 William Huynh
William Huynh Thanuja Dharmadasa1
Thanuja Dharmadasa1 Steve Vucic
Steve Vucic
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurol. , 04 January 2019
Sec. Neurodegeneration
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.01141
This article is part of the Research Topic Biomarkers and Clinical Indicators in Motor Neuron Disease View all 38 articles
The clinical diagnosis of amyotrophic lateral sclerosis (ALS) relies on determination of progressive dysfunction of both cortical as well as spinal and bulbar motor neurons. However, the variable mix of upper and lower motor neuron signs result in the clinical heterogeneity of patients with ALS, resulting frequently in delay of diagnosis as well as difficulty in monitoring disease progression and treatment outcomes particularly in a clinical trial setting. As such, the present review provides an overview of recently developed novel non-invasive electrophysiological techniques that may serve as biomarkers to assess UMN and LMN dysfunction in ALS patients.
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disease that was first described in the 1869 by Jean-Martin Charcot (1–3) although earlier detailed clinicopathological descriptions of a case of ALS, was published by Radcliffe and Clarke (4). Charcot postulated the importance of the upper motor neuron in its pathogenesis (3) and its associated degeneration of motor cortical Betz cells that has become a well-recognized pathological feature (5, 6). The diagnosis of classical amyotrophic lateral sclerosis (ALS) relies on the clinical identification of progressive dysfunction in both the cortical (“upper”, UMN) and spinal (“lower”, LMN) motor neurons involving multiple body regions, much of which is encompassed within the El Escorial criteria (7, 8). The clinical heterogeneity of ALS is a result of the variable mix of UMN and LMN signs (9), hence contributing to delay in diagnosis and difficulty in monitoring disease progression as well as treatment outcomes particularly in a clinical trial setting (6). As such, there is a critical need to devise objective biomarkers of disease progression in ALS that may facilitate both improvement in diagnosis as well as to provide meaningful outcome measures to monitor treatment (10).
The present review will provide an overview of recently developed neurophysiological biomarkers, with emphases on novel non-invasive electrophysiological techniques used to assess UMN and LMN dysfunction in ALS patients.
An important component in the diagnosis of ALS relies on clinical features of UMN involvement in the presence of progressive LMN weakness (11), but often these signs of UMN impairment may be underappreciated in a limb that is concurrently affected by LMN loss especially in early stages of ALS (6, 12, 13). Upper motor neuron signs may initially be absent in approximately 7–10% of ALS patients (6, 14). As such, objective UMN biomarkers may be critical for the diagnosis of ALS, as potential mimicking disorders such as multifocal motor neuropathy, Kennedy's disease and adult-onset spinal muscular atrophy (SMA), may present as pure LMN syndromes (6, 15, 16). Autopsy reports have also demonstrated UMN degeneration in 50–75% of patients with clinically pure LMN syndromes (5, 17, 18).
Since its original description more than 3 decades ago (19), Transcranial magnetic stimulation (TMS) has undergone significant evolution as a non-invasive technique for cortical stimulation, providing valuable insight into the functional integrity of brain pathways. Its main application has been in the investigation of complex neuronal networks of the primary motor cortex (M1), which is influenced by both inhibitory and excitatory mechanisms (20). Transcranial magnetic stimulation (TMS) biomarkers of cortical hyperexcitability appear to be useful biomakers of UMN dysfunction in ALS (21). In addition, TMS have provided insights into the underlying pathophysiological mechanisms in ALS, thereby allowing for the development of diagnostic and prognostic biomarkers in ALS (21).
The TMS technique utilizes a transient magnetic field to induce an electric current in the cortex (22). This magnetic field is generated through a stimulating coil held over a subject's head, which painlessly and non-invasively penetrates the skull without attenuation (Figure 1). Depending on stimulation intensity and coil type, the electromagnetic force can stimulate neurons at a depth of 1.5–3.0 cm beneath the scalp (23). There have been several theoretical models postulated to explain the exact effect of this electromagnetic field on biological tissue, with studies in both animals and humans conferring that TMS generates a corticomotoneuronal volley composed of direct (D) and indirect (I) waves occurs at intervals of 1.5–2.5 ms (24). Direct waves are thought to represent the activation of corticospinal axons and are only recruited at high intensities or with the TMS coil positioned such that induces currents in a lateral-medial direction. Indirect waves seem to be activated at lower intensities and are mediated by a more complex interaction between cortical excitatory and inhibitory neurons (25). TMS delivered over the primary motor cortex (M1) is thought to activate pyramidal neurons (Betz cells) trans-synaptically via I-waves (26), but the exact neural circuitries evoked remain to be determined. These complex neural circuits are critically dependent on both excitatory and inhibitory interneuronal systems, facilitated by cellular receptor and neurotransmitter interactions (27). Excitation is primarily mediated by glutamate/NMDA receptor interaction, while inhibition is facilitated by γ-aminobutyric acid (GABA)/GABAA/B receptor action (28).
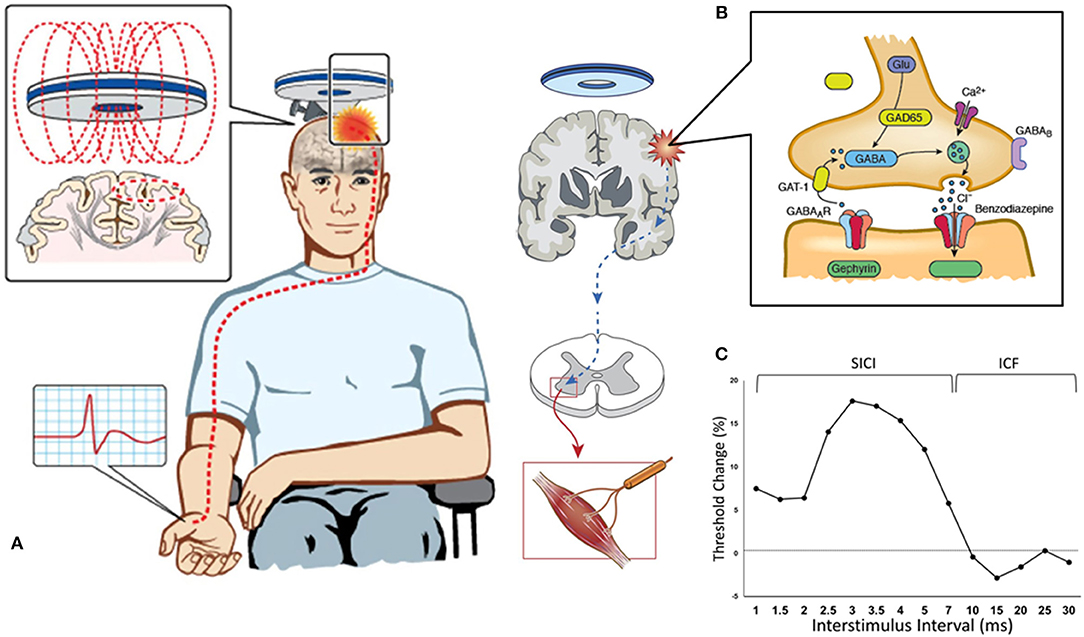
Figure 1. Paired-pulse threshold tracking transcranial magnetic stimulation (TMS). (A) TMS coil placed over the vertex activates the primary motor cortex and the response (motor evoked potential, MEP) is recorded from the contralateral abductor pollicis brevis muscle. (B) TMS parameters are mediated by a complex interplay between intraneural circuits and cortical output cells, with cortical interneurons mediating inhibition by activation of GABAergic synapses leading to influx of chloride anions (Cl−) and hyperpolarization of post-synaptic neurons. (C) Change in stimulus intensity required to achieve a target MEP of 0.2 mV (±20%) is used to quantify SICI (which is recorded with interstimulus intervals between 1–7 ms) and ICF (between 10–30 ms).
