
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol. , 10 October 2018
Sec. Multiple Sclerosis and Neuroimmunology
Volume 9 - 2018 | https://doi.org/10.3389/fneur.2018.00841
This article is part of the Research Topic Emerging Challenges in the Diagnosis and Treatment of Autoimmune Encephalitis View all 12 articles
 Gerda Ricken1
Gerda Ricken1 Carmen Schwaiger1Desiree De Simoni1Valerie Pichler1Julia Lang1Sarah Glatter1
Carmen Schwaiger1Desiree De Simoni1Valerie Pichler1Julia Lang1Sarah Glatter1 Stefan Macher2
Stefan Macher2 Paulus S. Rommer2
Paulus S. Rommer2 Petra Scholze3Helmut Kubista4
Petra Scholze3Helmut Kubista4 Inga Koneczny1
Inga Koneczny1 Romana Höftberger1*
Romana Höftberger1*This review provides an overview on different antibody test methods that can be applied in cases of suspected paraneoplastic neurological syndromes (PNS) and anti-neuronal autoimmune encephalitis (AIE) in order to explain their diagnostic value, describe potential pitfalls and limitations, and discuss novel approaches aimed at discovering further autoantibodies. Onconeuronal antibodies are well-established biomarkers for PNS and may serve as specific tumor markers. The recommended procedure to detect onconeuronal antibodies is a combination of indirect immunohistochemistry on fixed rodent cerebellum and confirmation of the specificity by line assays. Simplification of this approach by only using line assays with recombinant proteins bears the risk to miss antibody-positive samples. Anti-neuronal surface antibodies are sensitive and specific biomarkers for AIE. Their identification requires the use of test methods that allow the recognition of conformation dependent epitopes. These commonly include cell-based assays and tissue based assays with unfixed rodent brain tissue. Tissue based assays can detect most of the currently known neuronal surface antibodies and thus enable broad screening of biological samples. A complementary testing on live neuronal cell cultures may confirm that the antibody recognizes a surface epitope. In patients with peripheral neuropathy, the screening may be expanded to teased nerve fibers to identify antibodies against the node of Ranvier. This method helps to identify a novel subgroup of peripheral autoimmune neuropathies, resulting in improved immunotherapy of these patients. Tissue based assays are useful to discover additional autoantibody targets that play a role in diverse autoimmune neurological syndromes. Antibody screening assays represent promising avenues of research to improve the diagnostic yield of current assays for antibody-associated autoimmune encephalitis.
Autoimmune diseases in the brain may affect different parts of the nervous system including neurons, glial cells or components of the blood-brain barrier. The pathobiology can be predominantly driven by T-cells or B-cells that recognize cerebral antigens. The field of autoantibody mediated autoimmune diseases of the nervous system has been expanding in the recent years, propelled by the discovery of autoantibodies against synaptic or extrasynaptic membrane antigens that lead to a new approach in diagnosing and treating patients with suspected autoimmune neurological diseases (1). While autoimmune responses against intracellular antigens are mainly associated with paraneoplastic or idiopathic neurological syndromes with poor neurological outcome, patients with surface autoimmunity show substantial response to immunotherapy (1). Cell-mediated immune attack by T-cells resulting in progressive destruction of cells is a hallmark of paraneoplastic neurological syndromes (PNS) and may explain the limited response to immunotherapy (2). Although some pathogenic impact has been described for anti-amphiphysin antibodies (3), the mechanisms and functions of other autoantibodies that evolve in the context of classical paraneoplastic syndromes (so called onconeuronal antibodies) are still poorly understood and they are rather considered as an epiphenomenon. However, the antibodies indicate the paraneoplastic etiology of the associated neurological syndrome and may serve as biomarkers for recognizing an underlying malignancy (Table 1) (4). In contrast, autoantibodies against surface antigens may directly mediate the disease (e.g., by antigenic modulation or by recruitment of immune cells or components of the complement system), among the antibodies against neuronal membrane antigens, these effects are often reversible and explain the good response to immunotherapy. Autoantibodies against cell surface antigens on neurons and glial cells can be tumor associated but derive more frequently from an idiopathic origin (1). To date, more than 16 such autoimmune syndromes are known and are summarized in Table 2. These diseases occur worldwide in diverse ethnicities and cultures. Among the anti-neuronal surface antibodies, anti-NMDAR are probably the most common ones, followed by anti-LGI1 with a reported annual incidence of 0.83 per million in one Dutch study (7). Other antibodies seem to be less frequent or their incidence has to be defined in prospective experience. Many autoimmune neurological or demyelinating syndromes are currently considered as antibody negative despite some evidence that they are antibody-mediated. Among these are patients with suspected but yet unknown antigenic targets, and further studies are required to discover these. Nevertheless, a substantial fraction of seronegative patients may harbor known autoantibodies that could be detected with a more thorough testing strategy. The following review gives an overview of the most widely used test methods and their limitations in the detection of autoantibodies and provides an outlook on possible novel approaches that are able to broaden the spectrum of identifyable antibodies.
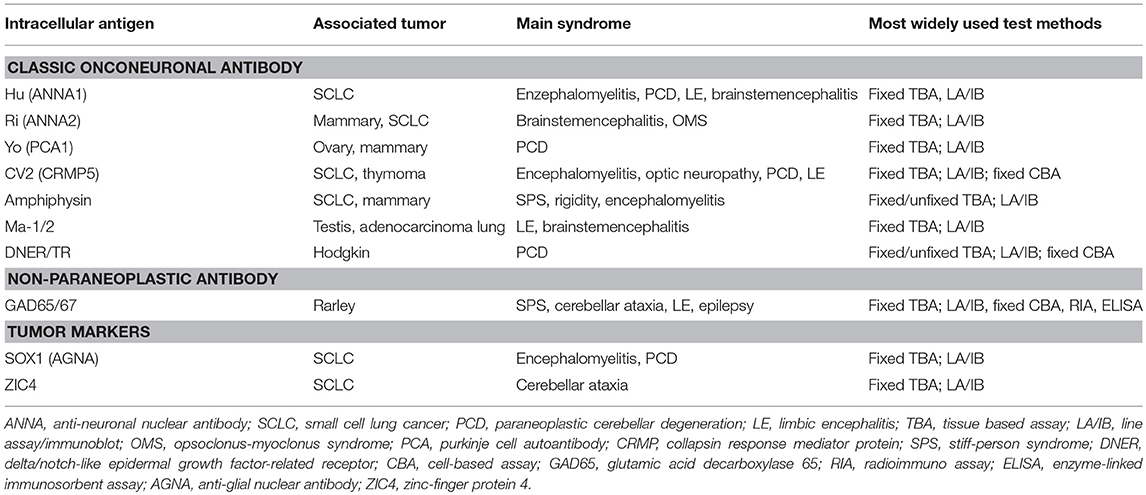
Table 1. Antibodies targeting intracellular antigens.
