 Adonis Sfera
Adonis Sfera Roberto Gradini
Roberto Gradini Michael Cummings2
Michael Cummings2 Eddie Diaz
Eddie Diaz Amy I. Price
Amy I. Price- 1Psychiatry, Loma Linda University, Loma Linda, CA, United States
- 2Patton State Hospital, San Bernardino, CA, United States
- 3Department of Pathology, La Sapienza University of Rome, Rome, Italy
- 4Evidence Based Medicine, University of Oxford, Oxford, United Kingdom
Alzheimer's disease, the most common form of dementia, is marked by progressive cognitive and functional impairment believed to reflect synaptic and neuronal loss. Recent preclinical data suggests that lipopolysaccharide (LPS)-activated microglia may contribute to the elimination of viable neurons and synapses by promoting a neurotoxic astrocytic phenotype, defined as A1. The innate immune cells, including microglia and astrocytes, can either facilitate or inhibit neuroinflammation in response to peripherally applied inflammatory stimuli, such as LPS. Depending on previous antigen encounters, these cells can assume activated (trained) or silenced (tolerized) phenotypes, augmenting or lowering inflammation. Iron, reactive oxygen species (ROS), and LPS, the cell wall component of gram-negative bacteria, are microglial activators, but only the latter can trigger immune tolerization. In Alzheimer's disease, tolerization may be impaired as elevated LPS levels, reported in this condition, fail to lower neuroinflammation. Iron is closely linked to immunity as it plays a key role in immune cells proliferation and maturation, but it is also indispensable to pathogens and malignancies which compete for its capture. Danger signals, including LPS, induce intracellular iron sequestration in innate immune cells to withhold it from pathogens. However, excess cytosolic iron increases the risk of inflammasomes' activation, microglial training and neuroinflammation. Moreover, it was suggested that free iron can awaken the dormant central nervous system (CNS) LPS-shedding microbes, engendering prolonged neuroinflammation that may override immune tolerization, triggering autoimmunity. In this review, we focus on iron-related innate immune pathology in Alzheimer's disease and discuss potential immunotherapeutic agents for microglial de-escalation along with possible delivery vehicles for these compounds.
Highlights
- LPS activates microglial TLRs, training these cells excessively and triggering the release of C1q, TNF-alpha, and IL-1 alpha, biomolecules associated with autoimmunity.
- A1 astrocytes, likely autoimmune, eliminate viable neurons and oligodendrocytes, contributing to Alzheimer's disease neuronal and synaptic loss.
- LPS, a danger signal, promotes intracellular iron sequestration, increasing the risk of ROS. LPS also promotes ferritinophagy, increasing the free intracellular iron levels.
- ROS and iron activate NLRP3 inflammasomes, generating prolonged inflammation which may override microglial tolerization, engendering autoimmune A1 astrocytes.
- Promoting tolerization and lowering training may de-escalate microglia, lowering the risk of neuronal loss and Alzheimer's disease.
Introduction
Alzheimer's disease (AD), the most common form of dementia, is marked by progressive memory impairment and functional decline believed to reflect synaptic and neuronal loss. Elimination of these structures may be carried out by an aggressive astrocytic phenotype, defined as A1. Preclinical studies show that LPS-activated microglia enable the conversion of trophic to A1 astrocytes by releasing several cytokines, including tumor necrosis factor alpha (TNF-alpha), interlukin-1 alpha (IL-1 alpha), and complement component C1q. These molecules alter astrocytic transcriptomes, promoting the neurotoxic A1 cells observed to engage in the phagocytosis of healthy neurons and oligodendrocytes, contributing to their loss (1). In this article we take the position that A1 astrocytes are autoimmune in nature, resulting from the prolonged microglial activation that overrides LPS tolerization.
LPS tolerization refers to the absence of an inflammatory response after repeated or prolonged exposure to this microbial endotoxin as re-challenged “tolerant” innate immune cells are incapable of immunological activation. This is relevant to AD because the constantly elevated brain LPS levels, detected in this disorder, fail to trigger microglial tolerization and lower neuroinflammation (2, 3).
Excess LPS in the brain microenvironment, a danger signal, is detected by microglial TLRs which activate the pathogen-mediated iron sequestration via hepcidin-ferroportin axis (4) (Figure 1). Indeed, recent neuroimaging studies detected iron-containing microglia in the hippocampi of AD patients, likely reflecting LPS-triggered iron sequestration (5).
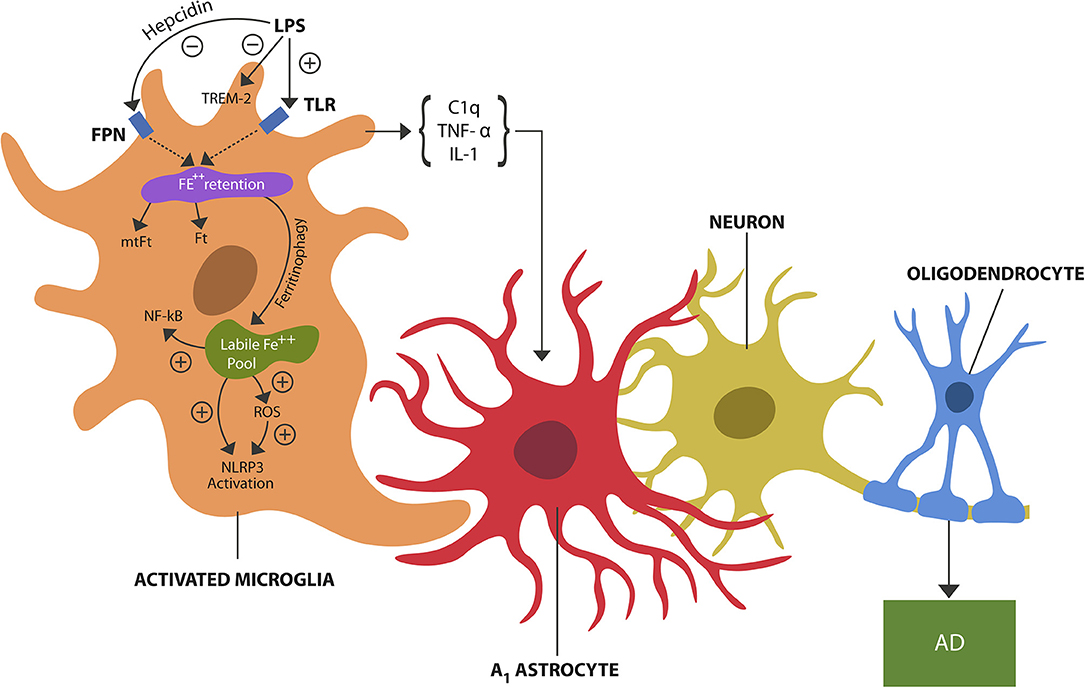
Figure 1. LPS, a danger signal, binds to TLRs of microglia, inducing pathogen-mediated intracellular iron sequestration. LPS also promotes astrocytic hepcidin synthesis which via FPN prevents iron egress. In addition, LPS inhibits TREM-2 expressed on microglial cells. Intracellular iron is stored in Ft and mtFt. Iron can dissociate from ferritins via ferritinophagy, augmenting the free iron (labile iron pool). Unbound iron directly and indirectly (via ROS) activates NLRP3 and NF-kB, inducing microglial activation (training). Microglial release of cytokines, including TNF-alpha, IL-1, and C1q induces A1 astrocytes shown to engage in the phagocytosis of viable neurons and oligodendrocytes, contributing to neuronal loss and possibly AD.
The source of brain LPS is unclear at this time, but it has been hypothesized that it may derive from the gastrointestinal (GI) tract microbial community. These bacteria were found to lie dormant in various tissues, possibly including the brain, for prolonged periods of time restrained by the lack of free iron (3, 6). Later in life, impaired iron homeostasis caused by aging and/or defective ferritinophagy may provide these microbes with enough metal to resume their growth and LPS shedding, engendering inflammatory neuropathology (6).
Iron is closely linked to immunity as it plays a key role in immune cells proliferation and maturation, but it is also indispensable to pathogens and malignancies which compete for its capture (discussed in The Trained Arm of Innate Immunity section). To safeguard iron, innate immune cells, including microglia and astrocytes, sequestrate it intracellularly bound to cytosolic and mitochondrial ferritin, an arrangement which can generate excessive ROS and inflammation when disrupted (4). Indeed, iron-ferritin dissociation was demonstrated in AD animal models in which LPS-induced ferritinophagy increased intracellular free iron, engendering neuroinflammation (7, 8). Moreover, iron-damaged mitochondria were shown to act as danger signals, activating the inflammatory cascade by igniting inflammasomes, including NLRP3 (9).
LPS-activated microglia release cytokines, including TNF-alpha, IL-1 alpha, and C1q, that have been associated with autoimmune inflammation, an association that led to the development of vaccines, including anti-TNF-alpha antibodies (for the treatment of autoimmune disorders) and anti-C1q antibody, currently in phase I trials for both AD and autoimmunity (10, 11).
In contrast to immune tolerization which may be an adaptive mechanism to prevent autoimmune inflammation, allowing the clearance of damaged self-components, immune training may serve the purpose of overriding immune anergy encountered in various pathologies, including sepsis and cancer (12, 13). Indeed, the peripheral and central innate immune cells are engaged in a balancing act as they must be sufficiently trained to effectively defend the host against pathogens and malignancies, while at the same time tolerant enough to accept self-components, food, the fetus and commensal microbes. Disruption of this fine equilibrium may either cause unchecked inflammation or excessive tolerance (12). Iron interferes with this balance by promoting activation and inflammation, a property exploited in oncology to train tumor-associated macrophages (TAMs) in fighting malignant cells (14). On the other hand, iron chelators oppose training, providing therapeutic benefit in AD. Indeed, deferiprone, currently in phase 2 clinical trials, may soon become the first FDA approved iron chelator indicated for AD (15). Along these lines, a recent preclinical study demonstrated that in response to peripherally applied inflammatory stimuli, microglia can undergo either activation (training) or tolerization, the former augmenting, the latter lowering neuroinflammation (13). This is important for AD because this study also found that training increases while tolerization decreases β-amyloid accumulation, suggesting that central tolerance can be promoted by peripheral stimulation, comprising a novel therapeutic strategy (13). Previous AD studies are in line with this model as it was demonstrated that immune trained subjects (positive for TNF-alpha) presented with memory loss, while the tolerized patients (TNF-alpha negative) lacked cognitive deterioration (16). Moreover, trained microglia and astrocytes were found able to undergo transcriptomic alterations, morphing into antigen presenting cells (APCs) that can establish immunologic synapses with infiltrating peripheral lymphocytes, further perpetuating neuroinflammation (17).
Traditionally, immunological memory has been associated with adaptive immunity, however novel studies indicate that innate immune cells, including microglia and astrocytes can “recall” prior vaccinations or infections (13). For example, β-glucan, a cell wall component of Candida Albicans and bacillus Calmette-Guerin (BCG), was shown to train monocytes, improving the outcome of sepsis-induced immune paralysis (18). Indeed, the development of treatments based on innate immune memory manipulation has been encouraged by the recent FDA approval of immune training-inducers, the BCG vaccine for the treatment and prophylaxis of urinary bladder carcinoma in situ (CIS) and muramyl tripeptide for osteosarcoma (19). Along these lines, innate immune memory manipulation may improve AD outcome by de-escalating microglia via peripherally-applied tolerizing stimuli (13). Since tolerized immunity, unlike immunosuppression, is antigen specific and natural, harnessing it may not only benefit AD but also cancer and autoimmune disorders (20, 21). The recent finding that altered endogenous molecules, including dysfunctional mitochondria, can trigger microglial training will likely result in novel AD strategies, including mitophagy-activating therapeutics (22).
In this article we look beyond iron chelators, focusing on lowering the consequences of iron pathology, especially microglial training and defective tolerization. The immunotherapeutic agents discussed here include inflammasome inhibitors, histone deacetylase inhibitors, activators of co-inhibitory receptors, acetylcholinesterase inhibitors and selective serotonin reuptake inhibitors.
Iron Homeostasis and the Innate Immune System
Iron is an indispensable but potentially toxic trace element and for this reason it has to be circulated throughout the body attached to its carrier protein, transferrin (Tf). Dietary iron is absorbed in the gut by crossing the enterocyte brush border membrane via divalent metal-ion transporter 1 (DMT-1). After conversion from ferrous to ferric iron by hephaestin and/or ceruloplasmin it is exported through the basolateral membrane via ferroportin (FPN) channels (23).
