
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurol. , 20 November 2018
Sec. Multiple Sclerosis and Neuroimmunology
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00946
This article is part of the Research Topic Peripheral Immunity in Parkinson's Disease: Emerging Role and Novel Target for Therapeutics View all 13 articles
 Alice J. White*
Alice J. White* Ruwani S. Wijeyekoon
Ruwani S. Wijeyekoon Kirsten M. Scott
Kirsten M. Scott Nushan P. GunawardanaShaista HayatI. H. SolimH. T. McMahon
Nushan P. GunawardanaShaista HayatI. H. SolimH. T. McMahon Roger A. Barker
Roger A. Barker Caroline H. Williams-Gray
Caroline H. Williams-GrayThe immune system is activated in Parkinson's Disease (PD), as evidenced by neuroinflammatory changes within the brain as well as elevated immune markers in peripheral blood. Furthermore, inflammatory cytokine levels in the blood are associated with disease severity and rate of progression. However, the factors driving this immune response in PD are not well established. We investigated cell-extrinsic factors in systemic immune activation by using α-synuclein monomers and fibrils, as well as bacterial toxins, to stimulate peripheral blood mononuclear cells (PBMCs) derived from 31 patients and age/gender-matched controls. α-synuclein monomers or fibrils resulted in a robust cytokine response (as measured by supernatant cytokine concentrations and mRNA expression in cultured cells) in both PD and control PBMCs, similar to that induced by bacterial LPS. We found no PD vs. control differences in cytokine production, nor in mRNA expression. Levels of endotoxin within the recombinant α-synuclein used in these experiments were very low (0.2–1.3EU/mL), but nonetheless we found that comparable levels were sufficient to potentially confound our cytokine concentration measurements for a number of cytokines. However, α-synuclein monomers increased production of IL-1β and IL-18 to levels significantly in excess of those induced by low-level endotoxin. In conclusion, this study: (i) highlights the importance of accounting for low-level endotoxin in antigen-PBMC stimulation experiments; (ii) indicates that cell-extrinsic factors may be a major contributor to immune activation in PD; and (iii) suggests that α-synuclein may play a role in inflammasome-related cytokine production in the periphery.
The immune system is known to be altered in Parkinson's disease (PD). Whilst some of these changes may be secondary phenomena, a growing body of evidence suggests that the immune system may play a contributory role in the primary progression of PD (1, 2). α-synuclein is the key protein implicated in the pathogenesis of PD, forming intracellular aggregates known as Lewy bodies (3). Fibrillar α-synuclein is the principal pathological form present in Lewy bodies (4), but the protein also exists in monomeric and oligomeric forms within the CNS, and all three may trigger a central immune response orchestrated by microglia (5–7). Mutations in α-synuclein are known to be associated with PD risk (8) and in-vitro studies of the behavior of monocytes and microglia stimulated with mutant α-synuclein monomers demonstrate increased cytotoxic immune responses in comparison to wild-type α-synuclein-exposed cells (9, 10). Components of α-synuclein have also been shown to lead to activation of T-lymphocytes and monocytes (11, 12), all of which suggests that α-synuclein may drive both a neuronal pathology and an inflammatory process in PD. Overproduction of cytokines in PD perpetuates the inflammatory response centrally and systemically (13). Serum cytokines [for example, IL-1ß, IL-2, IL-10, IFNγ, and TNF-α (14, 15)] and peripheral blood mononuclear cell (PBMC) cytokine production has been correlated with PD symptom severity (16, 17) and rate of disease progression (2). The question therefore arises: could this be driven by α-synuclein in the periphery?
Aberrant α-synuclein is distributed throughout peripheral organs, blood, interstitial and extracellular fluids in PD (18–20) and may act as a catalyst for activation of the peripheral immune system (1). Indeed, selected α-synuclein peptides stimulate a specific T-cell response in 40% of patients, via presentation by MHC alleles which are known to be genetically associated with PD risk (11). Fibrillar α-synuclein has been shown to act via Toll-like receptor (TLR) and inflammasome pathways in monocytes leading to IL-1β production (12).