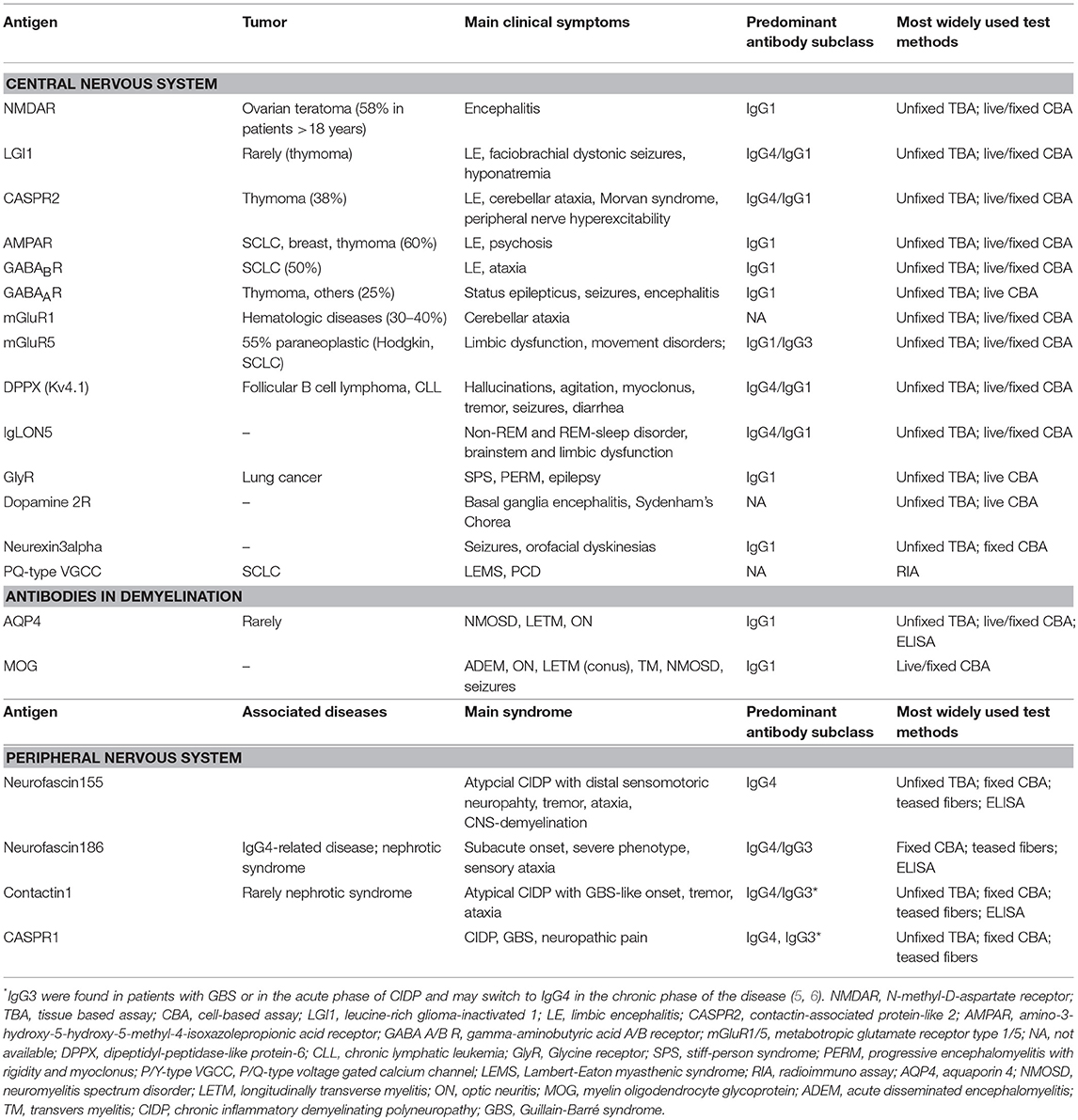
Table 2. Antibodies against surface antigens.
Since the 1980s, detailed clinical and immunological studies revealed several autoantibodies against intracellular antigens that are associated with specific paraneoplastic or idiopathic neurological syndromes (8–15). Antibodies directed against intracellular antigens usually recognize linear epitopes that can be detected by methods such as western blot analysis, line assays, enzyme-linked immunosorbent assay (ELISA), fixed tissue- (fixed TBA) or cell-based assays (CBA), or radioimmunoassay (RIA). In clinical laboratories, line assays and fixed TBAs are used most frequently. For line assays, purified recombinant proteins (e.g., paraneoplastic antigens such as Yo, Hu, Ri, CV2/CRMP5, and others) are applied on blot strips and incubated with the patient's serum or CSF. Line assays are commercially available and include most of the currently known well-characterized autoantibodies that are screened within one test run. The fixed TBAs use paraformaldehyde-fixed rodent (mouse or rat) or monkey tissue (cerebellum and enteric nervous system). The fixation is necessary for the intracellular antigen retrieval. Autoantibodies are defined as well-characterized if the serum or CSF produces a recognizable staining pattern in the fixed TBA (e.g., selective staining of Purkinje cells with Yo-positive patient's serum) (Figures 1A–J) and the antibody specificity is confirmed with the recombinant line assay (16).
