
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurol., 20 December 2018
Sec. Movement Disorders
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.01111
 Yun Peng1Wei Ye1
Yun Peng1Wei Ye1 Zhao Chen1Huirong Peng1Puzhi Wang1Xuan Hou1Chunrong Wang1Xin Zhou1Xiaocan Hou1Tianjiao Li1Rong Qiu2Zhengmao Hu3
Zhao Chen1Huirong Peng1Puzhi Wang1Xuan Hou1Chunrong Wang1Xin Zhou1Xiaocan Hou1Tianjiao Li1Rong Qiu2Zhengmao Hu3 Beisha Tang1,3,4,5,6,7,8
Beisha Tang1,3,4,5,6,7,8 Hong Jiang1,3,5,9*
Hong Jiang1,3,5,9*Objective: Variants in SYNE1 have been widely reported in ataxia patients in Europe, with highly variable clinical phenotype. Until now, no mutation of SYNE1 ataxia has been reported among the Chinese population. Our aim was to screen for SYNE1 ataxia patients in China and extend the clinicogenetic spectrum.
Methods: Variants in SYNE1 were detected by high-throughput sequencing on a cohort of 126 unrelated index patients with unexplained autosomal recessive or sporadic ataxia. Pathogenicity assessments of SYNE1 variants were interpreted according to the ACMG guidelines. Potential pathogenic variants were confirmed by Sanger sequencing. Clinical assessments were conducted by two experienced neurologists.
Results: Two Chinese families with variable ataxia syndrome were identified (accounting for 1.6%; 2/126), separately caused by the novel homozygous SYNE1 mutation (NM_033071.3: c.21568C>T, p.Arg7190Ter), and compound heterozygous SYNE1 mutation (NM_033071.3: c.18684G>A, p.Trp6228Ter; c.17944C>T, p.Arg5982Ter), characterized by motor neuron impairment, mental retardation and arthrogryposis.
Conclusions: SYNE1 ataxia exists in the Chinese population, as a rare form of autosomal recessive ataxia, with a complex phenotype. Our findings expanded the ethnic, phenotypic and genetic diversity of SYNE1 ataxia.
Mutations in SYNE1 were first discovered to cause autosomal recessive cerebellar ataxia among French-Canadian pedigrees in 2007, referred to as autosomal recessive cerebellar ataxia type 1 (ARCA1), also called spinocerebellar ataxia autosomal recessive 8 (SCAR8), which is described as an adult-onset, relatively pure cerebellar ataxia (1). Due to the large size of SYNE1, the most giant isoform (nesprin-1 giant or enaptin) of which consists of 146 exons, it is hard to screen SYNE1 gene by conventional Sanger sequencing in ataxia patients. Following recent progress in the development of high throughput sequencing technologies, variants in SYNE1 have been widely reported in ataxia patients from Europe, with other cases from South America, North America, Oceania, Africa, and Asia (2–18). These reported SYNE1 ataxia patients show a strong heterogeneity in clinical features and disease severity, ranging from a pure cerebellar ataxia to a complex multisystem disorder (19). Till now, only four Japanese and two Korean SYNE1 ataxia families were reported in Eastern Asia population, and no mutation of SYNE1 ataxia has been reported among the Chinese population. Here, we screened SYNE1 mutations among a cohort of 126 unrelated index patients with unexplained autosomal recessive or sporadic ataxia in China, by whole-exome sequencing or targeted panel sequencing. Our aim was to search for SYNE1 ataxia patients in China and describe their clinical characteristics.
A cohort of 126 unrelated index patients (31 autosomal recessive, 95 sporadic) were enrolled from the Department of Neurology, Xiangya Hospital of Central South University from 2013 to 2017 (Supplementary Material_Table S1). The age at onset (AAO) was 31.3 ± 14.9 (mean ± SD) ranging from 5 to 59 years. They were diagnosed with unexplained autosomal recessive or sporadic ataxia, in whom repeat expansion disorders including SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA10, SCA12, SCA17, SCA31, SCA36, DRPLA, and Friedreich's ataxia have been excluded. Patients (n = 89) enrolled from January 2013 to November 2016 were screened by targeted panel sequencing, and patients enrolled from December 2016 to December 2017 were screened by whole exome sequencing (n = 37). The decision of change in screening strategy (from targeted panel sequencing to whole exome sequencing) were made in December 2016, due to the increasing evidence of the priority of the whole exome sequencing in undiagnosed inherited and sporadic ataxias over the targeted panel sequencing (20). This study was approved by the Ethics Committee of Xiangya Hospital of Central South University. Written informed consent was obtained from all subjects engaged in this study.
Genomic DNA was extracted from EDTA-anticoagulated blood from index patients using a standard phenol–chloroform method. Additional samples were taken from affected or unaffected relatives to test for segregation analysis when necessary.
Thirty-seven patients were screened for SYNE1 mutations as part of a whole exome-sequencing study. Genomic DNA was fragmented into 250–300 bp length fragments with the use of sonication. The DNA fragments were then processed by end-repairing, A-tailing and adaptor ligation, a 4-cycle pre-capture PCR amplification, and enriched by xGen Exome Research Panel (Integrated DNA Technologies, Skokie, IL, USA). Paired-end sequencing (150 bp) was performed on Illumina HiSeq X-ten platform to provide a mean sequence coverage of more than 100 ×, with more than 95% of the target bases having at least 20 × coverage. The reads were mapped to the human reference genome (UCSC hg19) with the use of the BWA (version 0.7.15, http://bio-bwa.sourceforge.net) (21), duplicate sequence reads were removed by Picard (version 2.10.3; http://picard.sourceforge.net), and GATK (version 3.2, https://software). broadinstitute.org/gatk/was used to detect variants (22). Variants were annotated using Variant Effect Predictor (VEP) (23) and loaded into GEMINI (v0.20. 1) (24). Variants with minor allele frequency of 1% or higher in 1000 Genomes Project (1000 Genomes Phase 3), Genome Aggregation Database (gnomAD r2.0.2) (25) were excluded. Only non-synonymous variants, indels, and putative splice site variants were considered for further analysis. The nomenclature of variants based on the guidelines of the Human Genome Variation Society (HGVS), the DNA and protein changes were determined according to the SYNE1 reference sequence (RefSeq NM_033071.3).