Cortical hyperexcitability in ALS is heralded by reduced short-interval intracortical inhibition and CSP duration, in addition to increased intracortical facilitation and motor evoked potential amplitude (12, 29, 30). Furthermore, significant bilateral TMS abnormalities was observed in ALS patients at an early disease stage (31), consistent with previous studies that have reported functional abnormalities of the motor cortex as an early and specific feature of ALS, and preceding the onset of LMN dysfunction (6, 12, 29, 30, 32–34). More recent studies have demonstrated changes in TMS parameters indicative of cortical hyperexcitability, were more prominent over the dominant motor cortex and in particular, contralateral to the site of disease onset, suggesting a vulnerability of the dominant motor cortical neurons and supporting the importance of cortical processes in the pathophysiology of ALS as postulated first by Menon et al. (31).
The resting motor threshold (RMT) is a reflection of the ease with which corticomotoneurons are excited, hence the corticomotoneuronal membrane excitability, as well as the density of UMN projections onto motor neurons (35). Through the α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptors, RMT is influenced by the glutamatergic neurotransmitter system, such that excessive glutamate activity reduces RMT, and is susceptible to modulation by sodium channel blockers (28, 36). In ALS, the RMT is reduced early in the disease (indicative of cortical hyperexcitability) followed by progressive increase and eventual inexcitability with disease progression (32, 37–39). As motor threshold is modulated by glutamate activity (28), the reduced motor threshold supports the notion that cortical hyperexcitability being an early feature of ALS contributing to the ensuing lower motor neuron degeneration (21). The motor cortex is found to be inexcitable in approximately 20% of ALS patients and appears to be a late finding. In contrast, motor cortex inexcitability is a relatively frequent fidning in patients exhibiting the pure UMN phenotype termed primary lateral sclerosis [PLS] (40).
The central motor conduction time (CMCT) time is defined by the time interval between stimulation of the motor cortex and arrival of the corticospinal volleys at the spinal motor neurons, and is inferred from the motor evoked potential (MEP) onset latency (21). Prolongation of CMCT is an invariable finding in ALS being documented in 16–100% across different series (5, 21, 37, 41–44). In patients without clinically predominant UMN phenotypes, prolongation of CMCTs occurs in 50–71% of patients (41, 44). Although the mechanisms underlying CMCT prolongation are presently not fully elucidated, an increase in desynchronization of corticomotoneuronal volleys resulting from degeneration of the fastest conducting corticomotoneuronal fibers has been suggested (45, 46). Large discrepancies in sensitivity of this parameter reported by previous studies are likely attributable to technique-dependent variations associated with CMCT calculations.
The cortical silent period (CSP) refers to the interruption of voluntary electromyography activity in a target muscle after motor cortex stimulation (47), and the mechanisms that underly the CSP are complex but thought to be mediated primarily by the activation of inhibitory neurons acting via GABA-B receptors within the cortex (21, 48). The CSP duration has been consistently reduced in patients across all ALS phenotypes (21, 30, 32, 34, 43, 49–51). The decrease in CSP duration in ALS patients likely represent a combination of degeneration of inhibitory interneurons as well as GABAB-mediated receptor inhibition dysfunction (21).
In the paired-pulse paradigm, a conditioning stimulus (CS) precedes and is utilized to modulate the effect of a second test stimulus (TS). By varying the time interval between the paired pulses (the interstimulus interval, ISI) a number of parameters can be determined, using either a constant stimulus method [in which the CS and TS are kept at a constant level and MEP amplitude is evaluated (52)] or the threshold-tracking (TT) TMS protocol (53). TT-TMS was developed to overcome the marked MEP amplitude variability seen when utilizing the earlier protocol and uses a fixed MEP response which is tracked by a varying TS (53, 54). By applying a subthreshold (set at 70% RMT) conditioning stimulus at predetermined time intervals prior to a suprathreshold test stimulus, the threshold-tracking TMS technique allows the short-interval intracortical inhibition (SICI) and intracortical facilitation (ICF) to be investigated (53, 55) (Figure 1).
Reduction or absence of SICI, which is a biomarker of cortical interneuronal inhibitory GABAergic function, has been established as an early feature of ALS (Figures 2A,B), correlating with biomarkers of peripheral neurodegeneration and at times preceding the onset of LMN dysfunction in sporadic ALS cohorts [(31, 53), etc]. Although there were no significant differences in the degree of reduction observed between the sides of the motor cortices, there was a trend for more changes observed over the dominant motor cortex, particularly contralateral to the side of disease onset (31). The changes were also similar regardless of the severity of LMN dysfunction, or site of onset (bulbar or limb) (12, 21, 32, 56).
