 97:557–64.
5. Wu Y-H, Swaab DF. Disturbance and strategies for reactivation of the circadian rhythm system in aging and Alzheimer's disease. Sleep Med. (2007) 8:623–36. doi: 10.1016/j.sleep.2006.11.010
6. Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. (2013) 12:357–67. doi: 10.1016/s1474-4422(13)70044-9
7. Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. (2009) 66:1469–75. doi: 10.1001/archneurol.2009.269
8. Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. (2010) 6:131–44. doi: 10.1038/nrneurol.2010.4
9. Leruez S, Annweiller C, Etcharry-Bouyx F, Verny C, Beauchet O, Milea D. Les troubles visuels au cours de la maladie d'Alzheimer. J Français Ophtalmol. (2012) 35:308–11. doi: 10.1016/j.jfo.2011.11.003
10. Javaid FZ, Brenton J, Guo L, Cordeiro MF. Visual and ocular manifestations of Alzheimer's disease and their use as biomarkers for diagnosis and progression. Front Neurol. (2016) 7:55. doi: 10.3389/fneur.2016.00055
11. Granholm EL, Panizzon MS, Elman JA, Jak AJ, Hauger RL, Bondi MW, et al. Pupillary responses as a biomarker of early risk for Alzheimer's disease. J Alzheimer Dis. (2017) 56:1419–28. doi: 10.3233/jad-161078
12. Tomlinson BE, Irving D, Blessed G. Cell loss in the locus coeruleus in senile dementia of Alzheimer type. J Neurol Sci. (1981) 49:419–28. doi: 10.1016/0022-510X(81)90031-9
13. Scinto LF, Frosch M, Wu CK, Daffner KR, Gedi N, Geula C. Selective cell loss in Edinger-Westphal in asymptomatic elders and Alzheimer's patients. Neurobiol Aging. (2001) 22:729–36. doi: 10.1016/s0197-4580(01)00235-4
14. Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part II: physiological and pharmacological manipulations and pathological alterations of locus coeruleus activity in humans. Curr Neuropharmacol. (2008) 6:254–85. doi: 10.2174/157015908785777193
15. Hattar S, Liao H-W, Takao M, Berson DM, Yau K-W. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. (2002) 295:1065–70. doi: 10.1126/science.1069609
16. Hannibal J, Kankipati L, Strang CE, Peterson BB, Dacey D, Gamlin PD. Central projections of intrinsically photosensitive retinal ganglion cells in the macaque monkey. J Comp Neurol. (2014) 522:Spc1. doi: 10.1002/cne.23555
17. Szabadi E. Modulation of physiological reflexes by pain: role of the locus coeruleus. Front Integr Neurosci. (2012) 6:94. doi: 10.3389/fnint.2012.00094
18. Gooley JJ, Lu J, Fischer D, Saper CB. A broad role for melanopsin in nonvisual photoreception. J Neurosci. (2003) 23:7093–106. doi: 10.1523/JNEUROSCI.23-18-07093.2003
19. Lu J, Shiromani P, Saper CB. Retinal input to the sleep-active ventrolateral preoptic nucleus in the rat. Neuroscience. (1999) 93:209–14. doi: 10.1016/S0306-4522(99)00094-9
20. Najjar RP, Zeitzer JM. Chapter 2—anatomy and physiology of the circadian system. In: Miglis MG, editor. Sleep and Neurologic Disease. San Diego: Academic Press (2017). p. 29–53. Available online at: http://www.sciencedirect.com/science/article/pii/B9780128040744000029
21. Ohtsuka K, Asakura K, Kawasaki H, Sawa M. Respiratory fluctuations of the human pupil. Exp Brain Res. (1988) 71:215–7. doi: 10.1007/BF00247537
22. Bradley MM, Miccoli L, Escrig MA, Lang PJ. The pupil as a measure of emotional arousal and autonomic activation. Psychophysiology. (2008) 45:602–7. doi: 10.1111/j.1469-8986.2008.00654.x
23. Brink RL van den, Murphy PR, Nieuwenhuis S. Pupil diameter tracks lapses of attention. PLoS ONE. (2016) 11:e0165274. doi: 10.1371/journal.pone.0165274