However, other factors such as infections or translocation of bacterial toxins from the gut may also contribute to inflammation in PD (21, 22). For example, lipopolysaccharides (LPS) stimulate PBMCs via the TLR and inflammasome pathways to produce an inflammatory cytokine response (12), and several studies have investigated this response in PD albeit with inconsistent results. LPS-stimulated cytokine production has been reported to be elevated in PD compared to controls, along with the basal production of some cytokines (IL-1ß, IFNγ, and TNF-α) (16). A second study showed that production of IL-1ß, IL-6, and TNF-α is enhanced in PD PBMCs, while IL-2 is reduced (23). However, in another study, production of IFNγ by LPS-stimulated PBMCs was lower in patients than controls, while IL-6, IL-1α, and IL-1ß levels were no different, but decreasing concentrations correlated worsened disease severity (24).
Given this ambiguity in the literature and the absence of any study investigating both α-synuclein and LPS stimulation of PBMCs in PD patients, we sought to understand how stimulation by α-synuclein monomers, fibrils, and LPS affects PBMC cytokine production in PD patients and matched controls.
Early-stage PD patients (Hoehn and Yahr ≤2), fulfilling UK PD Brain Bank Criteria, aged 55–80, were recruited from the PD Research Clinic at the John van Geest Center for Brain Repair, Cambridge. A movement disorder accredited neurologist conducted clinical and neuropsychological assessments.
Age and gender matched control participants were recruited from the NIHR Cambridge Bioresource (http://www.cambridgebioresource.org.uk) and had no history of neurological disease, self-reported memory problems, or depression. Ethical approval was obtained from the East of England-Cambridge Central Research Ethics Committee (REC 03/303). Exclusion criteria were: other neurodegenerative disorders, chronic inflammatory or autoimmune disorders, current clinically significant infection or use of anti-inflammatory/immunomodulatory medications, surgery within the last month, or recent vaccinations. Data from this cohort also contributed to our previously published study (25).
PBMCs were extracted from venous blood by centrifugation over a Ficoll gradient, washed and cultured (37°C, 5% CO2) for 24 h in RPMI (Life Technologies) and 10% fetal calf serum (FCS, Sigma) in aliquots of 1 million cells per mL per well, either unstimulated, or with LPS (1 ng/mL), α-synuclein monomers (2 nmol/mL) or fibrils (2 nmol/mL). Supernatant was collected and stored at −80°C, and cultured PBMCs were washed and stored in RNA protect (Qiagen) at −80°C. Matched samples were processed in parallel.
Recombinant α-synuclein was produced by expression in E.coli Rosetta using human α-synuclein cDNA, and aggregates were confirmed on SDS-PAGE gel (Supplementary Methods and Supplementary Figure 1). Endotoxin levels were determined using LAL assays (Lonza Verviers SPRL, Belgium).
Cytokines (IFNγ, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13, TNF-α) were measured in culture supernatants using the Meso Scale Discovery (Rockville) platform V-Plex Pro-inflammatory panel 1 electrochemiluminescence assay. Secondary analyses were performed in a subset of samples/conditions to measure IL-18 (MSD U-PLEX Human IL-18 assay) and caspase-1 (Human caspase-1/ICE Quantikine ELISA kit, R&D Systems) as markers of inflammasome activation. Assays were run according to manufacturer's instructions. Supernatant samples were diluted 1:10 or 2:3 in the appropriate buffer and assayed in duplicate. Cytotoxicity post-culture was quantified with the Pierce LDH Cytotoxicity Assay Kit (ThermoFisher).
RNA was extracted from cultured PBMCs using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. RNA was reverse-transcribed using SuperScript™ III First-Strand Synthesis SuperMix for qRT-PCR (ThermoFisher Scientific). TaqMan Real-Time PCR was used for quantification of gene expression, and primers were IL-6 (HS00174131M1) and IL-1ß (HS001555410M1) (ThermoFisher Scientific). Assays were run in triplicate. Relative quantification was carried out on a QuantStudio 12K Flex Real-Time PCR machine and calculated using comparative cycle threshold (ΔΔCT method) relative to the housekeeping gene GAPDH, and a randomly selected endogenous control common to all plates.
Cytokine concentrations across antigens and PD status were compared using repeated-measures ANOVAs, and mRNA production using 2-way ANOVAs (GraphPad Prism version 7, SPSS version 25). Outliers were removed using Grubbs' tests (p < 0.05).