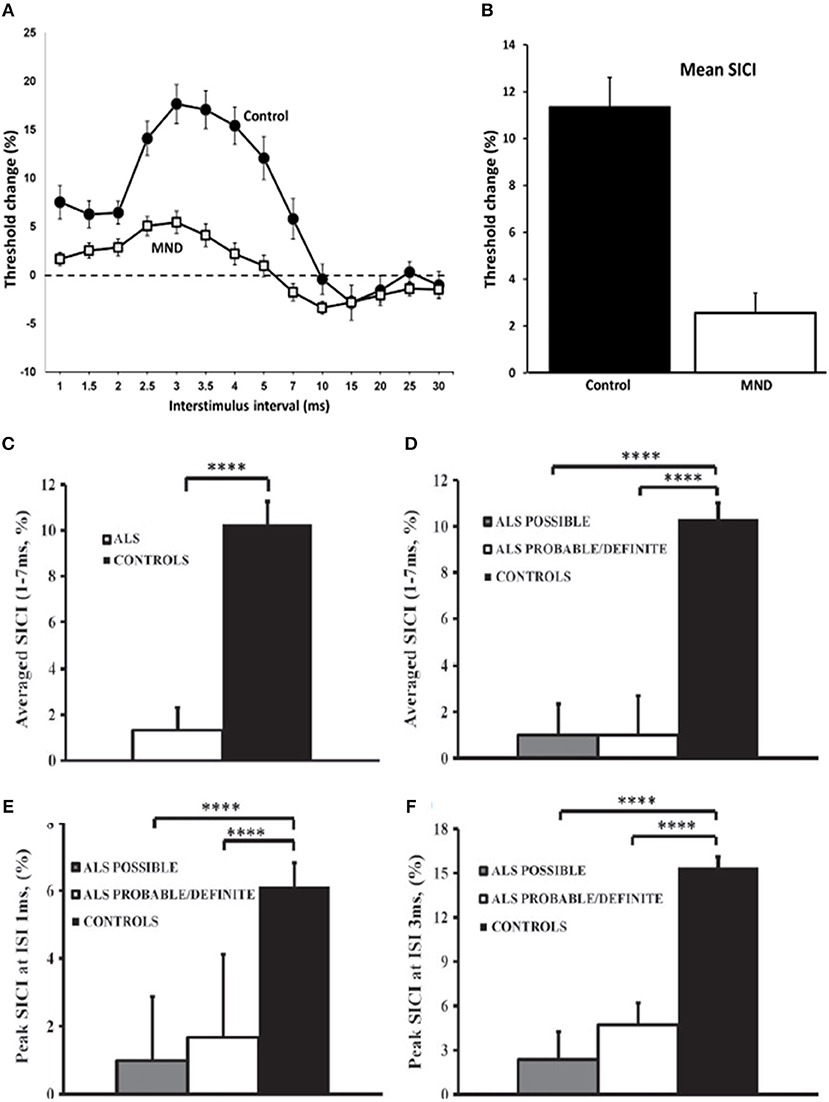
Figure 2. Cortical excitability in motor neuron disease (MND). Paired-pulse subthreshold conditioning transcranial magnetic stimulation demonstrating (A) reduction in short-interval intracortical inhibition (SICI, above dotted line) and intracortical facilitation (ICF, below dotted line) and (B) significant reductions in averaged SICI (between interstimulus intervals of 1–7 ms) in MND patients compared with controls (C) Averaged short-interval intracortical inhibition (SICI), between interstimulus interval (ISI) 1–7 ms, was significantly reduced in amyotrophic lateral sclerosis (ALS). (D) The reduction of averaged SICI was comparable in Awaji subgroups. Peak SICI at ISI (E) 1 ms, and (F) 3 ms was significantly reduced in Awaji subgroups. ****P < 0.0001. Reproduced with permission license no. 4457360494951 (1) and license no. 4457440155614 (12).
The reduction in SICI has been a widely reported feature present in both familial and sporadic forms of ALS with the alterations observed as an early feature (21, 30, 34, 57–62). Further to this, longitudinal assessments of asymptomatic SOD-1 mutation carriers have identified cortical hyperexcitability developing prior to the clinical onset of ALS, therefore suggesting that cortical hyperexcitability underlies the process of neurodegeneration in familial ALS (34).
The use of threshold-tracking TMS may be able to uncover UMN involvement in ALS phenotypes without clinically evident UMN signs such as the flail limb variant of ALS or progressive muscular atrophy (PMA). Moreover, this technique was able to reliably distinguish between ALS and other neurological mimic conditions including multifocal motor neuropathy, spinal muscular atrophy, Kennedy's disease, peripheral nerve hyperexcitability disorders, Hirayama disease, CIDP, lead neuropathy, hereditary spastic paraparesis, as well as hereditary motor neuropathy with pyramidal features (63–68).
SICI abnormalities using the threshold-tracking technique, appear to be the most robust diagnostic parameter that is indicative of UMN dysfunction in ALS patients (12, 29, 69). Using either an abnormal SICI or an inexcitable cortex, this TMS method demonstrated a sensitivity of approximately 73% and a specificity of 81% (69). Moreover, an absent SICI was associated with a 97% sensitivity (33). TMS abnormalities were observed in 77% of patients with ALS, with frequency of abnormalities that were similar across all Awaji diagnostic groups, using the established cut-off SICI of < 5.5% (63) resulting in 88% of Awaji-criteria possible patients being reclassified as Awaji-criteria probable or definite (12). More specifically, an abnormally reduced SICI was demonstrated in 56% of Awaji-criteria possible patients (12) (Figures 2C–F).
More recent studies have also documented increasing cortical hyperexcitability with advancing disease indicating that intracortical inhibitory neurons become progressively dysfunctional in ALS (Figure 3A) (70). Reduced SICI was also reported to be an independent prognostic biomarker in ALS patients within the first 2 years of disease onset (71) (Figure 3B). Separately, SICI was shown to partially normalize with treatment by riluzole (72), an anti-glutamatergic agent exhibiting modest clinical effectiveness in ALS (73, 74). Paralleling the clinical efficacy Riluzole, the modulating effects last about 3 months (75), and may be related to overexpression of efflux pumps located at the blood brain barrier during the disease course (76). Regardless of the underlying mechanisms, studies of riluzole have suggested a utility of threshold-tracking TMS in assessing biological effectiveness of compounds at an early stage of drug development. Taken together, these results suggest that non-invasive in vivo monitoring of cortical function and particularly, SICI may also be an effective biomarker used to monitor the effects of novel therapeutics in a clinical trial setting.
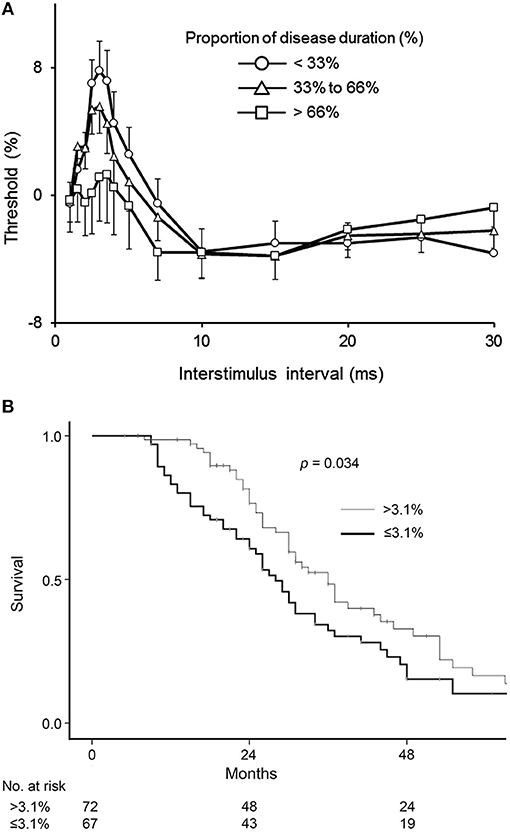
Figure 3. (A) Cortical excitability changes with disease progression. Patients were divided into three groups according to disease stage. The duration of the illness from onset to death was normalized between zero and one and expressed as a percentage (%), with data averaged by proportion of disease duration. Early stage (Circle) was defined as the proportion of disease duration < 33%, mid (Triangle) was 33–66%, and late (Square) was >66%. ALSFRS-R of patients in early stage was 42.3 ± 0.6, that in mid was 40.2 ± 0.7, and that in late was 34.8 ± 2.0. SICI at ISI 1–7 ms decreased with disease progression. Data are given as mean ± SE. Reproduced with permission license no. 4456860473754 (70). (B) Kaplan-Meier plots of survival probabilities according to averaged short-interval intracortical inhibition (SICI) values. Amyotrophic lateral sclerosis patients with a disease duration under 2 years were divided into 2 groups according to values in average SICI, interstimulus interval 1–7 ms. Patients with SICI ≤ 3.1% demonstrated reduced survival compared to patients with SICI >3.1% (p = 0.034). Estimated median survival was 28 months in patients with reduced SICI and 36 months in patients with higher SICI. Reproduced with permission license no. 4456870994973 (71).
