 Nattakarn Limphaibool
Nattakarn Limphaibool Piotr Iwanowski
Piotr Iwanowski- Department of Neurology, Poznan University of Medical Sciences, Poznan, Poland
Parkinson's Disease (PD) is a common neurodegenerative disorder manifesting as reduced facilitation of voluntary movements. Extensive research over recent decades has expanded our insights into the pathogenesis of the disease, where PD is indicated to result from multifactorial etiological factors involving environmental contributions in genetically predisposed individuals. There has been considerable interest in the association between neurological manifestations in PD and in inherited metabolic disorders (IMDs), which are genetic disorders characterized by a deficient activity in the pathways of intermediary metabolism leading to multiple-system manifestations. In addition to the parallel in various clinical features, there is increasing evidence for the notion that genetic mutations underlying IMDs may increase the risk of PD development. This review highlights the recent advances in parkinsonism in patients with IMDs, with the primary objective to improve the understanding of the overlapping pathogenic pathways and clinical presentations in both disorders. We discuss the genetic convergence and disruptions in biochemical mechanisms which may point to clues surrounding pathogenesis-targeted treatment and other promising therapeutic strategies in the future.
Introduction
Parkinson's disease (PD) is one of the most common neurodegenerative disorders, with a substantial, worldwide growth in prevalence in recent years. It is a debilitating disorder, manifesting as reduced facilitation of voluntary movements attributed to a progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the formation of intracellular inclusions containing aggregates of alpha-synuclein (a-syn) (1). Over the past decades, extensive research has expanded our insights into the pathogenesis of the disease. It has been suggested that PD's complex and multifactorial etiology results from environmental contributions in genetically predisposed individuals (2).
Inherited metabolic disorders (IMDs) are a complex and diverse group of genetic disorders characterized by the disruption of cellular biochemical functions and deficient activity in the pathways of intermediary metabolism. Consequential toxic substance accumulation or end-product deficiency leads to a multi-systemic clinical picture from liver dysfunction and cardiomyopathy to progression to encephalopathy, coma, or death if left untreated (3). The prevalence of newborn screening and implementation of advance treatment strategies have resulted in an expanding adult patient population, presenting with late neurological symptoms and movement disorders (4, 5). There has been considerable interest in the association between neurological manifestations and IMDs, as this can provide evidence regarding the underlying pathophysiology, potentially leading to advances in diagnosis and therapeutic strategies. This review highlights the recent developments in the convergence of parkinsonism in patients with IMDs, with the principal objective of improving the understanding of the overlapping pathogenesis and clinical features in both disorders (Tables 1, 2).
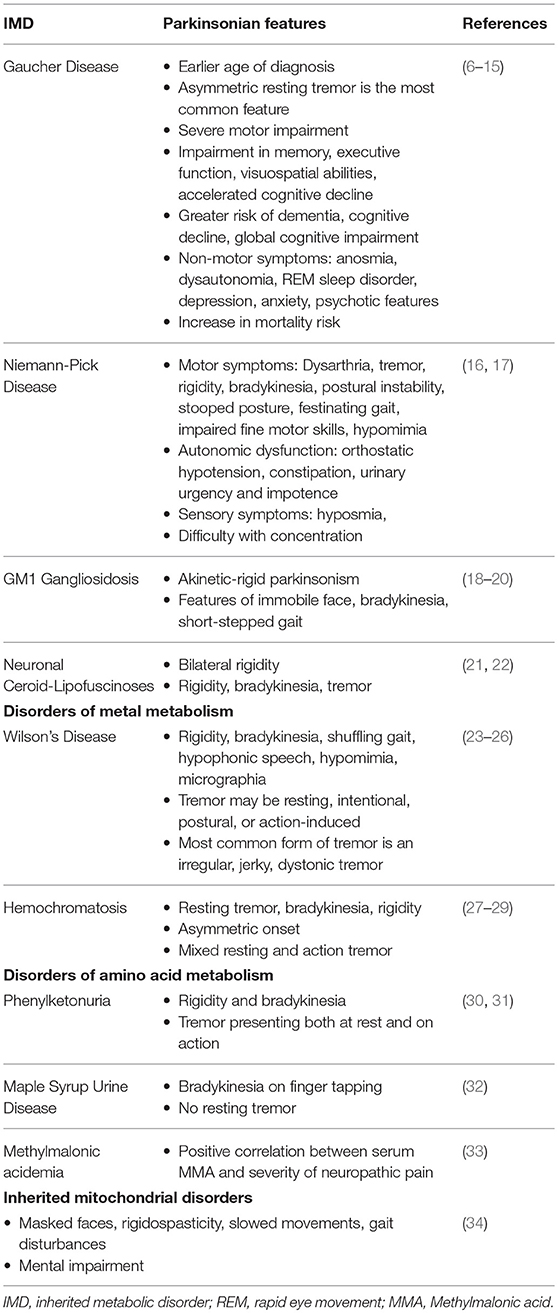
Table 1. Parkinsonian features in IMDs.
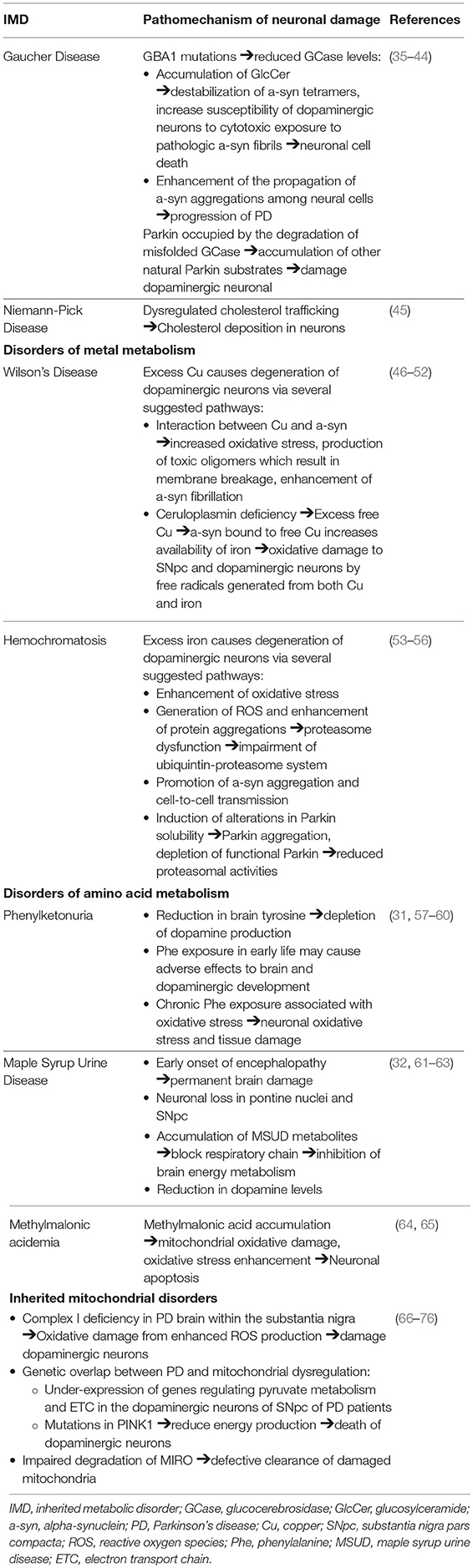
Table 2. Pathomechanism of neuronal damage contributing to parkinsonism in IMDs.