24. Lobato-Rincón L-L, Cabanillas-Campos M del C, Bonnin-Arias C, Chamorro-Gutiérrez E, Murciano-Cespedosa A, Sánchez-Ramos Roda C. Pupillary behavior in relation to wavelength and age. Front Hum Neurosci. (2014) 8:221. doi: 10.3389/fnhum.2014.00221
25. Hattar S, Lucas RJ, Mrosovsky N, Thompson S, Douglas RH, Hankins MW, et al. Melanopsin and rod-cone photoreceptive systems account for all major accessory visual functions in mice. Nature. (2003) 424:75–81. doi: 10.1038/nature01761
26. Kardon R, Anderson SC, Damarjian TG, Grace EM, Stone E, Kawasaki A. Chromatic pupil responses: preferential activation of the melanopsin-mediated versus outer photoreceptor-mediated pupil light reflex. Ophthalmology. (2009) 116:1564–73. doi: 10.1016/j.ophtha.2009.02.007
27. Kawasaki A, Crippa SV, Kardon R, Leon L, Hamel C. Characterization of pupil responses to blue and red light stimuli in autosomal dominant retinitis pigmentosa due to NR2E3 mutation. Invest Ophthalmol Vis Sci. (2012) 53:5562–9. doi: 10.1167/iovs.12-10230
28. Rukmini AV, Milea D, Baskaran M, How AC, Perera SA, Aung T, et al. Pupillary responses to high-irradiance blue light correlate with glaucoma severity. Ophthalmology. (2015) 122:1777–85. doi: 10.1016/j.ophtha.2015.06.002
29. Park JC, Moura AL, Raza AS, Rhee DW, Kardon RH, Hood DC. Toward a clinical protocol for assessing rod, cone, and melanopsin contributions to the human pupil response. Invest Ophthalmol Vis Sci. (2011) 52:6624–35. doi: 10.1167/iovs.11-7586
30. Craig LA, Hong NS, Kopp J, McDonald RJ. Selective lesion of medial septal cholinergic neurons followed by a mini-stroke impairs spatial learning in rats. Exp Brain Res. (2009) 193:29–42. doi: 10.1007/s00221-008-1592-5
31. Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol. (2000) 163:495–529. doi: 10.1006/exnr.2000.7397
32. Bartus RT, Dean RL, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. (1982) 217:408–14. doi: 10.1126/science.7046051
33. Hunter S. The rostral mesencephalon in Parkinson's disease and Alzheimer's disease. Acta Neuropathol. (1985) 68:53–8.
34. Morawski M, Hartlage-Rübsamen M, Jäger C, Waniek A, Schilling S, Schwab C, et al. Distinct glutaminyl cyclase expression in Edinger-Westphal nucleus, locus coeruleus and nucleus basalis Meynert contributes to pGlu-Abeta pathology in Alzheimer's disease. Acta Neuropathol. (2010) 120:195–207. doi: 10.1007/s00401-010-0685-y
35. Mavroudis IA, Manani MG, Petrides F, Petsoglou C, Njau SN, Costa VG, et al. Dendritic and spinal alterations of neurons from Edinger-Westphal nucleus in Alzheimer's disease. Folia Neuropathol. (2014) 52:197–204. doi: 10.5114/fn.2014.43791
36. Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part i: principles of functional organisation. Curr Neuropharmacol. (2008) 6:235–53. doi: 10.2174/157015908785777229
37. Bakes A, Bradshaw CM, Szabadi E. Attenuation of the pupillary light reflex in anxious patients. Br J Clin Pharmacol. (1990) 30:377–81. doi: 10.1111/j.1365-2125.1990.tb03787.x
38. Bitsios P, Szabadi E, Bradshaw CM. The inhibition of the pupillary light reflex by the threat of an electric shock: a potential laboratory model of human anxiety. J Psychopharmacol. (1996) 10:279–87. doi: 10.1177/026988119601000404
39. Theofilopoulos N, McDade G, Szabadi E, Bradshaw CM. Effects of reboxetine and desipramine on the kinetics of the pupillary light reflex. Br J Clin Pharmacol. (1995) 39:251–5. doi: 10.1111/j.1365-2125.1995.tb04444.x
40. Abercrombie ED, Jacobs BL. Microinjected clonidine inhibits noradrenergic neurons of the locus coeruleus in freely moving cats. Neurosci Lett. (1987) 76:203–8.