31 PD patients [mean disease duration 4.3 (1.1) years] and 31 controls (matched for age and gender) were included (Supplementary Table 1).
PBMCs were cultured with LPS (n = 31 case-control pairs), and α-synuclein monomers and fibrils (n = 19 case-control pairs). A subset of α-synuclein-cultured PBMCs were used for the gene expression assays based on RNA availability.
Stimulation with either LPS, α-synuclein monomers or fibrils led to robust cytokine stimulation compared to the unstimulated condition (p < 0.0001, RMANOVA, main effect of antigen). There was no main effect of patient vs. control status on cytokine production for any antigen (p > 0.05, RMANOVA; Figure 1). PBMC supernatant LDH levels were not different between α-synuclein, LPS, or unstimulated cultures. Expression of IL-6 and IL-1β was quantified by qRT-PCR, given that these cytokines showed the greatest PD-control differences on LPS stimulation in previous studies (16, 24). IL-6 and IL-1β expression were elevated in response to stimulation by both LPS and α-synuclein (2-way ANOVA and post-hoc Tukey's test p < 0.0001; Gene expression, relative units mean ± standard deviation(SD), IL-6: unstimulated PD = 2.18 ± 4.28, Control = 1.67 ± 2.82; α-synuclein PD = 929.72 ± 485.93, Control = 845.05 ± 419.67; LPS PD = 989.05 ± 968.66, Control = 810.00 ± 731.10, IL-1β: unstimulated PD = 2.99 ± 3.72, Control = 2.54 ± 3.76; α-synuclein PD = 207.01 ± 95.13, Control = 171.39 ± 60.10; LPS PD = 155.59 ± 116.50, Control = 136.77 ± 86.64). There was no main effect of disease status between PD and control groups (p > 0.05, two-way ANOVA).
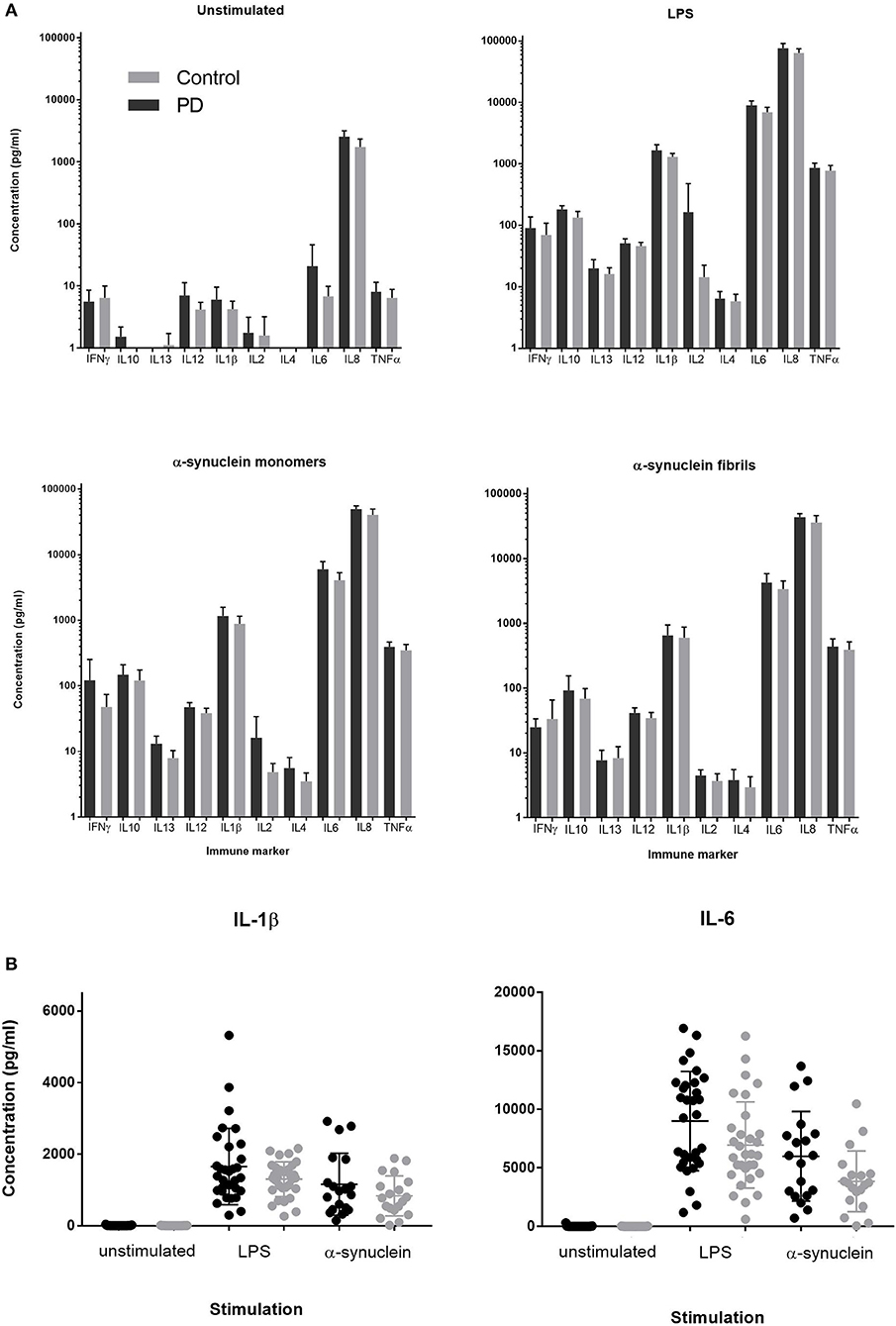
