 Khidhir Kamil
Khidhir Kamil Muhammad Dain Yazid2
Muhammad Dain Yazid2 Srijit Das
Srijit Das Jaya Kumar
Jaya Kumar
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 19 March 2019
Sec. Neurodegeneration
Volume 10 - 2019 | https://doi.org/10.3389/fneur.2019.00087
This article is part of the Research Topic Biomarkers in Neurology View all 17 articles
Demyelinating diseases represent a spectrum of disorders that impose significant burden on global economy and society. Generally, the prognosis of these diseases is poor and there is no available cure. In recent decades, research has shed some light on the biology and physiology of Schwann cells and its neuroprotective effects in the peripheral nervous system (PNS). Insults to the PNS by various infectious agents, genetic predisposition and immune-related mechanisms jeopardize Schwann cell functions and cause demyelination. To date, there are no effective and reliable biomarkers for PNS-related diseases. Here, we aim to review the following: pathogenesis of various types of peripheral demyelinating diseases such as Guillain-Barre syndrome, Chronic Inflammatory Demyelinating Polyradiculoneuropathy, Anti-Myelin Associated Glycoprotein Neuropathy, POEMS syndrome, and Charcot-Marie-Tooth disease; emerging novel biomarkers for peripheral demyelinating diseases, and Schwann cell associated markers for demyelination.
Peripheral demyelinating diseases (PDD) refer to a spectrum of disorders that involves substantial damage to axons and glial cells, particularly Schwann cells (SC) in the peripheral nervous system (PNS) (1). The incidence of these diseases is variable (2–4). Disease states are manifestations of damage against the myelin sheath caused by various inciting factors, such as infectious agents, auto-immune processes or genetic mutations (1, 5–7). Oxidative stress, the primary risk factor in many diseases (8), has also been implicated in demyelination disorders (9).
Schwann cells are principal glial cells in peripheral nerves that originate from the neural crest, which is a multipotent embryonic structure that also differentiates into other main glial subtypes of the PNS (10). SC development occurs through a series of embryonic and postnatal phases, which are tightly regulated by a number of cellular signaling pathways. During the early embryonic phase, neural crest cells differentiate into SC precursors that represent the first transitional stage in the SC lineage, that subsequently further differentiate into immature SC (10). At time of birth, these immature SC differentiate into either myelinating or non- myelinating SC that populates the mature nerve trunks and wrap around axons through a process known as myelination (10).
Myelination is a process whereby SC develops a multi-layered membrane called the myelin sheath around the axonal membrane (11). Mostly, larger axons (>1 um) are selected specifically by SC to form multiple internodes of the myelin sheath (12). Myelination begins with the establishment of a 1:1 relationship with the axon. At this level, the production of myelin structural proteins such as myelin protein zero (P0), peripheral myelin protein (PMP22), myelin basic protein (MBP) are increased along with lipid biosynthesis (11).
The myelin sheath is made of multiple sleeves of whitish lipoprotein plasma membranes of SC wrapped around the axon of a neuron in a spiral fashion (13). It is constituted of water, lipid and proteins that exist as segmented internodal structure around the axons (13). These internodes create insulation that facilitates propagation of action potentials by mean of saltatory conduction (jumping) at the node of Ranvier. Myelin sheath not only facilitates the conduction velocity of nerve impulse but also confers protection and nutritional support to axons. However, exposure to various factors such as autoimmunological insult, trauma, and injury to the nerve could trigger demyelination, and eventually neurodegeneration (14).
Demyelination describes the loss of the myelin sheath, where SC are being destroyed or unwrapped from axons (15). Demyelination causes neurological disability due to conduction block and axonal degeneration. Diagnosis of PDD depends on electrophysiological and cerebrospinal fluid (CSF) analysis. However, in some cases, no biomarkers are clinically available for diagnosis, disease monitoring and prognosis.
Guillain-Barre Syndrome (GBS) is an acute idiopathic autoimmune demyelinating disease of the PNS that is characterized by acute flaccid ascending neuromuscular paralysis (16). GBS is rare, with an incidence of 0.8–1.9/100,000 per annum across Europe and North America (17). Currently, the specific causative agent of GBS is unknown, but numerous theories have been proposed (18). Most cases of GBS are preceded by antecedent infections of several microbes of the gastrointestinal and upper respiratory tracts (19). Among those, 60% of GBS cases were related to autoantibodies, anti-monosialotetrahexosylganglioside- 1 (anti-GM1) and anti-ganglioside GD1a (anti-GD1a) associated with C. jejuni infection (20). Other microbes involved include M. pneumoniae, cytomegalovirus, Epstein-Barr virus, varicella zoster virus, and influenza virus (21–24). Apart from infections, some GBS cases are results of trauma, surgical interventions, treatment with monoclonal antibodies and vaccination (rare) (20).
The most frequent GBS variant is acute inflammatory demyelinating polyradiculopathy (AIDP); other axonal variants include acute motor axonal neuropathy (AMAN), acute motor sensory axonal neuropathy (AMSAN), Miller-Fisher syndrome and oropharyngeal weakness (25, 26). In AIDP, there are areas of segmental demyelination with inflammatory infiltrates such as lymphocytes and macrophages (27). Due to the observation of a high incidence of GBS after a preceding infection, it has been theorized that molecular mimicry plays a role in triggering an autoimmune response against peripheral nerve tissues. The presence of anti-GM1 and anti-GD1a antibodies in the serum suggest that ganglioside-like moieties carried by lipooligosaccharides found in the bacterial wall of C. jejuni has cross-reactivity against neural tissues of the PNS (5) (Figure 1). It has also been found that patients treated with gangliosides for pain and neuropathy in the early 1990s later developed GBS (28). Gangliosides, axo-glial junctional proteins, neurofascin and gliomedin at nodes of Ranvier could contribute toward the autoimmunity seen in GBS (29).
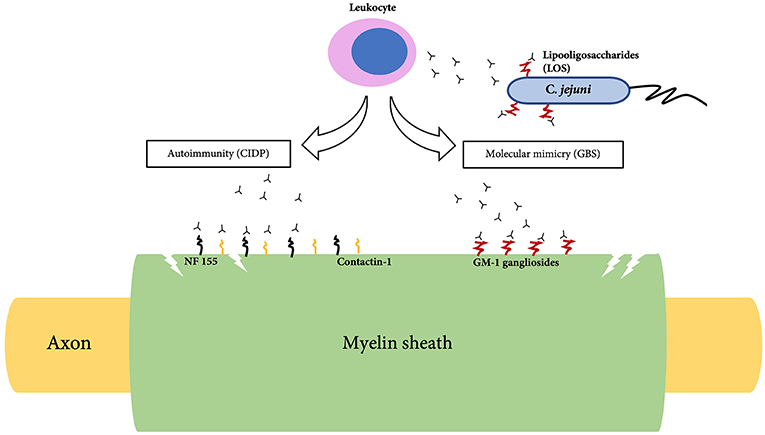
Figure 1. Pathogenesis of Guillain-Barre syndrome (GBS) and Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).
The clinical manifestations of GBS include acute ascending fairly symmetric paralysis and paresthesia, choking and difficulty in breathing over the course of hours to several days (2). Involvement of the respiratory muscles in GBS may require the need for artificial ventilation (30). Some patients also experienced autonomic dysfunctions such as cardiac arrhythmia, arterial hypotension, gastrointestinal dysmotility, urinary retention, and abnormal sweating (31). Management of GBS is mostly supportive (20). Affected patients would require comprehensive assisted respiratory ventilation with monitoring for cardiac arrhythmia and bed-bound complications such as ventilator-associated pneumonia, thromboembolism and infections (32). Plasma exchange and intravenous immunoglobulin (IVIG) have been shown in large randomized trials to be beneficial (33). Overall, most cases of GBS have good prognosis with functional recovery within 12 months after disease onset (34). However, some patients do suffer from residual deficits (35).
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is an acquired immune mediated demyelinating disease of the PNS characterized by progressive loss of motor and sensory functions (36). CIDP sometimes is quite similar to GBS, with the distinction that its clinical course is chronic with relapses (37). The onset is insidious and occurs more commonly in older age individuals (38, 39).