41. Bitsios P, Szabadi E, Bradshaw CM. The effects of clonidine on the fear-inhibited light reflex. J Psychopharmacol. (1998) 12:137–45. doi: 10.1177/026988119801200204
42. Clifford JM, Day MD, Orwin JM. Reversal of clonidine induced miosis by the alpha 2-adrenoreceptor antagonist RX 781094. Br J Clin Pharmacol. (1982) 14:99–101. doi: 10.1111/j.1365-2125.1982.tb04941.x
43. Joshi S, Li Y, Kalwani RM, Gold JI. Relationships between pupil diameter and neuronal activity in the locus coeruleus, colliculi, and cingulate cortex. Neuron. (2016) 89:221–34. doi: 10.1016/j.neuron.2015.11.028
44. Kelly SC, He B, Perez SE, Ginsberg SD, Mufson EJ, Counts SE. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer's disease. Acta Neuropathol Commun. (2017) 5:8. doi: 10.1186/s40478-017-0411-2
45. Prettyman R, Bitsios P, Szabadi E. Altered pupillary size and darkness and light reflexes in Alzheimer's disease. J Neurol Neurosurg Psychiatry. (1997) 62:665–8.
46. Najjar RP, Teikari P, Cornut P-L, Knoblauch K, Cooper HM, Gronfier C. Heterochromatic flicker photometry for objective lens density quantification. Invest Ophthalmol Vis Sci. (2016) 57:1063–71. doi: 10.1167/iovs.15-18642
47. Teikari P, Najjar RP, Knoblauch K, Dumortier D, Cornut P-L, Denis P, et al. Refined flicker photometry technique to measure ocular lens density. J Opt Soc Am A. (2012) 29:2469–78. doi: 10.1364/JOSAA.29.002469
48. Freund PR, Watson J, Gilmour GS, Gaillard F, Sauvé Y. Differential changes in retina function with normal aging in humans. Doc Ophthalmol. (2011) 122:177–90. doi: 10.1007/s10633-011-9273-2
49. Gerth C, Garcia SM, Ma L, Keltner JL, Werner JS. Multifocal electroretinogram: age-related changes for different luminance levels. Graefes Arch Clin Exp Ophthalmol. (2002) 240:202–8. doi: 10.1007/s00417-002-0442-6
50. Song H, Chui TYP, Zhong Z, Elsner AE, Burns SA. Variation of cone photoreceptor packing density with retinal eccentricity and age. Invest Ophthalmol Vis Sci. (2011) 52:7376–84. doi: 10.1167/iovs.11-7199
51. Rukmini AV, Milea D, Aung T, Gooley JJ. Pupillary responses to short-wavelength light are preserved in aging. Sci Rep. (2017) 7:43832. doi: 10.1038/srep43832
52. Najjar RP, Chiquet C, Teikari P, Cornut P-L, Claustrat B, Denis P, et al. Aging of non-visual spectral sensitivity to light in humans: compensatory mechanisms? PLoS ONE. (2014) 9:e85837. doi: 10.1371/journal.pone.0085837
53. Daneault V, Vandewalle G, Hébert M, Teikari P, Mure LS, Doyon J, et al. Does pupil constriction under blue and green monochromatic light exposure change with age? J Biol Rhythms. (2012) 27:257–64. doi: 10.1177/0748730412441172
54. Esquiva G, Lax P, Pérez-Santonja JJ, García-Fernández JM, Cuenca N. Loss of melanopsin-expressing ganglion cell subtypes and dendritic degeneration in the aging human retina. Front Aging Neurosci. (2017) 9:79. doi: 10.3389/fnagi.2017.00079