Eighty-nine patients were screened for SYNE1 variants as part of a targeted exon-capture strategy (Agilent SureSelect kit) coupled with multiplexing and high-throughput sequencing of 211 genes causing ataxia, hereditary spastic paraplegia and other related neurological disorders. The Agilent system consists in a capture of the genomic regions harboring the exons, based on the hybridization of complementary designed biotinylated cRNA oligonucleotides. Reads mapping, variant calling, and annotation followed the same method as described above in Whole-exome sequencing.
All the truncating SYNE1 variants were first taken into consideration as it has been established as a main mutation type in ataxia patients. Missense variants were taken into consideration when it meet all the following criteria as Synofzik et al. (8) suggested: (i) segregated in trans with a truncating SYNE1 variant; (ii) was absent or rare in public exome databases (dbSNP, 1000Genomes project, NHLBI ESP6500, ExAc, and gnomAD); (iii) located in highly conserved positions of the N-terminal actin-binding domain (codon 1–289); and (iv) predicted to be damaging by at least two out of three in silico algorithms [Mutation Taster (26); SIFT (27); and PolyPhen-2 (28)].
Sanger sequencing was performed to validate the putative pathogenic variants, allowing segregation analyses where possible (see Supplementary Material_Table S2 for primers employed).
For individuals with putative SYNE1 pathogenic variants, the detailed clinical data were obtained and a clinical examination and evaluation was performed by two experienced neurologists, including the Scale for Assessment and Rating of Ataxia (SARA), Inventory of Non-ataxia Symptoms (INAS), Mini Mental State Examination (MMSE), Montreal Cognitive Assessment (MoCA), Wechsler Intelligence Scale for Children (WISC) or Wechsler Adult Intelligence Scale (WAIS) were performed according to the age of patient at examination. Laboratory tests were operated, including routine blood test, liver and kidney function test, blood glucose test, blood lipid test, ceruloplasmin test and vitamin test (vitamin A/vitamin B1/vitamin B2/vitamin B9/vitamin B12/vitamin C/vitamin D/vitamin E), and myocardial enzyme. MRI scanning of the whole brain, electroencephalography (EEG), electromyography (EMG), nerve conduction study (NCS), electrocardiograph (ECG) were conducted in all the patients with putative SYNE1 pathogenic variants, except for that one patient (Patient II-2 of Family 2 in our study) did not receive EEG examination.
The proband (III-1) was a 16 year old boy from a non-consanguineous family with healthy parents (Figure 1A). He presented with weakness of upper and lower limbs, as well as hand muscle atrophy at the age 10. His medical history was insignificant. In the following time, muscle atrophy gradually spread to the forearm, upper arm, shoulder and pelvic girdles, thigh, crus, and foot muscles. At age 16, he showed an ataxic gait, upper limb ataxia, occasional diplopia, slurred speech, and cognitive decline. Neurological examinations revealed clinical signs of cerebellar ataxia, brisk tendon reflexes, and Babinski signs in lower limbs (Table 1). Muscle atrophy was mostly severe in the thenar muscles and interosseus muscles of hands (Figure 2A), and shoulder girdle muscle (Figure 2B). Bilateral pes cavus (Figure 2C) and mild ankle arthrogryposis were found. Serum CK level was 467 U/L (normal range, 50–310). MRI revealed diffuse cerebellar atrophy (Figure 2D). EEG showed increased slow waves, paroxysmal sharp-slow wave in the frontal and temporal region (Figure 2I). The motor nerve conduction velocities were normal and compound muscle action potentials amplitude decreased (Table 1). The sensory nerve conduction velocities and the sensory nerve action potentials were within the normal range (Table 1). F waves with increased amplitudes were found in the left tibial nerve (Supplementary Material_Figure S1).
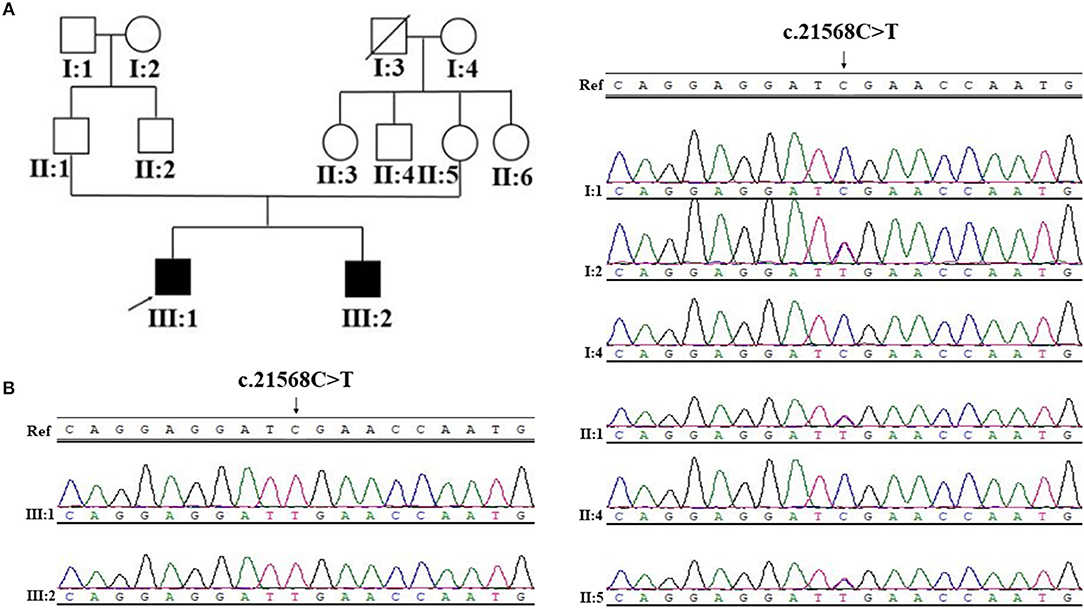
Figure 1. Pedigree structure, and Sanger sequencing of SYNE1 variant (c.21568C>T) of Family 1. (A) Pedigree structure. (B) Segregation of SYNE1 variant identified in subjects of Family 1, whose DNA sample were available. Ref: reference sequence in NCBI.