Novel studies have demonstrated that gram-negative commensal gut microbes are able to generate more than one type of LPS, thus activating or inhibiting innate immune responses (24). For example, Escherichia coli LPS was found to be an activator of innate immunity, while LPS derived from Bacteroides dorei was linked to immune tolerance (25). Interestingly, Bacteroides fragilis LPS (BF-LPS) was shown to contribute to autoimmune pathology by selectively inhibiting innate immune tolerization (26). Since the host and microbes share a common iron pool, its availability is directly linked to the survival and growth of the LPS-shedding bacteria.
Iron enters the CNS together with Tf (iron-Tf complex) by binding to transferrin receptor-1 (TFR-1) expressed by the endothelial cells of brain capillaries. After crossing into the cytosol of these cells and satisfying their metabolic needs, excess iron is stored in the cytosolic and mitochondrial ferritin. The remaining iron is exported via FPN into the extracellular space (27).
One of the key roles of the innate immune system is pathogen-mediated intracellular iron sequestration to deny it to bacteria and malignancies which are dependent on this biometal for growth (discussed in The Trained Arm of Innate Immunity section). Iron sequestration is initiated in response to exogenous and endogenous danger signals, consisting of pathogen-associated molecular patterns (PAMPs) or, damage-associated molecular patterns (DAMPs), respectively. For example, LPS, the cell wall component of gram-negative bacteria, is a PAMP that activates microglial TLR-4. In contrast, the endogenously produced ROS is a DAMP that activates the cytosolic inflammasome NOD-like receptor pyrin domain-containing-3 (NLRP3) (28). Iron, itself and/or iron-induced ROS were shown to activate NLRP3 inflammasomes, engendering neuroinflammation (9) (Figure 1).
Iron enters neurons via DMT-1 and TFR-1 receptors but can only exit these cells through FPN, a receptor controlled by hepcidin, the master regulator of iron metabolism. Hepcidin is biosynthesized in the liver, but since only a small fraction of this this peptide hormone crosses the brain-blood barrier (BBB), the brain produces its own hepcidin. High hepcidin levels are associated with intracellular iron accumulation, as this biomolecule binds to FPN, internalizing this receptor (29). LPS and pro-inflammatory cytokines stimulate hepcidin production, inducing hypoferremia, a mechanism involved in the anemia of chronic inflammation pathogenesis (30). Interestingly, astrocytes were shown to induce neuronal death in response to LPS-activated microglia and to synthesize hepcidin via the IL-6/STAT3 pathway, likely linking hepcidin to A1 astrocytes (31). In addition, iron activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), a transcription factor involved in innate immune training and inflammation (27).
Microglia are yolk-sac-derived myeloid cells, comprising up to 20% of the total brain glial cell population (32). Their main function consists of brain parenchyma surveillance, searching for endogenous or exogenous danger signals to which they respond by activation (33). Another major function of microglia, assisted by astrocytes, consists of tolerization or acceptance of self-molecules, food, commensal microbes, and the fetus. This task is accomplished by phagocytosis via prompt clearance of molecular debris, including dead or dying cells that were shown to trigger inflammation and autoimmunity when allowed to accumulate (34). Taken together, disruption of phagocytosis can lead to unchecked inflammation, impaired immune tolerance and autoantibodies against self-components. Indeed, increased levels of autoantibodies were documented in AD, suggesting that autoimmunity may play a role in this disorder (35).
The Trained Arm of Innate Immunity
Pathogen-mediated iron sequestration is an effective mechanism that protects the host against bacteria and malignancies at the risk of inflammation and ROS triggered by increased intracellular iron (36). To lower this risk, iron must be properly stored into the cytosolic and mitochondrial ferritin, a process that may be disrupted by inflammation and age-related pathology. In the CNS, pathogen-mediated iron sequestration is initiated upon detection of PAMPs and/or DAMPs by microglia and astrocyte, followed by the release acute phase proteins, including hepcidin and lipocalin-2 (LCN-2), to internalize iron (29, 37).
Aging augments the expression of hepcidin and LCN-2, which along with impaired ferritinophagy, increases the risk of free intracellular iron and prolonged inflammation via NLRP3 inflammasomes activation (16, 38). Both hepcidin and LCN-2 have been associated with neurotoxicity and AD, despite some studies describing them as neuroprotective (39–42). These findings are contradictory in appearance only because hepcidin and LCN-2 may have both beneficial and detrimental effects, depending on the phase of inflammation. For example, during the acute phase, iron sequestration may be adaptive as it is denied to pathogens, limiting infection and inflammation. On the other hand, with chronic inflammation and increasing age, intracellular iron may become detrimental as it activates NLRP3 likely overwhelming immune tolerization and triggering neuronal loss. In addition, iron also activates NF-kB implicated in tolerization disruption and autoimmunity when in excess or inappropriately activated (43) (Figure 1). Iron may impair immune tolerance directly by altering microglial secretome, enhancing the release of TNF-alpha, IL-1, and C1q, cytokines that promote A1 astrocytes (1). Indeed, the pathological phagocytosis that A1 cells engage in may reflect the loss of neuronal and oligodendrocytic tolerization. Interestingly, a different study reported A1 astrocytes' upregulation in healthy elderly individuals, linking these cells to the chronic inflammation of aging or inflammaging, considered by some an AD prodrome (44, 45). Moreover, a novel study found that glucagon-like peptide-1 receptor (GLP1R) agonists can block the conversion of trophic to A1 astrocytes, suggesting that Diabetes mellitus type 2 treatments, including liraglutide, may benefit inflammaging and AD (46). Interestingly, liraglutide was documented to lower inflammatory responses by decreasing the expression of NLRP3, suggesting that this drug may function as an inflammasome inhibitor (47).
Inflammasomes are cytosolic multiprotein complexes activated in innate immune cells in response to PAMPs or DAMPs (16, 48). Inflammation is ignited by the inflammasome assembly with caspase1-induced maturation of interleukin-1β and IL-18 which in turn activate multiple inflammatory pathways (49) (Figure 2). NLRP3 inflammasomes, abundantly expressed by microglia and astrocytes, may play a major role in AD as they can be activated by beta amyloid, iron and ROS, maintaining chronic inflammation (48, 50, 51). Indeed, continuous immune training in inflammaging and AD was associated with perpetually elevated NLRP1 and NLRP3 mRNA levels (52–54). Similarly, chronic NLRP3 activation was associated with atherosclerosis and Western diets, linking metabolism and chronic inflammation (55, 56).
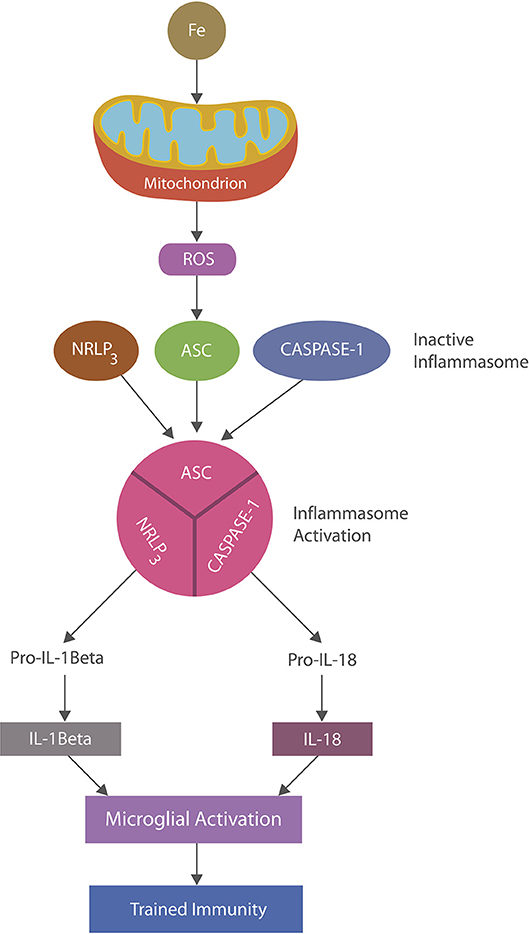
Figure 2. NLRP3 inflammasomes activation: iron-damaged mitochondria, acting as a DAMP, directly or via ROS triggers inflammation by activating NLRP3 inflammasomes. Inflammasomes are comprised of a sensor, the nucleotide-binding oligomerization domain (NOD), in this case the (NOD)-like receptor protein 3 (NLRP3), apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and caspase-1 which, upon stimulation, assemble and cleave premature IL-1 beta and IL-18 into their biologically active forms, activating microglia (trained immunity).
Recently the mitochondrion was identified as the interface connecting immunity and metabolism as this organelle contains the mammalian target of rapamycin (mTOR), a protein involved in shifting the metabolic gears from oxidative phosphorylation (OXPHOS), characterizing tolerized immunity, to aerobic glycolysis, marking immune training (11). Iron has been demonstrated to induce mitochondrial damage, probably lowering OXPHOS and prompting NLRP3 inflammasome assembly and microglial training (57, 58) (Figure 2). In fact, iron-disrupted OXPHOS may leave aerobic glycolysis as the only metabolic option for microglia and a possible source of neuroinflammation in AD. Along these lines, a recent study described a direct cross-talk between mTOR and iron metabolism, suggesting a connection between this biometal and aerobic glycolysis (59, 60). Mitophagy or elimination of damaged mitochondria (including that which is iron-induced) was reported to inhibit NLRP3 and lower immune training in AD models, pointing to mitophagy as a potential AD treatment (61–64). Interestingly, liraglutide, a GLP-1R agonist, was shown to promote mitophagy in addition to, or because of NLRP3 inhibition, indicating a potential “off label” use of this drug (65).
A recent preclinical study linked the NLRP3 component, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), with the prion-like seeding of beta amyloid (66). Similarly, in a previous study, ASC was reported as capable of migrating through the extracellular space (67). Another novel study found a link between NLRP3 inflammasomes and prion disease (68). Furthermore, multiple studies over the past decade established a close connection between various forms of prion diseases and autoimmunity, suggesting a common denominator (69, 70). Interestingly, iron was associated with both prion disease and autoimmunity, and chelation was found therapeutic in CNS autoimmune disorders, including MS and experimental autoimmune encephalomyelitis (EAE) (71–74). For example, brain iron dyshomeostasis was demonstrated in prion disease patients, while another study found a higher incidence of autoimmune disorders in individuals with hemochromatosis (71, 74). A preliminary study found that deferiprone, a BBB-crossing iron chelator, was therapeutic in MS patients (73). Moreover, some iron chelators were found beneficial in AD and Parkinson's disease (PD), possibly pointing to the role of autoimmune inflammation in these disorders (75–77). Indeed, over the past decade, several studies have associated autoimmunity with AD as serum and CSF autoantibodies against β-amyloid, tau, ceramide and adenosine triphosphate (ATP) synthase were detected in this disorder (35, 78–82). Interestingly, these molecules were also associated with iron and NLRP3, making it worthwhile to search for ASC autoantibodies in AD (83–87).
The Tolerized Arm of Innate Immunity
Effective protection from pathogens and malignant cells is an important function of the innate immune system, but an equally important task is protection against autoimmune inflammation by maintaining immunologic tolerance to self-structures, food, the fetus, and commensal bacteria. Tolerized immunity or absence of immune responses to repeated or prolonged LPS stimulation was originally described in rodents which survived the administration of a lethal endotoxin dose after previous exposure to sublethal concentrations (88). The adaptive role of this mechanism in the CNS may include “tolerating” elevated levels of LPS demonstrated in the brains of aging individuals and AD patients (2). Indeed, aging and AD have been associated with increased gram-negative bacteria titers in the GI tract and gums (89). For example, individuals with AD were found to have three times higher LPS blood levels compared to controls (90). Interestingly, novel studies have shown that B. fragilis generates a structurally different LPS (BF-LPS), that selectively inhibits immune tolerization, possibly contributing to the pathogenesis of both autoimmune disorders and AD (25, 26).
Aside from the LPS response, brain tolerized immunity may serve the purpose of inhibiting inflammation during phagocytosis of apoptotic cells, synapses, or damaged endogenous molecules, a process we define as tolerant phagocytosis (91, 92). Preclinical studies have shown that disruption of this pathway and accumulation of molecular debris activates autoimmune inflammation, inflicting neuronal damage (93, 94). A novel study has reported that LPS down-regulates the expression of the triggering receptor expressed on myeloid cells 2 (TREM2), the microglial receptor associated with immune tolerance (95) (Figure 1). Apolipoprotein E (ApoE) is a TREM2 ligand and their interaction may be necessary for tolerant phagocytosis (96–98). Indeed, TREM-2 blockade was demonstrated to exacerbate EAE and MS, linking this receptor to the CNS autoimmune disorders (99, 100).