55. Berisha F, Feke GT, Trempe CL, McMeel JW, Schepens CL. Retinal abnormalities in early Alzheimer's disease. Invest Ophthalmol Vis Sci. (2007) 48:2285–9. doi: 10.1167/iovs.06-1029
56. Chan VTT, Sun Z, Tang S, Chen LJ, Wong A, Tham CC, et al. Spectral-domain OCT measurements in Alzheimer's disease: a systematic review and meta-analysis. Ophthalmology. (2018) 126:497–510. doi: 10.1016/j.ophtha.2018.08.009
57. Cunha JP, Proença R, Dias-Santos A, Almeida R, Águas H, Alves M, et al. OCT in Alzheimer's disease: thinning of the RNFL and superior hemiretina. Graefes Arch Clin Exp Ophthalmol. (2017) 255:1827–35. doi: 10.1007/s00417-017-3715-9
58. Alexandrov PN, Pogue A, Bhattacharjee S, Lukiw WJ. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer's disease. Neuroreport. (2011) 22:623–7. doi: 10.1097/wnr.0b013e3283497334
59. Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, et al. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage. (2011) 54 (Suppl. 1):S204–17. doi: 10.1016/j.neuroimage.2010.06.020
60. Koronyo Y, Biggs D, Barron E, Boyer DS, Pearlman JA, Au WJ, et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer's disease. JCI Insight. (2017) 2:93621. doi: 10.1172/jci.insight.93621
61. Tsai Y, Lu B, Ljubimov AV, Girman S, Ross-Cisneros FN, Sadun AA, et al. Ocular changes in TgF344-AD rat model of Alzheimer's disease. Invest Ophthalmol Vis Sci. (2014) 55:523–34. doi: 10.1167/iovs.13-12888
62. Lucas RJ, Hattar S, Takao M, Berson DM, Foster RG, Yau K-W. Diminished pupillary light reflex at high irradiances in melanopsin-knockout mice. Science. (2003) 299:245–7. doi: 10.1126/science.1077293
63. Obara EA, Hannibal J, Heegaard S, Fahrenkrug J. Loss of melanopsin-expressing retinal ganglion cells in severely staged glaucoma patients. Invest Ophthalmol Vis Sci. (2016) 57:4661–7. doi: 10.1167/iovs.16-19997
64. Kim JY, Jeong AR, Chin HS, Kim NR. Melatonin levels in patients with primary open-angle glaucoma with high or low intraocular pressure. J Glaucoma. (2019) 28:154–60. doi: 10.1097/IJG.0000000000001130
65. Gubin DG, Malishevskaya TN, Astakhov YS, Astakhov SY, Cornelissen G, Kuznetsov VA, et al. Progressive retinal ganglion cell loss in primary open-angle glaucoma is associated with temperature circadian rhythm phase delay and compromised sleep. Chronobiol Int. (2019) 36:564–77. doi: 10.1080/07420528.2019.1566741
66. Kankipati L, Girkin CA, Gamlin PD. The post-illumination pupil response is reduced in glaucoma patients. Invest Ophthalmol Vis Sci. (2011) 52:2287–92. doi: 10.1167/iovs.10-6023
67. Feigl B, Mattes D, Thomas R, Zele AJ. Intrinsically photosensitive (melanopsin) retinal ganglion cell function in glaucoma. Invest Ophthalmol Vis Sci. (2011) 52:4362–7. doi: 10.1167/iovs.10-7069