Figure 1. (A) Supernatant cytokine concentration produced by PD and control PBMCs cultured for 24 h in media containing LPS (1ng/ml), α-synuclein fibrils, or α-synuclein monomers (1 nmol = 27.3 ng/ml). (B) Individual level data for IL-1β and IL-6 concentrations in post-culture supernatant in PD cases and matched controls for direct comparison between unstimulated, LPS and α-synuclein monomer stimulation for two key inflammatory cytokines. Error bars represent SEM.
Given that α-synuclein produced a similar magnitude of cytokine response to LPS, we examined the α-synuclein for the presence of any associated endotoxin. Despite procedures to remove contaminating endotoxin as detailed in the methods, endotoxin concentrations in samples at 2 nmol/mL were 0.2–1.3EU/mL on testing multiple aliquots (Lonza). LPS (1 ng/mL) contained >10 EU/mL. To ascertain whether the levels of contaminating endotoxin were sufficient to confound cytokine measurements in the α-synuclein cultures, we used six endotoxin standard dilutions (0, 0.1, 0.26, 0.64, 1.6, and LPS>10EU/mL) to stimulate PBMCs using otherwise identical conditions (PD n = 5, Control n = 4, age = 68.9 (not different from previous cohort) and compared supernatant cytokine concentrations with data obtained in our initial experiments (Figure 2). An endotoxin concentration of 1.6EU/mL produced similar levels of cytokine to α-synuclein (monomers or fibrils) for most cytokines thus suggesting a possible confounding effect of endotoxin. However, for IL-1β and IL-18, monomeric α-synuclein had a significantly greater effect than 1.6EU/mL endotoxin (p = 0.01), an EU level in excess of the measured level of contaminating endotoxin. A similar pattern was seen for α-synuclein fibrils compared to endotoxin at 1.6EU/mL, but this did not reach statistical significance. Despite the excess production of inflammasome-related cytokines IL-1β and IL-18 by PBMCs stimulated with α-synuclein monomers, there was no corresponding increase in caspase-1 secretion (p > 0.05, Figure 2D).