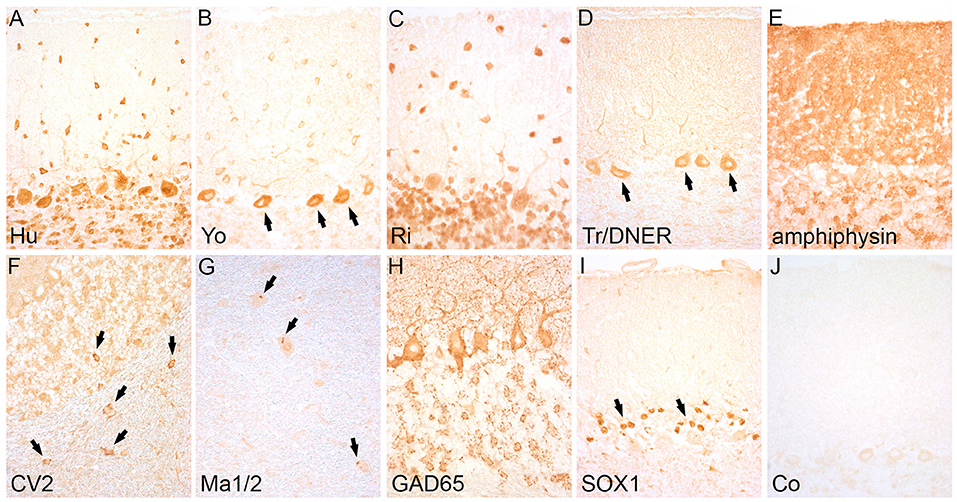
Figure 1. Staining pattern of antibodies targeting intracellular antigens. Indirect immunohistochemistry (avidin-biotin peroxidase method) on rat cerebellum shows a specific staining pattern of intracellular antibodies: (A) Anti-Hu-antibodies label the cytoplasm and nuclei of Purkinje and granule cells. (B) Anti-Yo antibodies show labeling of the cytoplasm of Purkinje cells (arrows) and stellate and basket cells in the molecular layer. (C) Anti-Ri-antibodies show the same staining pattern like Hu-antibodies in the cerebellum (differentiation is possible by staining enteric neurons of the gut that are positive with anti-Hu but negative with anti-Ri-antibodies). (D) Anti-Tr/DNER antibodies strongly label the Purkinj cell somata and dendrites (arrows). (E) Anti-amphiphysin antibodies show an intensive synaptic staining pattern in the molecular layer of the cerebellum. (F) Anti-CV2-antibodies mark a subgroup of oligodendrocytes in the cerebellar cortex and white matter (arrows). (G) Anti-Ma1/2-antibodies show a dot-like staining pattern in large neurons of the brainstem (arrows). (H) Anti-GAD65-antibodies display a dot-like staining of the base of Purkinje cells and a rosette-like staining pattern in the granular layer of the cerebellar cortex (I) Anti-SOX1-antibodies stain the nuclei of Bergmann glia in the cerebellar cortex (arrows). (J) Serum of a healthy control remains negative. Magnification: (A–J): x400.
To provide highest sensitivity and specificity for onconeuronal antibody testing, it is recommended to combine a fixed TBA and a line assay (16). Line assays may be more sensitive in some patients than indirect immunohistochemistry (17), in addition they can help to specify the onconeuronal antibody. Using the TBA alone has the disadvantage that concomitant antibodies such as anti-nuclear antibodies may mask the immunohistochemical staining pattern. On the other hand, commercial line assays may sometimes produce reactivity in control sera without reported cancer (18) and the clinical significance is unclear. Moreover, a recent study reported that the use of commercial line assays with recombinant protein harbors the risk to miss autoantibodies as it has been shown in 4 out of 53 patients with CV2/CRMP5-antibodies (19). It was hypothesized that the epitope repertoire of the CV2 antibodies that were missed in the line assay may be different from the typical CV2 antibodies (19).
Importantly, the fixed TBA is also able to detect rare antibodies such as for example anti-protein-kinase Cgamma (PKCgamma) (20), anti-carbonic anhydrase-related protein VIII (CARP VIII) (21) or anti-rhoGTPase-activating protein 26 (ARGHAP26) (22) that bind intracellular proteins highly expressed in Purkinje cells and were originally identified in patients presenting with subacute autoimmune cerebellar ataxia. Currently, the detection of these antibodies is only possible with in-house assays and the results of the TBA can either be confirmed with in-house immunoblots or fixed cell-based assays. The PKCgamma, CARP VIII and ARGHAP26 are potentially paraneoplastic antibodies and a positive antibody-test should prompt tumor search. Recently, a novel astrocytic IgG autoantibody targeting glial fibrillary acidic protein (GFAP) has been identified in the CSF and serum of 16 patients with relapsing steroid-responsive meningoencephalitis with or without myelitis and was clinically characterized in a series of 102 patients (23, 24). The antibody was identified in the TBA showing an immunofluorescence staining of a subpopulation of astrocytes confined to pia, subpia, midbrain foci, periventricular region and rostral migratory stream and subsequently characterized in the fixed CBA as GFAP-specific. An underlying tumor can be found in 22% of patients, which include teratoma, carcinoid, salivary pleomorphic adenoma, prostate carcinoma, and melanoma. Some patients may have coexisting antibodies such as anti-NMDAR or aquaporin-4 (AQP4) antibodies, which may indicate an underlying teratoma. Although the antigen is intracellularly located, patients show good response to immunotherapy. Future investigations are necessary to clarify the role of antibodies in disease evolution, give insight into T-cell antigen specificities, and reveal possible genetic factors.
The glutamic acid decarboxylase (GAD) is an enzyme that catalyzes the transformation of glutamate into gamma-amino-butyric-acid (GABA). Two isoforms have been described, the 65 and the 67 kDa isoform. Both can be found in GABAergic neurons in the brain, the 65 kDa isoform is additionally expressed in islet cells of the pancreas. Low titers of GAD65 antibodies can occur in about 1% of healthy controls and in up to 80% of patients with diabetes mellitus type I (25). Currently available commercial test methods focus on the detection of the GAD65 isoform and include ELISA, radioimmunoassay, TBA, and line assays. The ELISA and RIA are more sensitive than TBA or line assays and can detect very low titers of GAD65, however, only high titers (usually >2,000 U/ml) are considered to be associated with autoimmune neurological disorders including stiff-person syndrome, ataxia, epilepsy, limbic encephalitis, and other syndromes (25). It has long been believed that screening for GAD65 antibodies is sufficient for identifying patients with GAD-autoimmunity. Interestingly, a recent study with GAD65-antibody positive patients with neurological disorders reported that GAD67 antibodies were present in the CSF even if the serum was negative for GAD67 antibodies, indicating an intrathecal antibody synthesis (26). Later it has been shown that few patients harbor antibodies only against the GAD67 isoform. The clinical picture of patients with GAD67 antibodies in serum and/or CSF is currently believed to be indistinguishable from the phenotype associated with GAD65 antibodies but the patients would be missed if GAD65 specific assays are employed such as line assays or RIA (27). Currently GAD67 can only be detected by in-house assays that either use TBA, in-house immunoblots or fixed cell-based assays.