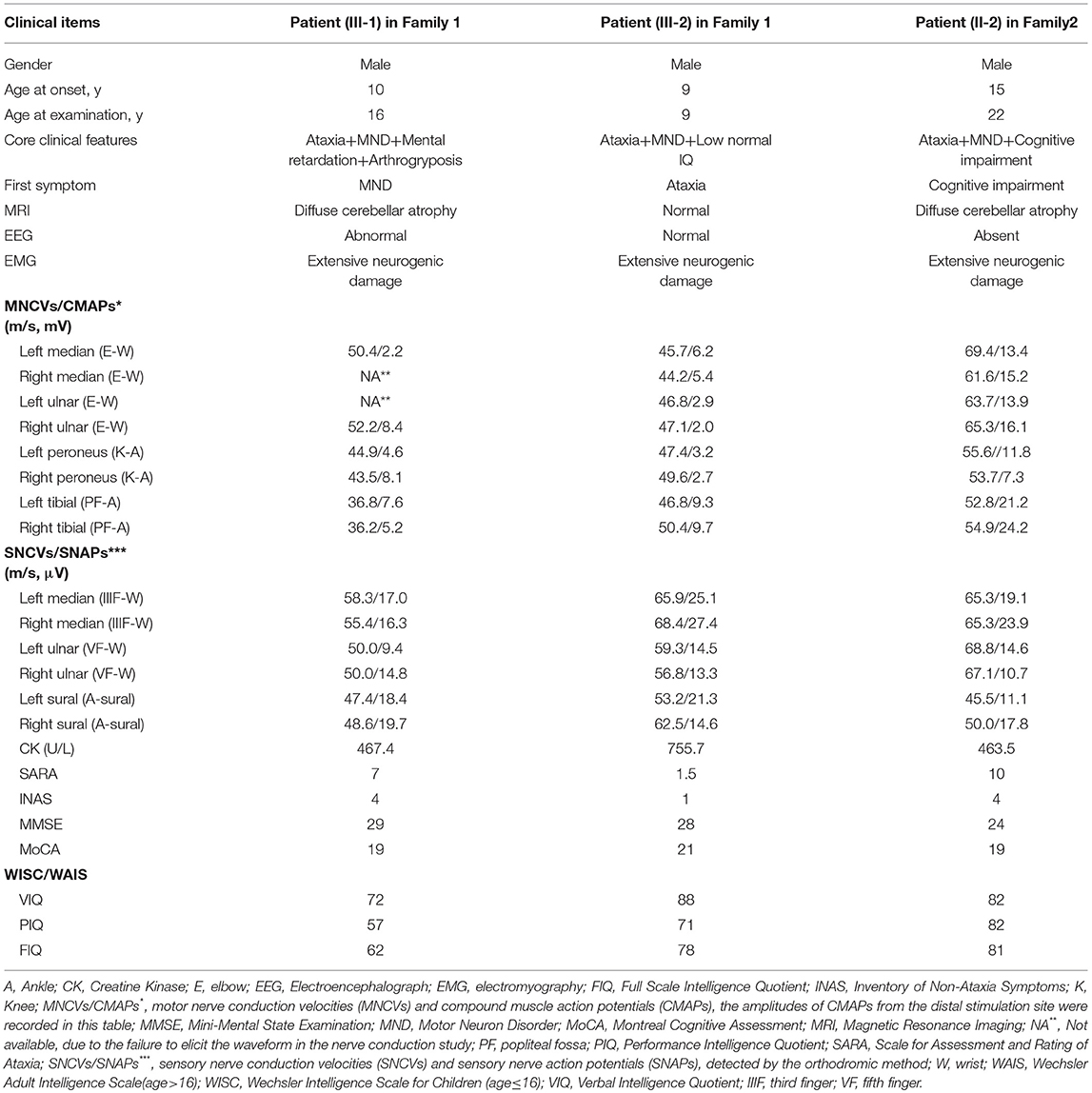
Table 1. Clinical characteristics of SYNE1 ataxia patients in this study.