Objective assessment of LMN dysfunction, utilizing neurophysiological techniques, appear to be more sensitive than clinical assessments (77, 78). Conventional neurophysiological techniques, such as nerve conduction studies which measure the compound muscle action potential (CMAP) amplitude, may be relatively insensitive in assessing LMN degeneration due to the process of reinnervation (79).
As such, various methods to approximate the number of motor units innervating individual muscles, including motor unit number estimation (MUNE), and motor unit number index (MUNIX), may potentially represent valuable biomarkers of LMN degeneration. Since the development of the first MUNE technique in 1971 (80), there have been numerous other MUNE techniques introduced (81–85). The basic principle of MUNE techniques is the dividing of the maximal CMAP amplitude by the average surface-recorded motor unit potential (86). The original MUNE technique utilized incremental stimulation whereby the stimulus intensity at one point on the nerve was gradually increased from subthreshold until 10 increments in the motor response was recorded, but this technique relied on the assumption that the smallest recorded potential using the surface electrode over a target muscle following minimal stimulation represented a single motor unit potential (Figure 4A). Consequently, the variance in the result MUNE was considerable and resulted not uncommonly in artificially lower MUNE counts (86, 88).
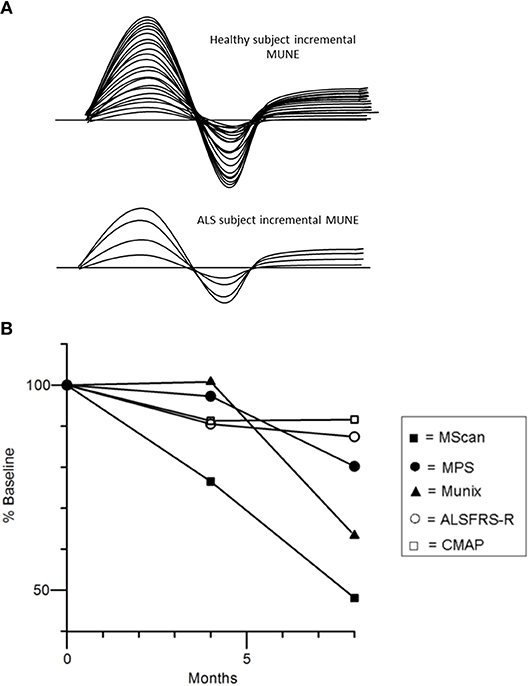
Figure 4. (A) Incremental MUNE in healthy and ALS subjects, demonstrating a large number of “steps” with increasing stimulus intensity consistent with a large number of functioning motor units within measured muscle, whilst there were only four steps in the ALS subject indicating only four functional motor units remaining in muscle. (B) Percentage changes in MUNE values (geometric means) and mean ALSFRS-R and CMAP amplitude at 4 and 8 months. Reproduced with permission license no. 4457481173441 (87).
The motor unit index (MUNIX) technique is a method designed to express the number of functioning motor units within a muscle as an index, instead of providing a direct measure of their absolute numbers. It is based on patients performing a voluntary contraction at various intensity levels and surface interference patterns being captured and decomposed to obtain a normalized motor unit size, which is then in turn divided into the maximal CMAP value to obtain the MUNIX (86, 89, 90).
Recent studies using different MUNE methods have demonstrated potential utility for assessing disease progression in ALS patients as reflected by a progressive linear decline in MUNE counts (87, 91–94). Interestingly, a recently developed MUNE technique, termed MScan, appeared to be the most sensitive MUNE method in detecting ALS disease progression (Figure 4B) (87). Additionally, MUNIX was able to detect disease progression in presymptomatic muscles in ALS (95, 96), and changes longitudinally in these muscle groups appeared more sensitive to those changes in the revised ALS Functional Rating Scale (ALSFRS-R) (93).
The neurophysiological index (NI) is a potential electrophysiological biomarker in assessing lower motor neuron loss in ALS (97). Using a simple formula, The NI has the advantage of using routine CMAP amplitude, F-wave frequency, and distal motor latency of the ulnar-nerve innervated abductor digit minimi (ADM) muscle and is more sensitive than the CMAP amplitude alone in demonstrating longitudinal lower motor neuron loss in ALS. NI was able to detect motor neuron loss in muscles of the presymptomatic limb in ALS patients as well as successfully tracking disease progression, demonstrating continued loss of functional motor units during this presymptomatic period, when weakness, atrophy, or fasciculations were not detectable to both patients and evaluating clinicians (78). The validation of NI as a clinically meaningful parameter in disease progression of ALS patients was also demonstrated longitudinally in the symptomatic muscles of patients that correlated with their ALSFRS-R decline (97, 98). Additionally, NI was able to detect deterioration that occurred over a short period of 4 weeks in ALS patients, hence enabling the utility of this index in a clinical trial setting (77). NI has favorable reproducibility and low intraindividual variability but amongst its limitations, the index is only restricted to the ADM muscle (which is less affected compared to other intrinsic hand muscles such as the APB and FDI, in keeping with the split hand pattern of wasting and weakness) (99) and requires persistent F-waves (that can be frequently absent in ALS) (78).
The split-hand sign is documented as an early and specific clinical feature in patients with ALS that is not characteristic in other commonly encountered clinical mimics (99, 100). It refers to the preferential wasting and weakness of the thenar complex muscles (APB and FDI) with relative preservation of the hypothenar muscle, ADM (99), and appeared to have a cortical origin with the corticomotoneuronal input to the thenar complex in ALS patients preferentially affected (101, 102). This clinical observation provided an opportunity to develop a simple neurophysiological biomarker to aid the diagnosis of ALS using conventional nerve conduction studies. The split-hand index (SI) was derived by multiplying the CMAP amplitude of the APB muscle by the FDI CMAP amplitude and then dividing the product by the ADM CMAP amplitude. It was demonstrated that a reduction in the split-hand index was consistent across ALS phenotypes but appeared most pronounced in those with limb-onset, and that a cut-off value ≤ 5.2 reliably differentiated between ALS and other neurological disorders (103).
Electrical impedance myography (EIM) is a novel non-invasive form of testing to provide quantitative information on neuromuscular disorders that may be useful and reliable in assessing longitudinally the severity of a disease process (104–107). EIM utilizes a small, high-frequency electrical current applied across two electrodes positioned over a muscle, and the resulting surface voltages are measured between a second pair of electrodes, from which the resistive and capacitive properties of the tissue are obtained (86, 105). The advantage is that this technique does not rely on inherent electrical activity of the tissue (which conventional neurophysiological techniques do), but rather on how the tissue impacts the applied current, rendering the technique sensitive to structural and compositional changes in muscle such as denervation, reinnervation, myofiber atrophy and fat replacement within the muscle that occur in ALS (104). EIM values have been shown to correlate with standard clinical approaches including handheld dynamometry and MUNE (106, 107), and may be able to provide more than a five-fold reduction in sample size requirements for ALS clinical therapeutic trials over standard outcome measures such as the ALS functional rating scale-revised (ALSFRS-R) (108). Although EIM can detect changes early in the disease course of ALS as well as in clinically unaffected muscle groups (105), a limitation of EIM is that identified changes may not be able to differentiate ALS from other neuromuscular conditions (109, 110).