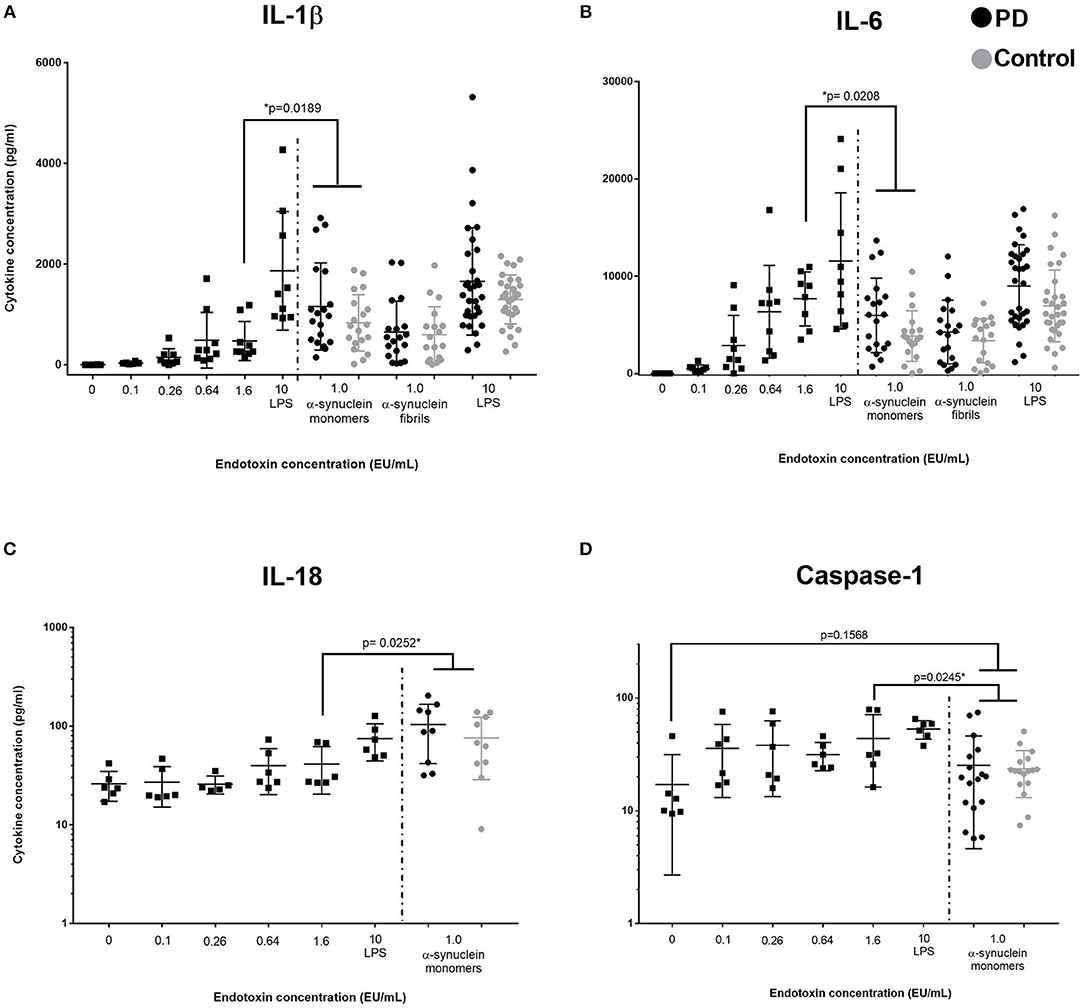
Figure 2. Comparison of low concentration endotoxin and α-synuclein stimulation of PBMCs. Squares represent cytokine concentrations from PBMC cultures with varying concentrations of endotoxin (A, B: n = 9, 5 PD and 4 controls, C,D: n = 6, 3PD and 3 controls, assayed in duplicate; left of dashed line). Circles represents PD (black) and control (gray) concentrations in post-culture supernatants from the original assays cultured with α-synuclein monomers, fibrils, or LPS (α-synuclein conc: 1 nmol = 27.3 ng/ml, LPS conc: 1 ng/ml). P-values indicates comparison between 1.6 EU/ml (comparable to endoxin level in our α-synuclein preparation) and grouped PD/control cohort stimulated by α-synuclein monomers (No PD/control differences were observed in post-culture cytokine concentrations) Bars represent mean value and first and third quartiles. (A) Data suggest that IL-1β production is stimulated by α-synuclein in excess of stimulation by the equivalent value of endotoxin present as a contamiant. (B) IL-6 (and all other measured cytokines) do not show this increased production. Data indicates that endotoxin is the primary driver of elevated IL-6 concentration, rather tha α-synuclein. (C) IL-18 prodution is also significantly increased in response to α-synuclein stimulation, compared to stimulation with an endotoxin concentration comparable to contaminating levels. (D) Caspase-1 levels are not significantly increased by α-synuclein stimulation compared to the unstimulated condition.