Inherited Metabolic Disorders
Lysosomal Storage Diseases
Gaucher Disease
Gaucher Disease (GD), a prevalent inherited lysosomal storage disease (LSD), is caused by deficient glucocerebrosidase (GCase) activity due to mutations in the coding gene (GBA). The resulting accumulation of glucosylceramide (GlcCer) in lysosomes of reticuloendothelial cells leads to complex systemic manifestations involving the liver, spleen, bone marrow, and to progressive neurodegenerative diseases (77). The three major clinical types are classified by the involvement of primary central nervous system (CNS) disease. Type 1 GD, the most common variant, has a non-neuronopathic presentation characterized by the presence of hepatosplenomegaly, orthopedic complications, and blood disorders. Types 2 and 3 are characterized by the presence of primary neurologic disease distinguished by their rapid vs. gradual progressive course, respectively (78). PD is present in 4% of type 1 GD. Even in cases which the PD diagnostic criteria are not met, over 20% of GD patients were found to display at least one parkinsonian finding (79).
Evidence linking GBA mutations to the clinical characteristics of PD, including disease phenotype, progression, and prognosis, has been extensively documented. At the point of diagnosis, the clinical presentation of GD patients with PD can be almost identical to that of idiopathic PD (6). The most commonly presenting feature is an asymmetric resting tremor, although postural instability and gait difficulties are also relatively frequent (7). However, GBA carrier status has a significant impact on the natural history of PD. Patients reported an earlier age of symptom onset and severe motor impairment (8–10). Genetic polymorphism has been shown to influence motor progression (6). There is a higher prevalence of dementia and a distinct pattern of cognitive deficits, characterized by greater impairment in memory, executive function and visuospatial abilities (8, 9, 11, 12). The accelerated cognitive decline is indicated to relate the GBA mutation severity (6, 9, 11–14). Other non-motor clinical features are also highly prevalent, with the most common being anosmia and dysautonomia, as well as REM sleep disorder, depression, anxiety, and psychotic features presenting as hallucinations (8, 9, 15). A 2-fold increase in mortality risk in GBA-carriers has also been found (9).
Clinical reports of an association between GD and parkinsonism instigated further investigation into the mechanistic and genetic overlap between the two syndromes. The fundamental finding that a GBA1 gene mutation is a significant risk factor for PD development is of significant interest (10, 80, 81). Mutations in GBA1 amongst PD patients are common and reported as the most important risk factor yet discovered for PD, present in 3.5–6.7% of the PD population (6, 10, 82, 83). It is reported as the first finding of a genetic locus shown to influence motor progression in PD, possibly leading to its role as a prognostic marker in the future (6). Most analyses have focused on the most common pathogenic mutations in GBA, N370S and L444P variants, which represents up to 50% of the genetic alterations (10, 13, 80, 84, 85). In fact, it is reported that over 90% of GD-PD patients carry at least one N370S mutation (86). L444P mutation has recently been found to have a highly detrimental effect, responsible suspending the production and destabilization of GCase (35). Carriers with severe GBA mutation have been found to have a higher risk of PD development, with earlier onset of symptoms (87). For example, patients with the GBA mutation 84insG, a severe mutation leading to decreased GCase enzyme activity, are at higher risk of developing PD compared to milder mutations such as N370S (36).
The overexpression of the poorly functional GBA1, leading to reduced GCase levels and subsequent accumulation of its lipid substrate GlcCer, is found to be linked with increased a-syn accumulation and subsequent neuronal cell death. The accumulation of GlcCer contributes to the destabilization of a-syn tetramers and increase the susceptibility of human dopaminergic neurons to the cytotoxic exposure to pathologic a-syn fibrils (37–41). It was also demonstrated that GBA1 dysfunction and GCase depletion enhances the propagation of a-syn aggregations among neural cells, which is associated with the progression of PD (42). The pathologic implication of reduced GCase levels was supported by the finding that the direct administration of GCase to the brain resulted in a reduction of a-syn levels and an improvement of parkinsonian symptoms (38, 88–90).
Conversely, reduced levels of GCase were also associated with increased a-syn in idiopathic PD patients without GBA mutations, indicating that a-syn can independently inhibit GCase enzyme activity (36, 91–93). A-syn's contribution to decreased GCase activity is through its disruption of GCase and other lysosomal hydrolases trafficking to lysosomes, further contributing to lysosomal dysfunction and neuronal dyshomeostasis (38). These findings support the “gain of function” hypothesis whereby mutated GCase promotes a-syn aggregation, accelerating lewy body formation and neuronal loss (94). In summary, the possible mechanism indicated to underlie a-syn accumulation and PD development is the perpetual cycle in which GCase depletion increases the level of a-syn accumulation, which in turn inhibits the regular function of GCase and causes further aggregation of a-syn (40).
An alternate theory is the association between heterozygote GBA mutations and dysregulations in the protein degradation systems, which have been implicated as a common mechanism underlying neurodegenerative diseases. PARK2 gene mutations encoding parkin, the E3 ubiquitin ligase, has been identified as a significant cause for an autosomal recessive form of PD (43). Parkin is involved in the mediation of the proteasomal degradation of misfolded mutant GCase. Parkin was suggested to be occupied by ubiquitination of misfolded GCase in GD carriers, causing accumulation of other natural Parkin substrates, which is detrimental to dopaminergic neuronal survival, contributing to their demise and subsequent PD development (44). Dopaminergic cells expressing mutant GCase are particularly susceptible to apoptotic stimuli (95). Further investigation is necessary to unravel the mechanism of the bidirectional relationship between GBA1 mutation and a-syn and the effect of GBA mutations in misfolded protein accumulation leading to cell damage.
Niemann-Pick Disease
Niemann-Pick Disease (NPD), a neurovisceral LSD, presents with a broad clinical spectrum ranging from a rapidly fatal disorder in neonates to a chronic neurodegenerative disease later in life. The two distinct metabolic abnormalities include, firstly, a deficiency in the enzyme sphingomyelinase (ASM) resulting from mutations in the SMPD1 gene accounting for types A and B NPD. Growing evidence suggests they represent opposite ends of the clinical spectrum with regard to the disease severity and neurologic involvement. The second category accounting for NPD type C (NP-C) involves a dysfunction in cholesterol transport due to NPC1 and NPC2 gene mutations (96, 97). NP-C is of particular interest in this discussion due to parkinsonism symptoms reported among heterozygeous carriers of NPC1 gene mutations and the association between impaired cholesterol metabolism in the brain and neurodegeneration (16, 98).
Presentations of parkinsonian symptoms have been reported in several cases of patients with mutated NPC1, encoding a lysosomal cholesterol transporter that regulates vesicular trafficking. Parkinsonian symptoms are less frequent manifestations compared to the more prevalent deep brain signs such as cerebellar ataxia and supranuclear opthalmoplegia in NP-C (99). Presenting motor features include dysarthria, tremor, rigidity, bradykinesia, postural instability, as well as stooped posture and festinating gait, impaired fine motor skills, and hypomimia. Autonomic dysfunction, including orthostatic hypotension, constipation, urinary urgency, and impotence, was also present along and sensory symptoms of hyposmia and difficulty in concentration (16, 17).
Alterations in the autophagy-lysosome pathway were suggested as a contributory pathophysiological factor for PD in NP-C. Dysregulations in cholesterol trafficking resulting in increased cholesterol deposition in neuronal cells have been observed in both NP-C and in PD (45). Evidence supporting the positive correlation between high plasma cholesterol level and increased risk of PD development includes: (1) higher cholesterol derivatives in brains and plasma of PD individuals (100, 101), (2) elevated levels of oxidized cholesterol metabolites increasing levels of a-syn and accelerates its aggregation (102–104) (3) cholesterol inhibitors, such as statins, reduce the levels of a-syn in neuronal cells whereas exogenous cholesterol supplementation to neurons increase a-syn aggregation and reduces neuron growth (105). (4) In vivo evidence of increased vulnerability of midbrain dopaminergic neurons and exacerbation of PD symptoms to hypercholesterolemia (106).