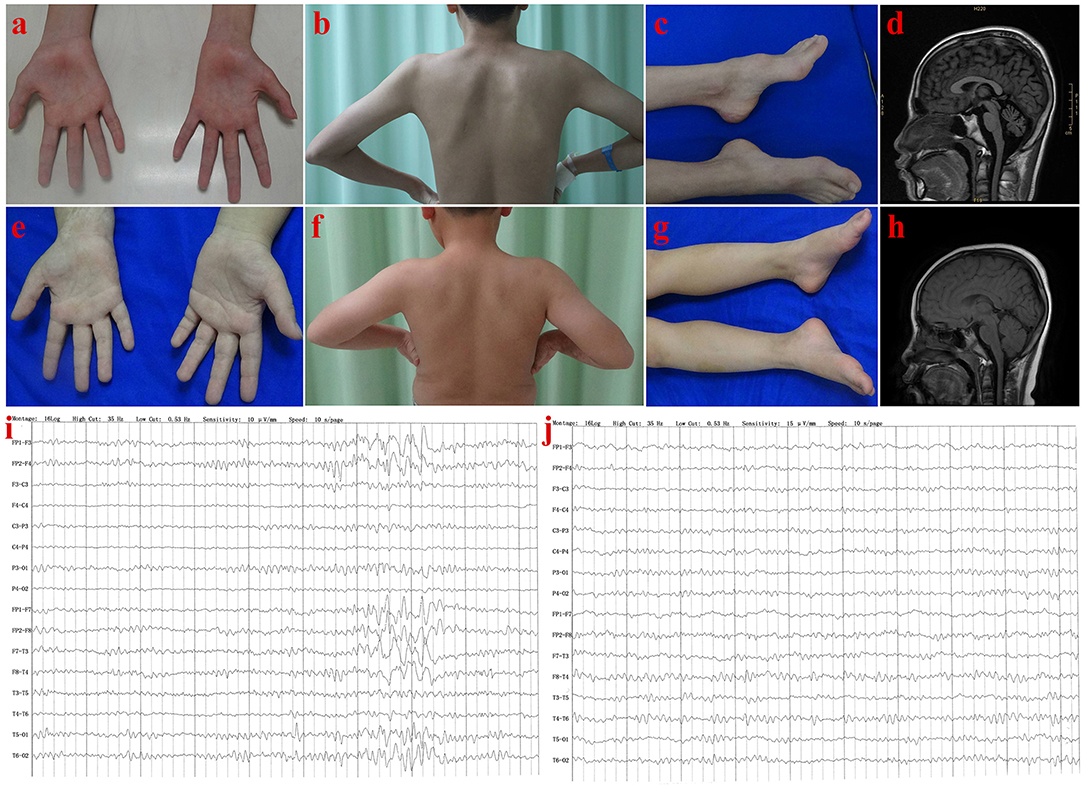
Figure 2. Clinical features of patients in Family 1. Patient III-1: Muscle atrophy in the thenar muscle, interosseous muscle of hands (A), and shoulder girdle muscle (B); bilateral pes cavus (C); diffuse cerebellar atrophy on T1-weighted MRI (D); EEG showed increased slow waves, paroxysmal sharp-slow wave in the frontal and temporal region (I). Patient III-2: No muscle atrophy was found in hand (E), and shoulder girdle muscle (F), with normal appearance of feet (G); T1-weighted MRI of the brain (H), and EEG (J) were normal.
Needle EMG revealed very little or no spontaneous activity (fibrillation, positive sharp wave, and fasciculation potentials) in all investigated muscles (musculus quadratus labii inferioris, sternocleidomastoid, paravertebral muscle, biceps, abductor digiti minimi, tibialis anterior muscle). Large amplitude, long duration motor unit potentials, as well as reduced recruitment were shown in the muscles mentioned above. In summary, this patient (III-1) showed an obvious motor neuron disorder phenotype as its initial and dominant symptom, which mimicked juvenile-onset amyotrophic lateral sclerosis (ALS), accompanying with relatively slight cerebellar ataxia, mental retardation (FIQ < 70), and ankle arthrogryposis (Table 1).
The younger brother (III-2) of the index patient was 9 years old. He reported no subjective symptoms at first. It was only when we told him the abnormalities in his routine neurological examination, he recalled that he showed difficulty in standing alone by one foot when playing games in the past 3 months. Physical examinations showed mild abnormalities in tandem step test, finger nose test of left hand, and normal muscle volume. EMG and NCV results were similar to his brother (III-1). The main clinical features were summarized in Table 1 and shown in Figure 2. In summary, this patient (III-2) showed mild ataxia and subclinical motor neuron disorder (summarized in Table 1).
The proband (II-2) was a 22 year old man from a non-consanguineous family with healthy parents and elder sister (Figure 3A). He firstly reported cognitive decline at the age 15, especially for the difficulty in study. At age 20, he developed a spastic-ataxic gait, followed by slurred speech. Neurological examinations revealed clinical signs of cerebellar ataxia, increased muscle tension in lower limbs, hyperreflexia in upper and lower limbs, positive pathologic reflexes, ankle clonus, with normal muscle strength, and volume (Table 1 and Figure 3). MRI of the brain showed diffuse cerebellar atrophy (Figure 3B). NCS demonstrated normal sensory nerve conduction and motor nerve conduction (Table 1). Needle EMG revealed very little or no spontaneous activity (fibrillation, positive sharp wave, and fasciculation potentials) in multiple muscles (musculus quadratus labii inferioris, sternocleidomastoid, paravertebral muscle, biceps, abductor digiti minimi, tibialis anterior muscle). Large amplitude, long duration motor unit potentials, as well as reduced recruitment were shown in these muscles. In summary, this patient (II-2) shows as a mild ataxia phenotype, and subclinical motor neuron disorder, with cognitive decline.
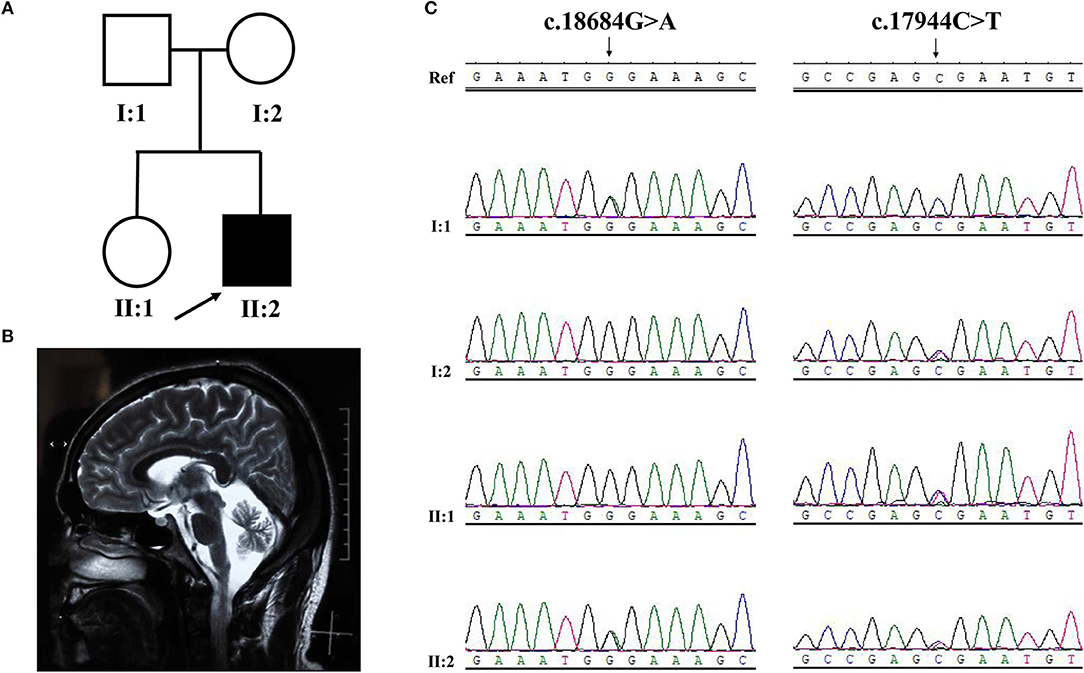
Figure 3. Pedigree structure, clinical features and Sanger sequencing of SYNE1 variant of subjects in Family 2. (A) Pedigree structure. (B) Diffuse cerebellar atrophy on T2-weighted MRI. (C) Segregation of SYNE1 variants identified in subjects of Family 2, whose DNA sample were available. Ref: reference sequence in NCBI.