We found that PBMCs collected from both PD patients and age/gender-matched controls stimulated by α-synuclein (both monomeric and fibrillary) produced a robust inflammatory cytokine response. The response was similar in magnitude to LPS stimulation as assessed by both cytokine concentrations in culture supernatant and mRNA expression. Whilst this response may have been confounded by low levels of endotoxin in the α-synuclein preparation, the response of the IL1-β and IL-18 is greater than this low level endotoxin effect, which suggests that α-synuclein may have a specific independent effect on inflammasome-related pathways. Interestingly, it has previously been shown that α-synuclein fibrils (produced from a strain of E. Coli with strongly reduced endotoxicity) stimulate the NLRP3 inflammasome pathway in monocytes to produce IL-1β, in addition to the TLR pathway that is activated by bacterial endotoxin (12). In our study, we found that α-synuclein monomers had a more pronounced effect on IL-1β production than fibrils, but comparison between studies is difficult given the likely variability in aggregate size according to the methodology used to prepare fibrils. We found no significant increase in the PBMC supernatant levels of the inflammasome pathway mediator caspase-1 with α-synuclein stimulation, suggesting that α-synuclein may be acting via caspase-1 independent inflammasome pathways in this setting (26).
In contrast to previous studies (16, 23, 24), there was no evidence of PD-control difference in cytokine production or mRNA expression. Notably, our case-control pairs were well-matched for age and gender and processed in parallel to eliminate variation that may have confounded previous studies. Hence, our data do not support a differential effect of PBMC stimulation in PD cases vs. controls, irrespective of the stimulating antigen. The lack of any patient vs. control differences in cytokine production in response to PBMC stimulation suggests raised levels of inflammatory markers in the serum in PD may relate more to levels of exogenous stimulating antigens or other cytokine sources, rather than to intrinsic properties of the peripheral mononuclear cells. Additionally, oligomeric α-synuclein species might contribute to inflammation in PD but this has not been specifically tested in this study.
The generation of α-synuclein for experimental use typically involves producing recombinant protein in E.coli, which invariably leads to endotoxin contamination of the protein product; contamination which can be removed to some extent by cleaning methods, but may remain at low levels and confound cellular processes with sensitivity to endotoxin (27). Our data confirms that even very low level endotoxin levels can have a significant confounding effect. A previous study found that α-synuclein-derived peptides drive specific T-cell responses in PD (11), but it is unclear whether the presence of associated endotoxin had been entirely excluded in these experiments. However, it may be relevant to further study co-stimulation with endotoxin and α-synuclein, given that endotoxin may act synergistically with α-synuclein in TLR stimulation (12), as has been shown in α-synuclein-primed murine microglia (28). Furthermore, endotoxin may influence the conformation of α-synuclein, with different LPS β-sheet content driving alterations in fibril density and changes in associated behavioral phenotypes in animal models (29). However, these mechanisms are not well understood in patients.
A limitation of this study is that the assessment of the PBMC response to varying endotoxin concentrations was undertaken in an independent sub-sample. However, subjects included were similar in age and disease status and the measured cytokine concentrations had minimal between-subject variation suggesting that the responses were representative.
In conclusion, our data suggest that even low levels of endotoxin can confound the measurement of immune cell responses to α-synuclein in-vitro and future studies should consider endotoxin quantification. α-synuclein may have independent effects on production of inflammasome-related cytokines, which may perpetuate the immune response in PD. Furthermore, PD and control PBMCs behaved similarly in the face of stimulation in our study which suggests that cell-extrinsic factors may be an important contributor to the chronic inflammation which has been observed in PD. The nature of these agents remains to be fully determined but both α-synuclein and bacterial endotoxins may play a critical role.
This study was carried out in accordance with the recommendations of the Health Research Authority East of England-Cambridge Central Research Ethics Committee with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the East of England-Cambridge Central Research Ethics Committee (REC 03/303).