The immune system primarily attacks and damages the myelin sheath of the PNS followed by segmental demyelination and axonal degeneration (6). Histological findings of CIDP demonstrate thin myelin sheath with short internodes described as onion bulbs. Demyelination is indicated by the slow nerve conduction velocity suggestive of conduction block (6). Recently evidence of autoimmunity toward neurofascin-155 (NF155) and contactin-1 (CNTN1) in some patients have been reported. (40, 41) (Figure 1). NF155 is an adhesion molecule that is expressed at paranodes of glial side which interacts with CNTN1, a key axonal adhesion molecule (42). This interaction is essential for the formation of paranodal septate-like junction and loss of this junction is associated with slow conduction (42).
Symptoms of CIDP develop slowly but progressive and neurological deficits peak after 8 weeks of disease onset (36). Typical symptoms are tingling/numbness of the extremities due to the association of large nerve fibers, symmetrical weakness and paresthesia of legs and arms, loss of reflex, fatigue, ataxia and limb incoordination (6). Treatment with oral glucocorticoids usually produce a favorable response (43). Apart from that, plasmapheresis and IVIG are also effective (36).
Anti-Myelin Associated Glycoprotein (MAG) neuropathy is a demyelinating polyneuropathy associated with IgM monoclonal gammopathy towards MAG in peripheral nerves (44). MAG is a type I transmembrane glycoprotein l presents in peri-axonal SC and oligodendroglial membranes of myelin sheaths that central in glial-axon interaction and maintenance of axonal function (45). Loss of MAG compromises the myelin sheath integrity and axonal function. MAG contains a carbohydrate epitope shared with other glycoconjugates that serve as primary antigenic targets for IgM paraproteins (44). Injection of serum containing IgM anti-MAG paraproteins into chickens causes segmental demyelination and conduction block (46).
The disease is also described as progressive mild to moderate distal muscle weakness; along with progressive sensory ataxia and frequent tremors (47). The clinical course is generally benign, with minimal functional deterioration manifested over time (47). As the symptoms of anti-MAG neuropathy usually are minimal and do not interfere with the patient's daily activities initially; management at this stage comprises of supportive care such as exercise and balance training. However, patients with sensorimotor weakness should be treated. Steroids, IVIG and plasmapheresis are rarely effective. Rituximab, a monoclonal antibody against CD20 surface antigen is promising (48).
POEMS syndrome is a rare paraneoplastic syndrome with demyelinating neuropathy (49). Emprical data on POEMS syndrome is deficient owing to the complexity and multisystemic nature of its clinical manifestations. It is usually associated with an underlying plasma cell neoplasm (50). POEMS syndrome commonly presents in the fifth to sixth decade (49). The pathogenesis of POEMS syndrome is not well understood, but several hypotheses have been proposed. High serum level of vascular endothelial growth factor (VEGF) is detected in POEMS patients, whereas low levels are often reported upon successful treatment (50). The pathological assessment does not reveal inflammatory infiltrates or immunoglobulin deposition within the nerves; instead, there is endothelial cell hypertrophy with extended process, reduced luminal diameter, and disrupted tight junction that could cause leakage (51). Excessive VEGF secreted by plasma cells is thought to cause endothelial proliferation and subsequent leaky vessels that compromise blood flow (51).
POEMS is an acronym of its multiorgan features: Polyneuropathy, Organomegaly, Endocrinopathy, M protein, and Skin changes. The polyneuropathy involves both sensory and motor systems (52). Patients usually begin to experience sensory abnormalities described as tingling, paresthesia and coldness in the feet, along with touch, pressure, and proprioception disturbances (52). Motor symptoms then develop, including symmetrical severe weakness on extremities progressing distally with gradual spreading to proximal (52). Nerve conduction studies, as well as nerve biopsies show evidence of demyelination and axonal loss (49). Hepatomegaly is commonly reported, but splenomegaly and lymphadenopathy are not frequent. Endocrinopathy, usually gonadal dysfunction is noted by testicular atrophy and gynecomastia and diabetes mellitus. The M-protein of IgG or IgA is commonly detected. Skin changes include hyperpigmentation and hypertrichosis. Other additional features that are not included in the acronym are sometimes present. These include peripheral edema, effusion in body cavities such as ascites, sclerotic bone lesions, Castleman's disease, elevated intracranial pressure, papilledema, fatigue, renal failure and clubbing (50). However, not all features are necessarily required for the diagnosis.
Treatment with high dose chemotherapy and autologous peripheral blood stem cell transplant are the first line therapy. Alternative therapeutics are including corticosteroids, low-dose alkylator therapy and radiation therapy. The median survival for patients with POEMS syndrome is about 13.8 years. Mortality usually results from cardiorespiratory failure, infection and renal failure. Supportive care such as physical and occupational therapy should be in line with the treatment to improve outcome and quality of life. Some patients might even require assisted ventilation due to respiratory muscle weakness (49).
Charcot Marie Tooth disease (CMT) is a rare hereditary neurological disorder affecting the peripheral nerves (7). Although CMT is rare, it is the most commonly inherited form of neuropathy affecting approximately 1 in 2,500 people (53). The majority of CMT have an autosomal dominant inheritance but X-linked and autosomal recessive pattern also exist (54). The gene abnormalities in CMT disrupt the structure and functions of Schwann cell and peripheral nerve axons. Several subtypes of CMT have been identified: mutations of genes encoding myelin-related proteins such as PMP22, P0, and connexin 32 are classified as demyelinating subtype (54); mutations of proteins involved in axonal transport such as mitofusin-2(MFN2), ganglioside-induced differentiation- associated protein 1 (GDAP1), heat shock factor binding protein 1 are classified as axonal neuropathies subtype (54). Next-generation gene sequencing involving 17,000 samples with neuropathy identified the prevalence of specific mutations as such: 78.6% involve PMP22 mutations, 6.7% involve GJB1, 5.3% involve P0, and 4.3% involve MFN2 (55).
The most common form of CMT, CMT1A is a result of duplications of the PMP22 gene that increased expression of PMP22 structural protein (56). CMT1B on the other hand, involves mutations of P0 gene that result in misfolding and retention of a mutant P0 protein intracellularly (54). This condition triggers the activation of unfolded protein response which later lead to cell apoptosis (57). CMT2A involve mutations of MFN2 which aids in the fusion of mitochondria (54, 58). Typically, the progression of CMT is slow. The neuropathy of CMT could affect both motor and sensory nerves. Patients may experience distal muscle weakness, foot drop that formed pes cavus, scoliosis and hammer toes. Respiratory insufficiency is rare, but possible. Neuropathic pain and fatigue have been reported in several cases.
At present, the management of PDD is central upon synthetic drugs and natural products (59). However, this disease remains underdiagnosed owing to lack of reliable biomarkers and a disease specific-diagnostic criteria.
Table 1 summarized the pathogenesis, clinical features and management of PDD.