68. Nissen C, Sander B, Milea D, Kolko M, Herbst K, Hamard P, et al. Monochromatic pupillometry in unilateral glaucoma discloses no adaptive changes subserved by the ipRGCs. Front Neurol. (2014) 5:15. doi: 10.3389/fneur.2014.00015
69. Adhikari P, Zele AJ, Thomas R, Feigl B. Quadrant field pupillometry detects melanopsin dysfunction in glaucoma suspects and early glaucoma. Sci Rep. (2016) 6:33373. doi: 10.1038/srep33373
70. Najjar RP, Sharma S, Atalay E, Rukmini AV, Sun C, Lock JZ, et al. Pupillary responses to full-field chromatic stimuli are reduced in patients with early-stage primary open-angle glaucoma. Ophthalmology. (2018) 125:1362–1371. doi: 10.1016/j.ophtha.2018.02.024
71. Kawasaki A, Herbst K, Sander B, Milea D. Selective wavelength pupillometry in Leber hereditary optic neuropathy. Clin Experiment Ophthalmol. (2010) 38:322–4. doi: 10.1111/j.1442-9071.2010.02212.x
72. Moura ALA, Nagy BV, La Morgia C, Barboni P, Oliveira AGF, Salomão SR, et al. The pupil light reflex in Leber's hereditary optic neuropathy: evidence for preservation of melanopsin-expressing retinal ganglion cells. Invest Ophthalmol Vis Sci. (2013) 54:4471–7. doi: 10.1167/iovs.12-11137
73. Loo JL, Singhal S, Rukmini AV, Tow S, Amati-Bonneau P, Procaccio V, et al. Multiethnic involvement in autosomal-dominant optic atrophy in Singapore. Eye. (2017) 31:475–80. doi: 10.1038/eye.2016.255
74. La Morgia C, Ross-Cisneros FN, Sadun AA, Hannibal J, Munarini A, Mantovani V, et al. Melanopsin retinal ganglion cells are resistant to neurodegeneration in mitochondrial optic neuropathies. Brain. (2010) 133 (Pt 8):2426–38. doi: 10.1093/brain/awq155
75. Fotiou DF, Brozou CG, Haidich A-B, Tsiptsios D, Nakou M, Kabitsi A, et al. Pupil reaction to light in Alzheimer's disease: evaluation of pupil size changes and mobility. Aging Clin Exp Res. (2007) 19:364–71. doi: 10.1007/bf03324716
76. Fotiou DF, Stergiou V, Tsiptsios D, Lithari C, Nakou M, Karlovasitou A. Cholinergic deficiency in Alzheimer's and Parkinson's disease: evaluation with pupillometry. Int J Psychophysiol. (2009) 73:143–9. doi: 10.1016/j.ijpsycho.2009.01.011
77. Frost S, Kanagasingam Y, Sohrabi H, Bourgeat P, Villemagne V, Rowe CC, et al. Pupil response biomarkers for early detection and monitoring of Alzheimer's disease. Curr Alzheimer Res. (2013) 10:931–9. doi: 10.2174/15672050113106660163
78. Bittner DM, Wieseler I, Wilhelm H, Riepe MW, Müller NG. Repetitive pupil light reflex: potential marker in Alzheimer's disease? J Alzheimers Dis. (2014) 42:1469–77. doi: 10.3233/jad-140969
79. Granholm E, Morris S, Galasko D, Shults C, Rogers E, Vukov B. Tropicamide effects on pupil size and pupillary light reflexes in Alzheimer's and Parkinson's disease. Int J Psychophysiol. (2003) 47:95–115. doi: 10.1016/s0167-8760(02)00122-8
80. Ferrario E, Molaschi M, Villa L, Varetto O, Bogetto C, Nuzzi R. Is videopupillography useful in the diagnosis of Alzheimer's disease? Neurology. (1998) 50:642–4.