Autoantibodies directed against surface antigens often recognize conformation dependent epitopes and their detection depends on methods that preserve the three-dimensional structure of the antigen such as CBA or unfixed/postfixed TBA. In clinical laboratories, CBAs are used most frequently. The CBA consists of human or murine cells that are transfected with human complementary DNA (cDNA) and express the target antigen on their surface. Sera or CSF from patients are evaluated for the presence of antibodies by binding to these expressed antigens. CBAs are commercially available and either offered as set that allows screening of several autoantibodies within one test run [e.g., combined testing of NMDAR, AMPAR, GABA(B)R, LGI1, CASPR2, and DPPX] or as individual tests (e.g. IgLON5). The unfixed/postfixed TBAs use rodent (mouse or rat) brain tissue that contains the hippocampus and cerebellum. Sera or CSF from patients are evaluated for the presence of antibodies by binding to the rodent brain tissue and subsequently visualized either via an avidin-biotin method and light microscopy or immunofluorescence. This approach has been successful in discovering most of the autoantibodies described in the past decade. The TBAs for testing surface antibodies are commercially available or can be produced in-house and can be used as screening tool or to confirm the results of the CBA.
One of the first neurological autoimmune diseases that were defined by the presence of pathogenic surface autoantibodies was myastenia gravis associated with anti-acetylcholine receptor antibodies (AChR) (28). Later, surface antibodies to the P/Q type voltage-gated calcium channel (PQ-type VGCC) were identified in patients with Lambert-Eaton myasthenic syndrome (LEMS) (29, 30). Both antibodies were discovered by using RIA assays in which the antigens were labeled with 125I-specific neurotoxins and precipitated with patient's antibodies (31). Synthetic peptide binding studies in LEMS patients demonstrated that three epitope regions of the external linker peptides S5-S6 of domain II and IV of the alpha-1A subunit of the PQ-type VGCC were essential for creating reactivity in 9/12 patients. These epitopes are considered to be linear and test methods that lack correct membrane topology are suitable for their detection (32). In contrast, other pathogenic surface antibodies mostly recognize conformational epitopes and test methods that measure antibodies against linear or refolded epitopes often produce contradictory results, including variable frequencies of seropositivity in patients with diverse clinical syndromes and healthy controls. The RIA may give false positive results due to two issues: (1) the availability of intracellular epitopes may pick up irrelevant antibodies. For example only 56% of the serum samples that were tested positive in a RIA for voltage-gated potassium channel (VGKC) complex antibodies contained antibodies against the extracellular domain of LGI1 or CASPR2, while a considerable amount of LGI1/CASPR2-negative samples were directed against cytosolic epitopes of the VGKC (33, 34). (2) False positive results may also derive from the presence of autoantibodies against the 125I-neurotoxin itself (33) and false negative results may derive from an overlap of the antibody binding epitope with the binding site for the 125I-neurotoxin, a known phenomenon in mysthenia gravis and AChR antibodies (35–37). These difficulties emphasize the importance of test validation with different screening methods that ensure the exclusive recognition of the conformational epitope of the respective antigen and excludes interference with confounding components in the assay such as neurotoxins. The CBA allows the screening for conformation-dependent antibodies and enables the unequivocal identification of a specific surface antibody. The sensitivity of the CBA can be increased with different strategies such as 1. Using live CBAs instead of fixed cells as fixation methods may damage some epitopes (see Table 1), 2. Clustering of the antigen at high densitiy for example by co-transfecting clustering proteins such as rapsyn in the clustered AChR antibody assay (38) or 3. Increasing the number of recognizable antigens by adding further subunits of a receptor such as e.g., the gamma2 subunit of the GABA(A)R (39). The disadvantage of live CBAs is that they are technically demanding and time-consuming and their use is limited to specialized centers. The commercial CBAs are used by most clinical laboratories, however, not all antibodies can be tested with this method so far, either because the antibodies were only recently discovered and commercial CBAs may not be (readily) available, or the development of commercial assays is challenging due to methodological issues or the lack of sufficient numbers of positive controls. Another method that allows the screening for conformation-dependent antibodies is the unfixed/postfixed TBA. The TBA is a highly sensitive test method and can be used for initial screening and subsequent confirmation of positives by an antigen-specific CBA, may help to confirm the result of the CBA in case of doubtful results and is able to identify novel antibodies. A systematic comparison of the sensitivity and specificity of TBA and CBA was performed in a single-center study for anti-NMDAR antibodies and found an equal sensitivitiy for TBA and CBA (100%) in CSF, while in serum TBA was more sensitive (91.6%) than fixed CBA (86.8%) (40). Multicenter studies will be necessary to compare different assays for more target antigens and to evaluate assay reliability and reproducibility.
Some patients with autoimmune neurological syndromes remain antibody negative despite some evidence that they are antibody-mediated. Unfixed/postfixed TBAs can detect most of the currently known surface antibodies involved in autoimmune encephalitis such as NMDAR, AMPAR, LGI1, CASPR2, GABA(B)R, GABA(A)R, mGluR1, mGluR5, DPPX, Tr/DNER, Neurexin3alpha, and IgLON5 (41). In addition, anti-glial antibodies such as AQP4 antibodies can be detected (42). One limitation may be that the unfixed TBA is based on rodent brain, and antibodies that recognize only human-specific epitopes may not be detected. This is the case in most patients with anti-myelin-oligodendrocyte-glycoprotein (MOG)-antibodies (43). Furthermore, some limitations in the detection by unfixed/postfixed TBA have been described for autoantibodies against the dopamine2 receptor (D2R), glycine receptor (GlyR), and P/Q-type VGCC that are only poorly visible with this technique (41) As a consequence, it is recommended to use specific CBAs (GlyR, MOG, D2R) (44–46) or a RIA (P/Q-type VGCC) (47) for the detection of these antibodies. A potential limitation may be that some antibodies require the use of live CBAs and fixation-dependent staining protocols are inappropriate to reveal a specific antigenic epitope (see Table 2).
Anti-AQP4- and anti-MOG-antibodies are autoantibodies against glial cells that are associated with a specific spectrum of demyelinating diseases. Anti-AQP4-antibodies were the first antibodies with a clearly defined target that were identified in patients with demyelinating diseases (48) and now serve as biomarker for the diagnosis of patients with neuromyelitis optica spectrum disorders (NMOSD) (49). The incidence of AQP4-antibody positive NMO ranges from 0.05 to 0.4 per 100,000 (50). The antibodies were originally discovered by using indirect immunofluorescence on rodent brain tissue showing a characteristic staining pattern of astrocyte end feet around blood vessels, along the pial surfaces and Virchow-Robin spaces (48, 51). Meanwhile, the standard for most clinical laboratories for testing AQP4-antibodies is the use of CBAs either in form of commercially available fixed CBAs with the AQP4-M1 isoform or in-house live CBAs using the AQP4-M23 isoform. A large multicenter study systematically compared different AQP4 assays including CBAs, TBAs, flow cytometry, and ELISA and found the CBA as most sensitive and specific test method, with some benefit of using the AQP4-M23 isoform and additionally described high sensitivity and specificity for immunohistochemistry and flow cytometry in some specialized centers (52). Recently, the search for novel antibodies in demyelinating diseases by using monoclonal recombinant antibodies from patients with NMOSD revealed an anti-endothelial cell antibody against the endoplasmic reticulum chaperon GRP78 that may compromise the blood-brain barrier (53). Further studies will be necessary to clarify a potential role in initiating the inflammatory cascade and disease activity of NMOSD.