AW acquisition of data, analysis and interpretation of data, drafting of the manuscript. RW acquisition of data, critical revision of the manuscript. KS acquisition of data, critical revision of the manuscript. NG preparation of α-synuclein species, acquisition of data, critical revision of the manuscript. HM preparation of α-synuclein species, acquisition of data, critical revision of the manuscript. SH acquisition of data, critical revision of the manuscript. IS acquisition of data, critical revision of the manuscript. RB study supervision, critical revision of the manuscript. CW-G study design and supervision, critical revision of the manuscript.
Grant funding from the Academy of Medical Sciences UK, the Rosetrees Trust, the Stevenage Biosciences Catalyst and Addenbrooke's Charitable Trust contributed to this work. The research was also supported by the NIHR Cambridge Biomedical Research Center (Cambridge University Hospitals NHS Trust/University of Cambridge). CH Williams-Gray is supported by a fellowship from the Medical Research Council. AJ White was funded by Homerton College. RS Wijeyekoon was supported by a fellowship from Addenbrooke's Charitable Trust. KM Scott is supported by a fellowship from the Wellcome Trust. R A Barker is supported by the Wellcome-MRC Cambridge Stem Cell Institute and is an NIHR Senior Investigator.
The funding bodies had no role in the design of the study, the collection, analysis, and interpretation of data or the writing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge the participation of all our patient volunteers, and NIHR Cambridge BioResource volunteers, and we thank the NIHR Cambridge BioResource center and staff for their contribution. We thank the National Institute for Health Research and NHS Blood and Transplant. We also acknowledge the support of the Core Biochemical Assay Laboratory at Cambridge University Hospitals.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2018.00946/full#supplementary-material
1. Mosley RL, Hutter-Saunders JA, Stone DK, Gendelman HE. Inflammation and adaptive immunity in Parkinson's Disease. Cold Spring Harb Perspect Med. (2012) 2:a009381. doi: 10.1101/cshperspect.a009381
2. Williams-Gray CH, Wijeyekoon R, Yarnall AJ, Lawson RA, Breen DP, Evans JR, Cummins GA, et al. Serum immune markers and disease progression in an incident Parkinson's disease cohort (ICICLE-PD). Mov Disord. (2016) 31:995–1003. doi: 10.1002/mds.26563
3. Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature (1997) 388:839–40. doi: 10.1038/42166
4. Wakabayashi K, Tanji K, Mori F, Takahashi H. The lewy body in Parkinson's disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathol Off J Jpn Soc Neuropathol. (2007) 27:494–506. doi: 10.1111/j.1440-1789.2007.00803.x
5. Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J Off Publ Fed Am Soc Exp Biol. (2005) 19:533–42. doi: 10.1096/fj.04-2751com
6. Lee S-J. Origins and effects of extracellular α-synuclein: implications in Parkinson's Disease. J Mol Neurosci. (2008) 34:17–22. doi: 10.1007/s12031-007-0012-9
7. Sanchez-Guajardo V, Barnum CJ, Tansey MG, Romero-Ramos M. Neuroimmunological processes in Parkinson's disease and their relation to α-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN NEURO (2013) 5:113–39. doi: 10.1042/AN20120066
8. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science (1997) 276:2045–7.