These findings suggest that the defect in the metabolic pathways in NP-C could reveal pathogenic overlaps associated with aberrant a-syn aggregation and neurodegeneration in PD (45). However, controversial findings report a correlation between plasma cholesterol level, statin therapy, and the risk of PD development (107–110). Reports supporting the notion that NPC1 variants are risk factors for PD also remain unclear, necessitating further clarification through extensive population genetic studies (16, 111).
Parkinsonism in Lysosomal Storage Diseases
The significance of GBA1 mutations in PD development has led to further insights into the pathomechanism centered around the dysregulation of the autophagic-lysosomal pathway. The effects of PD mutations and a-syn aggregations which converge on the lysosomal degradation system indicates the significance of protein dyshomeostasis in the mediation of neurodegeneration in PD. Apart from the important connection with GD which have been extensively explored, associations between PD and other LSDs including GM1—(18–20) and GM2-gangliosidosis (112–114), neuronal ceroid lipofuscinosis (21, 22), and metachromatic leukodystrophy (115, 116) have been described, although these are not as well-documented due to the rarity of these conditions.
Remarkably, recent emerging data have suggested that the lysosomal connection in PD likely extends far beyond GBA1. A recent genome-wide association study (GWAS) found that several of the identified PD risk genes play an integral role in lysosomal biology and autophagy (117). Conversely, over half of PD cases in a cohort study were found to present with at least one damaging variant in the LSD gene. These results are consistent with the previous genetic analyses, revealing an important connection between genetic factors responsible for LSD and PD risk (118, 119). These findings indicate the possibility that LSD gene polymorphism may lead to lysosomal dysfunction, resulting in the accumulation of toxic substrates and increasing PD susceptibility (118).
Disorders of Metal Metabolism
Wilson's Disease
Wilson's Disease (WD), an inborn error of copper (Cu) metabolism, is caused by a mutation in the copper-transporting gene ATP7B. This autosomal recessive disease is characterized by excessive copper deposition, primarily within hepatocytes and in the CNS (120). Movement disorders, including tremor, choreiform movements, parkinsonism, and rigid dystonia are the characteristic neurological presentation of WD (121). The “wing-beating” tremor, a swinging movement of the upper extremity, in combination with dysarthria, is a classic observation, strongly suggesting a WD diagnosis (23, 24).
Parkinsonian symptoms of WD includes rigidity, bradykinesia, shuffling gait, hypophonic speech, hypomimia, and micrographia. The tremor observed may be resting, intentional, postural, or action-induced, though the most common form of tremor in WD is an irregular, jerky, dystonic tremor (23–25). Dysarthria is commonly observed as a result of parkinsonism or other neurological disorder including cerebellar and pyramidal dysfunctions in WD (26).
The indicated underlying pathomechanism of parkinsonian manifestations amongst heterozygotes for WD describes the process of prolonged free Cu and iron accumulation leading to oxidative damage in the basal ganglia. This stems from the frequent observation of Parkinson symptoms among WD patients as well as the reduced level of ceruloplasmin and Cu dyshomeostasis reported in PD patients (46). Cu dyshomeostasis in PD patients has been linked to mutations in the gene ATP7B, indicating a genetic overlap between the two disorders (47). Recent studies have found the interaction between Cu and a-syn to be involved in multiple pathways leading to neuronal damage, including increased oxidative stress, the production of toxic oligomers which eventually result in membrane breakage and enhanced a-syn fibrillation (47–49).
Ceruloplasmin deficiency leads to the binding between a-syn and excess free Cu, which acts as a ferrireductase and increases the availability of iron. The increase in iron deposition, occurring as a result may be involved in the underlying pathophysiology of PD onset and development (50, 51). The accumulation of both excess copper and iron may contribute to free radical generation and oxidative damage to the SNpc and the oxidative labile dopaminergic neurons (52). Evidence of nigro-striatal dopaminergic deficit can be found in WD subjects with neurological symptoms (120).
Further research is necessary to explore the role of copper, ceruloplasmin, and iron dyshomeostasis in PD and the possibility of an underlying heterozygote WD in some PD cases, in which help in an early diagnosis and in establishing promising therapeutic strategies.
Hemochromatosis
Hemochromatosis is an autosomal recessive disorder which leads to abnormal iron deposition in multiple organs, including the brain, leading to the development of neurodegenerative diseases. Rare reports of parkinsonian syndrome in hereditary hemochromatosis describes characteristic symptoms of resting tremor, bradykinesia, rigidity, and an asymmetric onset. Mixed resting and action tremor were also reported in one patient case (27–29).
Iron is essential in the maintenance of normal neurological function through its role in oxidative metabolism and the synthesis of neurotransmitters and myelin (122). As previously discussed, the dysregulation of iron metabolism has been suggested to contribute to the risk of PD development. This is further evidenced through the observation of altered iron levels in the SNpc of PD patients through MRI findings and postmortem samples (27, 123–125). Increased intracellular iron promotes the degeneration of dopaminergic neurons via several suggested pathways, in addition to the enhancement of oxidative stress as previously described (53). Iron dysregulation has been suggested to play a role in the dysfunction of the ubiquitin-proteasome system. The generation of reactive oxygen species (ROS) and enhancement of protein aggregations exaggerates proteasome dysfunction, resulting in the impairment of the ubiquitin-proteasome system, leading to neuronal injury (54). Iron has also been studied to promote a-syn aggregation and cell-to-cell transmission by inhibiting transcription factor EB (TFEB)-mediated autophagosome-lysoosme fusion (55). Iron induces alterations in Parkin solubility, resulting in its intracellular aggregation. The consequential depletion in functional Parkin leads to reduced proteasomal activities and ultimately, increased cell death (56).
Furthermore, recent data has demonstrated the neuroprotective role of iron chelators against dopamine neuronal degeneration and suggests its therapeutic potential for PD treatment (126, 127). Although the theoretical risk of increased parkinsonism exists with disorders of iron regulation, the association between hemochromatosis and its genetic mutations, levels of iron, and PD has conflicting results, necessitating further extensive population and genetic studies (27, 128, 129).
Disorders of Amino Acid Metabolism
Phenylketouria
Phenylketouria (PKU), a prevalent inherited defect in amino acid metabolism, is characterized by an underlying deficiency in phenylalanine hydroxylase, the hepatic enzyme that catalyzes the hydroxylation of phenylalanine (Phe) to tyrosine (Tyr) (130). The resulting accumulation of Phe leads to a neurological sequela of mental retardation, epilepsy, spastic paraparesis, and late-onset neuropsychological impairment in untreated PKU patients (131). Delayed neurological syndromes, including tremor and dystonia, are indicated to result from the hypomyelination in the CNS (30). Reports of parkinsonism, although exceedingly rare, have been documented as delayed manifestations amongst PKU patients (31). Presentations include the classical rigidity and bradykinesia, in addition to atypical tremor symptoms which presented both at rest and, at times, more prominently on action (30, 31).
It has been suggested that signs of parkinsonism may be secondary to the depletion of dopamine activity in the brain, a result of the reduction of brain Tyr, a dopamine precursor, as a biochemical consequence of PKU (31, 57, 58). This is supported by a report of decreased symptoms of parkinsonism after levodopa administration in a patient suffering from PKU (31). Further evidence has shown decreased dopamine levels in the frontal cortex of PKU subjects where cerebral dopamine deficiency has been implicated in the underlying pathophysiology of executive functioning deficits (132). On a different note, the exposure to high serum Phe during early life may result in adverse effects on early brain growth, including the development of brain dopamine. Chronic exposure to high Phe concentrations has been associated with oxidative stress, which may promote tissue damage and the elicit the onset and progression of neurological impairment in PKU (59, 60).