Anti-MOG antibodies were defined in patients with acute disseminated encephalomyelitis (ADEM), uni- or bilateral optic neuritis, transverse myelitis, longitudinally extensive transverse myelitis, and neuromyelitis optica. In children, one third of patients with an acute demyelinating syndrome are MOG-antibody positive (44, 54, 55). Most of the patient's antibodies recognize a human-specific epitope and TBAs based on rodent tissue are not suitable for their detection. Human MOG-antibodies were recently tested on human brain tissue and 88% of samples showed a staining of white matter (56), this approach could provide a promising screening tool in the search for novel antibodies. Currently anti-MOG-antibodies are either tested with commercial or in-house live CBAs employing HEK cells transfected with full-length human MOG (57–59). Further multicenter studies of different assays will be necessary to compare the sensitivity and specificity and identify difficulties in different test methods.
In patients with autoimmunity that primarily affects nervous tissue outside the CNS, the TBA can be expanded to the respective target region. For example, in patients with peripheral neuropathy, screening on teased sciatic nerve fiber preparations from rodents can detect antibodies against proteins in the node of Ranvier (Figures 2A–C) (5, 60, 61). The node of Ranvier is a highly specialized structure that is important for the saltatory conduction of impulses in myelinated nerve fibers. A large number of adhesion molecules are involved in the formation of the axon-myelin junctions and compartmentalization of voltage-gated potassium channels and serve as potential target for autoimmunity (62). Autoimmune diseases associated with antibodies against proteins in the paranodal region of the node of Ranvier are subsumed as paranodopathies and define an exciting group of autoimmune peripheral neuropathies clinically presenting as atypical chronic inflammatory demyelinating polyneuropathy (CIDP) or Guillain-Barré-syndrome (GBS) that may benefit from treatment with rituximab (Table 2) (63). Based on results of teased nerve fiber screening, it is supposed that up to 40% of CIDP patients harbor antibodies against components of the myelin or the axon (64). Some antibodies such as anti-neurofascin155 or anti-contactin1 are detectable in teased nerve fibers and in the unfixed TBA (hippocampus and molecular layer of cerebellum) (6, 61, 65), while others may only be detectable in teased nerve fibers (5).
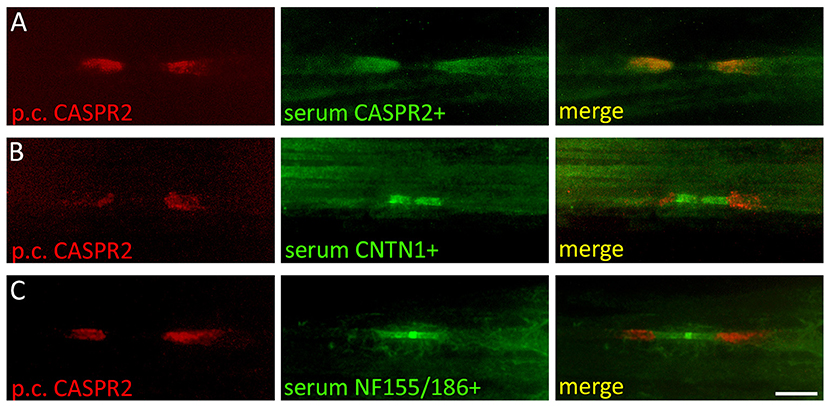
Figure 2. Screening of autoantibodies on teased nerve fibers in patients with peripheral neuropathies. Rat sciatic nerve fibers were immunostained with a polyclonal rabbit anti-CASPR2 antibody (red) and serum from a patient with (A) anti-CASPR2 antibodies (green), (B) anti-contactin1 antibodies (green), and (C) anti-neurofascin155/186 antibodies (green). CASPR2 labels the juxtaparanodal region of the node of Ranvier, contactin1 the paranodal and neurofascin155/186 the paranodal and nodal region. CNTN1, contactin1; NF155/186, neurofascin155/186; Scale bar = 10 μm.
A complementary method to the screening of surface antibodies on tissue based assays are live cultures of neurons. These neurons can be used to identify a binding between an individual's antibody and a specific surface peptide on the intact neuronal membrane. A positive staining of the cells gives evidence that the detected autoantibody recognizes a surface antigen and is likely to play a pathogenic role in the disease (41). This method is used in research laboratories and may (1) help in the diagnostic procedure to differentiate between surface or intracellular reactivity in samples with doubtful results in the TBA and (2) can be used to identify the target antigen by performing immunoprecipitation of the patient's serum together with the cell culture and subsequently identify the co-precipitated target antigen by mass spectrometry. Rat hippocampal neurons are the most frequently used cell culture system for the visualization of anti-neuronal surface antibodies, however, not all neuronal surface proteins are expressed in these cells and the absence of binding should not necessarily imply the absence of surface reactivities. Moreover, anti-glial antibodies are not displayed. Alternatively, other neuronal or mixed glioneuronal cell cultures may be useful to demonstrate a neuronal or glial surface autoantibody. Anti-contactin1 and anti-CASPR1 antibodies were shown to label both rat hippocampal neurons and dorsal root ganglion cells (5, 66), in contrast, anti-AQP4-antibodies may only be detectable in glioneuronal cell cultures including rat retinal cell cultures that contain Mueller cells (Figures 3A–O) (67, 68). The screening of samples with suspected seronegative autoimmune encephalitis on different live cell cultures might enable to broaden the spectrum of identifyable antibodies and provide a promising approach for discovering novel autoantibodies against surface antigens.