81. Fotiou F, Fountoulakis KN, Tsolaki M, Goulas A, Palikaras A. Changes in pupil reaction to light in Alzheimer's disease patients: a preliminary report. Int J Psychophysiol. (2000) 37:111–20. doi: 10.1016/s0167-8760(00)00099-4
82. Fotiou D, Kaltsatou A, Tsiptsios D, Nakou M. Evaluation of the cholinergic hypothesis in Alzheimer's disease with neuropsychological methods. Aging Clin Exp Res. (2015) 27:727–33. doi: 10.1007/s40520-015-0321-8
83. Frost S, Robinson L, Rowe CC, Ames D, Masters CL, Taddei K, et al. Evaluation of cholinergic deficiency in preclinical alzheimer's disease using pupillometry. J Ophthalmol. (2017) 2017:7935406. doi: 10.1155/2017/7935406
84. Van Stavern GP, Bei L, Shui Y-B, Huecker J, Gordon M. Pupillary light reaction in preclinical Alzheimer's disease subjects compared with normal ageing controls. Br J Ophthalmol. (2018). doi: 10.1136/bjophthalmol-2018-312425. [Epub ahead of print].
85. Tales A, Troscianko T, Lush D, Haworth J, Wilcock GK, Butler SR. The pupillary light reflex in aging and Alzheimer's disease. Aging. (2001) 13:473–8.
86. Gamlin PDR, McDougal DH, Pokorny J, Smith VC, Yau K-W, Dacey DM. Human and macaque pupil responses driven by melanopsin-containing retinal ganglion cells. Vis Res. (2007) 47:946–54. doi: 10.1016/j.visres.2006.12.015
87. Mure LS, Cornut P-L, Rieux C, Drouyer E, Denis P, Gronfier C, et al. Melanopsin bistability: a fly's eye technology in the human retina. PLoS ONE. (2009) 4:e5991. doi: 10.1371/journal.pone.0005991
88. Sunderland T, Linker G, Mirza N, Putnam KT, Friedman DL, Kimmel LH, et al. Decreased beta-amyloid1-42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA. (2003) 289:2094–103. doi: 10.1001/jama.289.16.2094
89. Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. (2010) 31:1275–83. doi: 10.1016/j.neurobiolaging.2010.04.007
90. Hays MT, Watson EE, Thomas SR, Stabin M. MIRD dose estimate report no. 19: radiation absorbed dose estimates from (18)F-FDG. J Nucl Med. (2002) 43:210–4.
91. Santana P do C, Mourão AP, de Oliveira PMC, Bernardes FD, Mamede M, da Silva TA. Dosimetry of patients submitted to cerebral PET/CT for the diagnosis of mild cognitive impairment. Radiol Bras. (2014) 47:350–4. doi: 10.1590/0100-3984.2013.1800
92. McDonald RJ. Multiple combinations of co-factors produce variants of age-related cognitive decline: a theory. Can J Exp Psychol. (2002) 56:221–39. doi: 10.1037/h0087399
93. Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R, et al. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci USA. (1994) 91:11183–6. doi: 10.1073/pnas.91.23.11183
94. Frost SM, Kanagasingam Y, Sohrabi HR, Taddei K, Bateman R, Morris J, et al. Pupil response biomarkers distinguish amyloid precursor protein mutation carriers from non-carriers. Curr Alzheimer Res. (2013) 10:790–6. doi: 10.2174/15672050113109990154
95. O'Bryant SE, Humphreys JD, Smith GE, Ivnik RJ, Graff-Radford NR, Petersen RC, et al. Detecting dementia with the mini-mental state examination (MMSE) in highly educated individuals. Arch Neurol. (2008) 65:963–7. doi: 10.1001/archneur.65.7.963
96. Scinto LF, Daffner KR, Dressler D, Ransil BI, Rentz D, Weintraub S, et al. A potential noninvasive neurobiological test for Alzheimer's disease. Science. (1994) 266:1051–4.