Maple Syrup Urine Disease
Maple Syrup Urine Disease (MSUD), a disorder of branched-chain amino acid metabolism, manifests as toxic encephalopathy in early life. Treated adults commonly present with movement disorders, predominantly paroxysmal dystonia or ataxia. Patients with parkinsonism in MSUD present with bradykinesia on finger tapping with no resting tremor. These neurological symptoms were speculated to be secondary to the early cerebral damage and chronic reduction in levels of neurotransmitters (32).
Extensive neuronal loss in the pontine nuclei and SNpc in addition to various neurochemical changes were reported to contribute to the development of movement disorders (especially dystonia and ataxia) in MSUD (32). Free radical generation is found to be elicited in MSUD and is possibly involved in the pathophysiology of neurological dysfunction (61). Previous in vitro findings have indicated that accumulating metabolites caused the suppression of brain energy metabolism through the blockade of the respiratory chain, contributing to neurological impairment in patients (62). The depletion of dopamine levels and disruption of normal brain development which accompanied the presentation of limb dystonia and gait abnormalities in MSUD mice models has also been reported (63).
Methylmalonic Acidemia
Methylmalonic acidemia is an autosomal recessive disorder of branched chain amino acid metabolism, caused by deficient activity of methylmalonyl-coenzyme A mutase. While primarily recognized as a pediatric condition, significant multi-systemic morbidity may be present in patients who reach adulthood (133). Among other neurological manifestations, movement disorders including dystonia, chorea, myoclonus, and tremor have been reported (134, 135). Although parkinsonian symptoms are less well-documented in this disorder, it is important to discuss the role of methylmalonic acid (MMA) in neurological dysfunction. Han et al. reported the toxic effects of MMA on neurons and its contribution to neuronal apoptosis (64). Mitochondrial oxidative damage and increase oxidative stress has also been reported in the involvement of neuronal apoptosis caused by MMA in the pathoetiology of PD (64, 65). Furthermore, a positive correlation has been found between serum MMA and the severity of neuropathic pain in amongst PD patients, suggesting its use as a potential marker in assessing PD peripheral neuropathy (33).
Inherited Mitochondrial Disorders
Mitochondrial diseases are a heterogeneous group of disorders with multi-systemic involvement, from musculoskeletal to cardiac to gastrointestinal presentations. The spectrum of mitochondrial pathologies has been reported to result from mutations in the mitochondrial DNA which led to impaired oxidative phosphorylation and dysfunction of the mitochondrial respiratory chain (136). Growing evidence supports the role of mitochondrial dysfunction in the pathogenesis of both the sporadic and familial forms of the PD. Mitochondrial disorders should be particularly suspected in PD patients with extra-cerebral manifestations. Signs and symptoms of parkinsonism reported among these patients include masked faces, mental impairment, rigidospasticity, slowed movements, and gait disturbances (34). Finsterer et al. reported the observation of parkinsonism in 12% of patients with mitochondrial disorders (137). Another study found parkinsonism to be the most common movement disorder among patients with mitochondrial disease, presenting in 43% of research subjects (138). Conversely, 6% of patients with PD have been reported to present with phenotypic features of mitochondrial disorders during the course of their disease (34, 137).
The role of mitochondrial dysfunction in the pathomechanism of PD was initially observed through the findings of induced parkinsonism following the administration of the neurotoxin 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP), an inhibitor of the mitochondrial respiratory chain (MRC) complex I (139, 140). This led to findings of complex I deficiency in the PD brain within the substantia nigra and other regions such as the frontal cortex (66, 141). The inhibition of MRC complex I resulted in enhanced production of ROS and increased oxidative stress, ultimately leading to cellular damage of the dopaminergic neurons in the SNpc (142).
Genetic evidence has further supported the common pathway between PD and the maintenance of mitochondrial function in dopaminergic neurons (143). A GWAS has identified genes sets which regulate pyruvate metabolism and the electron transport chain (ETC) which are under-expressed in the dopaminergic neurons of the SNpc of PD patients (67). Mutations or polymorphisms in several genes (including a-syn, parkin, PINK1, LRRK2, DJ-1) have implicated mitochondrial dysfunction as a prominent cause for PD pathogenesis (65, 68–74). For example, a recent study has shown that mutations in the PINK1 gene, resulting in a shortage of energy production, was implicated as an underlying cause of dopaminergic neuronal damage and subsequent movement difficulties associated with PD (75).
In addition to the oxidative stress and genetic polymorphism which has long been a focus in PD research, recent studies suggest that the disruption in the breakdown of MIRO (a protein connecting the outer mitochondrial membrane to microtubules) may contribute to the impaired process in the cleranace of damaged mitochondria. This reduced degradation of MIRO and the delay in the dysfunctional mitochondrial clearance appears to be a common characteristic of familial and sporadic PD (76).
Conclusion and Future Directions
The clinical convergence between PD and IMDs has instigated further research into the overlapping genetic and biochemical dysregulations between the two conditions. The discovery of shared pathways of an unbalanced homeostasis continues to expand our understanding of the clinical implications of oxidative stress and aberrant protein accumulation on the process of neurodegeneration. Despite the substantial progress that has been made, much work remains to determine the validity and strength of the proposed link between IMDs and PD. Further genetic and biochemical studies may provide insights into risk factors that synergistically favor the development of parkinsonism in particular subsets of IMDs patients. The better understanding of how these factors interact and contribute to the development of parkinsonism may point to clues surrounding pathogenesis-targeted treatment. This includes the possibility of a prognostic marker to establish prompt diagnoses, leading to the availability of genetic counseling and promising therapeutic strategies which may alleviate the onset and progression of this disorder.