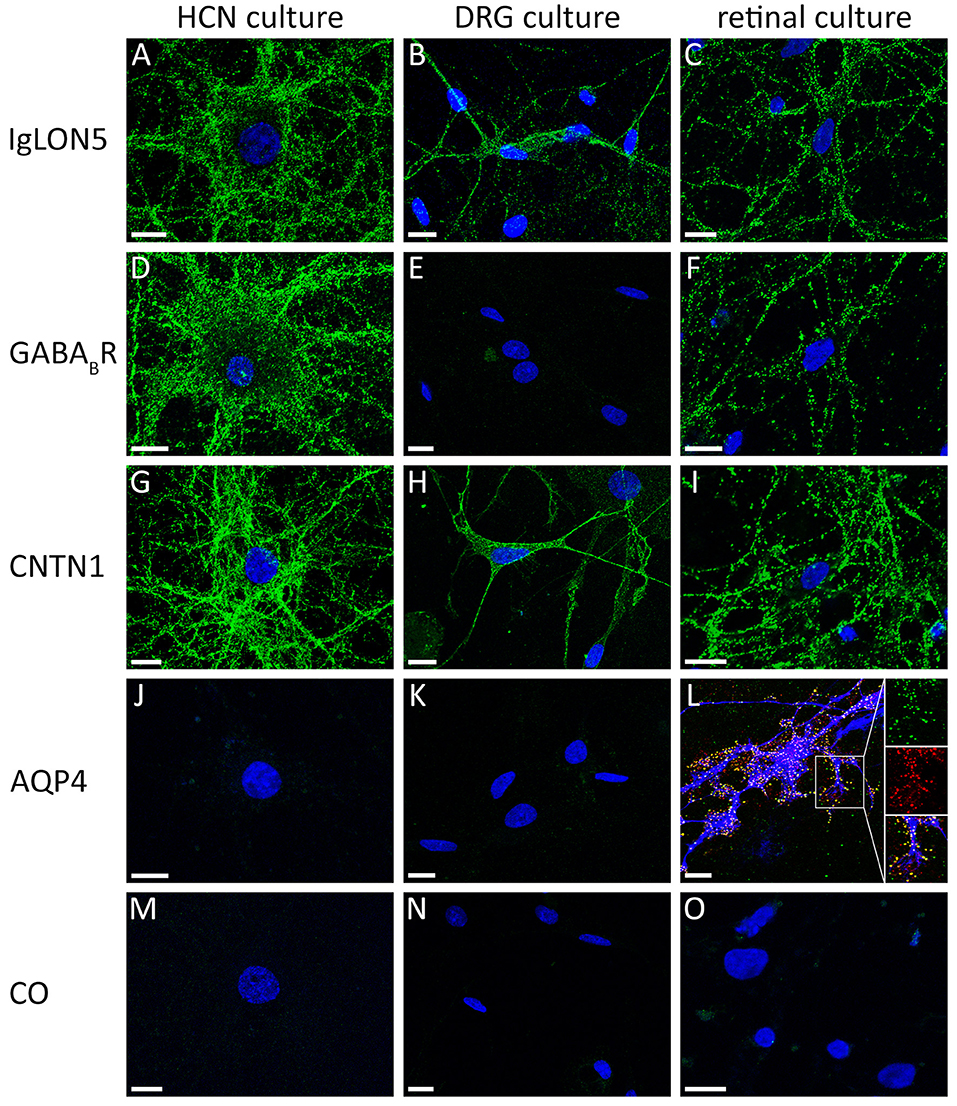
Figure 3. Comparison of reactivity of different antibodies against cell surface antigens on different primary neuronal and glioneuronal cell cultures. (A) The serum of a patient with anti-IgLON5 antibodies shows an intensive labeling of live nonpermeabilized rat hippocampal neurons, (B) rat dorsal root ganglion cells (DRGs), and (C) dissociated rat retinal cell culture. In contrast, (D) the serum of a patient with anti-GABA(B)R antibodies labels hippocampal neurons but not (E) DRGs. (F) The retinal cell culture is strongly GABA(B)R positive. (G–I) A serum of a patient with anti-contactin1 antibodies labels all three types of cell cultures. A serum of a patient with (J) anti-aquaporin4 antibodies is negative on hippocampal neurons and (K) DRGs, but (L) labels the end feet membranes of GFAP-positive Müller cells (red: rabbit polyclonal anti-AQP4 antibody; green: serum of a patient with AQP4 antibodies; blue: mouse monoclonal anti-GFAP antibody). (M–O) A healthy control is negative. HCN, hippocampal neurons; DRG, dorsal root ganglion cells; retinal culture, dissociated rat retinal cell culture; CNTN1, contactin1; AQP4, aquaporin-4; CO, healthy control; Scale bar = 10 μm.
A critical step to successfully detect anti-neuronal or anti-glial antibodies is the combined testing of serum and CSF in an individual patient. This has several reasons. First, the detectability of specific antibodies may differ between serum and CSF. Some antibodies may be easier identifyable in CSF than serum, for example antibodies against the NMDAR, GABA(B)R or AMPAR. In a study of 577 patients with anti-NMDAR encephalitis, in one out of 7 patients antibodies were only detectable in CSF and testing restricted to serum would have misdiagnosed the patients as seronegative (69). Other autoimmunities may present with a substantial systemic autoantibody production such as patients with GAD65 antibodies, but they may additionally harbor antibodies against the GAD67 isoform in CSF and few cases were described with a restricted autoimmunity to GAD67. It will be important to collect more cases with exclusive GAD67 reactivity to see whether they present specific neurological features. Finally, some antibodies are more prevalent in serum than in CSF. These are for example anti-AQP4 or anti-MOG-antibodies (44). ADEM may be an important differential diagnosis for anti-neuronal autoimmune encephalitis and the testing for anti-MOG-antibodies only in CSF may lead to false negative results and delay in diagnosis.
Second, serum and CSF might harbor different sets of antibodies and in this constellation the antibodies in CSF may correlate better with the neurological symptoms than those in serum, as it has been shown in a study of patients with GABA(A)R antibodies (70).
Third, testing of serum and CSF may have methodological implications. It has been shown that testing of antibodies only in serum harbors the risk for increased background and unspecific cross-reactivity that may result in contradictory test interpretations (71, 72). To avoid misinterpretations or delay in diagnosis the testing of both serum and CSF is recommended (73).