Amyotrophic lateral sclerosis remains a devastating neurodegenerative disorder with a poor prognosis, much of which is attributable to frequent delays in diagnosis, an incomplete understanding of the underlying pathophysiological mechanisms, and the current lack of effective disease-modifying treatment available. As such, there is a critical need to devise accurate and reliable biomarkers to address the above shortfalls in current ALS management. The current review has presented recent developments in novel neurophysiological biomarkers that are able to effectively interrogate upper and lower motor neuron dysfunction and characterize their change over time with disease progression, thereby exhibiting the potential to improve diagnosis, as well as facilitating in the prognosis and monitoring of the effects of future therapeutic agents in a clinical trial setting.
WH drafted manuscript and made all necessary edits and revisions as well as obtained required copyright permissions for figures. TD, SV, and MK revised and edited manuscript.
This work was supported by funding to ForeFront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neuron disease, from the National Health and Medical Research Council (NHMRC) (1132524, 1095127, 1060992). WH was supported by the Beryl Bayley Fellowship of Motor Neuron Disease Research Institute of Australia. MK is a NHMRC Practitioner Fellow (#1156093).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Simon NG, Huynh W, Vucic S, Talbot K, Kiernan MC. Motor neuron disease: current management and future prospects. Intern Med J. (2015) 45:1005–13. doi: 10.1111/imj.12874
2. Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. (2001) 2:806–19. doi: 10.1038/35097565
3. Charcot J, Joffroy A. Deus cas d'atrophie musculaire progressive avec lesions de la substance grise et des faisceaux antéro-latérale. Arch Physiol. (1869) 2:354–67.
4. Radcliffe CB, Clarke JL. An important case of paralysis and muscular atrophy, with disease of the nervous centres. Br Foreign Med Chir Rev. (1862) 30:215–25.
5. Kaufmann P, Pullman SL, Shungu DC, Chan S, Hays AP, Del Bene ML, et al. Objective tests for upper motor neuron involvement in amyotrophic lateral sclerosis (ALS). Neurology (2004) 62:1753–7. doi: 10.1212/01.WNL.0000125182.17874.59
6. Huynh W, Simon NG, Grosskreutz J, Turner MR, Vucic S, Kiernan MC. Assessment of the upper motor neuron in amyotrophic lateral sclerosis. Clin Neurophysiol. (2016) 127:2643–60. doi: 10.1016/j.clinph.2016.04.025
7. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. (1994) 124:S96–107.
8. Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:293–9. doi: 10.1080/146608200300079536
9. Sabatelli M, Zollino M, Luigetti M, Grande AD, Lattante S, Marangi G, et al. Uncovering amyotrophic lateral sclerosis phenotypes: clinical features and long-term follow-up of upper motor neuron-dominant ALS. Amyotroph Lateral Scler. (2011) 12:278–82. doi: 10.3109/17482968.2011.580849
10. Kiernan M, Vucic S, Cheah B, Turner M, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet (2011) 277:942–55. doi: 10.1016/S0140-6736(10)61156-7
11. De Carvalho M. Testing upper motor neuron function in amyotrophic lateral sclerosis: the most difficult task of neurophysiology. Brain (2012) 135:2581–2. doi: 10.1093/brain/aws228
12. Geevasinga N, Menon P, Yiannikas C, Kiernan MC, Vucic S. Diagnostic utility of cortical excitability studies in amyotrophic lateral sclerosis. Eur J Neurol. (2014) 21:1451–7. doi: 10.1111/ene.12422
13. Swash M. Why are upper motor neuron signs difficult to elicit in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatr. (2012) 83:659–62. doi: 10.1136/jnnp-2012-302315
14. Rocha AJ, Maia Junior AC. Is magnetic resonance imaging a plausible biomarker for upper motor neuron degeneration in amyotrophic lateral sclerosis/primary lateral sclerosis or merely a useful paraclinical tool to exclude mimic syndromes? A critical review of imaging applicability in clinical routine. Arq Neuropsiquiatr. (2012) 70:532–9. doi: 10.1590/S0004-282X2012000700012
15. Simon NG, Turner MR, Vucic S, Al-Chalabi A, Shefner J, Lomen-Hoerth C, et al. Quantifying disease progression in amyotrophic lateral sclerosis. Ann Neurol. (2014) 76:643–57. doi: 10.1002/ana.24273
16. Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, De Carvalho M, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. (2013) 12:310–22. doi: 10.1016/S1474-4422(13)70036-X
17. Ince PG, Evans J, Knopp M, Forster G, Hamdalla HH, Wharton SB, et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology (2003) 60:1252–8. doi: 10.1212/01.WNL.0000058901.75728.4E
18. Lawyer T Jr, Netsky MG. Amyotrophic lateral sclerosis. AMA Arch Neurol Psychiatr. (1953) 69:171–92. doi: 10.1001/archneurpsyc.1953.02320260029002
19. Barker A, Jalinous R, Freeston I. Non-invasive magnetic stimulation of human motor cortex. Lancet (1985) 1:1106–7. doi: 10.1016/S0140-6736(85)92413-4
20. Vucic S, Kiernan M. Transcranial magnetic stimulation for the assessment of neurodegenerative disease. Neurotherapeutics (2017) 14:91–106. doi: 10.1007/s13311-016-0487-6
21. Vucic S, Kiernan MC. Utility of transcranial magnetic stimulation in delineating amyotrophic lateral sclerosis pathophysiology. Handb Clin Neurol. (2013) 116:561–75. doi: 10.1016/B978-0-444-53497-2.00045-0
22. Rossini P, Burke D, Chen R, Cohen L, Daskalakis Z, Di Lorio R, et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord, roots and peripheral nerves: basic principles and procedures for routine clinical and research application. An updated report from an IFCN Committee. Clin Neurophysiol. (2015) 126:1071–107. doi: 10.1016/j.clinph.2015.02.001
23. Roth B, Basser P. A model of the stimulation of a nerve fiber by electromagnetic induction. IEEE Trans Biomed Eng. (1990) 37:588–97. doi: 10.1109/10.55662
24. Di Lazzaro V, Oliviero A, Profice P, Saturno E, Pilato F, Insola A, et al. Comparison of descending volleys evoked by transcranial magnetic and electric stimulation in conscious humans. Electroencephalogr Clin Neurophysiol. (1998) 109:5. doi: 10.1016/S0924-980X(98)00038-1
25. Di Lazzaro V, Profice P, Ranieri F, Capone F, Dileone M, Oliviero A, et al. I-wave origin and modulation. Brain Stimul. (2012) 5:512–25. doi: 10.1016/j.brs.2011.07.008
26. Chen R, Cros D, Curra A, Di Lazzaro V, Lefaucheur J, Magistris M, et al. The clinical diagnostic utility of transcranial magnetic stimulation: report of an IFCN committee. Clin Neurophysiol. (2008) 119:504–32. doi: 10.1016/j.clinph.2007.10.014
27. Robinson D. Implications of neural networks for how we think about brain function. Behav Brain Sci. (1992) 13:644–55.