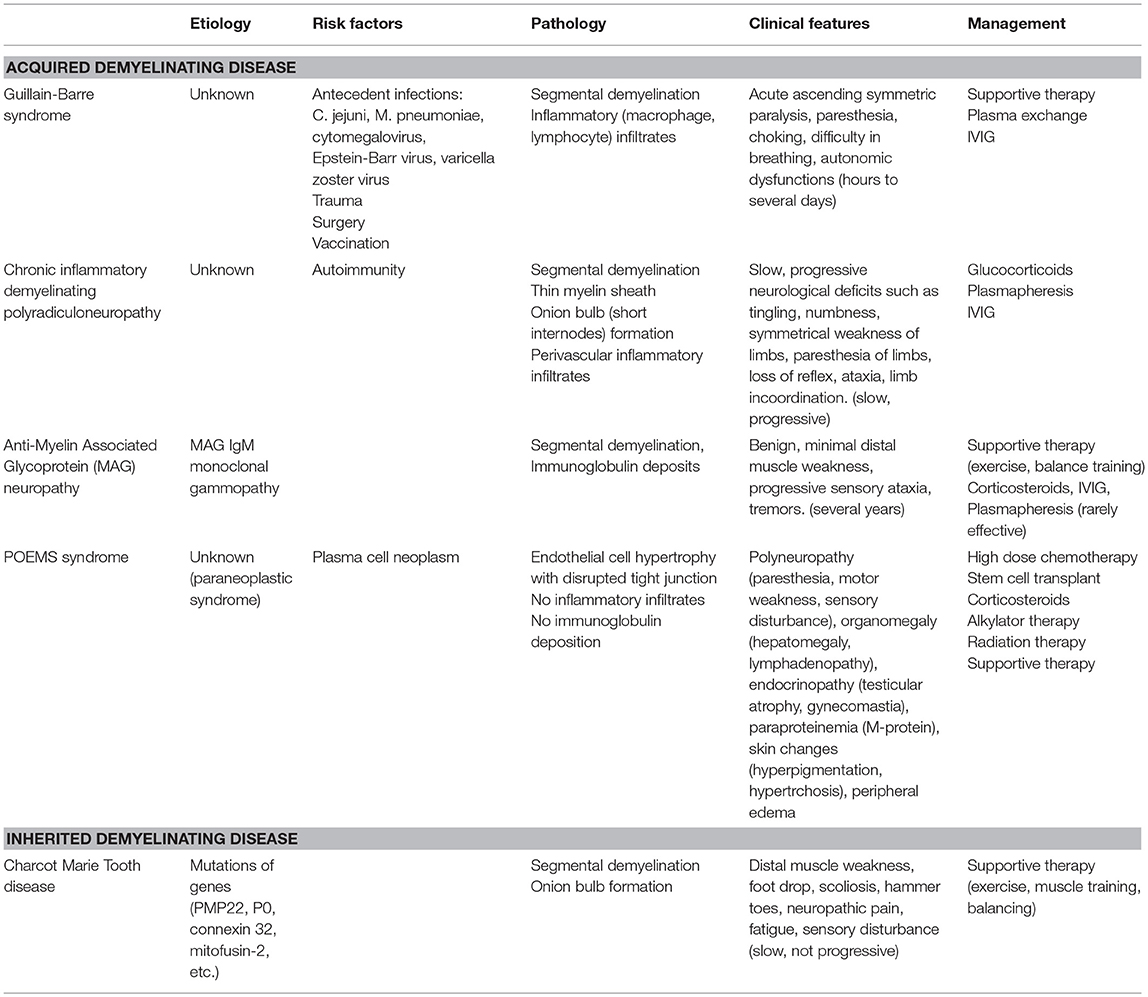
Table 1. Pathogenesis, clinical features and management of various types of Peripheral Demyelinating diseases.
According to the National Institute of Health Biomarkers Definitions Working Group, biological marker (biomarker) is defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention” (60). Biomarkers serve as an important clinical tool in disease diagnosis, therapeutic response and prognosis (60).
To identify the recent progress in biomarkers of PDD, we have conducted a literature search from SCOPUS search engine and database on biomarkers of distinct PDD from the year 2014–2018. Only original articles and in the English language were selected to be included in the review (Table 2).

Table 2. Recent biomarkers of Peripheral Demyelinating diseases.
Traditionally, the diagnosis of GBS has relied on clinical features such as electrodiagnostic studies and CSF analysis. Testing for serum IgG antibodies to gangliosides Q1b (GQ1b) is available and useful for the diagnosis of GBS variants (Miller Fisher syndrome) with 85 to 90 percent sensitivity although it is not routinely indicated1. Biomarkers for GBS have negligible clinical value, low sensitivity/specificity and costly. Furthermore, the laboratory measurement standard for these biomarkers also has not been established and studies using different methods have been plagued by inconsistent findings (71).
At present, novel biomarkers are being explored for better prognosis of GBS patients. The likes of neutrophil-lymphocyte ratio (NLR) and platelet-lymphocyte ratio (PLR) has received much attention as novel prognostic biomarkers of inflammation. Analysis of NLR and PLR levels of 62 GBS patients prior to-and following intravenous immunoglobulin treatment (IVIG) revealed NLR as a better prediction tool for the acute period of AIDP (major variants of GBS) with 83% sensitivity and 93% specificity. Whereas, PLR only showed 74% sensitivity and 70% specificity (61). NLR also studied for its correlation with the degree of weakness in several muscles assessed through the Medical Research Council (MRC) score (62). High levels of NLR were seen in the lower MRC score upon admission and increased baseline disability among GBS patients (62).
More recent findings indicate monocyte-lymphocyte ratio (MLR) along NLR as a better prognostic marker for GBS (63). NLR and MLR are significantly higher in GBS patients compared to healthy controls. Moreover, NLR and MLR are found to be tremendously increased in severe group (63). On the other hand, Piccolo, a multidomain zinc finger protein that is involved in synaptic active zones and synaptic vesicle trafficking was shown to present in sera of GBS patients. High serological levels of Piccolo were associated with better outcomes in GBS patients (65).
Ganglionic nicotinic acetylcholine receptors (gAChR) are nicotinic receptors that assist synaptic transmission in peripheral autonomic ganglia. Over the last decades, the autoantibodies toward gAChR have been associated with autoimmune dysautonomia (72). Several GBS patients experience autonomic dysfunctions such as cardiac arrhythmia and urinary retention (31). Detection of autoantibodies against gAChR could measure the risk of developing debilitating autonomic dysfunction. High level of α3 or β4 subunits of gAChR was detected in 13.6% of GBS patients with autonomic symptoms through luciferase immunoprecipitation (66).
Proteomic analysis of the CSF was used to isolate potential biomarker target specific to CIDP (73). Unfortunately, the disease specificity of the identified protein was low. The captured proteins include transferrin, proapolipoprotein, retinal binding protein and transthyretin (73). Several autoantibodies, including NF155, CNTN1, and contactin-1/contactin-associated protein 1 (CNTN1/CASPR1) complex also have been associated with the pathogenesis of CIDP (67).
Autoantibodies against ganglioside antibodies, myelin protein (P0, PMP22), nodal proteins (NF155, CNTN1, CNTN1/CASPR1, CNTN2/CASPR2 complex, neuronal cell adhesion molecule (NCAM)), gliomedin and two subunits of sodium channel at nodes of Ranvier (NavB1, NavB2) have been investigated in CIDP patients. Among these patients, 11 expressed anti-ganglioside antibody reactivity (anti-GM1, anti-GD1b), 4 reacted to CNTN1 (6.2%), 3 reacted to NF155 (6.2%), 1 against CNTN1/CASPR1 complex (1.5%) and 1 against PMP22 (67).
Current literature also associates reduced sialylation of IgG-Fc with increased clinical severity of CIDP. Sialylation and galactosylation of IgG-Fc were significantly lower in CIDP patients. Treatment with IVIG, on the other hand, increased the levels of sialylated IgG-Fc and concurrently attenuated the disease severity (68).
The diagnosis of Anti-MAG Neuropathy is through the detection of autoantibodies against MAG. Early findings from Latov's laboratory detected anti-MAG IgM in the sera of almost more than half of the anti-MAG patients (74). Some anti-MAG IgM also co-reacted with acidic glycolipid in the ganglioside fraction of the peripheral nerves, GM1, GD1a and were identified as sulfoglucuronyl glycosphingolipid (SGPG) (69). In clinically indistinguishable anti-MAG neuropathy without seropositivity of MAG and SGPG, the IgM may react to gangliosides such as GD1b, GT1b, and GQ1b (75, 76). Anti-MAG titers could also be correlated with prognosis of the neuropathy. High baseline and increasing anti-MAG titer correlated with higher chances of recurrence (77).
VEGF, an angiogenic factor, is markedly elevated in POEMS syndrome patients and also referred as one of the major criteria for diagnosis of POEMS syndrome (50). Clinical improvement and prolonged relapse-free survival were reported among patients with normalized serum level of VEGF (78).
Diagnostic markers for CMT disease are usually determined through genetic testing. Once the suspicion of CMT disease is made through clinical judgement and electrophysiological studies.
Diagnosis is confirmed through testing for mutations on PMP22 (CMT1A, most common), GJB1 (CMTX1), P0 (CMT1B), and MFN2 (CMT2A).
During demyelination, various components of myelin and axon are being released as a result of the damage toward the Schwann cells. Myelin sheath consists of approximately 70 % of lipids such as gangliosides, phospholipids, sphingomyelin and 30% of proteins such as myelin [P0, PMP22, myelin protein 2 (P2)] and nodal proteins (neurofascin, gliomedin, contactin) (Figure 2) (Table 3) (13). These components can be detected in CSF, serum and even peripheral nerve biopsies (71), which can potentially indicate the degree of demyelination during the disease progress and also the efficacy of the treatment as reflected by the degree of remyelination.