A recent pivotal study established the existence of an ApoE-TREM-2 axis which under normal circumstances engenders tolerized or homeostatic microglial phenotypes (101). In contrast, a dysfunctional ApoE-TREM-2 axis was associated with deficient tolerization and unopposed inflammation. For example, ApoE4 isoform, linked to late onset AD, presents with poor TREM-2 binding, leading to the activation of NLRP3 and immune training instead of tolerization (102, 103).
In AD, the tolerized arm of innate immunity appears to be impaired as persistently elevated LPS brain levels fail to induce tolerance, probably triggering the autoimmune elimination of neurons and oligodendrocytes via A1 astrocytes (1, 2, 98, 104). Moreover, LPS elevation in AD was linked to iron as dormant microbes, including B. fragilis, may survive in the CNS in an inactive state limited by the lack of free iron (6). During aging, impaired iron homeostasis provides these microbes with enough metal to resume their growth and LPS shedding, possibly explaining the LPS upregulation observed in AD (105). Indeed, iron may facilitate B. fragilis resuscitation in the brain as this microbe was shown to require iron or heme for surviving outside of the GI tract (106). In addition, iron-induced endothelial cells' damage may open the BBB (a property exploited in the treatment of gliomas), allowing microbial passage (107, 108). On the other hand, iron chelators were demonstrated to inhibit B. fragilis growth, lowering LPS brain levels, perhaps explaining some of the beneficial effects of these compounds in AD (109). In addition, LPS, not only promotes ferritinophagy, but also the astrocytic expression of hepcidin via IL6/STAT3, further augmenting the cellular free iron pool along with the chances of dormant bacterial resuscitation (31).
At the molecular level, tolerized immunity was shown to be associated with microglial TREM-2 receptors, interacting with anti-inflammatory molecules, including ApoE, and complement components C1q/C3 (110–112) (Figure 3). In addition, tolerized immunity was shown to be modulated by iron-dependent hypoxia-inducible factor 1α (HIF-1α), suggesting another tolerization target (113).
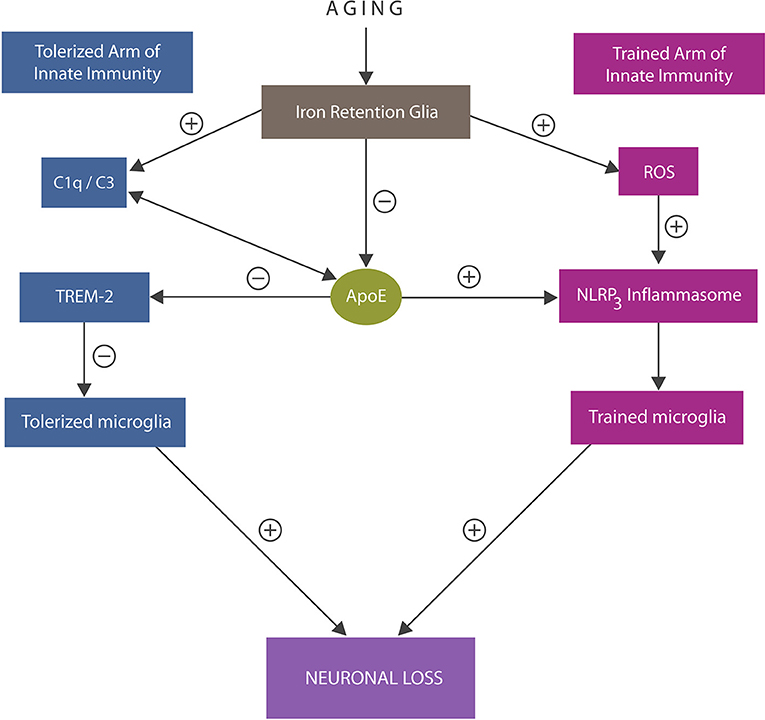
Figure 3. An overview of the trained and tolerized arm of innate immunity. Trained arm: excess iron and ROS trigger activation of NLRP3 inflammasomes with excessively trained microglia and neuroinflammation that may override tolerization. In addition, iron lowers ApoE expression and alters C1q and C3, further impairing tolerization via ApoE -TREM-2 axis. Both excessive immune training or insufficiently tolerized microglia may trigger neuronal loss. ApoE4 isoform binding C1q triggers inappropriate elimination of healthy synapses by astrocytes, likely by promoting the A1 cells.
Under physiological circumstances C1q and C3 facilitate phagocytosis by tagging dysfunctional synapses for engulfment by microglia and astrocytes (114, 115). In addition, C1q enhances immune tolerance and inhibits NLRP3-induced immune activation (116). Pathologically, deficient C1q was found in over 90% of systemic lupus erythematosus (SLE) patients, linking this protein to autoimmune inflammation (117). In contrast, ApoE4-induced C1q upregulation was associated with inappropriate elimination of healthy synapses by astrocytes, connecting this phenomenon to autoimmunity and the A1 cells (118). Indeed, the development of anti-C1q antibodies may comprise a novel therapeutic strategy for both AD and autoimmunity (10).
Iron disrupts tolerized immunity by lowering ApoE expression in microglia, impairing the ApoE-TREM-2 axis and amyloid plaque phagocytosis (119–121). Indeed, ApoE-immunoreactive microglia were found closely associated with senile plaques, suggesting a key role in AD pathology (122). In addition, iron disrupts tolerized immunity by upregulating C3 and altering the C1q expression, likely preventing their ApoE binding (123, 124). Indeed, C3 upregulation was linked to increased beta amyloid and phosphorylated tau, linking once more iron to AD pathogenesis (110, 125) (Figure 3). Other recent studies connected iron-upregulated C3 with the A1 astrocytes by demonstrating high complement levels in astrocyte-derived exosomes obtained from AD patients (114, 126).
Aside from ApoE and C1q/C3, iron inhibits α-secretase or ADAM-10, the enzyme involved in the generation of soluble TREM-2 (sTREM-2), linking again this biometal to autoimmunity (102, 123, 124, 127–129) (Figure 4).
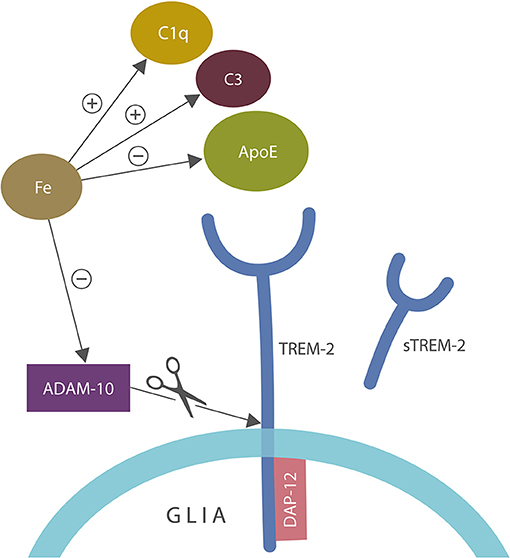
Figure 4. A closer view of microglial ApoE-TREM-2 axis: ApoE, C1q, and C3 engender tolerant phagocytosis (lack of inflammatory response during the clearance of damaged self-components). Iron disrupts this process by promoting prolonged inflammation that overrides immune tolerization. In addition, iron impairs tolerization by altering C1q and ApoE along with C3 upregulation. Iron also inhibits alpha secretase (ADAM-10) which cleaves tolerant TREM-2 receptor into sTREM-2, likely impairing phagocytosis. DAP-12 is a TREM-2 adapter, anchoring this receptor into the cell membrane. ADAM-10 is the same enzyme that cleaves amyloid precursor protein (APP), preventing the formation of amyloid-beta peptide.
Adenosine-triphosphate (ATP) synthase, the mitochondrial enzyme involved in ATP biosynthesis, is a newly identified AD target, especially because extracellular ATP (eATP) is a potent NLRP3 activator associated with autoimmune disorders (81–85). This is relevant as preclinical studies show iron-associated augmentation of F1 and F0 components of ATP synthase with loss of mitochondrial ATP, possibly suggesting ATP displacement to the extracellular compartment (130). In addition, iron-binding protein, lactoferrin clears eATP, presenting with a beneficial role in AD (see the section on exosomes and Inflammasomes). Furthermore, blockade of purinergic receptors P2Y1 was associated with improved cognition in AD models, while P2X7 inhibition was found beneficial in autoimmune disorders, linking eATP and iron to impaired immune tolerance (87, 130, 131).
The Innate-Adaptive Continuum
Novel preclinical studies indicate a close cooperation between the innate and adaptive immune systems via NLRP3 inflammasomes and IL-1β (132, 133). Microglia and astrocytes communicate extensively with peripheral lymphocytes, inducing their activation and CNS migration (10, 134). IL-1β is known for increasing BBB permeability, facilitating the infiltration of peripheral cytotoxic T-cells (CTCs) into the CNS (135). In addition, iron is known for altering the BBB by inflicting endothelial cells damage, facilitating peripheral cells access to the CNS (107). In addition, peripheral lymphocytes may access the brain via the newly discovered CNS-draining lymphatics (17).
The newly arrived cells were shown to activate NLRP3, attracting more blood-borne immune cells into the brain (136). On the other hand, tolerized glia may orchestrate the CNS import of T-regs or the newly identified regulatory B1a cells which in turn lower microglial training (137–140). In addition, B-cells have been known to facilitate the recruitment of T-regs to the CNS, augmenting their immunosuppressive activities (141).
T-regs, the preservers of immune tolerance, were found to delay AD progression, linking this condition to autoimmune inflammation (142, 143). The anti-inflammatory IL-10 released by tolerized microglia and T-regs was demonstrated to negatively regulate iron metabolism, possibly explaining the beneficial role of Tregs in AD (144). In addition, IL-4 also regulates iron metabolism and promotes immune tolerance (145). Indeed, iron has been shown to facilitate the CNS infiltration of peripheral CTCs, while iron chelation produces the opposite effect (146). Furthermore, IL-10 was demonstrated to inhibit aerobic glycolysis, the metabolic driver of trained immunity, promoting the tolerant OXPHOS (144, 147, 148). Interestingly, a recent preclinical study discovered a CNS population of cerebral T-regs, documenting that aside from blood borne, the brain maintains its own tolerogenic lymphocyte population (139). Other novel studies demonstrated the existence of various T-regs subpopulations, expressing different co-inhibitory T-cell receptors, including the T-cell immunoglobulin and ITIM Domain (TIGIT) that regulates immunity exclusively via iron-lowering IL-10, linking this receptor to iron downregulation (149, 150).
Co-inhibitory receptors, including cytotoxic T-lymphocyte associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), lymphocyte-activation protein 3 (Lag-3), and TIGIT play an essential role in the prevention of autoimmune disorders by promoting tolerized responses (151, 152). A growing body of evidence reveals that dysfunctional co-inhibitory receptors may contribute to AD neuroinflammation as under normal circumstances they function to inhibit inflammation-generating co-stimulatory signals (153). For example, upregulation of PD-1 expressed on both microglia and T-regs, was found beneficial in AD animal models (154). Another study demonstrated that non-PD-1 expressing T-regs were upregulated in mild cognitive impairment (MCI), demonstrating the beneficial role of PD-1 receptors in preventing the development of full-blown AD (155). Indeed, tolerized microglia also express PD-1, activating T-regs and probably facilitating their CNS import (156, 157).
These findings suggest that other co-inhibitory receptors, including TIGIT, may activate T-regs. Interestingly, a recent study found elevated TIGIT in the peripheral blood of healthy elderly individuals (158). As upregulated A1 astrocytes were also found in normal aging, TIGIT and A1 may comprise novel AD risk markers (45). In addition, as TIGIT modulates immunity via the iron-lowering IL-10, this receptor may be elevated in normal aging in a compensatory manner in response to iron overload (149). TIGIT is yet to be demonstrated on glial cells, however like PD-1, it may be a coordinator of adaptive and immune responses.
Taken together, this data suggests that innate and adaptive immunity operate on a continuum as trained immunity may activate CTCs, while tolerized immunity the T-regs, therefore targeting trained and tolerized immunity may lower the CNS infiltration of peripheral lymphocytes.