Author Contributions
NL Project administration, Supervision, Visualization, Writing review, and editing. PI Supervision, Writing review, and editing. MH and KP writing review and editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers (2017) 3:17013. doi: 10.1038/nrdp.2017.13
2. Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson's disease: a review of the evidence. Eur J Epidemiol. (2011) 26:S1–58. doi: 10.1007/s10654-011-9581-6
3. Ezgu F. Inborn errors of metabolism. Adv Clin Chem. (2016) 73:195–250. doi: 10.1016/bs.acc.2015.12.001
4. Boyer SW, Barclay LJ, Burrage LC. Inherited metabolic disorders: aspects of chronic nutrition management. Nutr Clin Pract. (2015) 30:502–10. doi: 10.1177/0884533615586201
5. Sanderson S, Green A, Preece MA, Burton H. The incidence of inherited metabolic disorders in the West Midlands, UK. Arch Dis Child. (2006) 91:896–9. doi: 10.1136/adc.2005.091637
6. Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, et al. Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community-based incident cohort. Brain (2013) 136(Pt 2):392–9. doi: 10.1093/brain/aws318
7. Chetrit EB, Alcalay RN, Steiner-Birmanns B, Altarescu G, Phillips M, Elstein D, et al. Phenotype in patients with Gaucher disease and Parkinson disease. Blood Cells Mol Dis. (2013) 50:218–21. doi: 10.1016/j.bcmd.2012.11.011
8. Lopez G, Kim J, Wiggs E, Cintron D, Groden C, Tayebi N, et al. Clinical course and prognosis in patients with Gaucher disease and parkinsonism. Neurol Genet. (2016) 2:e57. doi: 10.1212/NXG.0000000000000057
9. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson's disease: the mutation matters. Ann Neurol. (2016) 80:662–73. doi: 10.1002/ana.24777
10. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. (2009) 361:1651–61. doi: 10.1056/NEJMoa0901281
11. Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM, et al. GBA variants are associated with a distinct pattern of cognitive deficits in Parkinson's disease. Mov Disord. (2016) 31:95–102. doi: 10.1002/mds.26359
12. Alcalay RN, Caccappolo E, Mejia-Santana H, Tang M, Rosado L, Orbe Reilly M, et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology (2012) 78:1434–40. doi: 10.1212/WNL.0b013e318253d54b
13. Seto-Salvia N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A, et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson's disease course. Mov Disord. (2012) 27:393–9. doi: 10.1002/mds.24045
14. Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, et al. Specifically neuropathic Gaucher's mutations accelerate cognitive decline in Parkinson's. Ann Neurol. (2016) 80:674–85. doi: 10.1002/ana.24781
15. Goker-Alpan O, Lopez G, Vithayathil J, Davis J, Hallett M, Sidransky E. The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch Neurol. (2008) 65:1353–7. doi: 10.1001/archneur.65.10.1353
16. Kluenemann HH, Nutt JG, Davis MY, Bird TD. Parkinsonism syndrome in heterozygotes for niemann Pick C1. J Neurol Sci. (2013) 335:219–20. doi: 10.1016/j.jns.2013.08.033
17. Josephs KA, Matsumoto JY, Lindor NM. Heterozygous Niemann-Pick disease type C presenting with tremor. Neurology (2004) 63:2189–90. doi: 10.1212/01.WNL.0000145710.25588.2F
18. Roze E, Paschke E, Lopez N, Eck T, Yoshida K, Maurel-Ollivier A, et al. Dystonia and parkinsonism in GM1 type 3 gangliosidosis. Mov Disord. (2005) 20:1366–9. doi: 10.1002/mds.20593
19. Muthane U, Chickabasaviah Y, Kaneski C, Shankar SK, Narayanappa G, Christopher R, et al. Clinical features of adult GM1 gangliosidosis: report of three Indian patients and review of 40 cases. Mov Disord. (2004) 19:1334–41. doi: 10.1002/mds.20193
20. Yoshida K, Oshima A, Sakuraba H, Nakano T, Yanagisawa N, Inui K, et al. GM1 gangliosidosis in adults: clinical and molecular analysis of 16 Japanese patients. Ann Neurol. (1992) 31:328–32. doi: 10.1002/ana.410310316
21. van Diggelen OP, Thobois S, Tilikete C, Zabot MT, Keulemans JL, van Bunderen PA, et al. Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: first adult-onset patients of a childhood disease. Ann Neurol. (2001) 50:269–72. doi: 10.1002/ana.1103
22. Aberg L, Liewendahl K, Nikkinen P, Autti T, Rinne JO, Santavuori P. Decreased striatal dopamine transporter density in JNCL patients with parkinsonian symptoms. Neurology (2000) 54:1069–74. doi: 10.1212/WNL.54.5.1069
23. Machado A, Chien HF, Deguti MM, Cancado E, Azevedo RS, Scaff M, et al. Neurological manifestations in Wilson's disease: Report of 119 cases. Mov Disord. (2006) 21:2192–6. doi: 10.1002/mds.21170
24. Bandmann O, Weiss KH, Kaler SG. Wilson's disease and other neurological copper disorders. Lancet Neurol. (2015) 14:103–113. doi: 10.1016/S1474-4422(14)70190-5
25. Das SK, Ray K. Wilson's disease: an update. Nat Clin Pract Neurol. (2006) 2:482–93. doi: 10.1038/ncpneuro0291
26. Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study. Gut (2007) 56:115–20. doi: 10.1136/gut.2005.087262
27. Girotra T, Mahajan A, Sidiropoulos C. Levodopa responsive Parkinsonism in patients with hemochromatosis: case presentation and literature review. Case Rep Neurol Med. (2017) 2017:5146723. doi: 10.1155/2017/5146723
28. Nielsen JE, Jensen LN, Krabbe K. Hereditary haemochromatosis: a case of iron accumulation in the basal ganglia associated with a parkinsonian syndrome. J Neurol Neurosurg Psychiatry (1995) 59:318–21. doi: 10.1136/jnnp.59.3.318
29. Costello DJ. Concurrent hereditary haemochromatosis and idiopathic Parkinson's disease: a case report series. J Neurol Neurosurg Psychiatry (2004) 75:631–3. doi: 10.1136/jnnp.2003.027441
30. Evans AH, Costa DC, Gacinovic S, Katzenschlager R, O'Sullivan J D, Heales S, et al. L-Dopa-responsive Parkinson's syndrome in association with phenylketonuria: in vivo dopamine transporter and D2 receptor findings. Mov Disord. (2004) 19:1232–6. doi: 10.1002/mds.20146
31. Velema M, Boot E, Engelen M, Hollak C. Parkinsonism in phenylketonuria: a consequence of dopamine depletion? JIMD Rep. (2015) 20:35–8. doi: 10.1007/8904_2014_386
32. Carecchio M, Schneider SA, Chan H, Lachmann R, Lee PJ, Murphy E, et al. Movement disorders in adult surviving patients with maple syrup urine disease. Mov Disord. (2011) 26:1324–8. doi: 10.1002/mds.23629
33. Park JS, Park D, Ko PW, Kang K, Lee HW. Serum methylmalonic acid correlates with neuropathic pain in idiopathic Parkinson's disease. Neurol Sci. (2017) 38:1799–804. doi: 10.1007/s10072-017-3056-9
34. Finsterer J. Parkinson's syndrome and Parkinson's disease in mitochondrial disorders. Mov Disord. (2011) 26:784–91. doi: 10.1002/mds.23651
35. Thirumal Kumar D, Eldous HG, Mahgoub ZA, George Priya Doss C, Zayed H. Computational modelling approaches as a potential platform to understand the molecular genetics association between Parkinson's and Gaucher diseases. Metab Brain Dis. (2018). doi: 10.1007/s11011-018-0286-3. [Epub ahead of print].
36. Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain (2015) 138(Pt 9):2648–58. doi: 10.1093/brain/awv179
37. Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, et al. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc Natl Acad Sci USA. (2018) 115:798–803. doi: 10.1073/pnas.1700465115
38. Wong YC, Krainc D. Lysosomal trafficking defects link Parkinson's disease with Gaucher's disease. Mov Disord. (2016) 31:1610–8. doi: 10.1002/mds.26802
39. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurology. (2012) 11:986–98. doi: 10.1016/S1474-4422(12)70190-4
40. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell (2011) 146:37–52. doi: 10.1016/j.cell.2011.06.001
41. Migdalska-Richards A, Wegrzynowicz M, Rusconi R, Deangeli G, Di Monte DA, Spillantini MG, et al. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain (2017) 140:2706–21. doi: 10.1093/brain/awx221
42. Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat Commun. (2014) 5:4755. doi: 10.1038/ncomms5755
43. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. (2008) 183:795–803. doi: 10.1083/jcb.200809125
44. Ron I, Rapaport D, Horowitz M. Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease. Hum Mol Genet. (2010) 19:3771–81. doi: 10.1093/hmg/ddq292
45. Liu JP, Tang Y, Zhou S, Toh BH, McLean C, Li H. Cholesterol involvement in the pathogenesis of neurodegenerative diseases. Mol Cell Neurosci. (2010) 43:33–42. doi: 10.1016/j.mcn.2009.07.013
46. Johnson S. Is Parkinson's disease the heterozygote form of Wilson's disease: PD = 1/2 WD? Med Hypotheses (2001) 56:171–3. doi: 10.1054/mehy.2000.1134
47. Villar-Pique A, Rossetti G, Ventura S, Carloni P, Fernandez CO, Outeiro TF. Copper(II) and the pathological H50Q alpha-synuclein mutant: environment meets genetics. Commun Integr Biol. (2017) 10:e1270484. doi: 10.1080/19420889.2016.1270484
48. Carboni E, Lingor P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson's disease. Metallomics (2015) 7:395–404. doi: 10.1039/C4MT00339J
49. Dell'Acqua S, Pirota V, Anzani C, Rocco MM, Nicolis S, Valensin D, et al. Reactivity of copper-alpha-synuclein peptide complexes relevant to Parkinson's disease. Metallomics (2015) 7:1091–102. doi: 10.1039/C4MT00345D
50. Jin L, Wang J, Zhao L, Jin H, Fei G, Zhang Y, et al. Decreased serum ceruloplasmin levels characteristically aggravate nigral iron deposition in Parkinson's disease. Brain (2011) 134(Pt 1):50–8. doi: 10.1093/brain/awq319
51. Bharucha KJ, Friedman JK, Vincent AS, Ross ED. Lower serum ceruloplasmin levels correlate with younger age of onset in Parkinson's disease. J Neurol. (2008) 255:1957–62. doi: 10.1007/s00415-009-0063-7
52. Montes S, Rivera-Mancia S, Diaz-Ruiz A, Tristan-Lopez L, Rios C. Copper and copper proteins in Parkinson's disease. Oxid Med Cell Longev. (2014) 2014:147251. doi: 10.1155/2014/147251
53. Medeiros MS, Schumacher-Schuh A, Cardoso AM, Bochi GV, Baldissarelli J, Kegler A, et al. Iron and oxidative stress in Parkinson's disease: an observational study of injury biomarkers. PLoS ONE (2016) 11:e0146129. doi: 10.1371/journal.pone.0146129
54. Li XP, Xie WJ, Zhang Z, Kansara S, Jankovic J, Le WD. A mechanistic study of proteasome inhibition-induced iron misregulation in dopamine neuron degeneration. Neurosignals (2012) 20:223–36. doi: 10.1159/000332954
55. Xiao Y, Chen X, Huang S, Li G, Mo M, Zhang L, et al. Iron promotes alpha-synuclein aggregation and transmission by inhibiting TFEB-mediated autophagosome-lysosome fusion. J Neurochem. (2018) 145:34–50. doi: 10.1111/jnc.14312
56. Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, et al. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum Mol Genet. (2005) 14:3885–97. doi: 10.1093/hmg/ddi413
57. Shnitko TA, Taylor SC, Stringfield SJ, Zandy SL, Cofresi RU, Doherty JM, et al. Acute phenylalanine/tyrosine depletion of phasic dopamine in the rat brain. Psychopharmacology (2016) 233:2045–54. doi: 10.1007/s00213-016-4259-0
58. Daelman L, Sedel F, Tourbah A. Progressive neuropsychiatric manifestations of phenylketonuria in adulthood. Rev Neurol (2014) 170:280–7. doi: 10.1016/j.neurol.2013.09.012
59. Sirtori LR, Dutra-Filho CS, Fitarelli D, Sitta A, Haeser A, Barschak AG, et al. Oxidative stress in patients with phenylketonuria. Biochim Biophys Acta. (2005) 1740:68–73. doi: 10.1016/j.bbadis.2005.02.005
60. Sitta A, Barschak AG, Deon M, Barden AT, Biancini GB, Vargas PR, et al. Effect of short- and long-term exposition to high phenylalanine blood levels on oxidative damage in phenylketonuric patients. Int J Dev Neurosci. (2009) 27:243–7. doi: 10.1016/j.ijdevneu.2009.01.001
61. Barschak AG, Sitta A, Deon M, de Oliveira MH, Haeser A, Dutra-Filho CS, et al. Evidence that oxidative stress is increased in plasma from patients with maple syrup urine disease. Metab Brain Dis. (2006) 21:279–86. doi: 10.1007/s11011-006-9030-5
62. Sgaravatti AM, Rosa RB, Schuck PF, Ribeiro CAJ, Wannmacher CMD, Wyse ATS, et al. Inhibition of brain energy metabolism by the α-keto acids accumulating in maple syrup urine disease. Biochim Biophys Acta (2003) 1639:232–8. doi: 10.1016/j.bbadis.2003.09.010
63. Zinnanti WJ, Lazovic J, Griffin K, Skvorak KJ, Paul HS, Homanics GE, et al. Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain (2009) 132(Pt 4):903–18. doi: 10.1093/brain/awp024
64. Han L, Wu S, Han F, Gu X. Insights into the molecular mechanisms of methylmalonic acidemia using microarray technology. Int J Clin Exp Med. (2015) 8:8866–79.
65. Wang G, Pan J, Chen SD. Kinases and kinase signaling pathways: potential therapeutic targets in Parkinson's disease. Prog Neurobiol. (2012) 98:207–21. doi: 10.1016/j.pneurobio.2012.06.003
66. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. (1990) 54:823–7. doi: 10.1111/j.1471-4159.1990.tb02325.x
67. Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, et al. PGC-1 , A potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med. (2010) 2:52ra73. doi: 10.1126/scitranslmed.3001059
68. Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. (2006) 103:10793–8. doi: 10.1073/pnas.0602493103
69. Park J, Lee G, Chung J. The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun. (2009) 378:518–23. doi: 10.1016/j.bbrc.2008.11.086
70. Ordonez DG, Lee MK, Feany MB. alpha-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron (2018) 97:108–24 e6. doi: 10.1016/j.neuron.2017.11.036
71. Bir A, Sen O, Anand S, Khemka VK, Banerjee P, Cappai R, et al. alpha-Synuclein-induced mitochondrial dysfunction in isolated preparation and intact cells: implications in the pathogenesis of Parkinson's disease. J Neurochem. (2014) 131:868–77. doi: 10.1111/jnc.12966
72. Howlett EH, Jensen N, Belmonte F, Zafar F, Hu X, Kluss J, et al. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson's disease. Hum Mol Genet. (2017) 26:4340–51. doi: 10.1093/hmg/ddx320
73. Hao LY, Giasson BI, Bonini NM. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc Natl Acad Sci USA. (2010) 107:9747–52. doi: 10.1073/pnas.0911175107
74. Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, et al. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. (2012) 21:1931–44. doi: 10.1093/hmg/dds003
75. Tsai PI, Lin CH, Hsieh CH, Papakyrikos AM, Kim MJ, Napolioni V, et al. PINK1 phosphorylates MIC60/Mitofilin to control structural plasticity of mitochondrial crista junctions. Mol Cell. (2018) 69:744–56.e6. doi: 10.1016/j.molcel.2018.01.026
76. Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, et al. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson's disease. Cell Stem Cell. (2016) 19:709–24. doi: 10.1016/j.stem.2016.08.002
77. Baris HN, Cohen IJ, Mistry PK. Gaucher disease: the metabolic defect, pathophysiology, phenotypes and natural history. Pediatr Endocrinol Rev. (2014) 12(Suppl. 1):72–81.