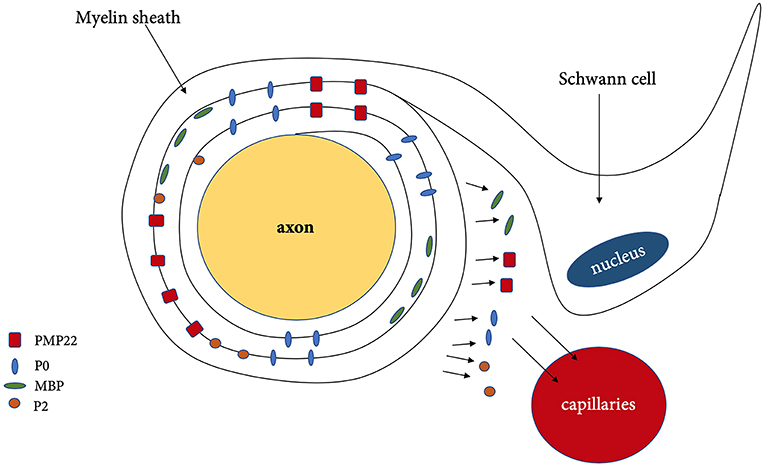
Figure 2. Demyelination and released of myelin associated protein. PMP22, peripheral myelin protein 22; P0, myelin protein zero; MBP, myelin basic protein; P2, myelin protein 2.
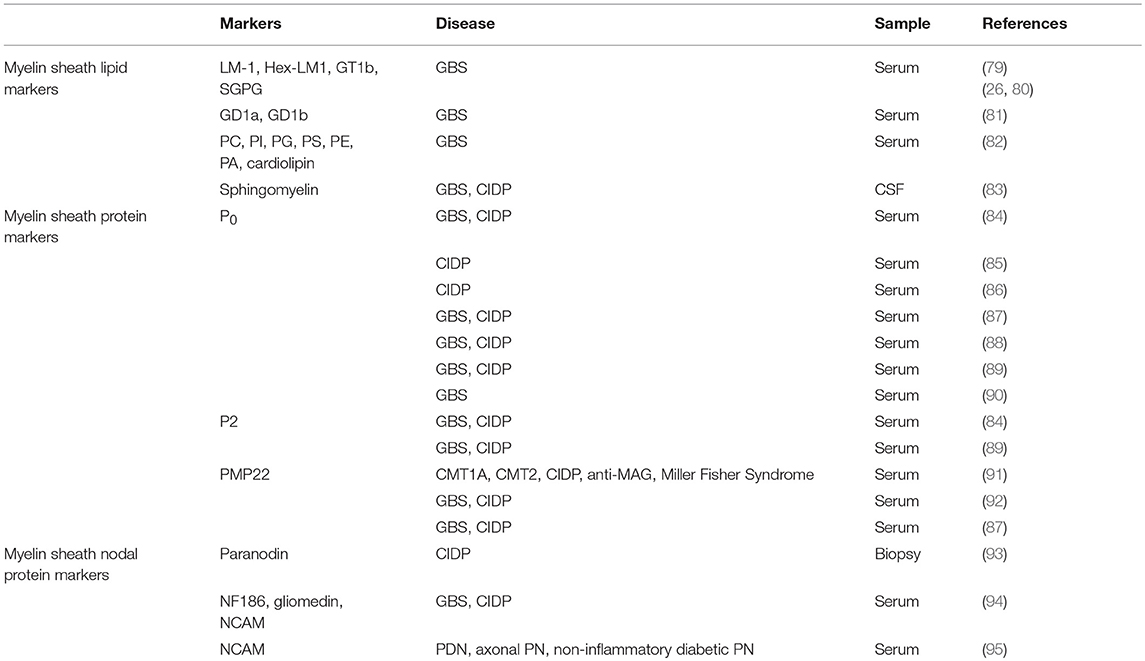
Table 3. List of myelin associated biomarkers.
Glycolipids such as gangliosides are one of the major lipid components of the myelin sheath. Situated in the plasma membrane with the hydrophilic carbohydrate moiety exposed extracellularly, gangliosides have a greater propensity for autoimmune reaction, especially in GBS, CIDP, and anti- MAG neuropathy (26). Thus, detection of antibody towards gangliosides such as anti-LM1, anti-Hex-LM1, anti-GT1b, anti-SGPG, anti-galactocerebroside in some GBS patients could serve as a diagnostic tool. In parallel to this, the presence of antibodies towards ganglioside complex (GD1a/GD1b, GD1b/GT1b) in serum has been associated with better prognosis of GBS in terms of disease severity (81).
Association of autoimmune diseases such as systematic lupus erythematosus, scleroderma with GBS (96) led researchers (Nakos et al) to ascertain the role of few antiphospholipid antibodies which include phosphatidic acid (PA), cardiolipin, phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidylserine (PS), phosphatidylinositol (PI), phosphatidylglyecerol (PG), and cardiolipin. The antibody levels were shown to decrease following 1-day treatment with γ-globulin (IVIG) and increased 2 days following cessation of treatment (82). This indicates that serum levels of anti-PI and anti-cardiolipin antibodies may be useful to monitor the response of the patient toward treatment with IVIG in GBS patients (82).
Sphingomyelin, another myelin-enriched lipid was reported to be significantly higher among PDD patients (GBS and CIDP) (83). The ability to distinguish between demyelinating variant from axonal variant supports the connotation that sphingomyelin is specific biomarker for peripheral myelin breakdown. In addition, the techniques used to detect and quantify sphingomyelin were also reliable, cost-effective with good sensitivity and specificity (83).
Although the lipid/protein ratio and lipid constituents of myelin sheath in both CNS and PNS are similar, the distribution and type of myelin proteins in PNS are different. Myelin sheath consists of two compartments, compact myelin (dense area around axon) and non-compact myelin (Figure 3). The compact myelin consists of intraperiod line and the major dense line (MDL). The myelin- specific protein in PNS includes P0, PMP22, and P2.
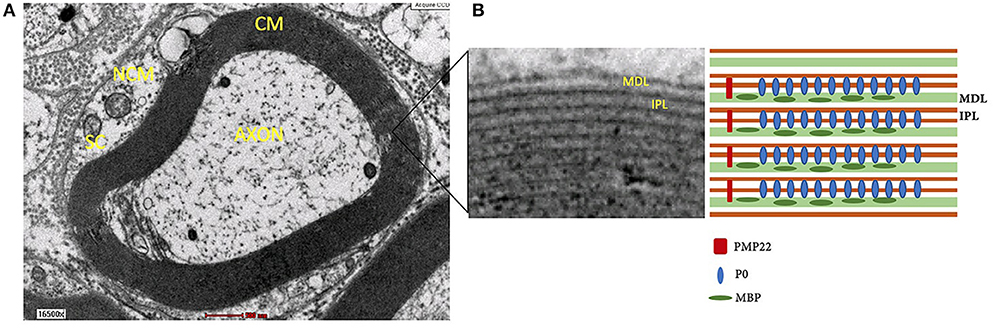
Figure 3. (A) Structure of myelin sheath of Schwann cell around axon under electron microscope. (B) Compact myelin that consist of major dense line and intraperiod line. NCM, non-compact myelin; CM, compact myelin; SC, Schwann cell; MDL, major dense line; IPL, intraperiod line; P0, myelin protein zero; MBP, myelin basic protein; PMP22, peripheral myelin protein 22.
P0 is a transmembrane glycoprotein that stabilizes the intraperiod line through homophilic binding to another P0 protein (97). Knockout P0 mice were shown to undergo severe hypomyelination and also demonstrated thin, non-compacted myelin sheath with axonal degeneration. In addition, the mice also exhibited tremors, convulsion and deficits in motor coordination (98). P2 protein, also participate in fusion of the MDL in compact myelin (99). PMP22 is a transmembrane protein (100, 101) that is synthesized by Schwann cells and makes up 2–5% myelin protein (102).