Antibodies in human plasma belong to different isotypes according to their type of heavy chains and include IgG, IgA, IgM, IgE, and IgD. The IgG is the most abundant antibody isotype and can be classified into four subclasses IgG1, 2, 3, and 4. Pathogenic mechanisms in anti-neuronal autoimmune encephalitis were mainly associated with antibodies of the IgG isotype that can have different effects on the targeted antigen. The IgG1-3 subclasses may alter the synaptic structure by cross-linking and internalization of the receptor such as in anti-NMDAR (74) or anti-AMPAR encephalitis (75), serve as antagonist of baclofen in anti-GABA(B)R autoimmunity (41), or reduce the amount of receptor at the synapse such as in anti-GABA(A)R autoimmunity (70). In contrast, antibodies of the IgG4 subclass mainly seem to mechanically interfere between the receptor-ligand interaction resulting in the blockade of protein-protein interaction (76). Recently, antibodies of the IgA and IgM isotype against the NMDAR were found in up to 22% of patients with different neurological diseases and in healthy controls by using fixed CBAs and it was hypothesized that the symptomatic relevance of the antibodies is related to a compromised blood-brain barrier that allows access to the brain (77–81). Moreover, it was demonstrated that NMDAR antibodies regardless of the clinical presentation of the donor (healthy or ill) and immunoglobulin class could provoke receptor internalization in human-induced pluripotent stem cell-derived neurons and reduced the glutamate-evoked currents in NMDAR expressing Xenopus oocytes (79). However, the functional significance of IgA and IgM NMDAR antibodies and their ability to internalize the NMDAR could not be confirmed in a subsequent study using CBAs, unfixed TBAs, and immunostaining of live primary hippocampal neurons (82). Since robust association with anti-NMDAR encephalitis was only shown for IgG antibodies, the antibody testing in clinical practice should be focused on the IgG antibodies.
The expanding field of antibody-mediated autoimmunity allows the identification of a vast range of neuronal and glial autoantibodies, which enables a more precise diagnosis of specific syndromes and disease subtypes. It is important to know that testing for onconeuronal antibodies requires other methods (line assays, fixed TBAs) than surface antibodies (CBAs and unfixed/postfixed TBAs). The highest sensitivity and specificity of a test result can be achieved by cross-validation with different test methods and the combined testing of serum and CSF samples. Test results should always be interpreted in context with the clinical presentation. In case of an unexpected positive or negative result, re-testing of the sample or performing confirmatory tests might be considered. The screening for surface antibodies on unfixed TBA can detect a large number of anti-neuronal and some anti-glial antibodies with some limitation for anti-GlyR, anti-D2R, and anti-MOG-antibodies. In patients with peripheral neuropathies, the screening can be expanded to teased nerve fibers to detect antibodies against proteins of the node of Ranvier. Moreover, the staining of primary cultures of neurons or glioneuronal cell cultures may give evidence that the detected autoantibody recognizes a surface antigen and enables the characterization of novel surface antibodies. The accurate and rapid detection of autoantibodies in CSF and serum may initiate immunotherapies to improve patients outcome.
GR and RH have access to all the data and take responsibility for the data, accuracy of the data analysis, design and conceptualization of the review. CS, DD, VP, JL, SG, SM, PR, PS, HK, and IK: analysis and interpretation of the data, drafting, and revising the manuscript for intellectual content.
RH received speaker honoraria from Euroimmun. The Medical University of Vienna receives payments for antibody assays and antibody validation assays (aquaporin-4 and other anti-neuronal and anti-glial antibodies) organized by Euroimmun (Lübeck, Germany).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was partly supported by the Medical Scientific Fund of the Mayor of the City of Vienna, Project 15022, the Jubiläumsfonds der Österreichischen Nationalbank, Project 16919, and the Austrian Science Fund (FWF): I 3334-B27. IK was supported by an Erwin Schrödinger Fellowship by the Austrian Science Fund (FWF): J 3545-B13.
AMPAR,amino-3-hydroxy-5-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; CASPR1/2, contactin-associated protein-like 1/2; CNTN1, contactin1; D2R, dopamine-2 receptor; DPPX, dipeptidyl-peptidase-like protein-6; GABA A/B R, gamma-aminobutyric acid A/B receptor; GAD, glutamic acid decarboxylase; GlyR, Glycine receptor; LGI1, leucine-rich glioma-inactivated 1; mGluR1/5, metabotropic glutamate receptor type 1/5; NF155, neurofascin155; NMDAR, N-methyl-D-aspartate receptor; P/Q-type VGCC, P/Q-type voltage-gated calcium channel; VGKC, voltage-gated potassium channel.
1. Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med. (2018) 378:840–51. doi: 10.1056/NEJMra1708712
2. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain (2012) 135(Pt 5):1622–38. doi: 10.1093/brain/aws082
3. Werner C, Pauli M, Doose S, Weishaupt A, Haselmann H, Grünewald B, et al. Human autoantibodies to amphiphysin induce defective presynaptic vesicle dynamics and composition. Brain (2016) 139(Pt 2):365–79. doi: 10.1093/brain/awv32
4. Graus F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol. (2010) 257:509–17. doi: 10.1007/s00415-009-5431-9
5. Doppler K, Appeltshauser L, Villmann C, Martin C, Peles E, Krämer HH, et al. Auto-antibodies to contactin-associated protein 1 (Caspr) in two patients with painful inflammatory neuropathy. Brain (2016) 139(Pt 10):2617–30. doi: 10.1093/brain/aww189
6. Doppler K, Appeltshauser L, Wilhelmi K, Villmann C, Dib-Hajj SD, Waxman SG, et al. Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J Neurol Neurosurg Psychiatry (2015) 86:720–8. doi: 10.1136/jnnp-2014-309916
7. van Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, de Bruijn MA, et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology (2016) 87:1449–56. doi: 10.1212/WNL.0000000000003173