Microglial Reprograming: Trained and Tolerized Therapies
The recent FDA approval of immune training-inducers prompted research for similar therapies for cancer and neurodegeneration (18). Immune stimulatory vaccines, comprised of antigen and pro-inflammatory molecules, activate TLRs or NLRP3, inducing immune activation. These therapeutics have been utilized to train APCs and reverse cancer or sepsis-induced T cell anergy. Iron as an inducer of trained immunity has been utilized to activate TAMs and several nanoparticles are currently being designed for its delivery (14). On the other hand, the development of specific tolerized therapies, including “inverse vaccines,” capable of improving pathology without causing generalized immunosuppression, has been vexing (20). The same is true regarding the first-generation AD vaccines, like AN1793, based on anti-Aβ1-42 antibodies which was abandoned due to adverse effects (159). However, despite these draw backs, a new generation of AD vaccines, including anti-C1q mentioned above, are currently being developed. Since neuroinflammation represents the predominant pathology in many neurodegenerative and even psychiatric disorders, de-escalating iron-trained microglia may comprise an effective treatment strategy. Indeed, the anti-inflammatory iron chelator deferiprone is currently in phase 2 trials for AD (15, 160). Deferiprone may have a dual mechanism of action, lowering trained immunity and augmenting tolerization, suggesting that simultaneously targeting both arms of innate immunity may yield better therapeutic outcomes than addressing each one in isolation. Another dual-mechanism-of-action compound, hydroxamic acid, is a promising histone deacetylase-6 inhibitor (HDAC6i) which presents with the added benefit of being an iron chelator (161, 162). Histone deacetylases are enzymes involved in the removal of acetyl groups from histone proteins, altering the chromatin landscape and gene expression. HDACs have been associated with AD pathogenesis, while some HDACis were found to improve cognition (163, 164). In addition, selective HDAC inhibitors were shown to suppress LPS-trained microglia via IL-6 and TNF-α, suggesting a therapeutic potential in AD neuroinflammation (165). Moreover, HDACs 1 and 2 inhibitors are augmenters of microglial β amyloid phagocytosis, linking these compounds to the immuno-metabolic reprograming via OXPHOS augmentation (165, 166).
Inflammasome Inhibitors
NLRP3 inhibition to lower immune training and the subsequent neuroinflammation is an emerging therapeutic strategy in AD and traumatic brain injury (TBI) (167–169).
Purinergic receptor blockers, ASC inhibitors, diarylsulphonylurea inhibitors, β-sulfonyl nitrile compounds, and caspase-1 blockers are the broad categories of recently developed NLRP3 inhibitors (170, 171). The most studied compound, MCC950 is a diarylsulphonylurea inhibitor, a potent anti-inflammatory drug and a facilitator of β amyloid clearance in AD animal models (172). Another recently developed molecule, OLT1177, is a β-sulfonyl nitrile which inhibits NLRP3 by limiting ROS generation after LPS challenge. This compound is currently in phase I clinical trials.
Suppressing NLRP3 by ASC inhibition is an attractive AD strategy as ASC was recently found to disseminate β amyloid throughout the CNS (66, 172). Interestingly, saxagliptin, a dipeptidyl peptidase-4 (DPP-4) inhibitor, which increases plasma GLP-1 concentration, appears to selectively inhibit ASC, suggesting a role in preventing the spread of AD pathology (173). This is significant since GLP-1R agonists were found to inhibit the conversion of trophic into A1 astrocytes, probably linking ASC to this neurotoxic phenotype (1, 46). Moreover, Tranilast, an anti-allergic drug derived from tryptophan, was also reported to selectively inhibit ASC, suggesting a preventive potential in AD (174).
Of Exosomes and Inflammasomes
Exosomes are (50–150 nm) extracellular nanovesicles released by most cell types, including neurons and glia, which can carry molecular cargos and trigger functional changes in target cells. Exosomes cross the BBB, facilitating the transfer of inflammation as they can carry a variety of peripheral molecules, including inflammasome-derived proteins (175, 176).
Exosomes, involved in innate immune cells' cross talk, participate in microglial training or tolerization by augmenting or lowering neuroinflammation, depending on their cells of origin (17, 177, 178). Novel studies have found that neuronal exosomes have beneficial effects, while astrocytes-derived exosomes (ADE) are mostly detrimental, inducing neurodegeneration. Neuronal exosomes present with the adaptive function of clearing β amyloid by delivering it to microglia for phagocytosis. For this reason, pharmacological facilitation of neuronal exosomes' synthesis could be developed into an AD therapy (176). For example, designer neuronal exosomes were recently developed and utilized in animal models of Parkinson's disease (170). Interestingly, microglial cells were shown to release exosomes that were previously reported to originate from B cells and dendritic cells, suggesting pro-inflammatory roles (179). CNS autoimmune disorders have been associated with exosomes transporting histocompatibility complex (MHC) class II proteins released by interferon-γ activated microglia (180).
ADE were shown to promote the conversion of trophic to A1 astrocytes by releasing exosomes containing complement components C1q and C3 (126, 181–183). Preventing ADE release may comprise another potential AD strategy. Moreover, a novel study has demonstrated that excessive interstitial ATP can facilitate the generation of A1 astrocytes, suggesting clearance of ATP from the extracellular space (ECS) as a potential AD treatment modality (174). Interestingly, the natural iron chelator, lactoferrin, was found to facilitate the ECS removal of ATP, indicating a novel therapeutic application (184). Indeed, a recent study suggested that engineered lactoferrin-containing exosomes could accomplish this goal (185).
Histone Deacetylases Inhibitors (HDACi)
Innate immune cells are endowed with an epigenetically regulated memory of previous antigen encounters, suggesting that epigenome manipulation may comprise an efficient reprograming strategy in these cells (17). Acetylation and deacetylation of DNA and histone proteins are the most studied epigenetic mechanisms regulating innate immune cells. Indeed, HDACi were shown to lower the LPS-induced microglial training, promoting tolerization (165). For example, valproic acid, a HDACi utilized in the treatment of bipolar disorder and epilepsy, was shown to downregulate the mammalian target of rapamycin (mTOR), restoring OXPHOS, the driver of immune tolerance (11, 186–188). Furthermore, HDACi were recently shown to augment astrocytic ApoE secretion (189). Since ApoE inhibits NLRP3 activation, HDAC inhibition may have a potential therapeutic value in the fight against AD neuroinflammation (Figure 3).
HDACi are facilitators of ferritin storage, lowering intracellular free iron and the risk of NLRP3 activation (161, 190). In addition, HDACi reverse the iron-induced augmentation of glycogen synthase kinase-3β (GSK3β), a pro-inflammatory enzyme implicated in AD (191, 192). GSK-3β inhibitors, including lithium, were shown to promote immune tolerance, suggesting a therapeutic role in lowering iron levels (193–195). Interestingly, the tolerizing receptor TREM-2 is a negative regulator of GSK-3β, further associating this protein with decreasing iron-induced neuroinflammation (191, 196).
Co-inhibitory T-cell Receptors in AD
Dysfunctional co-inhibitory receptors may contribute to AD neuroinflammation as under normal circumstances, they block co-stimulatory signals, lowering inflammation (153). For example, stimulation of PD-1 receptors, expressed on both microglia and T-regs, was found beneficial in AD animal models (154). On the other hand, increased number of non-PD-1 expressing T-regs in MCI, shows the importance of this co-inhibitory receptor in AD prevention (155).
T cell activation by APCs is crucial for mounting adequate adaptive immune responses. In the CNS, microglia and astrocytes can become APCs, engendering immunologic synapses with peripheral T cells (10). The proliferation, differentiation, and migration of these cells is triggered by the interplay between co-stimulatory and co-inhibitory signals which fine tune T cell responses (197). For example, the co-stimulatory receptors promote effector T-cells function, while co-inhibitory receptors function to inhibit co-stimulatory signals, promoting T-regs (6).
Activation of co-inhibitory receptors, including CTLA-4, PD-1, LAG-3, TIM-3, and TIGIT, induce immune tolerance, therefore their stimulation would be beneficial in autoimmune disorders (198). On the other hand, their blockade was shown to train TAMs in a manner similar to iron and eliminate cancer cells (199). Interestingly, TIGIT competes with the co-stimulatory signal CD226 for a common ligand CD155, suggesting that promoting the TIGIT-CD155 axis may lower intracellular iron as TIGIT operates exclusively via the iron-lowering IL-10 (200). Indeed, IL-10 or CD-155 loaded exosomes may enhance the co-inhibitory receptor TIGIT, lowering microglial intracellular iron. In addition, the CD226 genetic variant, Gly307Ser, was associated with several autoimmune disorders, suggesting that CD 226 inhibition may facilitate tolerized immunity and benefit the treatment of these conditions as well (201, 202).
Tolerized Immunity and Acetylcholinesterase Inhibitors
Acetylcholinesterase inhibitors (AChEIs) are cholinergic compounds widely utilized in AD. AChEIs, including donepezil, rivastigmine, and galantamine function by inhibiting cholinesterase, the acetylcholine-degrading enzyme, increasing its synaptic availability in the cholinergic tracts. Some of these drugs may be modulators of trained or tolerized immunity as they oppose iron-induced NLRP3 activation and the inhibition of ADAM-10, the protective α-secretase, in charge of cleaving both TREM-2 and APP (203–205) (Figure 4). In addition, α7- and α9 nicotinic acetylcholine receptor (nAChR) agonists were found to lower autoimmune inflammation, by augmenting tolerization (206, 207). Moreover, activation of α7nAChRs was found to reverse post-surgical cognitive impairment, suggesting that AChEIs-linked cognitive enhancement may be due to their anti-inflammatory actions (208). On the other hand, a recent study demonstrated that iron and ROS can inhibit α7nAChRs signaling, likely disrupting tolerized immunity (209, 210). Interestingly, conjugation of an AChEI, such as galantamine, with the natural iron chelator, lactoferrin, was recently proposed as an AD therapy (211).
Tolerized Immunity and Selective Serotonin Reuptake Inhibitors (SSRIs)
Serotonin reuptake inhibitors (SSRIs) are widely prescribed antidepressant drugs that block 5-hydroxytryptamine reuptake in the presynaptic neurons of brain serotonergic tracts, augmenting the synaptic availability of this neurotransmitter. However, these compounds were found to also lower trained immune responses. For example, a recent study indicated that the SSRI, fluoxetine reduces the central and peripheral levels of IL-1β by inhibiting NLRP3 (212, 213).
Another group demonstrated the existence of a cross talk between the brain innate immunity and serotonin signaling, possibly linking major depressive disorder with NLRP3 activation (214). Along the same lines, it was recently shown that indoleamine 2,3-dioxygenase-1 (IDO-1), an enzyme associated with tryptophan catabolism, is abundantly expressed by microglia and astrocytes, connecting serotonergic signaling with tolerized immunity (215). IDO-1 induces immune tolerance by processing tryptophan to L-kynurenine, an inhibitor of immune cells' proliferation and maturation (216). IDO-1 is known for orchestrating mother's immune acceptance of the fetus, however some bacteria and cancers were found to exploit this pathway (217, 218). Indeed, an IDO vaccine to block cancer-induced immune tolerance is currently under development (219). Moreover, IDO-1 contributes to the biosynthesis of quinolinic acid (QUIN), a neurotoxic tryptophan catabolite that was linked to AD pathogenesis (220). Interestingly, iron-QUIN complexes were demonstrated in this neurodegenerative disorder, indicating that QUIN toxicity may be iron-related (221, 222). QUIN was demonstrated to activate microglia and astrocytes, resulting in neuronal death, possibly by inducing A1 cells (223–225).
Conclusions
The healthy immune system must keep trained and tolerized immunity in balance to ensure effective defenses against pathogens and malignancies, while at the same time accepting the self-components, food, the fetus, and commensal microbes. Iron interferes with this process as it triggers a competition between the host and pathogens. Both biological systems have developed mechanisms to capture iron and deny it to the opposing side. The innate immune cells sequestrate iron to restrict its availability to pathogens, but elevated intracellular iron increases the risk of mitochondrial damage, ROS generation, and autoimmune inflammation.
Aging and iron are associated with increased permeability of both GI and brain microvessels, allowing gut microbes, including B. fragilis, to migrate into the CNS and shed LPS, disrupting immune tolerization. Manipulation of trained and tolerized immunity, especially via exosomes and nanoparticles loaded with iron chelators could represent future AD strategies (76). Indeed, iron-oxide loaded exosomes for magnetic hyperthermia have been successfully used in oncology (226).