Myriad studies have reported the development of antibodies toward P0, PMP22, and P2, especially among GBS and CIDP patients (84, 86–88, 92). The antibodies toward P0 were only present in small proportion of patients with GBS and CIDP and therefore were not useful as a diagnostic test (84). Antibodies toward P0 were only detected in 22% of CIDP and 19% of GBS patient (88). Other studies also reported lower levels of P0 antibodies using various techniques including ELISA, Immunoblot, and Western blot (85, 86, 89). In contrary, some studies reported absence of immune response or no significant response toward P0 (87, 90).
Compared to P0 antibodies, detection of P2 antibodies was reported to be more common in some cases of GBS and CIDP (84). Subsequent studies revealed trace level of IgG to anti-P2 in GBS patients during the peak stage of the disease. The detected trace level of P2 in most patients with GBS and CIDP across all stages of the diseases were within the same range as in the control group (84, 89). Therefore, P2 is not sensitive and specific biomarkers for demyelinating diseases.
PMP22 was detected in 70% of CMT1 and 60% of CMT2 cases. Surprisingly, no significant difference in the immune response of PMP22 from healthy donors and patients with acquired neuropathies (91). Similarly, another study also reported antibodies against PMP22 in 52% of GBS and 35% of CIDP patients (92). Contrary to these findings, absence of an immune response to PMP22 P0 and Cx32 proteins were reported in GBS and CIDP patients (87). These discrepancies were probably due to different usage of PMP22 antigen and also different patient groups that might reflect distinct immunoreactivity toward PMP22.
In a myelinated fiber, there are multiple internodes of Schwann cells, which are separated by the node of Ranvier. A detailed and magnified look into the boundaries of the node of Ranvier with Schwann cell internode further shows detailed compartment that viewed the node, paranode and juxtraparanode (Figure 4). Nodal proteins such as contactin, neurofascin and NCAM were identified at these regions and they took part in the formation of the septate-like junction that closely in contact with axons (67).
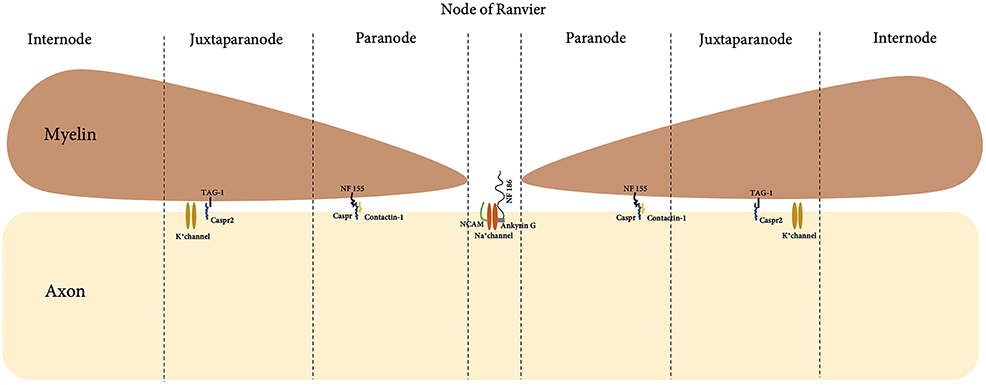
Figure 4. Anatomical compartment of the node of Ranvier and myelin.
Abnormalities of the nodal proteins were reported in the pathogenesis of CIDP (103). Paranodin, a nodal protein involved in axoglial contacts was tested through immunostaining of biopsies from patients with CIDP. Evaluation of positively-stained paranodin biopsies led to the correct diagnosis of CIDP in 70% of the reported cases (93). Further laboratory investigations revealed IgG autoimmunity towards myelin nodal proteins, neurofascin-186 (NF186), gliomedin, contactin and NCAM in 43% of GBS (n = 100) and 30% of CIDP (n = 50) patients (94). In addition, passive transfer of anti-gliomedin IgG to Lewis rat induced progressive neuropathy characterized by conduction defect and demyelination of spinal nerves (104). This has led the authors to suggest that myelin nodal proteins may play a role in induction of demyelination. Furthermore, clinical remission seen in these animals was parallel with the gradual decrease of IgG titers (94), suggesting that levels of antibody titers toward gliomedin could serve as a biomarker for disease remission.
Another study by Niezgoda et al. compared the level of serum NCAM in peripheral demyelinating neuropathy (PDN), axonal polyneuropathy (PN), non-inflammatory diabetic PN and healthy controls. In the study, Overall Neuropathy Limitation Scale (ONLS) and electrophysiological analysis comprises of motor and sensory studies were employed for clinical assessment. Significant increase in NCAM was seen among PDN group (n = 40 GBS, 29 CIDP, 11 Multifocal Motor Neuropathy) compared to other groups. In patients with PDN but not PN and non-inflammatory diabetic PN, serum NCAM levels had a high positive correlation to ONLS and negative correlation to motor conduction velocity. Thus, it has been concluded that NCAM detection could serve as a specific marker for peripheral nerve immune-mediated demyelination and sensitive marker for peripheral nerve involvement (95).
Poor prognosis remained a dilemma in the management of the PDDs. Many people remain underdiagnosed or diagnosed when disease is already advanced. Moreover, the biomarkers that currently being used develop late in the disease process. Therefore, understanding the physiology of myelination and SC biology is vital to help further delineate the mechanisms involved in the pathogenesis of PDD. Autoantibodies against several types of gangliosides, phospholipids, glycoproteins, and nodal proteins have been shown to be present in numerous PDDs. However, their serum levels are yet to be correlated with a clinical course, and prognosis. A reliable biomarker should be sensitive and specific. Can peripheral autoantibodies that develop against the myelination-associated proteins or lipids in the early stage of PDDs be appropriate biomarker candidates? Future studies should explore this premise to discover time-sensitive biomarkers for early detection of PDDs.
KK and JK performed the literature search and drafted the manuscript. MY, RI, and SD reviewed and finalized the manuscript. The figures were designed by KK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by Universiti Kebangsaan Malaysia (UKM) research grant, FF-2017-486. The author would like to thank Tissue Engineering Center, UKM for electron microscope image (Figure 3).
PDD, Peripheral Demyelinating Disease; SC, Schwann Cell; PNS, Peripheral Nervous System; CNS, Central Nervous System; P0, Protein Zero; PMP22, Peripheral Myelin Protein 22; MBP, Myelin basic protein; CSF, Cerebrospinal fluid; GBS, Guillain Barre Syndrome; GM-1, Monosialotetrahexosylganglioside-1; GD1a, Ganglioside GD1a; EBV, Epstein Barr virus. AIDP, Acute inflammatory demyelinating polyradiculopathy; AMAN, Acute motor axonal neuropathy; AMSAN, Acute motor sensory axonal neuropathy; LOS, Lipooligosaccharides; IVIG, Intravenous immunoglobulin; CIDP, Chronic inflammatory demyelinating polyradiculoneuropathy; NF155, Neurofascin-155; CNTN1, Contactin-1; MAG, Myelin Associated Glycoprotein; VEGF, Vascular endothelial growth factor; CMT, Charcot Marie Tooth; GDAP1, Ganglioside-induced differentiation-associated protein 1; MFN2, Mitofusin-2; GQ1b, Gangliosides Q1b; NLR, Neutrophil-lymphocyte ratio; PLR, Platelet-lymphocyte ratio; MRC, Medical Research Council; MLR, Monocyte-lymphocyte ratio; gAChR, Ganglionic nicotinic acetylcholine receptor; CNTN1/CASPR1, Contactin-1/contactin-associated protein-1; NCAM, Neuronal cell adhesion molecule; NavB1/2, Sodium channel at node of Ranvier ½; SGPG, Sulfoglucuronyl glycosphingolipid; ELISA, Enzyme-linked immunosorbent assay; P2, Protein 2; PC, phosphatidylcholine; PI, phosphatidylinositol; PG, phosphatidylglycerol; PS, phosphatidylserine; PE, phosphatidylethanolamine; PA, phosphatidic acid; MDL, Major denseline; PDN, Peripheral Demyelinating neuropathy; PN, Polyneuropathy; GJB1, Gap junction beta 1.
1. ^Francine J Vriesendorp, MD Guillain-Barré syndrome in adults: Clinical features and diagnosis Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. http://www.uptodate.com (Accessed on May 09, 2018).