Among the 126 patients, 2 index patients were identified separately with the homozygous SYNE1 variant (c.21568C>T, p.Arg7190Ter; Family 1) and compound heterozygous SYNE1 variants (c.18684G>A, p.Trp6228Ter; c.17944C>T, p.Arg5982Ter; Family 2) in high-throughput screening.
Sanger sequence identifies the segregation of the novel SYNE1 truncating variant (c.21568C>T, p.Arg7190Ter) in family I (Figure 1B). This variant was absent or extremely rare in the population database (Table 2), and was not found in disease database (Clin Var, OMIM, HGMD, and literatures in PubMed till to Nov 6, 2018). It was predicted as disease causing by Mutation Taster (26), a high position-specific conservation score by GERP (30), and a likelihood of high pathogenicity by CADD (31) (Table 2). According to ACMG guidelines (29), the novel SYNE1 mutation (c.21568C>T) is categorized to be the disease “pathogenic variant” because it belongs to PVS1 (non-sense mutation and loss of function of SYNE1 has been validated to cause SYNE1 ataxia), PM2 (extremely low population frequency if recessive), PP1 (segregation with phenotype in family), and PP4 (phenotype of this patient coincide with previously reported SYNE1 related patients).
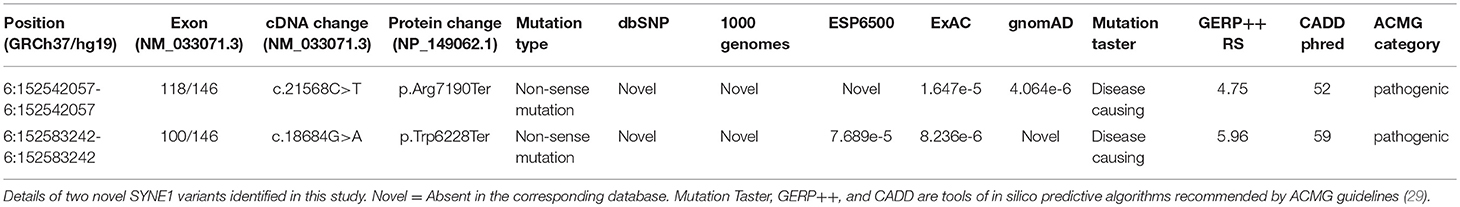
Table 2. Details of the two novel SYNE1 variants found in this study.
Sanger sequence identified the segregation of SYNE1 variants (c.18684G>A; c.17944C>T) in Family 2 (Figure 3C). The variant (c.17944C>T; p.Arg5982Ter), has been reported to cause SYNE1 ataxia previously (8). The novel truncating variant (c.18684G>A; p.Trp6228Ter) was extremely rare or absent in population database, absent in disease database (Clin Var, OMIM, HGMD, and literatures in PubMed), and predicted as disease causing by Mutation Taster, a high position-specific conservation score by GERP, and a likelihood of high pathogenicity by CADD (Table 2). Similar to above, the novel SYNE1 variant (c.18684G>A) should also be categorized to be the disease “pathogenic variant” because it belongs to PVS1, PM2, PP1, PP4, as well as PM3 (for recessive disorders, detected in trans with a pathogenic variant) according to ACMG guidelines.
The recessive spinocerebellar ataxias (ARCAs or SCARs) are a complex group of neurodegenerative diseases with significant genetic and clinical heterogeneity, as well as regional and ethnic differences. Previous studies showed that SYNE1 ataxia accounted for 5.3% (23/434), 6% (7/116), 2% (4/196), 0.9% (1/110) separately in four large independent cohorts of recessive and sporadic ataxia patients (7–10). In this study, we identified 2 unrelated index ataxia patients with novel SYNE1 truncating variants, accounting for 1.6% (2/126) in this Chinese cohort of recessive and sporadic ataxia patients. This is the first report of SYNE1 ataxia in China.
The recognition of phenotype of SYNE1 ataxia has been going on for a long time. SYNE1 ataxia was firstly described as an adult-onset, relatively pure cerebellar ataxia in 2007. In 2013, it was first reported as another form of early onset ataxia (age at onset: 6) with motor neuron disorder in Japan, which mimicked the phenotype of juvenile-onset amyotrophic lateral sclerosis (ALS) (2). Gradually, a growing number of ataxia patients with a wide range of extra-cerebellar neurological and non-neurological dysfunctions, have been found with pathogenic recessive SYNE1 mutation, in different cohorts of ataxia patients from Europe (8–10, 14). Reported extra-cerebellar neurological and non-neurological dysfunctions included motor neuron disorder, mental retardation and cognitive impairment, depression, strabismus, ophthalmoparesis, bulging eyes, dystonia, reduced vibration sense, urge incontinence, kyphosis, scoliosis, pseudarthrosis clavicula, pes cavus, achilles tendon contractures, respiratory distress, macroglossia, sacral cyst, malrotation colon, splanchnectopia et al. Due to the above findings, Synofzik et al. proposed a new correct recognition of SNYE1 ataxia as multisystemic ataxia in 2016 (10).