Tolerogenic immunization also known as “inverse vaccines,” can for example, utilize CD-155 loaded exosomes to enhance the co-inhibitory receptor TIGIT and lower AD neuroinflammation (227). In addition, nanoparticles containing NLRP3-inhibitors, HDACi or ASC blockers may soon become parts of the AD armamentarium.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Activated microglia induce neurotoxic reactive astrocytes via Il-1α, TNFα, and C1q. Nature 541:481–7. doi: 10.1038/nature21029
2. Zhan X, Stamova B, Sharp FR. Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer's disease brain: a review. Front Aging Neurosci. (2018) 10:42. doi: 10.3389/fnagi.2018.00042
3. Zhao Y, Cong L, Lukiw WJ. Lipopolysaccharide (LPS) accumulates in neocortical neurons of Alzheimer's Disease (AD) brain and impairs transcription in human neuronal-glial primary co-cultures. Front Aging Neurosci. (2017) 9:407. doi: 10.3389/fnagi.2017.00407
4. Abreu R, Quinn F, Giri PK. Role of the hepcidin-ferroportin axis in pathogen-mediated intracellular iron sequestration in human phagocytic cells. Blood Adv. (2018) 2:1089–100. doi: 10.1182/bloodadvances.2017015255
5. Zeineh MM, Chen Y, Kitzler HH, Hammond R, Vogel H, Rutt BK. Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol Aging (2015) 36:2483–500. doi: 10.1016/j.neurobiolaging.2015.05.022
6. Kell DB, Pretorius E. No effects without causes: the Iron Dysregulation and Dormant Microbes hypothesis for chronic, inflammatory diseases. Biol Rev Cambridge Philos Soc. (2018) 93:1518–57. doi: 10.1111/brv.12407
7. Biasiotto G, Di Lorenzo D, Archetti S, Zanella I. Iron and neurodegeneration: is ferritinophagy the link? Mol Neurobiol. (2016) 53:5542–74. doi: 10.1007/s12035-015-9473-y
8. François A, Terro F, Quellard N, Fernandez B, Chassaing D, Janet T, et al. Impairment of autophagy in the central nervous system during lipopolysaccharide-induced inflammatory stress in mice. Mol Brain (2014) 7:56. doi: 10.1186/s13041-014-0056-z
9. Nakamura K, Kawakami T, Yamamoto N, Tomizawa M, Fujiwara T, Ishii T, et al. Activation of the NLRP3 inflammasome by cellular labile iron. Exp Hematol. (2016) 44:116–24. doi: 10.1016/j.exphem.2015.11.002
10. Lansita JA, Mease KM, Qiu H, Yednock T, Sankaranarayanan S, Kramer S. Nonclinical development of ANX005: a humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int J Toxicol. (2017) 36:449–62. doi: 10.1177/1091581817740873
11. Chatzantoni K, Mouzaki A. Anti-TNF-alpha antibody therapies in autoimmune diseases. Curr Top Med Chem. (2006) 6:1707–14. doi: 10.2174/156802606778194217
12. Arts RJW, Joosten LAB, Netea MG. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front Immunol. (2018) 9:298. doi: 10.3389/fimmu.2018.00298
13. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
14. Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, et al. Iron induces anti-tumor activity in tumor-associated macrophages. Front Immunol. (2017) 8:1479. doi: 10.3389/fimmu.2017.01479
15. Cummings J, Lee G, Ritter A, Zhong K. Alzheimer's disease drug development pipeline: 2018. Alzheimer's Dementia. (2018) 4:195–214. doi: 10.1016/j.trci.2018.03.009
16. Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology (2009) 73:768–74. doi: 10.1212/WNL.0b013e3181b6bb95
17. Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to tango. Front Immunol. (2017) 8:1905. doi: 10.3389/fimmu.2017.01905
18. Novakovic B, Habibi E, Wang SY, Arts RJ, Davar R, Megchelenbrink W, et al. β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell (2016) 167:1354–68e14. doi: 10.1016/j.cell.2016.09.034
19. Mourits VP, Wijkmans JC, Joosten LA, Netea MG. Trained immunity as a novel therapeutic strategy. Curr Opin Pharmacol. (2018) 41:52–8. doi: 10.1016/j.coph.2018.04.007
20. Kishimoto TK, Maldonado RA. Nanoparticles for the induction of antigen-specific immunological tolerance. Front Immunol. (2018) 9:230. doi: 10.3389/fimmu.2018.00230
21. Arbore G, Kemper C. A novel “complement–metabolism–inflammasome axis” as a key regulator of immune cell effector function. Eur J Immunol. (2016) 46:1563–73. doi: 10.1002/eji.201546131
22. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
23. Jiang B, Liu G, Zheng J, Chen M, Maimaitiming Z, Chen M, et al. Hephaestin and ceruloplasmin facilitate iron metabolism in the mouse kidney. Sci Rep. (2016) 6:39470. doi: 10.1038/srep39470
24. D'Hennezel E, Abubucker S, Murphy LO, Cullen TW. Total lipopolysaccharide from the human gut microbiome silences toll-like receptor signaling. mSystems (2017) 2:e00046-17. doi: 10.1128/mSystems.00046-17
25. Vatanen T, Kostic AD, d'Hennezel E, Siljander H, Franzosa EA, Yassou M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell (2016) 165:842–53. doi: 10.1016/j.cell.2016.04.007
26. Lukiw WJ. Bacteroides fragilis lipopolysaccharide and inflammatory signaling in Alzheimer's disease. Front Microbiol. (2016) 7:1544. doi: 10.3389/fmicb.2016.01544
27. Ashraf A, Clark M, So PW. The aging of iron man. Front Aging Neurosci. (2018) 10:65. doi: 10.3389/fnagi.2018.00065
28. Abais JM, Xia M, Zhang Y, Boini KM, Li P-L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. (2015) 22:1111–29. doi: 10.1089/ars.2014.5994
29. Nairz M, Haschka D, Demetz E, Weiss G. Iron at the interface of immunity and infection. Front Pharmacol. (2014) 5:152. doi: 10.3389/fphar.2014.00152
30. Ganz T, Nemeth E. Regulation of iron acquisition and iron distribution in mammals. Biochim Biophys Acta (2006) 1763:690–9. doi: 10.1016/j.bbamcr.2006.03.014
31. You L-H, Yan C-Z, Zheng B-J, Ci YZ, Chang SY, Yu P, et al. Astrocyte hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death Dis. (2017) 8:e2676. doi: 10.1038/cddis.2017.93
32. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. (2012) 122:1164–71. doi: 10.1172/JCI58644
33. Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Exp Neurol. (2014) 258:5–16. doi: 10.1016/j.expneurol.2014.01.001
34. Yin J, Valin KL, Dixon ML, Leavenworth JW. The role of microglia and macrophages in CNS homeostasis, autoimmunity, and cancer. J Immunol Res. (2017) 2017:5150678. doi: 10.1155/2017/5150678
35. Wu J, Li L. Autoantibodies in Alzheimer's disease: potential biomarkers, pathogenic roles, and therapeutic implications. J Biomed Res. (2016) 30:361–72. doi: 10.7555/JBR.30.20150131
36. Johnson EE, Wessling-Resnick M. Iron metabolism and the innate immune response to infection. Microbes Infect. (2012) 14:207–16. doi: 10.1016/j.micinf.2011.10.001
37. Xiao X, Yeoh BS, Vijay-Kumar M. Lipocalin 2: An emerging player in iron homeostasis and inflammation. Annu Rev Nutr. (2017) 37:103–30. doi: 10.1146/annurev-nutr-071816-064559
38. Erdei J, Tóth A, Balogh E, Nyakundi BB, Bányai E, Ryffel B, et al. Induction of NLRP3 inflammasome activation by heme in human endothelial cells. Oxid Med Cell Longev. (2018) 2018:4310816. doi: 10.1155/2018/4310816
39. Sternberg Z, Hu Z, Sternberg D, Waseh S, Quinn JF, Wild K, et al. Serum hepcidin levels, iron dyshomeostasis and cognitive loss in Alzheimer's disease. Aging Dis. (2017) 8:215–27. doi: 10.14336/AD.2016.0811
40. Song J, Kim OY. Perspectives in lipocalin-2: emerging biomarker for medical diagnosis and prognosis for Alzheimer's disease. Clin Nutr Res. (2018) 7:1–10. doi: 10.7762/cnr.2018.7.1.1
41. Xing C, Wang X, Cheng C, Montaner J, Mandeville E, Leung W, et al. Neuronal production of lipocalin-2 as a help-me signal for glial activation. Stroke (2014) 45:2085–92. doi: 10.1161/STROKEAHA.114.005733
42. Zhou Y-F, Zhang C, Yang G, Qian ZM, Zhang MW, Ma J, et al. Hepcidin protects neuron from hemin-mediated injury by reducing iron. Front Physiol. (2017) 8:332. doi: 10.3389/fphys.2017.00332
43. Herrington FD, Carmody RJ, Goodyear CS. Modulation of NF-κB signaling as a therapeutic target in autoimmunity. J Biomol Screen. (2016) 21:223–42. doi: 10.1177/1087057115617456
44. Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, et al. Inflammaging as a prodrome to Alzheimer's disease. J Neuroinflamm. (2008) 5:51. doi: 10.1186/1742-2094-5-51
45. Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA. (2018) 115:E1896–905. doi: 10.1073/pnas.1800165115
46. Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med. (2018) 24:931–8. doi: 10.1038/s41591-018-0051-5
47. Chen A, Chen Z, Xia Y, Lu D, Yang X, Sun A, et al. Liraglutide attenuates NLRP3 inflammasome-dependent pyroptosis via regulating SIRT1/NOX4/ROS pathway in H9c2 cells. Biochem Biophys Res Commun. (2018) 499:267–72. doi: 10.1016/j.bbrc.2018.03.142
48. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature (2013) 493:674–8. doi: 10.1038/nature11729
49. Jo E-K, Kim JK, Shin D-M, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. (2016) 13:148–59. doi: 10.1038/cmi.2015.95
50. Song L, Pei L, Yao S, Wu Y, Shang Y. NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci. (2017) 11:63. doi: 10.3389/fncel.2017.00063
51. Yin J, Zhao F, Chojnacki JE, Fulp J, Klein WL, Zhang S, et al. NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer's disease. Mol Neurobiol. (2018) 55:1977–87. doi: 10.1007/s12035-017-0467-9
52. Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Mol Neurodegener. (2016) 11:23. doi: 10.1186/s13024-016-0088-1
53. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and 'Garb-aging'. Trends Endocrinol Metab. (2017) 28:199–212. doi: 10.1016/j.tem.2016.09.005
54. Cordero MD, Williams MR, Ryffel B. AMP-activated protein kinase regulation of the NLRP3 inflammasome during aging. Trends Endocrinol Metab. (2018) 29:8–17. doi: 10.1016/j.tem.2017.10.009
55. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell (2018) 172:162–75.e14. doi: 10.1016/j.cell.2017.12.013
56. Leentjens J, Bekkering S, Joosten LAB, Netea MG, Burgner DP, Riksen NP. Trained innate immunity as a novel mechanism linking infection and the development of atherosclerosis. Circ Res. (2018) 122:664–9. doi: 10.1161/CIRCRESAHA.117.312465
57. Nakahira K, Haspel JA, Rathinam VA, Lee S-J, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune response by inhibiting NALP3 inflammasome-mediated mitochondrial DNA release. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
58. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
59. Bayeva M, Khechaduri A, Puig S, Chang HC, Patial S, Blackshear PJ, et al. mTOR regulates cellular iron homeostasis through tristetraprolin. Cell Metab. (2012) 16:645–57. doi: 10.1016/j.cmet.2012.10.001