28. Ziemann U, Reis J, Schwenkreis P, Rosanova M, Strafella A, Badawy R, et al. TMS and drugs revisited (2014). Clin Neurophysiol. (2015) 126:1847–68. doi: 10.1016/j.clinph.2014.08.028
29. Menon P, Kiernan MC, Vucic S. Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin Neurophysiol. (2015) 126:803–9. doi: 10.1016/j.clinph.2014.04.023
30. Vucic S, Kiernan MC. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain (2006) 129:2436–46. doi: 10.1093/brain/awl172
31. Menon P, Geevasinga N, Van Den Bos M, Yiannikas C, Kiernan MC, Vucic S. Cortical hyperexcitability and disease spread in amyotrophic lateral sclerosis. Eur J Neurol. (2017) 24:816–24. doi: 10.1111/ene.13295
32. Grieve SM, Menon P, Korgaonkar MS, Gomes L, Foster S, Kiernan MC, et al. Potential structural and functional biomarkers of upper motor neuron dysfunction in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2015) 17:85–92. doi: 10.3109/21678421.2015.1074707
33. Vucic S, Ziemann U, Eisen A, Hallett M, Kiernan MC. Transcranial magnetic stimulation and amyotrophic lateral sclerosis: pathophysiological insights. J Neurol Neurosurg Psychiatr. (2013) 84:1161–70. doi: 10.1136/jnnp-2012-304019
34. Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain (2008) 131:1540–50. doi: 10.1093/brain/awn071
35. Rossini PM, Berardelli A, Deuschl G, Hallett M, Maertens De Noordhout AM, Paulus W, et al. Applications of magnetic cortical stimulation. The International Federation of Clinical Neurophysiology. Electroencephalogr Clin Neurophysiol Suppl. (1999) 52:171–85.
36. Di Lazzaro V, Oliviero A, Profice P, Pennisi MA, Pilato F, Zito G, et al. Ketamine increases human motor cortex excitability to transcranial magnetic stimulation. J Physiol. (2003) 547:485–96. doi: 10.1113/jphysiol.2002.030486
37. Triggs WJ, Menkes D, Onorato J, Yan RS, Young MS, Newell K, et al. Transcranial magnetic stimulation identifies upper motor neuron involvement in motor neuron disease. Neurology (1999) 53:605–11. doi: 10.1212/WNL.53.3.605
38. Mills KR, Nithi KA. Corticomotor threshold is reduced in early sporadic amyotrophic lateral sclerosis. Muscle Nerve (1997) 20:1137–41.
39. Eisen A, Pant B, Stewart H. Cortical excitability in amyotrophic lateral sclerosis: a clue to pathogenesis. Can J Neurol Sci. (1993) 20:11–6. doi: 10.1017/S031716710004734X
40. Geevasinga N, Menon P, Sue CM, Kumar KR, Ng K, Yiannikas C, et al. Cortical excitability changes distinguish the motor neuron disease phenotypes from hereditary spastic paraplegia. Eur J Neurol. (2015) 22:826–31, e57–8. doi: 10.1111/ene.12669
41. Miscio G, Pisano F, Mora G, Mazzini L. Motor neuron disease: usefulness of transcranial magnetic stimulation in improving the diagnosis. Clin Neurophysiol. (1999) 110:975–81. doi: 10.1016/S1388-2457(99)00030-9
42. Mills KR, Nithi KA. Peripheral and central motor conduction in amyotrophic lateral sclerosis. J Neurol Sci. (1998) 159:82–7. doi: 10.1016/S0022-510X(98)00148-8
43. Pouget J, Trefouret S, Attarian S. Transcranial magnetic stimulation (TMS): compared sensitivity of different motor response parameters in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1:S45–9. doi: 10.1080/14660820052415817
44. Schulte-Mattler WJ, Muller T, Zierz S. Transcranial magnetic stimulation compared with upper motor neuron signs in patients with amyotrophic lateral sclerosis. J Neurol Sci. (1999) 170:51–6. doi: 10.1016/S0022-510X(99)00201-4
45. Eisen A, Entezari-Taher M, Stewart H. Cortical projections to spinal motoneurons: changes with aging and amyotrophic lateral sclerosis. Neurology (1996) 46:1396–404. doi: 10.1212/WNL.46.5.1396
46. Kohara N, Kaji R, Kojima Y, Mills KR, Fujii H, Hamano T, et al. Abnormal excitability of the corticospinal pathway in patients with amyotrophic lateral sclerosis: a single motor unit study using transcranial magnetic stimulation. Electroencephalogr Clin Neurophysiol. (1996) 101:32–41. doi: 10.1016/0013-4694(95)00166-2
47. Cantello R, Gianelli M, Civardi C, Mutani R. Magnetic brain stimulation: the silent period after the motor evoked potential. Neurology (1992) 42:1951–9. doi: 10.1212/WNL.42.10.1951
48. Ziemann U, Netz J, Szelenyi A, Homberg V. Spinal and supraspinal mechanisms contribute to the silent period in the contracting soleus muscle after transcranial magnetic stimulation of human motor cortex. Neurosci Lett. (1993) 156:167–71. doi: 10.1016/0304-3940(93)90464-V
49. Desiato MT, Caramia MD. Towards a neurophysiological marker of amyotrophic lateral sclerosis as revealed by changes in cortical excitability. Electroencephalogr Clin Neurophysiol. (1997) 105:1–7. doi: 10.1016/S0924-980X(96)96582-0
50. Uozumi T, Tsuji S, Murai Y. Motor potentials evoked by magnetic stimulation of the motor cortex in normal subjects and patients with motor disorders. Electroencephalogr Clin Neurophysiol. (1991) 81:251–6. doi: 10.1016/0168-5597(91)90010-U
51. Prout AJ, Eisen AA. The cortical silent period and amyotrophic lateral sclerosis. Muscle Nerve (1994) 17:217–23. doi: 10.1002/mus.880170213
52. Ziemann U, Rothwell J, Ridding M. Interaction between intracortical inhibition and facilitation in human mortor cortex. J Physiol. (1996) 496:873–81. doi: 10.1113/jphysiol.1996.sp021734
53. Vucic S, Howells J, Trevillion L, Kiernan M. Assessment of cortical excitability using threshold tracking techniques. Muscle Nerve (2006) 33:477–86. doi: 10.1002/mus.20481
54. Kiers L, Cros D, Chiappa KH, Fang J. Variability of motor potentials evoked by transcranial magnetic stimulation. Electroencephalogr Clin Neurophysiol. (1993) 89:415–23. doi: 10.1016/0168-5597(93)90115-6
55. Kujirai T, Caramia MD, Rothwell JC, Day BL, Thompson PD, Ferbert A, et al. Corticocortical inhibition in human motor cortex. J Physiol. (1993) 471:501–19. doi: 10.1113/jphysiol.1993.sp019912
56. Turner MR, Kiernan MC, Leigh PN, Talbot K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. (2009) 8:94–109. doi: 10.1016/S1474-4422(08)70293-X
57. Vucic S, Winhammar JM, Rowe DB, Kiernan MC. Corticomotoneuronal function in asymptomatic SOD-1 mutation carriers. Clin Neurophysiol. (2010) 121:1781–5. doi: 10.1016/j.clinph.2010.02.164