The present Chinese study provided new evidence supporting this suggestion. All the three ataxia patients shared a common character of ataxia syndrome, with motor neuron disorder, and mental retardation or cognitive decline. It is noteworthy that the patient III-1 in Family 1 showed a mild arthrogryposis in ankle, in addition to symptoms mentioned above. This is the second reported SYNE1 ataxia case combined with arthrogryposis (information about the previously reported patient available in Summary patient ID 52, Supplementary Material_Table S3). EEG abnormalities in SYNE1 ataxia had never been reported before. In this study, increased slow wave activity and paroxysmal sharp-slow waves in the frontal and temporal region were showed in the EEG of patient (III-1) in Family 1. The common pathologies contributing to EEG abnormalities, including seizure, cerebrovascular diseases, traumatic brain injury, central nervous system infections, central nervous demyelination, have been excluded by the clinical observation. Accordingly, the EEG abnormality in this patient, should be attributed to the cerebral cortex impairment caused by SYNE1 mutation.
Another noteworthy finding in this study was EMG results, which may be beneficial for the differentiation diagnosis from juvenile ALS. Notwithstanding the different progression stage of these three patients, they shared the same characters in the Needle EMG: spontaneous activity (fibrillation, positive sharp wave, and fasciculation potentials) were very few or absent, but large amplitude, long duration motor unit potentials and reduced recruitment were very common. These results were different from typical EMG characters of juvenile ALS, which are obvious signs of denervation (positive sharp waves and fibrillation potentials) and fasciculation potentials, in combination with signs of reinnervation (large amplitude, long duration motor unit potentials, and reduced recruitment) in multiple regions (32). In contrast, EMG results in three patients in our study, revealed no signs of denervation, but only signs of reinnervation. These differences in EMG may be attributed to a slower pathological process of motor neurons in SYNE1 ataxia, compared with juvenile ALS. Due to the similar clinical presentation, which includes both upper and lower motor neuron dysfunction, it is hard to make an accurate differential diagnosis between juvenile ALS and SYNE1 ataxia, especially when the ataxia symptoms are very slight. There is a reason to assume that, the difference in EMG characters may add valuable information to the differential diagnosis between juvenile ALS and SYNE1 ataxia. Research of EMG in more SYNE1 ataxia patients is warranted to elucidate this hypothesis in the future.
The correlation between genotype and phenotype has not been clearly elucidated in SYNE1 related diseases. Except for SYNE1 ataxia, SYNE1 variants have been found to cause other Mendelian disease, including Emery-Dreifuss muscular dystrophy 4 (EDMD4) (33), and arthrogryposis multiplex congenital (AMC) (34). In addition, it also has been associated with some other phenotype or complex diseases, including congenital muscular dystrophy (CMD), dilated cardiomyopathy (DCM), autism (AUT), mental retardation (MTD), schizophrenia.
SYNE1 is one of the largest human gene consisting of 515, 716 bp, and 146 exons (35). SYNE1 encodes a member of the spectrin family of structural proteins that participate in a complex that links the nucleoskeleton to the cytoskeleton (LINC), and links the plasma membrane to the actin cytoskeleton (36). SYNE1 has multiple alternative start and termination sites, allowing the generation of alternating isoforms. The largest isoform (NM_182961) of SYNE1 contains and encodes a 27748–base pair messenger RNA, and translates into an 8797–amino acid protein, noted as full-length Nesprin-1 (Nesprin-1 giant), started with two N-terminal actin-binding regions that comprise tandem paired calponin-homology domain (CH), followed by 74 spectrin repeats (SRs), and ending with the C-terminal klarsicht domain (KASH; Figure 4). Apart from full-length nesprin-1, at least 16 KASH- containing, 14 CH-containing, and 6 Spectrin repeat (SR)-only SYNE1 isoforms were reported (37). Razafsky et al. (38) noted that Nesprin-1 giant, and one of the CH-containing isoforms devoid of the KASH (KLNes1g) are specifically expressed in the central nervous system, and KLNes1g is mostly abundantly expressed in the cerebellum. One of KASH-containing isoforms (nesprin-1α2), devoid of CH and lots of SRs, is a predominantly muscle-specific isoform (39).

Figure 4. All of the reported pathogenic SYNE1 variants, and their locations in the schematic diagram of Nesprin-1 and some of the major alternating isoforms of SYNE1. Locations of the variants found in the present study are denoted by the arrow head. Circle variants are missense variants, rectangles are non-sense variants, triangles are frameshift variants, and rhombus are splice site variants. Biallelic variants are shown in a solid color. ATX pure, patients with pure ataxia; ATX plus, ataxia patients with some other non-ataxia symptoms; AMC, arthrogryposis multiplex congenital; EDMD, Emery-Dreifuss muscular dystrophy; CMD, congenital muscular dystrophy; DCM, dilated cardiomyopathy; AUT, autism; MTD, mental retardation as the major syndrome.
To explore the correlation between genotype and phenotype, we summarized all reported patients with SYNE1 variant related phenotype (Figure 4, and Supplementary Material_Table S3).