60. Mleczko-Sanecka K, Roche F, Rita da Silva A, Call D, D'Alessio F, Ragab A, et al. Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood (2014) 123:1574–85. doi: 10.1182/blood-2013-07-515957
61. Sancho D, Enamorado M, Garaude J. Innate immune function of mitochondrial metabolism. Front Immunol. (2017) 8:527. doi: 10.3389/fimmu.2017.00527
62. Kim M-J, Yoon J-H, Ryu J-H. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep. (2016) 49:529–35. doi: 10.5483/BMBRep.2016.49.10.115
63. Song Y, Ding W, Xiao Y, Lu K. The progress of mitophagy and related pathogenic mechanisms of the neurodegenerative diseases and tumor. Neurosci J. (2015) 2015:543758. doi: 10.1155/2015/543758
64. Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, et al. Mitophagy and Alzheimer's disease: cellular and molecular mechanisms. Trends Neurosci. (2017) 40:151–66. doi: 10.1016/j.tins.2017.01.002
65. Qiao H, Ren H, Du H, Zhang M, Xiong X, Lv R. Liraglutide repairs the infarcted heart: the role of the SIRT1/Parkin/mitophagy pathway. Mol Med Rep. (2018) 17:3722–34. doi: 10.3892/mmr.2018.8371
66. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer's disease. Nature (2017) 552:355–61. doi: 10.1038/nature25158
67. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. ASC has extracellular and prionoid activities that propagate inflammation. Nat Immunol. (2014) 15:727–37. doi: 10.1038/ni.2913
68. Lai M, Yao H, Shah SZA, Wu W, Wang D, Zhao Y, et al. The NLRP3-caspase 1 inflammasome negatively regulates autophagy via TLR4-TRIF in prion peptide-infected microglia. Front Aging Neurosci. (2018) 10:116. doi: 10.3389/fnagi.2018.00116
69. Zhu BT. Human and animal spongiform encephalopathies are the result of chronic autoimmune attack in the CNS: a novel medical theory supported by overwhelming experimental evidence. Histol Histopathol. (2005) 20:575–92. doi: 10.14670/HH-20.575
70. Maat P, de Beukelaar JW, Jansen C, et al. Pathologically confirmed autoimmune encephalitis in suspected Creutzfeldt-Jakob disease. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e178. doi: 10.1212/NXI.0000000000000178
71. Singh A, Isaac AO, Luo X, Mohan ML, Cohen ML, Chen F, et al. Abnormal brain iron homeostasis in human and animal prion disorders. PLoS Pathog. (2009) 5:e1000336. doi: 10.1371/journal.ppat.1000336
72. Ghio AJ, Baker JF. Iron homoeostasis in rheumatic disease. Rheumatology (2009) 48:1339–44. doi: 10.1093/rheumatology/kep221
73. Sweeney ME, Slusser JG, Lynch SG, Benedict SH, Garcia SL, Rues L, et al. Deferiprone modulates in vitro responses by peripheral blood T cells from control and relapsing-remitting multiple sclerosis subjects. Int Immunopharmacol. (2011)11:1796–801. doi: 10.1016/j.intimp.2011.07.007
74. Mitchell KM, Dotson AL, Cool KM, Chakrabarty A, Benedict SH, LeVine SM. Deferiprone, an orally deliverable iron chelator, ameliorates experimental autoimmune encephalomyelitis. Mult Scler. (2007)13:1118–26. doi: 10.1177/1352458507078916
75. Bolognin S, Drago D, Messori L, Zatta P. Chelation therapy for neurodegenerative diseases. Med Res Rev. (2009) 29:547–70. doi: 10.1002/med.20148
76. Liu G, Men P, Perry G, Smith MA. Nanoparticle and iron chelators as a potential novel Alzheimer therapy. Methods Mol Biol. (2010) 610:123–44. doi: 10.1007/978-1-60327-029-8_8
77. Wang N, Jin X, Guo D, Tong G, Zhu X. Iron chelation nanoparticles with delayed saturation as an effective therapy for Parkinson disease. Biomacromolecules (2017) 18:461–74. doi: 10.1021/acs.biomac.6b01547
78. Lehrer S, Rheinstein PH. Is Alzheimer's disease autoimmune inflammation of the brain that can be treated with nasal nonsteroidal anti-inflammatory drugs? Am J Alzheimers Dis Other Demen. (2015) 30:225–7. doi: 10.1177/1533317514545478
79. Carter CJ. Alzheimer's disease: a pathogenetic autoimmune disorder caused by herpes simplex in a gene-dependent manner. Int J Alzheimer's Dis. (2010) 2010:140539. doi: 10.4061/2010/140539
80. Vacirca D, Delunardo F, Matarrese P, Colasanti T, Margutti P, Siracusano A, et al. Autoantibodies to the adenosine triphosphate synthase play a pathogenetic role in Alzheimer's disease. Neurobiol Aging (2012) 33:753–66. doi: 10.1016/j.neurobiolaging.2010.05.013
81. Dinkins MB, Dasgupta S, Wang G, Zhu G, He Q, Kong JN, et al. The 5XFAD mouse model of Alzheimer's disease exhibits an age-dependent increase in anti-ceramide IgG and exogenous administration of ceramide further increases anti-ceramide titers and amyloid plaque burden. J Alzheimer's Dis. (2015) 46:55–61. doi: 10.3233/JAD-150088
82. Maftei M, Thurm F, Schnack C, Tumani H, Otto M, Elbert T, et al. Increased levels of antigen-bound β-amyloid autoantibodies in serum and cerebrospinal fluid of Alzheimer's disease patients. PLoS ONE (2013) 8:e68996. doi: 10.1371/journal.pone.0068996
83. Scheiblich H, Schlütter A, Golenbock DT, Latz E, Martinez-Martinez P, Heneka MT. Activation of the NLRP3 inflammasome in microglia: the role of ceramide. J Neurochem. (2017) 143:534–50. doi: 10.1111/jnc.14225
84. Van Duijn S, Bulk M, van Duinen SG, Nabuurs RJA, van Buchem MA, van der Weerd L, et al. Cortical iron reflects severity of Alzheimer's disease. J Alzheimer's Dis. (2017) 60:1533–45. doi: 10.3233/JAD-161143
85. Liu L, Chan C. The role of inflammasome in Alzheimer's disease. Ageing Res Rev. (2014) 15:6–15. doi: 10.1016/j.arr.2013.12.007
86. Shen HH, Yang YX, Meng X, Luo XY, Li XM, Shuai ZW, et al. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmun Rev. (2018) 17:694–702. doi: 10.1016/j.autrev.2018.01.020
87. Yi Y-S. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J Physiol Pharmacol. (2018) 22:1–15. doi: 10.4196/kjpp.2018.22.1.1
88. Zhang R, Miller RG, Gascon R, Champion S, Katz J, Lancero M, et al. Circulating endotoxin and systemic immune activation in sporadic Amyotrophic Lateral Sclerosis (sALS). J Neuroimmunol. (2009) 206:121–4. doi: 10.1016/j.jneuroim.2008.09.017
89. Pritchard AB, Crean S, Olsen I, Singhrao SK. Periodontitis, microbiomes and their role in Alzheimer's disease. Front Aging Neurosci. (2017) 9:336. doi: 10.3389/fnagi.2017.00336
90. Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell (2016) 167:915–32. doi: 10.1016/j.cell.2016.10.027
91. Yoon KW. Dead cell phagocytosis and innate immune checkpoint. BMB Rep. (2017) 50:496–503. doi: 10.5483/BMBRep.2017.50.10.147
92. Fu R, Shen Q, Xu P, Luo JJ, Tang Y. Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol. (2014) 49:1422–34. doi: 10.1007/s12035-013-8620-6
93. Neniskyte U, Neher JJ, Brown GC. Neuronal death induced by nanomolar amyloid β is mediated by primary phagocytosis of neurons by microglia. J Biol Chem. (2011) 286:39904–13. doi: 10.1074/jbc.M111.267583
94. Neher JJ, Neniskyte U, Brown GC. Primary phagocytosis of neurons by inflamed microglia: potential roles in neurodegeneration. Front Pharmacol. (2012) 3:27. doi: 10.3389/fphar.2012.00027
95. Zhong L, Zhang Z-L, Li X, Liao C, Mou P, Wang T, et al. TREM2/DAP12 complex regulates inflammatory responses in microglia via the JNK signaling pathway. Front Aging Neurosci. (2017) 9:204. doi: 10.3389/fnagi.2017.00204
96. Atagi Y, Liu C-C, Painter MM, Chen XF, Verbeeck C, Zheng H, et al. Apolipoprotein E is a ligand for triggering receptor Expressed on myeloid cells 2 (TREM2). J Biol Chem. (2015) 290:26043–50. doi: 10.1074/jbc.M115.679043
97. Hsieh CL, Koike M, Spusta S, Niemi EC, Yenari M, Nakamura MC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. (2009) 109:1144–56. doi: 10.1111/j.1471-4159.2009.06042.x
98. Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, et al. TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron (2018) 97:1023–31.e7. doi: 10.1016/j.neuron.2018.01.031
99. Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, et al. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. (2007)37:1290–301. doi: 10.1002/eji.200636837
100. Öhrfelt A, Axelsson M, Malmeström C, Novakova L, Heslegrave A, Blennow K, et al. Soluble TREM-2 in cerebrospinal fluid from patients with multiple sclerosis treated with natalizumab or mitoxantrone. Mult Scler. (2016) 22:1587–95 doi: 10.1177/1352458515624558
101. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity (2017) 47:566–81.e9. doi: 10.1016/j.immuni.2017.08.008
102. Jendresen C, Årskog V, Daws MR, Nilsson LNG. The Alzheimer's disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J Neuroinflamm. (2017) 14:59. doi: 10.1186/s12974-017-0835-4
103. Wang R, Wang Y, Mu N, Lou X, Li W, Chen Y, et al. Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab Invest (2017) 97:922–34. doi: 10.1038/labinvest.2017.30
104. Beurel E. HDAC6 regulates LPS-tolerance in astrocytes. PLoS ONE (2011) 6:e25804. doi: 10.1371/journal.pone.0025804
105. Pretorius E, Bester J, Kell DB. A bacterial component to Alzheimer's-type dementia seen via a systems biology approach that links iron dysregulation and inflammagen shedding to disease. J Alzheimer's Dis. (2016) 53:1237–56. doi: 10.3233/JAD-160318
106. Rocha ER, Bergonia HA, Gerdes S, Jeffrey Smith C. Bacteroides fragilis requires the ferrous-iron transporter FeoAB and the CobN-like proteins BtuS1 and BtuS2 for assimilation of iron released from heme. Microbiologyopen (2018) 21:e00669. doi: 10.1002/mbo3.669
107. Won SM, Lee JH, Park UJ, Gwag J, Gwag BJ, Lee YB. Iron mediates endothelial cell damage and blood-brain barrier opening in the hippocampus after transient forebrain ischemia in rats. Exp Mol Med. (2011) 43:121–8. doi: 10.3858/emm.2011.43.2.020
108. Liu H, Zhang J, Chen X, Du XS, Zhang JL, Liu G, et al. Application of iron oxide nanoparticles in glioma imaging and therapy: from bench to bedside. Nanoscale (2016) 8:7808–26. doi: 10.1039/C6NR00147E
109. Rocha ER, de Uzeda M, Brock JH. Effect of ferric and ferrous iron chelators on growth of Bacteroides fragilis under anaerobic conditions. FEMS Microbiol Lett. (1991) 68:45–50. doi: 10.1111/j.1574-6968.1991.tb04567.x
110. Bonham LW, Desikan RS, Yokoyama JS, for the Alzheimer's Disease Neuroimaging Initiative. The relationship between complement factor C3, APOE ε4, amyloid and tau in Alzheimer's disease. Acta Neuropathol Commun. (2016) 4:65. doi: 10.1186/s40478-016-0339-y
111. Fraser DA, Pisalyaput K, Vogler EC, Tenner AJ. A role for complement components C1q and C3 in the clearance of apoptotic cells by microglia. FASEB J. (2008) 22(2 Suppl.):554–4.