97. FitzSimon JS, Waring SC, Kokmen E, McLaren JW, Brubaker RF. Response of the pupil to tropicamide is not a reliable test for Alzheimer disease. Arch Neurol. (1997) 54:155–9. doi: 10.1001/archneur.1997.00550140031009
98. Kurz A, Marquard R, Fremke S, Leipert KP. Pupil dilation response to tropicamide: a biological test for Alzheimer's disease? Pharmacopsychiatry. (1997) 30:12–5.
99. Loupe DN, Newman NJ, Green RC, Lynn MJ, WIlliams KK, Geis TC, et al. Pupillary response to tropicamide in patients with Alzheimer disease. Ophthalmology. (1996) 103:495–503. doi: 10.1016/S0161-6420(96)30666-0
100. Winn B, Whitaker D, Elliott DB, Phillips NJ. Factors affecting light-adapted pupil size in normal human subjects. Invest Ophthalmol Vis Sci. (1994) 35:1132–7.
101. Aarts MPJ, Rosemann ALP. Towards a uniform specification of light therapy devices for the treatment of affective disorders and use for non-image forming effects: radiant flux. J Affect Disorders. (2018) 235:142–9. doi: 10.1016/j.jad.2018.04.020
102. Bullough JD, Rea MS, Figueiro MG. Of mice and women: light as a circadian stimulus in breast cancer research. Cancer Causes Control. (2006) 17:375–83. doi: 10.1007/s10552-005-0574-1
103. Adhikari P, Pearson CA, Anderson AM, Zele AJ, Feigl B. Effect of age and refractive error on the melanopsin mediated post-illumination pupil response (PIPR). Sci Rep. (2015) 5:17610. doi: 10.1038/srep17610
104. Kankipati L, Girkin CA, Gamlin PD. Post-illumination pupil response in subjects without ocular disease. Invest Ophthalmol Vis Sci. (2010) 51:2764–9. doi: 10.1167/iovs.09-4717
105. Herbst K, Sander B, Lund-Andersen H, Broendsted AE, Kessel L, Hansen MS, et al. Intrinsically photosensitive retinal ganglion cell function in relation to age: a pupillometric study in humans with special reference to the age-related optic properties of the lens. BMC Ophthalmol. (2012) 12:4. doi: 10.1186/1471-2415-12-4
106. Sharma S, Baskaran M, Rukmini AV, Nongpiur ME, Htoon H, Cheng C-Y, et al. Factors influencing the pupillary light reflex in healthy individuals. Graefes Arch Clin Exp Ophthalmol. (2016) 254:1353–9. doi: 10.1007/s00417-016-3311-4
107. Rukmini AV, Milea D, Gooley JJ. Chromatic pupillometry methods for assessing photoreceptor health in retinal and optic nerve diseases. Front Neurol. (2019) 10:76. doi: 10.3389/fneur.2019.00076
108. Kardon R, Anderson SC, Damarjian TG, Grace EM, Stone E, Kawasaki A. Chromatic pupillometry in patients with retinitis pigmentosa. Ophthalmology. (2011) 118:376–81. doi: 10.1016/j.ophtha.2010.06.033
109. Lorenz B, Strohmayr E, Zahn S, Friedburg C, Kramer M, Preising M, et al. Chromatic pupillometry dissects function of the three different light-sensitive retinal cell populations in RPE65 deficiency. Invest Ophthalmol Vis Sci. (2012) 53:5641–52. doi: 10.1167/iovs.12-9974
110. Feigl B, Zele AJ, Fader SM, Howes AN, Hughes CE, Jones KA, et al. The post-illumination pupil response of melanopsin-expressing intrinsically photosensitive retinal ganglion cells in diabetes. Acta Ophthalmol. (2012) 90:e230–234. doi: 10.1111/j.1755-3768.2011.02226.x
111. Park JC, Chen Y-F, Blair NP, Chau FY, Lim JI, Leiderman YI, et al. Pupillary responses in non-proliferative diabetic retinopathy. Sci Rep. (2017) 7:44987. doi: 10.1038/srep44987
112. Dewey RB. Autonomic dysfunction in Parkinson's disease. Neurol Clin. (2004) 22 (Suppl. 3):S127–39. doi: 10.1016/j.ncl.2004.05.006
113. Perez-Lloret S, Barrantes FJ. Deficits in cholinergic neurotransmission and their clinical correlates in Parkinson's disease. NPJ Parkinsons Dis. (2016) 2:16001. doi: 10.1038/npjparkd.2016.1
114. Micieli G, Tassorelli C, Martignoni E, Pacchetti C, Bruggi P, Magri M, et al. Disordered pupil reactivity in Parkinson's disease. Clinical Autonomic Research. (1991) 1:55–8.