78. Chen M, Wang J. Gaucher disease: review of the literature. Arch Pathol Lab Med. (2008) 132:851–3. doi: 10.1043/1543-2165(2008)132[851:GDROTL]2.0.CO;2
79. Cherin P, Rose C, de Roux-Serratrice C, Tardy D, Dobbelaere D, Grosbois B, et al. The neurological manifestations of Gaucher disease type 1: the french observatoire on Gaucher disease (FROG). J Inherit Metab Dis. (2010) 33:331–8. doi: 10.1007/s10545-010-9095-5
80. Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med. (2004) 351:1972–7. doi: 10.1056/NEJMoa033277
81. Tayebi N. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab. (2003) 79:104–9. doi: 10.1016/S1096-7192(03)00071-4
82. Neumann J, Bras J, Deas E, O'Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain (2009) 132(Pt 7):1783–94. doi: 10.1093/brain/awp044
83. Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C, et al. Large-scale screening of the Gaucher's disease-related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum Mol Genet. (2011) 20:202–10. doi: 10.1093/hmg/ddq454
84. Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology (2007) 69:1270–7. doi: 10.1212/01.wnl.0000276989.17578.02
85. Mao XY, Burgunder JM, Zhang ZJ, An XK, Zhang JH, Yang Y, et al. Association between GBA L444P mutation and sporadic Parkinson's disease from Mainland China. Neurosci Lett. (2010) 469:256–9. doi: 10.1016/j.neulet.2009.12.007
86. Rosenbloom B, Balwani M, Bronstein JM, Kolodny E, Sathe S, Gwosdow AR, et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells Mol Dis. (2011) 46:95–102. doi: 10.1016/j.bcmd.2010.10.006
87. Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology (2008) 70:2277–83. doi: 10.1212/01.wnl.0000304039.11891.29
88. Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol. (2011) 69:940–53. doi: 10.1002/ana.22400
89. Rockenstein E, Clarke J, Viel C, Panarello N, Treleaven CM, Kim C, et al. Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson's disease. Hum Mol Genet. (2016) 25:2645–60. doi: 10.1093/hmg/ddw124
90. Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci USA. (2013) 110:3537–42. doi: 10.1073/pnas.1220464110
91. Moors TE, Paciotti S, Ingrassia A, Quadri M, Breedveld G, Tasegian A, et al. Characterization of brain lysosomal activities in GBA-related and sporadic parkinson's disease and dementia with lewy bodies. Mol Neurobiol. (2018). doi: 10.1007/s12035-018-1090-0. [Epub ahead of print].
92. Yap TL, Velayati A, Sidransky E, Lee JC. Membrane-bound alpha-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol Genet Metab. (2013) 108:56–64. doi: 10.1016/j.ymgme.2012.11.010
93. Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. (2012) 72:455–63. doi: 10.1002/ana.23614
94. Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med. (2011) 17:485–93. doi: 10.1016/j.molmed.2011.05.003
95. Bendikov-Bar I, Rapaport D, Larisch S, Horowitz M. Parkin-mediated ubiquitination of mutant glucocerebrosidase leads to competition with its substrates PARIS and ARTS. Orphanet J Rare Dis. (2014) 9:86. doi: 10.1186/1750-1172-9-86
96. Schuchman EH, Desnick RJ. Types A and B Niemann-Pick disease. Mol Genet Metab. (2017) 120:27–33. doi: 10.1016/j.ymgme.2016.12.008
97. Vanier MT. Niemann-Pick diseases. Handb Clin Neurol. (2013) 113:1717–21. doi: 10.1016/B978-0-444-59565-2.00041-1
98. Vance JE. Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis Model Mech. (2012) 5:746–55. doi: 10.1242/dmm.010124
99. Sévin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, et al. The adult form of Niemann-Pick disease type C. Brain (2007) 130(Pt 1):120–33. doi: 10.1093/brain/awl260
100. Cheng D, Jenner AM, Shui G, Cheong WF, Mitchell TW, Nealon JR, et al. Lipid pathway alterations in Parkinson's disease primary visual cortex. PLoS ONE (2011) 6:e17299. doi: 10.1371/journal.pone.0017299
101. Lee CY, Seet RC, Huang SH, Long LH, Halliwell B. Different patterns of oxidized lipid products in plasma and urine of dengue fever, stroke, and Parkinson's disease patients: cautions in the use of biomarkers of oxidative stress. Antioxid Redox Signal. (2009) 11:407–20. doi: 10.1089/ars.2008.2179
102. Bosco DA, Fowler DM, Zhang Q, Nieva J, Powers ET, Wentworth P Jr et al. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat Chem Biol. (2006) 2:249–53. doi: 10.1038/nchembio782
103. Rantham Prabhakara JP, Feist G, Thomasson S, Thompson A, Schommer E, Ghribi O. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on tyrosine hydroxylase and alpha-synuclein in human neuroblastoma SH-SY5Y cells. J Neurochem. (2008) 107:1722–9. doi: 10.1111/j.1471-4159.2008.05736.x
104. Marwarha G, Rhen T, Schommer T, Ghribi O. The oxysterol 27-hydroxycholesterol regulates alpha-synuclein and tyrosine hydroxylase expression levels in human neuroblastoma cells through modulation of liver X receptors and estrogen receptors–relevance to Parkinson's disease. J Neurochem. (2011) 119:1119–36. doi: 10.1111/j.1471-4159.2011.07497.x
105. Bar-On P, Crews L, Koob AO, Mizuno H, Adame A, Spencer B, et al. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson's disease. J Neurochem. (2008) 105:1656–67. doi: 10.1111/j.1471-4159.2008.05254.x
106. Paul R, Choudhury A, Kumar S, Giri A, Sandhir R, Borah A. Cholesterol contributes to dopamine-neuronal loss in MPTP mouse model of Parkinson's disease: involvement of mitochondrial dysfunctions and oxidative stress. PLoS ONE (2017) 12:e0171285. doi: 10.1371/journal.pone.0171285
107. Rozani V, Giladi N, El-Ad B, Gurevich T, Tsamir J, Hemo B, et al. Statin adherence and the risk of Parkinson's disease: a population-based cohort study. PLoS ONE (2017) 12:e0175054. doi: 10.1371/journal.pone.0175054
108. Liu G, Sterling NW, Kong L, Lewis MM, Mailman RB, Chen H, et al. Statins may facilitate Parkinson's disease: insight gained from a large, national claims database. Mov Disord. (2017) 32:913–7. doi: 10.1002/mds.27006
109. de Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum cholesterol levels and the risk of Parkinson's disease. Am J Epidemiol. (2006) 164:998–1002. doi: 10.1093/aje/kwj283
110. Huang X, Chen H, Miller WC, Mailman RB, Woodard JL, Chen PC, et al. Lower low-density lipoprotein cholesterol levels are associated with Parkinson's disease. Mov Disord. (2007) 22:377–81. doi: 10.1002/mds.21290
111. Zech M, Nubling G, Castrop F, Jochim A, Schulte EC, Mollenhauer B, et al. Niemann-Pick C disease gene mutations and age-related neurodegenerative disorders. PLoS ONE (2013) 8:e82879. doi: 10.1371/journal.pone.0082879
112. Bifsha P, Landry K, Ashmarina L, Durand S, Seyrantepe V, Trudel S, et al. Altered gene expression in cells from patients with lysosomal storage disorders suggests impairment of the ubiquitin pathway. Cell Death Differ. (2007) 14:511–23. doi: 10.1038/sj.cdd.4402013