1. Mehndiratta M, Gulati N. Central and peripheral demyelination. J Neurosci Rural Pract. (2014) 5:84. doi: 10.4103/0976-3147.127887
2. Sejvar JJ, Baughman AL, Wise M, Morgan OW. Population incidence of Guillain-Barré syndrome: a systematic review and meta-analysis. Neuroepidemiology. (2011) 36:123–33. doi: 10.1159/000324710
3. Hafsteinsdottir B, Olafsson E. Incidence and natural history of idiopathic chronic inflammatory demyelinating polyneuropathy: a population-based study in Iceland. Eur Neurol. (2016) 75:263–8. doi: 10.1159/000445884
4. Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol. (2011) 69:22–33. doi: 10.1002/ana.22166
5. Yuki N. Molecular mimicry and Guillain-Barré syndrome. Brain Nerve. (2015) 67:1341–6. doi: 10.11477/mf.1416200304
6. Soliven B. Chronic inflammatory demyelinating polyradiculoneuropathy In: Roos RP, Editor-in-Chief. MedLink Neurology. San Diego, CA: MedLink Corporation. Available online at: www.medlink.com (Accessed July 24, 2017).
7. Mathis S, Magy L, Vallat JM. Therapeutic options in Charcot–Marie–Tooth diseases. Expert Rev Neurother. (2015) 15:355–66. doi: 10.1586/14737175.2015.1017471
8. Kumar J, Teoh SL, Das S, Mahakknaukrauh P. Oxidative stress in oral diseases: understanding its relation with other systemic diseases. Front Physiol. (2017) 8:693. doi: 10.3389/fphys.2017.00693
9. O'sullivan SA, Velasco-Estevez M, Dev KK. Demyelination induced by oxidative stress is regulated by sphingosine 1-phosphate receptors. Glia. (2017) 65:1119–36. doi: 10.1002/glia.23148
10. Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. (2005) 6:671–82. doi: 10.1038/nrn1746
11. Salzer JL. Schwann cell myelination. Cold Spring Harb Perspect Biol. (2015) 7:a020529. doi: 10.1101/cshperspect.a020529
12. Woodhoo A, Sommer L. Development of the Schwann cell lineage: from the neural crest to the myelinated nerve. Glia. (2008) 56:1481–90. doi: 10.1002/glia.20723
13. Morell P, Quarles RH. Myelin formation, structure and biochemistry. In: Siegel GJ, editor. Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed. Lippincott-Raven (1999).
14. Stangel M, Hartung HP. Remyelinating strategies for the treatment of multiple sclerosis. Prog Neurobiol. (2002) 68:361–76. doi: 10.1016/S0301-0082(02)00105-3
16. Willison HJ, Jacobs BC, Van Doorn PA. Guillain-barre syndrome. Lancet. (2016) 388:717–27. doi: 10.1016/S0140-6736(16)00339-1
17. Sejvar JJ, Kohl KS, Gidudu J, Amato A, Bakshi N, Baxter R, et al. Guillain-Barré syndrome and Fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine. (2011) 29:599–612. doi: 10.1016/j.vaccine.2010.06.003
18. Jasti AK, Selmi C, Sarmiento-Monroy JC, Vega DA, Anaya JM, Gershwin ME. Guillain-Barré syndrome: causes, immunopathogenic mechanisms and treatment. Expert Rev Clin Immunol. (2016) 12:1175–89. doi: 10.1080/1744666X.2016.1193006
20. Lehmann HC, Hughes RA, Kieseier BC, Hartung HP. Recent developments and future directions in Guillain-Barré syndrome. J Peripher Nerv Syst. (2012) 17:57–70. doi: 10.1111/j.1529-8027.2012.00433.x
21. Meyer Sauteur PM, Huizinga R, Tio-Gillen AP, Roodbol J, Hoogenboezem T, Jacobs E, et al. Mycoplasma pneumoniae triggering the Guillain-Barré syndrome: a case-control study. Ann Neurol. (2016) 80:566–580. doi: 10.1002/ana.24755
22. Orlikowski D, Porcher R, Sivadon-Tardy V, Quincampoix JC, Raphaël JC, Durand MC, et al. Guillain–Barré syndrome following primary cytomegalovirus infection: a prospective cohort study. Clin Infect Dis. (2011) 52:837–44. doi: 10.1093/cid/cir074
23. Islam B, Islam Z, GeurtsvanKessel CH, Jahan I, Endtz HP, Mohammad QD, et al. Guillain-Barré syndrome following varicella-zoster virus infection. Eur J Clin Microbiol Infect Dis. (2018) 37:511–8. doi: 10.1007/s10096-018-3199-5
24. Tam CC, O'Brien SJ, Petersen I, Islam A, Hayward A, Rodrigues LC. Guillain-Barré syndrome and preceding infection with campylobacter, influenza and Epstein-Barr virus in the general practice research database. PLoS ONE. (2007) 2:e344. doi: 10.1371/journal.pone.0000344
25. Willison HJ, O'Hanlon GM. The immunopathogenesis of Miller Fisher syndrome. J Neuroimmunol. (1999) 100:3–12. doi: 10.1016/S0165-5728(99)00213-1
26. Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. (2002) 125:2591–625. doi: 10.1093/brain/awf272
27. Nakano Y, Kanda T. Pathology of Guillain–barré syndrome. Clin Exp Neuroimmunol. (2016) 7:312–9. doi: 10.1111/cen3.12342
28. Raschetti R, Maggini M, Popoli P, Caffari B, Da Cas R, Menniti-Ippolito F, et al. Gangliosides and Guillain-Barré syndrome. J Clin Epidemiol. (1995) 48:1399–405. doi: 10.1016/0895-4356(95)00557-9
29. Lonigro A, Devaux J. Disruption of neurofascin and gliomedin at nodes of Ranvier precedes demyelination in experimental allergic neuritis. Brain. (2009) 132:260–73. doi: 10.1093/brain/awn281
30. Durand MC, Porcher R, Orlikowski D, Aboab J, Devaux C, Clair B, et al. Clinical and electrophysiological predictors of respiratory failure in Guillain-Barré syndrome: a prospective study. Lancet Neurol. (2006) 5:1021–8. doi: 10.1016/S1474-4422(06)70603-2
31. Flachenecker P. Autonomic dysfunction in Guillain-Barré syndrome and multiple sclerosis. J Neurol. (2007) 254:II96–II101. doi: 10.1007/s00415-007-2024-3
32. Yuki N, Hartung HP. Guillain–Barré syndrome. N Engl J Med. (2012) 366:2294–304. doi: 10.1056/NEJMra1114525
33. Van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. (2008) 7:939–50. doi: 10.1016/S1474-4422(08)70215-1
34. González-Suárez I, Sanz-Gallego I, de Rivera FJR, Arpa J. Guillain-Barré syndrome: natural history and prognostic factors: a retrospective review of 106 cases. BMC Neurol. (2013) 13:95. doi: 10.1186/1471-2377-13-95
35. Ansar V, Valadi N. Guillain-Barré syndrome. Primary Care Clin Office Pract. (2015) 42:189–93. doi: 10.1016/j.pop.2015.01.001
36. Lewis RA. Chronic inflammatory demyelinating polyneuropathy. Neurol Clin. (2007) 25:71–87. doi: 10.1016/j.ncl.2006.11.003
37. Köller H, Kieseier BC, Jander S, Hartung HP. Chronic inflammatory demyelinating polyneuropathy. N Engl J Med. (2005) 352:1343–56. doi: 10.1056/NEJMra041347
38. Peltier AC, Donofrio PD. Chronic inflammatory demyelinating polyradiculoneuropathy: from bench to bedside. Semin Neurol. (2012) 32:187–95. doi: 10.1055/s-0032-1329194
39. Laughlin RS, Dyck PJ, Melton LJ III, Leibson C, Ransom J, Dyck PJ. Incidence and prevalence of CIDP and the association of diabetes mellitus. Neurology. (2009) 73:39–45 doi: 10.1212/WNL.0b013e3181aaea47
40. Miura Y, Devaux JJ, Fukami Y, Manso C, Belghazi M, Wong AHY, et al. Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain. (2015) 138:1484–91. doi: 10.1093/brain/awv054
41. Devaux JJ, Miura Y, Fukami Y, Inoue T, Manso C, Belghazi M, et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology. (2016) 10:1212. doi: 10.1212/WNL.0000000000002418
42. Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. (2001) 30:385–97. doi: 10.1016/S0896-6273(01)00296-3
43. Dyck PJ, O'Brien PC, Oviatt KF, Dinapoli RP, Daube JR, Bartleson JD, et al. Prednisone improves chronic inflammatory demyelinating polyradiculoneuropathy more than no treatment. Ann Neurol. (1982) 11:136–41. doi: 10.1002/ana.410110205
44. Herrendorff R, Hänggi P, Pfister H, Yang F, Demeestere D, Hunziker F, et al. Selective in vivo removal of pathogenic anti-MAG autoantibodies, an antigen- specific treatment option for anti-MAG neuropathy. Proc Natl Acad Sci USA. (2017) 114:E3689–98. doi: 10.1073/pnas.1619386114
45. Quarles RH. Myelin-associated glycoprotein (MAG): past, present and beyond. J Neurochem. (2007) 100:1431–48. doi: 10.1111/j.1471-4159.2006.04319.x
46. Tatum AH. Experimental paraprotein neuropathy, demyelination by passive transfer of human IgM anti-myelin-associated glycoprotein. Ann Neurol. (1993) 33:502–6. doi: 10.1002/ana.410330514
47. Rison RA, Beydoun SR. Paraproteinemic neuropathy: a practical review. BMC Neurol. (2016) 16:13. doi: 10.1186/s12883-016-0532-4
48. Dalakas MC. Pathogenesis and treatment of anti-MAG neuropathy. Curr Treat Opt Neurol. (2010) 12:71–83. doi: 10.1007/s11940-010-0065-x
49. Dispenzieri A, Gertz MA. Treatment options for POEMS syndrome. Expert Opin Pharmacother. (2005) 6:945–53. doi: 10.1517/14656566.6.6.945
50. Dispenzieri A. POEMS syndrome: update on diagnosis, risk-stratification, and management. Am J Hematol. (2015) 90:951–62. doi: 10.1002/ajh.24171
51. Scarlato M, Previtali SC, Carpo M, Pareyson D, Briani C, Del Bo R, et al. Polyneuropathy in POEMS syndrome: role of angiogenic factors in the pathogenesis. Brain. (2005) 128:1911–20. doi: 10.1093/brain/awh519
52. Soubrier MJ, Dubost JJ, Sauvezie BJ. POEMS syndrome: a study of 25 cases and a review of the literature. Am. J. Med. (1994) 97:543–53. doi: 10.1016/0002-9343(94)90350-6
53. Jani-Acsadi A, Krajewski K, Shy ME. Charcot-Marie-Tooth neuropathies: diagnosis and management. Semin Neurol. (2008) 28:185–94. doi: 10.1055/s-2008-1062264
54. Patzkó Á, Shy ME. Update on Charcot-Marie-tooth disease. Curr Neurol Neurosci Rep. (2011) 11:78–88. doi: 10.1007/s11910-010-0158-7
55. DiVincenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, et al. The allelic spectrum of Charcot–Marie–Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med. (2014) 2:522–9. doi: 10.1002/mgg3.106
56. Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogenduk JE, Baas F, et al. Duplication in chromosome 17p11. 2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). Neuromuscular Disord. (1991) 1:93–7. doi: 10.1016/0960-8966(91)90055-W
57. Pennuto M, Tinelli E, Malaguti M, Del Carro U, D'Antonio M, Ron D, et al. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. (2008) 57:393–405. doi: 10.1016/j.neuron.2007.12.021
58. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. (2008) 456:605. doi: 10.1038/nature07534
59. Kamil K, Kumar J, Yazid MD, Idrus RBH. Olive and its phenolic compound as the promising neuroprotective agent. Sains Malays. (2018) 47:2811–20. doi: 10.17576/jsm-2018-4711-24
60. Biomarkers Definitions Working Group, Atkinson Jr AJ, Colburn WA, DeGruttola VG, DeMets DL, Downing GJ, et al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Therap. (2001) 69:89–95. doi: 10.1067/mcp.2001.113989
61. Ozdemir HH. Analysis of the albumin level, neutrophil-lymphocyte ratio, and platelet-lymphocyte ratio in Guillain-Barré syndrome. Arq Neuropsiquiatr. (2016) 74:718–22. doi: 10.1590/0004-282X20160132
62. Sahin S, Cinar N, Karsidag S. Are cerebrospinal fluid protein levels and plasma neutrophil/lymphocyte ratio associated with prognosis of Guillain Barré syndrome. Neurol. Int. (2017) 9:7032. doi: 10.4081/ni.2017.7032
63. Huang Y, Ying Z, Quan W, Xiang W, Xie D, and Weng Y, et al. The clinical significance of neutrophil-to-lymphocyte ratio and monocyte-to-lymphocyte ratio in Guillain–Barré syndrome. Int J Neurosci. (2018) 128:729–35. doi: 10.1080/00207454.2017.1418342
64. Yamagishi Y, Suzuki H, Sonoo M, Kuwabara S, Yokota T, Nomura K, et al. Markers for Guillain-Barré syndrome with poor prognosis: a multi-center study. J Peripher Nerv Syst. (2017) 22:433–9. doi: 10.1111/jns.12234
65. Mateos-Hernández L, Villar M, Doncel-Pérez E, Trevisan-Herraz M, García-Forcada Á, Ganuza FR, et al. Quantitative proteomics reveals Piccolo as a candidate serological correlate of recovery from Guillain-Barré syndrome. Oncotarget. (2016) 7:74582. doi: 10.18632/oncotarget.12789
66. Nakane S, Higuchi O, Hamada Y, Maeda Y, Mukaino A, Sakai W, et al. Ganglionic acetylcholine receptor autoantibodies in patients with Guillain-Barre syndrome. J Neuroimmunol. (2016) 295:54–9. doi: 10.1016/j.jneuroim.2016.04.012
67. Querol L, Siles AM, Alba-Rovira R, Jáuregui A, Devaux J, Faivre-Sarrailh C, et al. Antibodies against peripheral nerve antigens in chronic inflammatory demyelinating polyradiculoneuropathy. Sci Rep. (2017) 7:14411. doi: 10.1038/s41598-017-14853-4
68. Wong AHY, Fukami Y, Sudo M, Kokubun N, Hamada S, Yuki N. Sialylated IgG-Fc: a novel biomarker of chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatr. (2015) 87:275–9. doi: 10.1136/jnnp-2014-309964
69. Dalakas MC. Advances in the diagnosis, immunopathogenesis and therapies of IgM-anti-MAG antibody-mediated neuropathies. Ther Adv Neurol Disord. (2018) 11:1756285617746640. doi: 10.1177/1756285617746640
70. Altinier S, Proko K, Zaninotto M, Ciubotaru D, Seguso M, Varagnolo M, et al. Free light chains and heavy/light chains in monitoring POEMS patients. Clin Chem Lab Med. (2016) 54:1065–71. doi: 10.1515/cclm-2015-0910
71. Wang Y, Sun S, Zhu J, Cui L, Zhang HL. Biomarkers of Guillain-Barré syndrome: some recent progress, more still to be explored. Mediators Inflamm. (2015) 2015:564098. doi: 10.1155/2015/564098
72. Vernino S, Low PA, Fealey RD, Stewart JD, Farrugia G, Lennon VA. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. (2000) 343:847–55. doi: 10.1056/NEJM200009213431204
73. Tumani H, Pfeifle M, Lehmensiek V, Rau D, Mogel H, Ludolph AC, et al. Candidate biomarkers of chronic inflammatory demyelinating polyneuropathy (CIDP): proteome analysis of cerebrospinal fluid. J Neuroimmunol. (2009) 214:109–12. doi: 10.1016/j.jneuroim.2009.06.012
74. Latov N, Braun PE, Gross RB, Sherman WH, Penn AS, Chess L. Plasma cell dyscrasia and peripheral neuropathy: identification of the myelin antigens that react with human paraproteins. Proc Natl Acad Sci USA. (1981) 78:7139–42. doi: 10.1073/pnas.78.11.7139
75. Daune GC, Farrer RG, Dalakas MC, Quarles RH. Sensory neuropathy associated with monoclonal immunoglobulin M to GD1b ganglioside. Ann Neurol. (1992) 31:683–5. doi: 10.1002/ana.410310621