9. Klegeris A, Pelech S, Giasson BI, Maguire J, Zhang H, McGeer EG, et al. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol Aging (2008) 29:739–52. doi: 10.1016/j.neurobiolaging.2006.11.013
10. Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Fernandez-Montesinos R, Caro M, Lachaud CC, et al. Glial innate immunity generated by non-aggregated alpha-synuclein in mouse: differences between wild-type and Parkinson's disease-linked mutants. PloS ONE (2010) 5:e13481. doi: 10.1371/journal.pone.0013481
11. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson's disease recognize α-synuclein peptides. Nature (2017) 546:656–61. doi: 10.1038/nature22815
12. Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, et al. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PloS ONE (2013) 8:e55375. doi: 10.1371/journal.pone.0055375
13. Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol. (2007) 150:963–76. doi: 10.1038/sj.bjp.0707167
14. Gao H-M, Hong J-S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. (2008) 29:357–65. doi: 10.1016/j.it.2008.05.002
15. Collins LM, Toulouse A, Connor TJ, Nolan YM. Contributions of central and systemic inflammation to the pathophysiology of Parkinson's disease. Neuropharmacology (2012) 62:2154–68. doi: 10.1016/j.neuropharm.2012.01.028
16. Reale M, Iarlori C, Thomas A, Gambi D, Perfetti B, Di Nicola M, et al. Peripheral cytokines profile in Parkinson's disease. Brain Behav Immun. (2009) 23:55–63. doi: 10.1016/j.bbi.2008.07.003
17. Banks WA, Erickson MA. The blood–brain barrier and immune function and dysfunction. Neurobiol Dis. (2010) 37:26–32. doi: 10.1016/j.nbd.2009.07.031
18. Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Döring F, Trenkwalder C, Schlossmacher MG. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol. (2011) 10:230–40. doi: 10.1016/S1474-4422(11)70014-X
19. Malek N, Swallow D, Grosset KA, Anichtchik O, Spillantini M, Grosset DG. Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson's disease - a systematic review. Acta Neurol Scand. (2014) 130:59–72. doi: 10.1111/ane.12247
20. El-Agnaf OMA, Salem SA, Paleologou KE, Curran MD, Gibson MJ, Court JA, et al. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson's disease. FASEB J Off Publ Fed Am Soc Exp Biol. (2006) 20:419–25. doi: 10.1096/fj.03-1449com
21. Houser MC, Tansey MG. The gut-brain axis: is intestinal inflammation a silent driver of Parkinson's disease pathogenesis? NPJ Park Dis. (2017) 3:3. doi: 10.1038/s41531-016-0002-0
22. Fang F, Wirdefeldt K, Jacks A, Kamel F, Ye W, Chen H. CNS infections, sepsis and risk of Parkinson's disease. Int J Epidemiol. (2012) 41:1042–9. doi: 10.1093/ije/dys052
23. Bessler H, Djaldetti R, Salman H, Bergman M, Djaldetti M. IL-1 beta, IL-2, IL-6 and TNF-alpha production by peripheral blood mononuclear cells from patients with Parkinson's disease. Biomed Pharmacother Biomedecine Pharmacother. (1999) 53:141–5. doi: 10.1016/S0753-3322(99)80079-1
24. Hasegawa Y, Inagaki T, Sawada M, Suzumura A. Impaired cytokine production by peripheral blood mononuclear cells and monocytes/macrophages in Parkinson's disease. Acta Neurol Scand. (2000) 101:159–164. doi: 10.1034/j.1600-0404.2000.101003159.x
25. Williams-Gray CH, Wijeyekoon RS, Scott KM, Hayat S, Barker RA, Jones JL. Abnormalities of age-related T cell senescence in Parkinson's disease. J Neuroinflam. (2018) 15:166. doi: 10.1186/s12974-018-1206-5
26. Afonina IS, Müller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
27. Schwarz H, Schmittner M, Duschl A, Horejs-Hoeck J. Residual endotoxin contaminations in recombinant proteins are sufficient to activate human CD1c+ dendritic cells. PloS ONE (2014) 9:e113840. doi: 10.1371/journal.pone.0113840
28. Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Lachaud CC, Guilliams T, Fernandez-Montesinos R, et al. Preconditioning of microglia by α-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PloS ONE (2013) 8:e79160. doi: 10.1371/journal.pone.0079160
Keywords: Parkinson's disease, immune system, alpha-synuclein, endotoxin, cytokines
Citation: White AJ, Wijeyekoon RS, Scott KM, Gunawardana NP, Hayat S, Solim IH, McMahon HT, Barker RA and Williams-Gray CH (2018) The Peripheral Inflammatory Response to Alpha-Synuclein and Endotoxin in Parkinson's Disease. Front. Neurol. 9:946. doi: 10.3389/fneur.2018.00946
Received: 09 July 2018; Accepted: 23 October 2018;
Published: 20 November 2018.
Edited by:
Cristoforo Comi, Università degli Studi del Piemonte Orientale, ItalyReviewed by:
Cintia Roodveldt, Centro Andaluz de Biología Molecular y Medicina Regenerativa (CABIMER), SpainCopyright © 2018 White, Wijeyekoon, Scott, Gunawardana, Hayat, Solim, McMahon, Barker and Williams-Gray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alice J. White, YWp3MjgzQGNhbS5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.