112. Baitsch D, Bock HH, Engel T, Telgmann R, Müller-Tidow C, Varga G, et al. Apolipoprotein E (Apoe) induces anti-inflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol. (2011) 31:1160–8. doi: 10.1161/ATVBAHA.111.222745
113. Nandal A, Ruiz JC, Subramanian P, Ghimire-Rijal S, Sinnamon RA, Stemmler TL, et al. Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. (2011) 14:647–57. doi: 10.1016/j.cmet.2011.08.015
114. Lian H, Litvinchuk A, Chiang AC-A, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of alzheimer's disease. J Neurosci. (2016) 36:577–89. doi: 10.1523/JNEUROSCI.2117-15.2016
115. Scott D, Botto M. The paradoxical roles of C1q and C3 in autoimmunity. Immunobiology (2016) 221:719–25. doi: 10.1016/j.imbio.2015.05.001
116. Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol. (2012) 188:5682–93. doi: 10.4049/jimmunol.1103760
117. Son M, Diamond B, Santiago-Schwarz F. Fundamental role of C1q in autoimmunity and inflammation. Immunol Res. (2015) 63:101–6. doi: 10.1007/s12026-015-8705-6
118. Chung W-S, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD, et al. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci USA. (2016) 113:10186–91. doi: 10.1073/pnas.1609896113
119. Zhao N, Liu CC, Qiao W, Bu G. Apolipoprotein E, receptors, and modulation of Alzheimer's disease. Biol Psychiatry (2018) 83:347–57. doi: 10.1016/j.biopsych.2017.03.003
120. Ulrich JD, Ulland TK, Mahan TE, Nyström S, Nilsson KP, Song WM, et al. ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med. (2018) 215:1047–58. doi: 10.1084/jem.20171265
121. Xu H, Perreau VM, Dent KA, Bush AI, Finkelstein DI, Adlard PA. Iron regulates apolipoprotein E expression and secretion in neurons and astrocytes. J Alzheimers Dis. (2016) 51:471–87. doi: 10.3233/JAD-150797
122. Uchihara T, Duyckaerts C, He Y, Kobayashi K, Seilhean D, Amouyel P, Hauw JJ. ApoE immunoreactivity and microglial cells in Alzheimer's disease brain. Neurosci Lett. (1995) 195:5–8. doi: 10.1016/0304-3940(95)11763-M
123. Li Y, Song D, Song Y, et al. Iron-induced local complement Component 3 (C3) up-regulation via non-canonical Transforming Growth Factor (TGF)-β signaling in the retinal pigment epithelium. J Biol Chem. (2015) 290:11918–34. doi: 10.1074/jbc.M115.645903
124. Dimitrov JD, Roumenina LT, Doltchinkova VR, Vassilev TL. Iron ions and haeme modulate the binding properties of complement subcomponent C1q and of immunoglobulins. Scand J Immunol. (2007) 65:230–9. doi: 10.1111/j.1365-3083.2006.01893.x
125. Shen Y, Yang L, Li R. What does complement do in Alzheimer's disease? Old molecules with new insights. Transl Neurodegener. (2013) 2:21. doi: 10.1186/2047-9158-2-21
126. Goetzl EJ, Schwartz JB, Abner EL, Jicha GA, Kapogiannis D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann Neurol. (2018) 83:544–52. doi: 10.1002/ana.25172
127. Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. The role of TREM2 in Alzheimer's disease and other neurodegenerative disorders. Lancet Neurol. (2018) 17:721–30. doi: 10.1016/S1474-4422(18)30232-1
128. Bodovitz S, Falduto MT, Frail DE, Klein WL. Iron levels modulate alpha-secretase cleavage of amyloid precursor protein. J Neurochem. (1995) 64:307–15. doi: 10.1046/j.1471-4159.1995.64010307.x
129. Silvestri L, Camaschella C. A potential pathogenetic role of iron in Alzheimer's disease. J Cell Mol Med. (2008) 12:1548–50. doi: 10.1111/j.1582-4934.2008.00356.x
130. Kim M, Kim J, Cheon CI, Cho DH, Park JH, Kim KI, Lee KY, Song E. Increased expression of the F(1)F(o) ATP synthase in response to iron in heart mitochondria. BMB Rep. (2008) 41:153–7. doi: 10.5483/BMBRep.2008.41.2.153
131. Goldberg J, Currais A, Prior M, Fischer W, Chiruta C, Ratliff E, et al. The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell (2018) 17:e12715. doi: 10.1111/acel.12715
132. Evavold CL, Kagan JC. How inflammasomes inform adaptive immunity. J Mol Biol. (2018) 430:217–37. doi: 10.1016/j.jmb.2017.09.019
133. Santarlasci V, Cosmi L, Maggi L, Liotta F, Annunziato F. IL-1 and T helper immune responses. Front Immunol. (2013) 4:182. doi: 10.3389/fimmu.2013.00182
134. Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol. (2018) 9:697. doi: 10.3389/fimmu.2018.00697
135. Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, et al. Interleukin-1β induces blood–brain barrier disruption by downregulating sonic hedgehog in astrocytes. PLoS ONE (2014) blood:e110024. doi: 10.1371/journal.pone.0110024
136. Yao Y, Chen S, Cao M, Fan X, Yang T, Huang Y, et al. Antigen-specific CD8+ T cell feedback activates NLRP3 inflammasome in antigen-presenting cells through perforin. Nat Commun. (2017) 8:15402. doi: 10.1038/ncomms15402
137. Ebner F, Brandt C, Thiele P, Richter D, Schliesser U, Siffrin V, et al. Microglial activation milieu controls regulatory T cell responses. J Immunol. (2013) 191:5594–602. doi: 10.4049/jimmunol.1203331
138. Trajkovic V, Vuckovic O, Stosic-Grujicic S, Miljkovic D, Popadic D, Markovic M, et al. Astrocyte-induced regulatory T cells mitigate CNS autoimmunity. Glia (2004) 47:168–79. doi: 10.1002/glia.20046
139. Xie L, Choudhury GR, Winters A, Yang S-H, Jin K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur J Immunol. (2015) 45:180–91. doi: 10.1002/eji.201444823
140. Miles K, Simpson J, Brown S, Cowan G, Gray D, Gray M. Immune tolerance to apoptotic self is mediated primarily by regulatory B1a cells. Front Immunol. (2017) 8:1952. doi: 10.3389/fimmu.2017.01952
141. Chien C-H, Chiang B-L. Regulatory T cells induced by B cells: a novel subpopulation of regulatory T cells. J Biomed Sci. (2017) 24:24. doi: 10.1186/s12929-017-0391-3
142. Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain (2016) 139(Pt 4):1237–51. doi: 10.1093/brain/awv408
143. Baek H, Ye M, Kang G-H, Lee C, Lee G, Choi DB, et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer's disease model. Oncotarget (2016) 7:69347–57. doi: 10.18632/oncotarget.12469
144. Huang P, Wang J, Lin X, Yang FF, Tan JH. Effects of IL-10 on iron metabolism in LPS-induced inflammatory mice via modulating hepcidin expression. Eur Rev Med Pharmacol Sci. (2017) 21:3469–75.
145. McCarthy RC, Sosa JC, Gardeck AM, Baez AS, Lee CH, Wessling-Resnick M. Inflammation-induced iron transport and metabolism by brain microglia. J Biol Chem. (2018) 293:7853–63. doi: 10.1074/jbc.RA118.001949
146. Bierer BE, Nathan DG. The effect of desferrithiocin, an oral iron chelator, on T-cell function. Blood (1990) 76:2052–9.
147. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science (2017) 356:513–9. doi: 10.1126/science.aal3535
148. Chang JS, Li YL, Lu CH, Owaga E, Chen WY, Chiou HY. Interleukin-10 as a potential regulator of hepcidin homeostasis in overweight and obese children: a cross-sectional study in Taiwan. Nutrition (2014) 30:1165–70. doi: 10.1016/j.nut.2014.02.021
149. Kurtulus S, Sakuishi K, Ngiow S-F, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. (2015) 125:4053–62. doi: 10.1172/JCI81187
150. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the co-inhibitory molecule TIGIT selectively inhibit pro-inflammatory Th1 and Th17 cell responses. Immunity (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
151. Patsoukis N, Weaver JD, Strauss L, Herbel C, Seth P, Boussiotis VA. Immunometabolic regulations mediated by coinhibitory receptors and their impact on T cell immune responses. Front Immunol. (2017) 8:330. doi: 10.3389/fimmu.2017.00330
152. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
153. Santos RR, Torres KC, Lima GS, Fiamoncini CM, Mapa FC, Pereira PA, et al. Reduced frequency of T lymphocytes expressing CTLA-4 in frontotemporal dementia compared to Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry (2014) 48:1–5. doi: 10.1016/j.pnpbp.2013.06.019
154. Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med. (2016) 22:135–7. doi: 10.1038/nm.4022
155. Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. (2010) 21:927–38. doi: 10.3233/JAD-2010-091696
156. Zhao S, Li F, Leak RK, Chen J, Hu X. Regulation of neuroinflammation through programed Death-1/programed death ligand signaling in neurological disorders. Front Cell Neurosci. (2014) 8:271. doi: 10.3389/fncel.2014.00271
157. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
158. Song Y, Wang B, Song R, Hao Y, Wang D, Li Y, et al. T-cell immunoglobulin and ITIM domain contributes to CD8+ T-cell immunosenescence. Aging Cell (2018) 17:e12716. doi: 10.1111/acel.12716
159. Fettelschoss A, Zabel F, Bachmann MF. Vaccination against Alzheimer disease: an update on future strategies. Hum Vaccines Immunother. (2014)10:847–51. doi: 10.4161/hv.28183
160. Zou C, Liu X, Xie R, Bao Y, Jin Q, Jia X, et al. Deferiprone attenuates inflammation and myocardial fibrosis in diabetic cardiomyopathy rats. Biochem Biophys Res Commun. (2017) 486:930–6. doi: 10.1016/j.bbrc.2017.03.127
161. Olson DE, Sleiman SF, Bourassa MW, Wagner FF, Gale JP, Zhang YL, et al. Hydroxamate-based histone deacetylase inhibitors can protect neurons from oxidative stress via an HDAC-independent catalase-like mechanism. Chem Biol. (2015) 22:439–445. doi: 10.1016/j.chembiol.2015.03.014
162. Hwang I, Lee E, Jeon SA, Yu JW. Histone deacetylase 6 negatively regulates NLRP3 inflammasome activation. Biochem Biophys Res Commun. (2015) 467:973–8. doi: 10.1016/j.bbrc.2015.10.033
163. Yang S, Zhang R, Wang G, Zhang Y. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer's disease. Transl Neurodegener. (2017) 6:19. doi: 10.1186/s40035-017-0089-1
164. Xu K, Dai X-L, Huang H-C, Jiang Z-F. Targeting HDACs: a promising therapy for Alzheimer's disease. Oxid Med Cell Longev. (2011) 2011:143269. doi: 10.1155/2011/143269
165. Durham BS, Grigg R, Wood IC. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J Neurochem. (2017)143:214–24. doi: 10.1111/jnc.14144
166. Datta M, Staszewski O, Raschi E, Frosch M, Hagemeyer N, Tay TL, et al. Histone deacetylases 1 and 2 regulate microglia function during development, homeostasis, and neurodegeneration in a context-dependent manner. Immunity (2018) 48:514–29.e6. doi: 10.1016/j.immuni.2018.02.016
167. Ren H, Kong Y, Liu Z, et al. Selective NLRP3 (pyrin domain–containing protein 3) inflammasome inhibitor reduces brain injury after intracerebral hemorrhage. Stroke (2018) 49:184–92. doi: 10.1161/STROKEAHA.117.018904
168. Xu X, Yin D, Ren H, Gao W, Li F, Sun D, et al. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis. (2018) 17:15–27. doi: 10.1016/j.nbd.2018.05.016
169. Qi Y, Klyubin I, Cuello AC, Rowan MJ. NLRP3-dependent synaptic plasticity deficit in an Alzheimer's disease amyloidosis model in vivo. Neurobiol Dis. (2018) 114:24–30. doi: 10.1016/j.nbd.2018.02.016
170. Redondo-Castro E, Faust D, Fox S, Baldwin AG, Osborne S, Haley MJ, et al. Development of a characterised tool kit for the interrogation of NLRP3 inflammasome-dependent responses. Sci Rep. (2018) 8:5667. doi: 10.1038/s41598-018-24029-3
171. Kwak S-B, Koppula S, In E-J, Sun X, Kim YK, Kim MK, et al. Artemisia extract suppresses NLRP3 and AIM2 inflammasome activation by inhibition of ASC phosphorylation. Mediat Inflamm. (2018) 2018:6054069. doi: 10.1155/2018/6054069
172. Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun. (2017) 61:306–16. doi: 10.1016/j.bbi.2016.12.014
173. Birnbaum Y, Bajaj M, Yang HC, Ye Y. Combined SGLT2 and DPP4 inhibition reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic nephropathy in mice with type 2 diabetes. Cardiovasc Drugs Ther. (2018) 32:135–45. doi: 10.1007/s10557-018-6778-x
174. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. (2018) 10:e8689. doi: 10.15252/emmm.201708689
175. Zhang Y, Liu F, Yuan Y, Jin C, Chang C, Zhu Y, et al. Inflammasome-derived exosomes activate NF-κB signaling in macrophages. J Proteome Res. (2017)16:170–8. doi: 10.1021/acs.jproteome.6b00599
176. Cai Z-Y, Xiao M, Quazi SH, Ke Z-Y. Exosomes: a novel therapeutic target for Alzheimer's disease? Neural Regener Res. (2018) 13:930–5. doi: 10.4103/1673-5374.232490
177. Li JJ, Wang B, Kodali MC, Chen C, Kim E, Patters BJ, et al. In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J Neuroinflamm. (2018) 15:8. doi: 10.1186/s12974-017-1038-8
178. Kouwaki T, Fukushima Y, Daito T, Sanada T, Yamamoto N, Mifsud EJ, et al. Extracellular vesicles including exosomes regulate innate immune responses to hepatitis B virus infection. Front Immunol. (2016) 7:335. doi: 10.3389/fimmu.2016.00335
179. Potolicchio I, Carven GJ, Xu X, Stipp C, Riese RJ, Stern LJ, Santambrogio L. Proteomic analysis of microglia-derived exosomes: metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. J Immunol. (2005) 175:2237–43. doi: 10.4049/jimmunol.175.4.2237
180. Fitzner D, Schnaars M, van Rossum D, Krishnamoorthy G, Dibaj P, Bakhti M, et al. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J Cell Sci. (2011) 124(Pt 3):447–58. doi: 10.1242/jcs.074088
181. Yuyama K, Igarashi Y. Exosomes as carriers of Alzheimer's amyloid-ß. Front Neurosci. (2017) 11:229. doi: 10.3389/fnins.2017.00229
182. Gangadaran P, Hong CM, Ahn B-C. An update on in vivo imaging of extracellular vesicles as drug delivery vehicles. Front Pharmacol. (2018) 9:169. doi: 10.3389/fphar.2018.00169
183. Xiao T, Zhang W, Jiao B, Pan C-Z, Liu X, Shen L. The role of exosomes in the pathogenesis of Alzheimer' disease. Transl Neurodegener. (2017) 6:3. doi: 10.1186/s40035-017-0072-x
184. Semenov DV, Kanyshkova TG, Akimzhanov AM, Buneva VN, Nevinsky GA. Interaction of human milk lactoferrin with ATP. Biochemistry (Mosc) (1998) 63:944–51.