58. Zanette G, Tamburin S, Manganotti P, Refatti N, Forgione A, Rizzuto N. Different mechanisms contribute to motor cortex hyperexcitability in amyotrophic lateral sclerosis. Clin Neurophysiol. (2002) 113:1688–97. doi: 10.1016/S1388-2457(02)00288-2
59. Ziemann U, Winter M, Reimers CD, Reimers K, Tergau F, Paulus W. Impaired motor cortex inhibition in patients with amyotrophic lateral sclerosis. Evidence from paired transcranial magnetic stimulation. Neurology (1997) 49:1292–8. doi: 10.1212/WNL.49.5.1292
60. Yokota T, Yoshino A, Inaba A, Saito Y. Double cortical stimulation in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatr. (1996) 61:596–600. doi: 10.1136/jnnp.61.6.596
61. Hanajima R, Ugawa Y, Terao Y, Ogata K, Kanazawa I. Ipsilateral cortico-cortical inhibition of the motor cortex in various neurological disorders. J Neurol Sci. (1996) 140:109–16. doi: 10.1016/0022-510X(96)00100-1
62. Geevasinga N, Menon P, Nicholson GA, Ng K, Howells J, Kril JJ, et al. Cortical function in asymptomatic carriers and patients with C9orf72 Amyotrophic Lateral Sclerosis. JAMA Neurol. (2015) 72:1268–74. doi: 10.1001/jamaneurol.2015.1872
63. Vucic S, Cheah BC, Yiannikas C, Kiernan MC. Cortical excitability distinguishes ALS from mimic disorders. Clin Neurophysiol. (2011) 122:1860–6. doi: 10.1016/j.clinph.2010.12.062
64. Vucic S, Cheah BC, Yiannikas C, Vincent A, Kiernan MC. Corticomotoneuronal function and hyperexcitability in acquired neuromyotonia. Brain (2010) 133:2727–33. doi: 10.1093/brain/awq188
65. Vucic S, Kiernan MC. Cortical excitability testing distinguishes Kennedy's disease from amyotrophic lateral sclerosis. Clin Neurophysiol. (2008) 119:1088–96. doi: 10.1016/j.clinph.2008.01.011
66. Vucic S, Nicholson GA, Kiernan MC. Cortical excitability in hereditary motor neuronopathy with pyramidal signs: comparison with ALS. J Neurol Neurosurg Psychiatr. (2010) 81:97–100. doi: 10.1136/jnnp.2008.157537
67. Farrar MA, Vucic S, Johnston HM, Kiernan MC. Corticomotoneuronal integrity and adaptation in spinal muscular atrophy. Arch Neurol. (2012) 69:467–73. doi: 10.1001/archneurol.2011.1697
68. Matamala JM, Geevasinga N, Huynh W, Dharmadasa T, Howells J, Simon NG, et al. Cortical function and corticomotoneuronal adaptation in monomelic amyotrophy. Clin Neurophysiol. (2017) 128:1488–95. doi: 10.1016/j.clinph.2017.05.005
69. Menon P, Geevasinga N, Yiannikas C, Howells J, Kiernan MC, Vucic S. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: a prospective study. Lancet Neurol. (2015) 14:478–84. doi: 10.1016/S1474-4422(15)00014-9
70. Shibuya K, Simon NG, Geevasinga N, Menon P, Howells J, Park SB, et al. The evolution of motor cortical dysfunction in amyotrophic lateral sclerosis. Clin Neurophysiol. (2017) 128:1075–82. doi: 10.1016/j.clinph.2017.03.004
71. Shibuya K, Park SB, Geevasinga N, Menon P, Howells J, Simon NG, et al. Motor cortical function determines prognosis in sporadic ALS. Neurology (2016) 87:513–20. doi: 10.1212/WNL.0000000000002912
72. Vucic S, Lin CS-Y, Cheah BC, Murray J, Menon P, Krishnan AV, et al. Riluzole exerts central and peripheral modulating effects in amyotrophic lateral sclerosis. Brain (2013) 136:1361–70. doi: 10.1093/brain/awt085
73. Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet (1996) 347:1425–31. doi: 10.1016/S0140-6736(96)91680-3
74. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. (1994) 330:585–91. doi: 10.1056/NEJM199403033300901
75. Geevasinga N, Menon P, Ng K, Van Den Bos M, Byth K, Kiernan MC, et al. Riluzole exerts transient modulating effects on cortical and axonal hyperexcitability in ALS. Amyotroph Lateral Scler Frontotemporal Degener. (2016) 17:1–9. doi: 10.1080/21678421.2016.1188961
76. Kuo MT. Redox regulation of multidrug resistance in cancer chemotherapy: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. (2009) 11:99–133. doi: 10.1089/ars.2008.2095
77. Cheah BC, Vucic S, Krishnan AV, Boland RA, Kiernan MC. Neurophysiological index as a biomarker for ALS progression: validity of mixed effects models. Amyotroph Lateral Scler. (2011) 12:33–8. doi: 10.3109/17482968.2010.531742
78. Escorcio-Bezerra ML, Abrahao A, Nunes KF, De Oliveira Braga NI, Oliveira ASB, Zinman L, et al. Motor unit number index and neurophysiological index as candidate biomarkers of presymptomatic motor neuron loss in amyotrophic lateral sclerosis. Muscle Nerve (2018) 58:204–12. doi: 10.1002/mus.26087
79. Felice KJ. A longitudinal study comparing thenar motor unit number estimates to other quantitative tests in patients with amyotrophic lateral sclerosis. Muscle Nerve (1997) 20:179–85.
80. McComas AJ, Fawcett PR, Campbell MJ, Sica RE. Electrophysiological estimation of the number of motor units within a human muscle. J Neurol Neurosurg Psychiatr. (1971) 34:121–31. doi: 10.1136/jnnp.34.2.121
81. Boe SG, Stashuk DW, Doherty TJ. Motor unit number estimation by decomposition-enhanced spike-triggered averaging: control data, test-retest reliability, and contractile level effects. Muscle Nerve (2004) 29:693–9. doi: 10.1002/mus.20031
82. Stashuk DW, Doherty TJ, Kassam A, Brown WF. Motor unit number estimates based on the automated analysis of F-responses. Muscle Nerve (1994) 17:881–90. doi: 10.1002/mus.880170807
83. Doherty TJ, Brown WF. The estimated numbers and relative sizes of thenar motor units as selected by multiple point stimulation in young and older adults. Muscle Nerve (1993) 16:355–66. doi: 10.1002/mus.880160404
84. Shefner JM. Motor unit number estimation in human neurological diseases and animal models. Clin Neurophysiol. (2001) 112:955–64. doi: 10.1016/S1388-2457(01)00520-X
85. Shefner JM, Cudkowicz ME, Brown RH Jr. Comparison of incremental with multipoint MUNE methods in transgenic ALS mice. Muscle Nerve (2002) 25:39–42. doi: 10.1002/mus.10000
86. Vucic S, Rutkove SB. Neurophysiological biomarkers in amyotrophic lateral sclerosis. Curr Opin Neurol. (2018) 31:640–7. doi: 10.1097/WCO.0000000000000593
87. Jacobsen AB, Bostock H, Tankisi H. Following disease progression in motor neuron disorders with 3 MUNE methods. Muscle Nerve (2018). doi: 10.1002/mus.26304. [Epub ahead of print].