In SYNE1 ataxia patients, causative variants are distributed across the entire gene, except for the KASH domain, and without obvious hot mutation spot (Figure 4). This distribution of variant location supports the hypothesis that the loss of function of KLNes1g isoform, which is devoid of the KASH domain of Nesprin-1 giant, underlies the molecular etiology of ARCA1 (38). No obvious difference of variant distribution was observed between patients with pure ataxia (named as ATX pure in Figure 4), and patients with ataxia plus some other non-ataxia symptoms (named as ATX plus in Figure 4). Three interesting non-ataxia features including arthrogryposis, EEG abnormalities, and motor neuron disorder, were observed in our study which deserves further investigation. The patient (III-1) in Family 1 is the second reported SYNE1 ataxia case with arthrogryposis. Coincidentally, the variants of the only two SYNE1 ataxia patients with arthrogryposis, were all located in a very close region (SR61-SR63) of SYNE1 protein. In addition, the SR61-SR63 is very close to the KASH domain, a vital domain involved in the pathogenesis of AMC (34). This may suggest that variants in SR61-SR63 of SYNE1 protein would increase the susceptibility to arthrogryposis. It is possible that SR61-SR63 is an important overlapping functional region for both nerve and muscle tissue. As EEG abnormalities in SYNE1 ataxia were only reported in our case [patient (III-1) in Family 1], it is still hard to evaluate the potential association between variant site and EEG abnormalities. The motor neuron disorder was the most frequent non-ataxia phenotype in SYNE1 ataxia. However, no hot mutation spot was observed in all reported SYNE1 ataxia patients with motor neuron disorders (Supplementary Material_Table S4). Nevertheless, more studies are needed to elucidate the potential correlation between variant sites and non-ataxia symptoms in SYNE1 ataxia.
In AMC, all the causative mutations were located in KASH domain or neighboring region of KASH, which would be expected to affect most of the KASH-containing isoforms, especially for Nesprin-1α2, one of the predominantly muscle-specific isoforms of SYNE1 (39, 40). In EDMD, CMD, DCM, AUT, MTD, Microcephaly, and Schizophrenia, relationship between variant location and phenotype was unremarkable. The pathological mechanism underlying the SYNE1 mutations responsible for phenotypic variations remains to be elucidated in future.
In conclusion, we reported the first two Chinese SYNE1 ataxia families caused by novel SYNE1 mutations, accompanied by motor neuron impairment, mental retardation, with or without arthrogryposis. Our findings expand the ethnic, phenotypic, and genetic diversity of SYNE1 ataxia.
This study was approved by the Ethics Committee of Xiangya Hospital, Central South University in China, which was in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. And written informed consent was obtained from all subjects. We also obtained written and informed consent from the patients who gave specific permission to publish the data.
YP and HJ: conceived and designed the experiments. YP, WY, and ZC: performed the experiments. YP, ZC, HP, and PW: analyzed the data. YP, XuH, CW, XZ, XiH, TL, RQ, ZH, BT, and HJ: contributed reagents, materials, analysis tools. YP and ZC: contributed to the writing of the manuscript. ZH and HJ: reviewed and critiqued the first manuscript.
This study was supported by National Key Research and Development Program of China (2016YFC0905100 and No. 2016YFC0901504 to HJ, 2016YFC1306000 to BT), the National Natural Science Foundation of China (No. 81471156, 81771231 to HJ; No. 81430023, 81130021 to BT; No.31401135 to RQ), Key Research and Development Program of Hunan Province (No. 2018SK2092 to HJ), Clinical Research Funds of Xiangya Hospital (No. 2014L03 to HJ), the Clinical and rehabilitation Funds of Peking University Weiming Biotech Group (No. xywm2015I10 to HJ), and Youth Foundation of Xiangya Hospital (No. 2017Q03 to ZC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like thanking patients and their family members for their generous participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2018.01111/full#supplementary-material
1. Gros-Louis F, Dupre N, Dion P, Fox MA, Laurent S, Verreault S, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. (2007) 39:80–5. doi: 10.1038/ng1927
2. Izumi Y, Miyamoto R, Morino H, Yoshizawa A, Nishinaka K, Udaka F, et al. Cerebellar ataxia with SYNE1 mutation accompanying motor neuron disease. Neurology (2013) 80:600–1. doi: 10.1212/WNL.0b013e3182815529
3. Noreau A, Bourassa CV, Szuto A, Levert A, Dobrzeniecka S, Gauthier J, et al. SYNE1 mutations in autosomal recessive cerebellar ataxia. JAMA Neurol. (2013) 70:1231–96. doi: 10.1001/jamaneurol.2013.3268
4. Fogel BL, Lee H, Deignan JL, Strom SP, Kantarci S, Wang X, et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. (2014) 71:1237–46. doi: 10.1001/jamaneurol.2014.1944
5. Ozoguz A, Uyan O, Birdal G, Iskender C, Kartal E, Lahut S, et al. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol Aging (2015) 36:1764–9. doi: 10.1016/j.neurobiolaging.2014.12.032
6. Keogh MJ, Steele H, Douroudis K, Pyle A, Duff J, Hussain R, et al. Frequency of rare recessive mutations in unexplained late onset cerebellar ataxia. J Neurol. (2015) 262:1822–7. doi: 10.1007/s00415-015-7772-x
7. Hamza W, Ali PL, Hamadouche T, Muller J, Drouot N, Ferrat F, et al. Molecular and clinical study of a cohort of 110 Algerian patients with autosomal recessive ataxia. BMC Med Genet. (2015) 16:36. doi: 10.1186/s12881-015-0180-3
8. Synofzik M, Smets K, Mallaret M, Di Bella D, Gallenmuller C, Baets J, et al. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain (2016) 139:1378–93. doi: 10.1093/brain/aww079