185. Malhotra H, Sheokand N, Kumar S, Chauhan AS, Kumar M, Jakhar P, et al. Exosomes: tunable nano vehicles for macromolecular delivery of transferrin and lactoferrin to specific intracellular compartment. J Biomed Nanotechnol. (2016) 12:1101–14. doi: 10.1166/jbn.2016.2229
186. Noh H, Seo H. Age-dependent effects of valproic acid in Alzheimer's disease (AD) mice are associated with nerve growth factor (NGF) regulation. Neuroscience (2014) 266:255–65. doi: 10.1016/j.neuroscience.2014.02.012
187. Chen X, Wang H, Zhou M, Li X, Fang Z, Gao H, et al. Valproic acid attenuates traumatic brain injury-induced inflammation in vivo: involvement of autophagy and the Nrf2/ARE signaling pathway. Front Mol Neurosci. (2018) 11:117. doi: 10.3389/fnmol.2018.00117
188. Nicolini C, Ahn Y, Michalski B, Rho JM, Fahnestock M. Decreased mTOR signaling pathway in human idiopathic autism and in rats exposed to valproic acid. Acta Neuropathol Commun. (2015) 3:3. doi: 10.1186/s40478-015-0184-4
189. Dresselhaus E, Duerr JM, Vincent F, Sylvain EK, Beyna M, Lanyon LF, et al. Class I HDAC inhibition is a novel pathway for regulating astrocytic apoE secretion. PLoS ONE (2018) 13:e0194661. doi: 10.1371/journal.pone.0194661
190. Wang W, Di X, Torti SV, Torti FM. FERRITIN H INDUCTION BY HISTONE DEACETYLASE INHIBITORS. Biochem Pharmacol. (2010) 80:316–24. doi: 10.1016/j.bcp.2010.04.008
191. Uranga RM, Giusto NM, Salvador GA. Iron-induced oxidative injury differentially regulates PI3K/Akt/GSK3beta pathway in synaptic endings from adult and aged rats. Toxicol Sci. (2009) 111:331–44. doi: 10.1093/toxsci/kfp152
192. Jellusova J, Cato MH, Apgar JR, Ramezani-Rad P, Leung CR, Chen C, et al. GSK3 is a metabolic checkpoint regulator in B cells. Nat Immunol. (2017) 18:303–12. doi: 10.1038/ni.3664
193. Nassar A, Azab AN. Effects of lithium on inflammation. ACS Chem Neurosci. (2014) 5:451–8. doi: 10.1021/cn500038f
194. Hill EV, Ng THS, Burton BR, Oakley CM, Malik K, Wraith DC. Glycogen synthase kinase-3 controls IL-10 expression in CD4+ effector T-cell subsets through epigenetic modification of the IL-10 promoter. Eur J Immunol. (2015) 45:1103–15. doi: 10.1002/eji.201444661
195. Leu SJ, Yang YY, Liu HC, Cheng CY, Wu YC, Huang MC, et al. Valproic acid and lithium meditate anti-inflammatory effects by differentially modulating dendritic cell differentiation and function. J Cell Physiol. (2017) 232:1176–86. doi: 10.1002/jcp.25604
196. Jiang Y, Li Z, Ma H, Cao X, Liu F, Tian A. Upregulation of TREM2 ameliorates neuroinflammatory responses and improves cognitive deficits triggered by surgical trauma in Appswe/PS1dE9 mice. Cell Physiol Biochem. (2018) 46:1398–411. doi: 10.1159/000489155
197. Fuertes Marraco SA, Neubert NJ, Verdeil G, Speiser DE. Inhibitory receptors beyond T cell exhaustion. Front Immunol. (2015) 6:310. doi: 10.3389/fimmu.2015.00310
198. Zhang Q, Vignali DA. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity (2016) 44:1034–51. doi: 10.1016/j.immuni.2016.04.017
199. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
200. Lowther D, Ramanan S, DeBartolo D, Park C, Duan X, Hafler D, et al. The TIGIT/CD226/CD155 axis is differentially expressed in MS and glioblastoma: implications for autoimmunity and tumor immune escape. Neurology (2015) 84(14 Suppl.):P4.043.
201. Chihara N. Dysregulated T cells in multiple sclerosis. Clin Exp Neuroimmunol. (2018) 9:20–9. doi: 10.1111/cen3.12438
202. Fang L, Jin J, Chen P, Wang N, Zeng H, Jin B, et al. CD226 deficiency improves cognitive functions and ameliorates anxiety-like behaviors in mice. Brain Behav. (2017). 7:e00871. doi: 10.1002/brb3.871
203. Ke P, Shao BZ, Xu ZQ, Chen XW, Wei W, Liu C. Activating α7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through regulation of β-arrestin-1. CNS Neurosci Ther. (2017) 23:875–84. doi: 10.1111/cns.12758
204. Báez-Pagán CA, Delgado-Vélez M, Lasalde-Dominicci JA. Activation of the macrophage α7 nicotinic acetylcholine receptor and control of inflammation. J Neuroimmune Pharmacol. (2015) 10:468–76. doi: 10.1007/s11481-015-9601-5
205. Peron R, Vatanabe IP, Manzine PR, Camins A, Cominetti MR. Alpha-secretase ADAM10 regulation: insights into Alzheimer's disease treatment. Pharmaceuticals (Basel). (2018) 11:E12. doi: 10.3390/ph11010012
206. Rothbard JB, Rothbard JJ, Soares L, Fathman CG, Steinman L. Identification of a common immune regulatory pathway induced by small heat shock proteins, amyloid fibrils, and nicotine. Proc Natl Acad Sci USA. (2018) 115:7081–6. doi: 10.1073/pnas.1804599115
207. Liu Q, Whiteaker P, Morley BJ, Shi F-D, Lukas RJ. Distinctive roles for α7*- and α9*-nicotinic acetylcholine receptors in inflammatory and autoimmune responses in the murine experimental autoimmune encephalomyelitis model of multiple sclerosis. Front Cell Neurosci. (2017) 11:287. doi: 10.3389/fncel.2017.00287
208. Chen K, Sun Y, Dong W, Zhang T, Zhou N, Yu W, et al. Activated A7nachr improves postoperative cognitive dysfunction and intestinal injury induced by cardiopulmonary bypass in rats: inhibition of the proinflammatory response through the Th17 immune response. Cell Physiol Biochem. (2018) 46:1175–88. doi: 10.1159/000489068
209. Cortes M, Cao M, Liu HL, Moore CS, Durosier LD, Burns P, et al. α7 nicotinic acetylcholine receptor signaling modulates the inflammatory phenotype of fetal brain microglia: first evidence of interference by iron homeostasis. Sci Rep. (2017) 7:10645. doi: 10.1038/s41598-017-09439-z
210. Gao Q, Liu YJ, Guan ZZ. Oxidative stress might be a mechanism connected with the decreased alpha 7 nicotinic receptor influenced by high-concentration of fluoride in SH-SY5Y neuroblastoma cells. Toxicol In Vitro (2008) 22:837–43. doi: 10.1016/j.tiv.2007.12.017
211. Akilo OD, Kumar P, Choonara YE, Pradeep P, du Toit LC, Pillay V. Hypothesis: apo-lactoferrin–Galantamine proteo-alkaloid conjugate for Alzheimer's disease intervention. J Cell Mol Med. (2018) 22:1957–63. doi: 10.1111/jcmm.13484
212. Du R-H, Tan J, Sun X-Y, Lu M, Ding J-H, Hu G. Fluoxetine Inhibits NLRP3 inflammasome activation: implication in depression. Int J Neuropsychopharmacol. (2016) 19:pyw037. doi: 10.1093/ijnp/pyw037
213. Lu Y, Xiao G, Luo W. Minocycline suppresses NLRP3 inflammasome activation in experimental ischemic stroke. Neuroimmunomodulation (2016) 23:230–8. doi: 10.1159/000452172
214. Ledo JH, Azevedo EP, Beckman D, Ribeiro FC, Santos LE, Razolli DS, et al. Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer's amyloid-β oligomers in mice. J Neurosci. (2016) 36:12106–16. doi: 10.1523/JNEUROSCI.1269-16.2016
215. Tronel C, Largeau B, Santiago Ribeiro MJ, Guilloteau D, Dupont AC, Arlicot N. Molecular targets for PET imaging of activated microglia: the current situation and future expectations. Int J Mol Sci. (2017) 18:E802. doi: 10.3390/ijms18040802
216. Jürgens B, Raberger J, Fuchs D, Heitger A. Indoleamine 2,3-dioxygenase in human hematopoietic stem cell transplantation. Int J Tryptophan Res. (2010) 3:77–90. doi: 10.4137/IJTR.S4076
217. Mbongue JC, Nicholas DA, Torrez TW, Kim N-S, Firek AF, Langridge WHR. The role of indoleamine 2, 3-dioxygenase in immune suppression and autoimmunity. Vaccines (2015) 3:703–29. doi: 10.3390/vaccines3030703
218. Guleria I, Sayegh MH. Maternal acceptance of the fetus: true human tolerance. J Immunol. (2007) 178:3345–51. doi: 10.4049/jimmunol.178.6.3345
219. Andersen MH, Svane IM. Indoleamine 2,3-dioxygenase vaccination. Oncoimmunology (2015) 4:e983770. doi: 10.4161/2162402X.2014.983770
220. Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer's disease hippocampus. Neuropathol Appl Neurobiol. (2005) 31:395–404. doi: 10.1111/j.1365-2990.2005.00655.x
221. Kubicova L, Hadacek F, Chobot V. Quinolinic acid: neurotoxin or oxidative stress modulator? Int J Mol Sci. (2013) 14:21328–38. doi: 10.3390/ijms141121328
222. Pláteník J, Stopka P, Vejrazka M, Stípek S. Quinolinic acid-iron(ii) complexes: slow autoxidation, but enhanced hydroxyl radical production in the Fenton reaction. Free Radic Res. (2001) 34:445–59. doi: 10.1080/10715760100300391
223. Ting KK, Brew BJ, Guillemin GJ. Effect of quinolinic acid on human astrocytes morphology and functions: implications in Alzheimer's disease. J Neuroinflamm. (2009) 6:36. doi: 10.1186/1742-2094-6-36
224. Gulaj E, Pawlak K, Bien B, Pawlak D. Kynurenine and its metabolites in Alzheimer's disease patients. Adv Med Sci. (2010) 55:204–11. doi: 10.2478/v10039-010-0023-6
225. Feng W, Wang Y, Liu ZQ, Zhang X, Han R, Miao YZ, et al. Microglia activation contributes to quinolinic acid-induced neuronal excitotoxicity through TNF-α. Apoptosis (2017) 22:696–709. doi: 10.1007/s10495-017-1363-5
226. Altanerova U, Babincova M, Babinec P, Benejova K, Jakubechova J, Altanerova V, et al. Human mesenchymal stem cell-derived iron oxide exosomes allow targeted ablation of tumor cells via magnetic hyperthermia. Int J Nanomed. (2017) 12:7923–36. doi: 10.2147/IJN.S145096
Keywords: inflammasomes, astrocytes, microglia, exosomes, trained immunity, tolerized immunity
Citation: Sfera A, Gradini R, Cummings M, Diaz E, Price AI and Osorio C (2018) Rusty Microglia: Trainers of Innate Immunity in Alzheimer's Disease. Front. Neurol. 9:1062. doi: 10.3389/fneur.2018.01062
Received: 09 August 2018; Accepted: 21 November 2018;
Published: 04 December 2018.
Edited by:
Isabella Zanella, Università degli Studi di Brescia, ItalyReviewed by:
Ilias Kounatidis, Diamond Light Source, United KingdomPo-Wah So, King's College London, United Kingdom
Copyright © 2018 Sfera, Gradini, Cummings, Diaz, Price and Osorio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adonis Sfera, ZHIuc2ZlcmFAZ21haWwuY29t