113. Inzelberg R, Korczyn AD. Parkinsonism in adult-onset GM2 gangliosidosis. Mov Disord. (1994) 9:375–7. doi: 10.1002/mds.870090325
114. Argov Z, Navon R. Clinical and genetic variations in the syndrome of adult GM2 gangliosidosis resulting from hexosaminidase A deficiency. Ann Neurol. (1984) 16:14–20. doi: 10.1002/ana.410160105
115. Martinelli P, Ippoliti M, Montanari M, Martinelli A, Mochi M, Giuliani S, et al. Arylsulphatase A (ASA) activity in parkinsonism and symptomatic essential tremor. Acta Neurol Scand. (1994) 89:171–4. doi: 10.1111/j.1600-0404.1994.tb01656.x
116. Antelmi E, Rizzo G, Fabbri M, Capellari S, Scaglione C, Martinelli P. Arylsulphatase A activity in familial parkinsonism: a pathogenetic role? J Neurol. (2014) 261:1803–9. doi: 10.1007/s00415-014-7425-5
117. Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. (2017) 49:1511–16. doi: 10.1038/ng.3955
118. Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain (2017) 140:3191–203. doi: 10.1093/brain/awx285
119. Clark LN, Chan R, Cheng R, Liu X, Park N, Parmalee N, et al. Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS ONE (2015) 10:e0125204. doi: 10.1371/journal.pone.0125204
120. Gasca-Salas C, Alonso A, Gonzalez-Redondo R, Obeso JA. Coexisting Parkinson's and Wilson's disease: chance or connection? Can J Neurol Sci. (2017) 44:215–8. doi: 10.1017/cjn.2016.327
121. Gouider-Khouja N. Wilson's disease. Parkinsonism Relat Disord. (2009) 15:S126–9. doi: 10.1016/S1353-8020(09)70798-9
122. Pinero DJ, Connor JR. Iron in the Brain: an important contributor in normal and diseased states. Neuroscientist (2000) 6:435–53. doi: 10.1177/107385840000600607
123. Mariani S, Ventriglia M, Simonelli I, Spalletta G, Bucossi S, Siotto M, et al. Effects of hemochromatosis and transferrin gene mutations on peripheral iron dyshomeostasis in mild cognitive impairment and Alzheimer's and Parkinson's diseases. Front Aging Neurosci. (2013) 5:37. doi: 10.3389/fnagi.2013.00037
124. Wang JY, Zhuang QQ, Zhu LB, Zhu H, Li T, Li R, et al. Meta-analysis of brain iron levels of Parkinson's disease patients determined by postmortem and MRI measurements. Sci Rep. (2016) 6:36669. doi: 10.1038/srep36669
125. Ramos P, Santos A, Pinto NR, Mendes R, Magalhaes T, Almeida A. Iron levels in the human brain: a post-mortem study of anatomical region differences and age-related changes. J Trace Elem Med Biol. (2014) 28:13–7. doi: 10.1016/j.jtemb.2013.08.001
126. Zhu W, Li X, Xie W, Luo F, Kaur D, Andersen JK, et al. Genetic iron chelation protects against proteasome inhibition-induced dopamine neuron degeneration. Neurobiol Dis. (2010) 37:307–13. doi: 10.1016/j.nbd.2009.09.024
127. Le W. Role of iron in UPS impairment model of Parkinson's disease. Parkinsonism Relat Disord. (2014) 20:S158–61. doi: 10.1016/S1353-8020(13)70038-5
128. Pichler I, Del Greco MF, Gogele M, Lill CM, Bertram L, Do CB, et al. Serum iron levels and the risk of Parkinson disease: a Mendelian randomization study. PLoS Med. (2013) 10:e1001462. doi: 10.1371/journal.pmed.1001462
129. Mostile G, Cicero CE, Giuliano L, Zappia M, Nicoletti A. Iron and Parkinson's disease: a systematic review and meta-analysis. Mol Med Rep. (2017) 15:3383–9. doi: 10.3892/mmr.2017.6386
130. Zschocke J. Phenylketonuria mutations in Europe. Hum Mutat. (2003) 21:345–56. doi: 10.1002/humu.10192
131. Perez-Duenas B, Valls-Sole J, Fernandez-Alvarez E, Conill J, Vilaseca MA, Artuch R, et al. Characterization of tremor in phenylketonuric patients. J Neurol. (2005) 252:1328–34. doi: 10.1007/s00415-005-0860-6
132. Boot E, Hollak CEM, Huijbregts SCJ, Jahja R, van Vliet D, Nederveen AJ, et al. Cerebral dopamine deficiency, plasma monoamine alterations and neurocognitive deficits in adults with phenylketonuria. Psychol Med. (2017) 47:2854–65. doi: 10.1017/S0033291717001398
133. Fraser JL, Venditti CP. Methylmalonic and propionic acidemias: clinical management update. Curr Opin Pediatr. (2016) 28:682–93. doi: 10.1097/MOP.0000000000000422
134. O'Shea CJ, Sloan JL, Wiggs EA, Pao M, Gropman A, Baker EH, et al. Neurocognitive phenotype of isolated methylmalonic acidemia. Pediatrics (2012) 129:e1541–51. doi: 10.1542/peds.2011-1715
135. Karimzadeh P, Jafari N, Ahmad Abadi F, Jabbedari S, Taghdiri MM, Nemati H, et al. Methylmalonic acidemia: diagnosis and neuroimaging findings of this neurometabolic disorder (An Iranian Pediatric Case Series). Iran J Child Neurol. (2013) 7:63–66.
136. Khan NA, Govindaraj P, Meena AK, Thangaraj K. Mitochondrial disorders: Challenges in diagnosis & treatment. Indian J Med Res. (2015) 141:13–26. doi: 10.4103/0971-5916.154489
137. Finsterer J. Parkinson syndrome as a manifestation of mitochondriopathy. Acta Neurol Scand. (2002) 105:384–9. doi: 10.1034/j.1600-0404.2002.01221.x
138. Martikainen MH, Ng YS, Gorman GS, Alston CL, Blakely EL, Schaefer AM, et al. Clinical, genetic, and radiological features of extrapyramidal movement disorders in mitochondrial disease. JAMA Neurol. (2016) 73:668. doi: 10.1001/jamaneurol.2016.0355
139. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science (1983) 219:979–80. doi: 10.1126/science.6823561
140. Vyas I, Heikkila RE, Nicklas WJ. Studies on the neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: inhibition of NAD-linked substrate oxidation by its metabolite, 1-methyl-4-phenylpyridinium. J Neurochem. (1986) 46:1501–7. doi: 10.1111/j.1471-4159.1986.tb01768.x
141. Parker WD Jr, Parks JK, Swerdlow RH. Complex I deficiency in Parkinson's disease frontal cortex. Brain Res. (2008) 1189:215–8. doi: 10.1016/j.brainres.2007.10.061
142. Leman G, Gueguen N, Desquiret-Dumas V, Kane MS, Wettervald C, Chupin S, et al. Assembly defects induce oxidative stress in inherited mitochondrial complex I deficiency. Int J Biochem Cell Biol. (2015) 65:91–103. doi: 10.1016/j.biocel.2015.05.017
Keywords: inherited metabolic disorders, lysosomal storage diseases, mitochondrial dysfunction, neurodegenerative disorder, Parkinson's disease
Citation: Limphaibool N, Iwanowski P, Holstad MJV and Perkowska K (2018) Parkinsonism in Inherited Metabolic Disorders: Key Considerations and Major Features. Front. Neurol. 9:857. doi: 10.3389/fneur.2018.00857
Received: 11 July 2018; Accepted: 24 September 2018;
Published: 12 October 2018.
Edited by:
Patrick Lewis, University of Reading, United KingdomReviewed by:
Eleanna Kara, UniversitätsSpital Zürich, SwitzerlandGeorgia Xiromerisiou, University of Thessaly, Greece
Copyright © 2018 Limphaibool, Iwanowski, Holstad and Perkowska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nattakarn Limphaibool, dG9uZ24ubGltQGdtYWlsLmNvbQ==