9. Wiethoff S, Hersheson J, Bettencourt C, Wood NW, Houlden H. Heterogeneity in clinical features and disease severity in ataxia-associated SYNE1 mutations. J Neurol. (2016) 263:1503–10. doi: 10.1007/s00415-016-8148-6
10. Mademan I, Harmuth F, Giordano I, Timmann D, Magri S, Deconinck T, et al. Multisystemic SYNE1 ataxia: confirming the high frequency and extending the mutational and phenotypic spectrum. Brain (2016) 139:e46. doi: 10.1093/brain/aww115
11. Algahtani H, Marzouk Y, Algahtani R, Salman S, Shirah B. Autosomal recessive cerebellar ataxia type 1 mimicking multiple sclerosis: a report of two siblings with a novel mutation in SYNE1 gene in a Saudi family. J Neurol Sci. (2017) 372:97–100. doi: 10.1016/j.jns.2016.11.036
12. Yucesan E, Ugur IS, Bilgic B, Gormez Z, Bakir GB, Sarac A, et al. SYNE1 related cerebellar ataxia presents with variable phenotypes in a consanguineous family from Turkey. Neurol Sci. (2017) 38:2203–7. doi: 10.1007/s10072-017-3049-8
13. Yoshinaga T, Nakamura K, Ishikawa M, Yamaguchi T, Takano K, Wakui K, et al. A novel frameshift mutation of SYNE1 in a Japanese family with autosomal recessive cerebellar ataxia type 8. Hum Genome Var. (2017) 4:17052. doi: 10.1038/hgv.2017.52
14. Coutelier M, Hammer MB, Stevanin G, Monin ML, Davoine CS, Mochel F, et al. Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. Jama Neurol. (2018) 75:591–9. doi: 10.1001/jamaneurol.2017.5121
15. Kim JS, Kim AR, Youn J, Lee C, Kim NS, Park WY, et al. Identifying SYNE1 ataxia and extending the mutational spectrum in Korea. Parkinsonism Relat Disord. (2018) S1353-8020(18)30360-2. doi: 10.1016/j.parkreldis.2018.08.009
16. Swan L, Cardinal J, Coman D. SYNE1-related autosomal recessive cerebellar ataxia, congenital cerebellar hypoplasia, and cognitive impairment. Clin Pract. (2018) 8:1071. doi: 10.4081/cp.2018.1071
17. Gama M, Piccinin CC, Rezende T, Dion PA, Rouleau GA, Franca JM, et al. Multimodal neuroimaging analysis in patients with SYNE1 Ataxia. J Neurol Sci. (2018) 390:227–30. doi: 10.1016/j.jns.2018.05.003
18. Sun M, Johnson AK, Nelakuditi V, Guidugli L, Fischer D, Arndt K, et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet Med. (2018). doi: 10.1038/s41436-018-0007-7 [Epub ahead of print].
19. Synofzik M, Nemeth AH. Recessive ataxias. Handb Clin Neurol. (2018) 155:73–89. doi: 10.1016/B978-0-444-64189-2.00005-6
20. Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, Appleton M, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain (2015) 138:276–83. doi: 10.1093/brain/awu348
21. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
22. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
23. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl variant effect predictor. Genome Biol. (2016) 17:122. doi: 10.1186/s13059-016-0974-4
24. Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. (2013) 9:e1003153. doi: 10.1371/journal.pcbi.1003153
25. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60, 706 humans. Nature (2016) 536:285–91. doi: 10.1038/nature19057
26. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods (2010) 7:575–6. doi: 10.1038/nmeth0810-575
27. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. (2012) 40:W452–7. doi: 10.1093/nar/gks539
28. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods (2010) 7:248–9. doi: 10.1038/nmeth0410-248
29. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
30. Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. (2010) 6:e1001025. doi: 10.1371/journal.pcbi.1001025
31. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
32. Agarwal S, Potocki L, Collier TR, Woodbury SL, Adesina AM, Jones J, et al. Utility of whole exome sequencing in evaluation of juvenile motor neuron disease. Muscle Nerve. (2016) 53:648–52. doi: 10.1002/mus.25030
33. Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, et al. Nesprin-1 and−2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. (2007) 16:2816–33. doi: 10.1093/hmg/ddm238
34. Attali R, Warwar N, Israel A, Gurt I, McNally E, Puckelwartz M, et al. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum Mol Genet. (2009) 18:3462–9. doi: 10.1093/hmg/ddp290
35. Shmueli O, Horn-Saban S, Chalifa-Caspi V, Shmoish M, Ophir R, Benjamin-Rodrig H, et al. GeneNote: whole genome expression profiles in normal human tissues. C R Biol. (2003) 326:1067–72. doi: 10.1016/j.crvi.2003.09.012
36. Cartwright S, Karakesisoglou I. Nesprins in health and disease. Semin Cell Dev Biol. (2014) 29:169–79. doi: 10.1016/j.semcdb.2013.12.010
37. Rajgor D, Shanahan CM. Nesprins: from the nuclear envelope and beyond. Expert Rev Mol Med. (2013) 15:e5. doi: 10.1017/erm.2013.6
38. Razafsky D, Hodzic D. A variant of Nesprin1 giant devoid of KASH domain underlies the molecular etiology of autosomal recessive cerebellar ataxia type I. Neurobiol Dis. (2015) 78:57–67. doi: 10.1016/j.nbd.2015.03.027
39. Zhang Q, Skepper JN, Yang F, Davies JD, Hegyi L, Roberts RG, et al. Nesprins: a novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. J Cell Sci. (2001) 114:4485–98.
Keywords: cerebellar ataxia, SYNE1, mutation, high-throughput nucleotide sequencing, Genotype–Phenotype
Citation: Peng Y, Ye W, Chen Z, Peng H, Wang P, Hou X, Wang C, Zhou X, Hou X, Li T, Qiu R, Hu Z, Tang B and Jiang H (2018) Identifying SYNE1 Ataxia With Novel Mutations in a Chinese Population. Front. Neurol. 9:1111. doi: 10.3389/fneur.2018.01111
Received: 16 September 2018; Accepted: 04 December 2018;
Published: 20 December 2018.
Edited by:
Giuseppe De Michele, University of Naples Federico II, ItalyReviewed by:
Cyril Goizet, Université de Bordeaux, FranceCopyright © 2018 Peng, Ye, Chen, Peng, Wang, Hou, Wang, Zhou, Hou, Li, Qiu, Hu, Tang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Jiang, amlhbmdob25nNzM4NjhAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.