88. Swash M. MUNIX in the clinic in ALS: MUNE comes of age. Clin Neurophysiol. (2017) 128:482–3. doi: 10.1016/j.clinph.2016.12.021
89. Nandedkar SD, Nandedkar DS, Barkhaus PE, Stalberg EV. Motor unit number index (MUNIX). IEEE Trans Biomed Eng. (2004) 51:2209–11. doi: 10.1109/TBME.2004.834281
90. Nandedkar SD, Barkhaus PE, Stalberg EV. Motor unit number index (MUNIX): principle, method, and findings in healthy subjects and in patients with motor neuron disease. Muscle Nerve (2010) 42:798–807. doi: 10.1002/mus.21824
91. Gooch CL, Shefner JM. ALS surrogate markers. MUNE. Amyotroph Lateral Scler Other Motor Neuron Disord. (2004) 5:104–7. doi: 10.1080/17434470410019889
92. Shefner JM, Watson ML, Simionescu L, Caress JB, Burns TM, Maragakis NJ, et al. Multipoint incremental motor unit number estimation as an outcome measure in ALS. Neurology (2011) 77:235–41. doi: 10.1212/WNL.0b013e318225aabf
93. Neuwirth C, Barkhaus PE, Burkhardt C, Castro J, Czell D, De Carvalho M, et al. Motor Unit Number Index (MUNIX) detects motor neuron loss in pre-symptomatic muscles in Amyotrophic Lateral Sclerosis. Clin Neurophysiol. (2017) 128:495–500. doi: 10.1016/j.clinph.2016.11.026
94. Neuwirth C, Barkhaus PE, Burkhardt C, Castro J, Czell D, De Carvalho M, et al. Tracking motor neuron loss in a set of six muscles in amyotrophic lateral sclerosis using the Motor Unit Number Index (MUNIX): a 15-month longitudinal multicentre trial. J Neurol Neurosurg Psychiatr. (2015) 86:1172–9. doi: 10.1136/jnnp-2015-310509
95. Fukada K, Matsui T, Furuta M, Hirozawa D, Matsui M, Kajiyama Y, et al. The Motor Unit Number Index of Subclinical Abnormality in Amyotrophic Lateral Sclerosis. J Clin Neurophysiol. (2016) 33:564–8. doi: 10.1097/WNP.0000000000000296
96. Escorcio-Bezerra ML, Abrahao A, De Castro I, Chieia MAT, De Azevedo LA, Pinheiro DS, et al. MUNIX: reproducibility and clinical correlations in Amyotrophic Lateral Sclerosis. Clin Neurophysiol. (2016) 127:2979–84. doi: 10.1016/j.clinph.2016.06.011
97. Swash M, De Carvalho M. The Neurophysiological Index in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. (2004) 5:108–10. doi: 10.1080/17434470410020067
98. De Carvalho M, Scotto M, Lopes A, Swash M. Quantitating progression in ALS. Neurology (2005) 64:1783–5. doi: 10.1212/01.WNL.0000162036.76024.AB
99. Wilbourn AJ. The “split hand syndrome”. Muscle Nerve (2000) 23:138. doi: 10.1002/(sici)1097-4598(200001)23:1<138::aid-mus22<3.0.co;2-7
100. Kuwabara S, Sonoo M, Komori T, Shimizu T, Hirashima F, Inaba A, et al. Dissociated small hand muscle atrophy in amyotrophic lateral sclerosis: frequency, extent, and specificity. Muscle Nerve (2008) 37:426–30. doi: 10.1002/mus.20949
101. Weber M, Eisen A, Stewart H, Hirota N. The split hand in ALS has a cortical basis. J Neurol Sci. (2000) 180:66–70. doi: 10.1016/S0022-510X(00)00430-5
102. Menon P, Kiernan MC, Vucic S. Cortical dysfunction underlies the development of the split-hand in amyotrophic lateral sclerosis. PLoS ONE (2014) 9:e87124. doi: 10.1371/journal.pone.0087124
103. Menon P, Kiernan MC, Yiannikas C, Stroud J, Vucic S. Split-hand index for the diagnosis of amyotrophic lateral sclerosis. Clin Neurophysiol. (2013) 124:410–6. doi: 10.1016/j.clinph.2012.07.025
104. Rutkove SB, Partida RA, Esper GJ, Aaron R, Shiffman CA. Electrode position and size in electrical impedance myography. Clin Neurophysiol. (2005) 116:290–9. doi: 10.1016/j.clinph.2004.09.002
105. Rutkove SB, Aaron R, Shiffman CA. Localized bioimpedance analysis in the evaluation of neuromuscular disease. Muscle Nerve (2002) 25:390–7. doi: 10.1002/mus.10048
106. Rutkove SB, Gregas MC, Darras BT. Electrical impedance myography in spinal muscular atrophy: a longitudinal study. Muscle Nerve (2012) 45:642–7. doi: 10.1002/mus.23233
107. Rutkove SB, Caress JB, Cartwright MS, Burns TM, Warder J, David WS, et al. Electrical impedance myography as a biomarker to assess ALS progression. Amyotroph Lateral Scler. (2012) 13:439–45. doi: 10.3109/17482968.2012.688837
108. Shefner JM, Rutkove SB, Caress JB, Benatar M, David WS, Cartwright MC, et al. Reducing sample size requirements for future ALS clinical trials with a dedicated electrical impedance myography system. Amyotroph Lateral Scler Frontotemporal Degener. (2018) 19:555–61. doi: 10.1080/21678421.2018.1510008
109. Rutkove S. Electrical impedance myography as a biomarker for ALS. Lancet Neurol. (2009) 8:226–7. doi: 10.1016/S1474-4422(09)70030-4
Keywords: amyotrophic lateral sclerosis, motor neuron disease, neurophysiological biomarkers, transcranial magnetic stimulation, cortical excitability
Citation: Huynh W, Dharmadasa T, Vucic S and Kiernan MC (2019) Functional Biomarkers for Amyotrophic Lateral Sclerosis. Front. Neurol. 9:1141. doi: 10.3389/fneur.2018.01141
Received: 07 November 2018; Accepted: 10 December 2018;
Published: 04 January 2019.
Edited by:
Peter Bede, Trinity College Dublin, IrelandReviewed by:
Foteini Christidi, National and Kapodistrian University of Athens Medical School, GreeceCopyright © 2019 Huynh, Dharmadasa, Vucic and Kiernan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William Huynh, d2lsbGlhbS5odXluaEBzeWRuZXkuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.