115. Jain S, Siegle GJ, Gu C, Moore CG, Ivanco LS, Jennings JR, et al. Autonomic insufficiency in pupillary and cardiovascular systems in Parkinson's disease. Parkinson Relat Disorders. (2011) 17:119–22. doi: 10.1016/j.parkreldis.2010.11.005
116. Joyce DS, Feigl B, Kerr G, Roeder L, Zele AJ. Melanopsin-mediated pupil function is impaired in Parkinson's disease. Sci Rep. (2018) 8:7796. doi: 10.1038/s41598-018-26078-0
117. Beach TG, Carew J, Serrano G, Adler CH, Shill HA, Sue LI, et al. Phosphorylated α-synuclein-immunoreactive retinal neuronal elements in Parkinson's disease subjects. Neurosci Lett. (2014) 571:34–8. doi: 10.1016/j.neulet.2014.04.027
118. Bodis-Wollner I, Kozlowski PB, Glazman S, Miri S. α-synuclein in the inner retina in Parkinson disease. Ann Neurol. (2014) 75:964–6. doi: 10.1002/ana.24182
119. Ferrari L, Huang S-C, Magnani G, Ambrosi A, Comi G, Leocani L. Optical coherence tomography reveals retinal neuroaxonal thinning in frontotemporal dementia as in Alzheimer's disease. J Alzheimers Dis. (2017) 56:1101–7. doi: 10.3233/jad-160886
120. Kim BJ, Irwin DJ, Song D, Daniel E, Leveque JD, Raquib AR, et al. Optical coherence tomography identifies outer retina thinning in frontotemporal degeneration. Neurology. (2017) 89:1604–11. doi: 10.1212/WNL.0000000000004500
121. DeBuc DC. The role of retinal imaging and portable screening devices in tele-ophthalmology applications for diabetic retinopathy management. Curr Diab Rep. (2016) 16:132. doi: 10.1007/s11892-016-0827-2
122. Roesch K, Swedish T, Raskar R. Automated retinal imaging and trend analysis—a tool for health monitoring. Clin Ophthalmol. (2017) 11:1015–20. doi: 10.2147/OPTH.S116265
123. Coughlan G, Coutrot A, Khondoker M, Minihane AM, Spiers H, Hornberger M. Impact of sex and APOE status on spatial navigation in pre-symptomatic Alzheimer's disease. bioRxiv. (2018). doi: 10.1101/287722. [Epub ahead of print].
Keywords: Alzheimer's disease, dementia, pupillary light response, chromatic pupillometry, melanopsin expressing intrinsically photosensitive retinal ganglion cells, Parkinson's disease, post -llumination pupil response, cholinergic deficit
Citation: Chougule PS, Najjar RP, Finkelstein MT, Kandiah N and Milea D (2019) Light-Induced Pupillary Responses in Alzheimer's Disease. Front. Neurol. 10:360. doi: 10.3389/fneur.2019.00360
Received: 12 November 2018; Accepted: 25 March 2019;
Published: 12 April 2019.
Edited by:
Andrew J. Zele, Queensland University of Technology, AustraliaReviewed by:
Shlomo Dotan, Hadassah Medical Center, IsraelCopyright © 2019 Chougule, Najjar, Finkelstein, Kandiah and Milea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Milea, ZGFuLm1pbGVhQHNuZWMuY29tLnNn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.