76. Quarles RH, Dalakas MC. Do anti-ganglioside antibodies cause human peripheral neuropathies? J Clin Invest. (1996) 97:1136–7. doi: 10.1172/JCI118526
77. Galassi G, Tondelli M, Ariatti A, Benuzzi F, Nichelli P, Valzania F. Long-term disability and prognostic factors in polyneuropathy associated with anti-myelin- associated glycoprotein (MAG) antibodies. Int J Neurosci. (2017) 127:439–47. doi: 10.1080/00207454.2016.1191013
78. Misawa S, Sato Y, Katayama K, Hanaoka H, Sawai S, Beppu M, et al. Vascular endothelial growth factor as a predictive marker for POEMS syndrome treatment response: retrospective cohort study. BMJ Open. (2015) 5:e009157. doi: 10.1136/bmjopen-2015-009157
79. Ilyas AA, Willison HJ, Quarles RH, Jungalwala FB, Cornblath DR, Trapp BD, et al. Serum antibodies to gangliosides in Guillain-Barré syndrome. Ann Neurol. (1988) 23:440–7. doi: 10.1002/ana.410230503
80. Ilyas AA, Mithen FA, Dalakas MC, Chen ZW, Cook SD. Antibodies to acidic glycolipids in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Sci. (1992) 107:111–21. doi: 10.1016/0022-510X(92)90217-9
81. Kaida K, Morita D, Kanzaki M, Kamakura K, Motoyoshi K, Hirakawa M. et al. Anti-ganglioside complex antibodies associated with severe disability in GBS. J Neuroimmunol. (2007) 182:212–8. doi: 10.1016/j.jneuroim.2006.09.013
82. Nakos G, Tziakou E, Maneta-Peyret L, Nassis C, Lekka ME. Anti- phospholipid antibodies in serum from patients with Guillain-Barré syndrome. Intensive Care Med. (2005) 31:1401–8. doi: 10.1007/s00134-005-2736-8
83. Capodivento G, Visigalli D, Garnero M, Fancellu R, Ferrara MD, Basit A, et al. Sphingomyelin as a myelin biomarker in CSF of acquired demyelinating neuropathies. Sci Rep. (2017) 7:7831. doi: 10.1038/s41598-017-08314-1
84. Khalili-Shirazi A, Atkinson P, Gregson N, Hughes RAC. Antibody responses to P0 and P2 myelin proteins in Guillain-Barré syndrome and chronic idiopathic demyelinating polyradiculoneuropathy. J Neuroimmunol. (1993) 46:245–51. doi: 10.1016/0165-5728(93)90255-W
85. Melendez-Vasquez C, Redford J, Choudhary PP, Gray IA, Maitland P, Gregson NA, et al. Immunological investigation of chronic inflammatory demyelinating polyradiculoneuropathy. J Neuroimmunol. (1997) 73:124–34. doi: 10.1016/S0165-5728(96)00189-0
86. Yan WX, Archelos JJ, Hartung HP, Pollard JD. P0 protein is a target antigen in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. (2001) 50:286–92. doi: 10.1002/ana.1129
87. Kwa MSG, Van Schaik IN, Brand A, Baas F, Vermeulen M. Investigation of serum response to PMP22, connexin 32 and P0 in inflammatory neuropathies. J Neuroimmunol. (2001) 116:220–5. doi: 10.1016/S0165-5728(01)00307-1
88. Allen D, Giannopoulos K, Gray I, Gregson N, Makowska A, Pritchard J, et al. Antibodies to peripheral nerve myelin proteins in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. (2005) 10:174–80. doi: 10.1111/j.1085-9489.2005.0010207.x
89. Inglis HR, Csurhes PA, McCombe PA. Antibody responses to peptides of peripheral nerve myelin proteins P0 and P2 in patients with inflammatory demyelinating neuropathy. J Neurol Neurosurg Psychiatry. (2007) 78:419–22. doi: 10.1136/jnnp.2006.106617
90. Makowska A, Pritchard J, Sanvito L, Gregson N, Peakman M, Hayday A, et al. Immune responses to myelin proteins in Guillain–Barré syndrome. J Neurol Neurosurg Psychiatry. (2008) 79:664–71. doi: 10.1136/jnnp.2007.123943
91. Ritz MF, Lechner-Scott J, Scott RJ, Fuhr P, Malik N, Erne B, et al. Characterisation of autoantibodies to peripheral myelin protein 22 in patients with hereditary and acquired neuropathies. J Peripher Nerv Syst. (2000) 5:239–9. doi: 10.1046/j.1529-8027.2000.00022-13.x
92. Gabriel CM, Gregson NA, Hughes RAC. Anti-PMP22 antibodies in patients with inflammatory neuropathy. J Peripher Nerv Syst. (2000) 5:239–9. doi: 10.1046/j.1529-8027.2000.00022-12.x
93. Cifuentes-Diaz C, Dubourg O, Irinopoulou T, Vigny M, Lachkar S, Decker L, et al. Nodes of ranvier and paranodes in chronic acquired neuropathies. PLoS ONE. (2011) 6:e14533. doi: 10.1371/journal.pone.0014533
94. Devaux JJ, Odaka M, Yuki N. Nodal proteins are target antigens in Guillain- Barré syndrome. J Peripher Nerv Syst. (2012) 17:62–71. doi: 10.1111/j.1529-8027.2012.00372.x
95. Niezgoda A, Michalak S, Losy J, Kalinowska-Łyszczarz A, Kozubski W. sNCAM as a specific marker of peripheral demyelination. Immunol Lett. (2017) 185:93–7. doi: 10.1016/j.imlet.2017.03.011
96. Paparounas K. Anti-GQ1b ganglioside antibody in peripheral nervous system disorders: pathophysiologic role and clinical relevance. Arch Neurol. (2004) 6:1013–6. doi: 10.1001/archneur.61.7.1013
97. Lemke G, Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell. (1985) 40:501–8. doi: 10.1016/0092-8674(85)90198-9
98. Giese KP, Martini R, Lemke G, Soriano P, Schachner M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. (1992) 71:565–76. doi: 10.1016/0092-8674(92)90591-Y
99. Uyemura K, Yoshimura K, Suzuki M, Kitamura K. Lipid binding activities of the P2 protein in peripheral nerve myelin. Neurochem Res. (1984) 9:1509–14. doi: 10.1007/BF00964676
100. Quarles RH. Myelin sheaths: Glycoproteins involved in their formation, maintenance and degeneration. Cell Mol Life Sci. (2002) 59:1851–71. doi: 10.1007/PL00012510
101. Rasband MN, Macklin WB. Myelin structure and biochemistry. In: Brady ST, Albers RW, Siegel GJ, Price DL, editors. Basic Neurochemistry, 8th ed. Waltham, MA: Academic Press (2012). p. 180–99. doi: 10.1016/B978-0-12-374947-5.00010
102. Snipes GJ, Suter U. Molecular anatomy and genetics of myelin proteins in peripheral nervous system. J Anat. (1995) 186:483–94.
103. Lim JP, Devaux J, Yuki N. Peripheral nerve proteins as potential autoantigens in acute and chronic inflammatory demyelinating polyneuropathies. Autoimmun Rev. (2014) 13:1070–8. doi: 10.1016/j.autrev.2014.08.005
Keywords: peripheral demyelinating disease, schwann cell, biomarker, Guillain-Barre syndrome, chronic inflammatory demyelinating polyradiculoneuropathy, anti-MAG neuropathy, POEMS syndrome, Charcot-Marie-Tooth disease
Citation: Kamil K, Yazid MD, Idrus RBH, Das S and Kumar J (2019) Peripheral Demyelinating Diseases: From Biology to Translational Medicine. Front. Neurol. 10:87. doi: 10.3389/fneur.2019.00087
Received: 31 October 2018; Accepted: 22 January 2019;
Published: 19 March 2019.
Edited by:
Wael M. Y. Mohamed, International Islamic University Malaysia, MalaysiaReviewed by:
Muthuraju Sangu, Universiti Sains Malaysia Health Campus, MalaysiaCopyright © 2019 Kamil, Yazid, Idrus, Das and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaya Kumar, amF5YWt1bWFyQHVrbS5lZHUubXk=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.