8. Graus F, Cordon-Cardo C, Posner JB. Neuronal antinuclear antibody in sensory neuronopathy from lung cancer. Neurology (1985) 35:538–43. doi: 10.1212/WNL.35.4.538
9. Solimena M, Folli F, Aparisi R, Pozza G, De Camilli P. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N Engl J Med. (1990) 322:1555–60. doi: 10.1056/NEJM199005313222202
10. Rosenfeld MR, Eichen JG, Wade DF, Posner JB, Dalmau J. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins. Ann Neurol. (2001) 50:339–48. doi: 10.1002/ana.1288
11. Pittock SJ, Lucchinetti CF, Lennon VA. Anti-neuronal nuclear autoantibody type 2: paraneoplastic accompaniments. Ann Neurol. (2003) 53:580–7. doi: 10.1002/ana.10518
12. Pittock SJ, Lucchinetti CF, Parisi JE, Benarroch EE, Mokri B, Stephan CL, et al. Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol. (2005) 58:96–107. doi: 10.1002/ana.20529
13. Honnorat J, Antoine JC, Derrington E, Aguera M, Belin MF. Antibodies to a subpopulation of glial cells and a 66 kDa developmental protein in patients with paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry (1996) 61:270–8. doi: 10.1136/jnnp.61.3.270
14. Peterson K, Rosenblum MK, Kotanides H, Posner JB. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology (1992) 42:1931–7. doi: 10.1212/WNL.42.10.1931
15. Graus F, Dalmau J, Valldeoriola F, Ferrer I, Rene R, Marin C, et al. Immunological characterization of a neuronal antibody (anti-Tr) associated with paraneoplastic cerebellar degeneration and Hodgkin's disease. J Neuroimmunol. (1997) 74:55–61. doi: 10.1016/S0165-5728(96)00205-6
16. Graus F, Delattre JY, Antoine JC, Dalmau J, Giometto B, Grisold W, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry (2004) 75:1135–40. doi: 10.1136/jnnp.2003.034447
17. Graus F, Dalmou J, Rene R, Tora M, Malats N, Verschuuren JJ, et al. Anti-Hu antibodies in patients with small-cell lung cancer: association with complete response to therapy and improved survival. J Clin Oncol. (1997) 15:2866–72. doi: 10.1200/JCO.1997.15.8.2866
18. Storstein A, Monstad SE, Haugen M, Mazengia K, Veltman D, Lohndal E, et al. Onconeural antibodies: improved detection and clinical correlations. J Neuroimmunol. (2011) 232:166–70. doi: 10.1016/j.jneuroim.2010.10.009
19. Sabater L, Saiz A, Dalmau J, Graus F. Pitfalls in the detection of CV2 (CRMP5) antibodies. J Neuroimmunol. (2016) 290:80–3. doi: 10.1016/j.jneuroim.2015.11.009
20. Hoftberger R, Kovacs GG, Sabater L, Nagy P, Racz G, Miquel R, et al. Protein kinase Cgamma antibodies and paraneoplastic cerebellar degeneration. J Neuroimmunol. (2013) 256:91–3. doi: 10.1016/j.jneuroim.2012.12.002
21. Hoftberger R, Sabater L, Velasco F, Ciordia R, Dalmau J, Graus F. Carbonic anhydrase-related protein VIII antibodies and paraneoplastic cerebellar degeneration. Neuropathol Appl Neurobiol. (2014) 40:650–3. doi: 10.1111/nan.12118
22. Doss S, Numann A, Ziegler A, Siebert E, Borowski K, Stöcker W, et al. Anti-Ca/anti-ARHGAP26 antibodies associated with cerebellar atrophy and cognitive decline. J Neuroimmunol. (2014) 267:102–4. doi: 10.1016/j.jneuroim.2013.10.010
23. Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. (2016) 73:1297–1307. doi: 10.1001/jamaneurol.2016.2549
24. Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol. (2017) 81:298–309. doi: 10.1002/ana.24881
25. Saiz A, Blanco Y, Sabater L, Gonzalez F, Bataller L, Casamitjana R, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain (2008) 131(Pt 10):2553–63. doi: 10.1093/brain/awn183
26. Gresa-Arribas N, Arino H, Martinez-Hernandez E, Petit-Pedrol M, Sabater L, Saiz A, et al. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS ONE (2015) 10:e0121364. doi: 10.1371/journal.pone.0121364
27. Guasp M, Sola-Valls N, Martinez-Hernandez E, Gil MP, Gonzalez C, Brieva L, et al. Cerebellar ataxia and autoantibodies restricted to glutamic acid decarboxylase 67 (GAD67). J Neuroimmunol. (2016) 300:15–7. doi: 10.1016/j.jneuroim.2016.09.019
28. Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology (1976) 26:1054–9. doi: 10.1212/WNL.26.11.1054
29. Vincent A, Lang B, Newsom-Davis J. Autoimmunity to the voltage-gated calcium channel underlies the Lambert-Eaton myasthenic syndrome, a paraneoplastic disorder. Trends Neurosci. (1989) 12:496–502. doi: 10.1016/0166-2236(89)90109-4
30. Lennon VA, Lambert EH. Autoantibodies bind solubilized calcium channel-omega-conotoxin complexes from small cell lung carcinoma: a diagnostic aid for Lambert-Eaton myasthenic syndrome. Mayo Clin Proc. (1989) 64:1498–504. doi: 10.1016/S0025-6196(12)65705-X
31. Vincent A, Roberts M, Willison H, Lang B, Newsom-Davis J. Autoantibodies, neurotoxins and the nervous system. J Physiol Paris (1995) 89:129–36. doi: 10.1016/0928-4257(96)80110-0
32. Parsons KT, Kwok WW. Linear B-cell epitopes in lambert-eaton myasthenic syndrome defined by cell-free synthetic peptide binding. J Neuroimmunol. (2002) 126:190–5. doi: 10.1016/S0165-5728(02)00063-2
33. Lang B, Makuch M, Moloney T, Dettmann I, Mindorf S, Probst C, et al. Intracellular and non-neuronal targets of voltage-gated potassium channel complex antibodies. J Neurol Neurosurg Psychiatry (2017) 88:353–61. doi: 10.1136/jnnp-2016-314758
34. van Sonderen A, Schreurs MW, de Bruijn MA, Boukhrissi S, Nagtzaam MM, Hulsenboom ES, et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology (2016) 86:1692–99. doi: 10.1212/WNL.0000000000002637
35. Besinger UA, Toyka KV, Homberg M, Heininger K, Hohlfeld R, Fateh-Moghadam A. Myasthenia gravis: long-term correlation of binding and bungarotoxin blocking antibodies against acetylcholine receptors with changes in disease severity. Neurology (1983) 33:1316–21. doi: 10.1212/WNL.33.10.1316
36. Saxena A, Stevens J, Cetin H, Koneczny I, Webster R, Lazaridis K, et al. Characterization of an anti-fetal AChR monoclonal antibody isolated from a myasthenia gravis patient. Sci Rep. (2017) 7:14426. doi: 10.1038/s41598-017-14350-8
37. Hara H, Hayashi K, Ohta K, Itoh N, Nishitani H, Ohta M. Detection and characterization of blocking-type anti-acetylcholine receptor antibodies in sera from patients with myasthenia gravis. Clin Chem. (1993) 39:2053–7.
38. Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP, et al. IgG1 antibodies to acetylcholine receptors in 'seronegative' myasthenia gravis. Brain (2008) 131(Pt 7):1940–52. doi: 10.1093/brain/awn092
39. Pettingill P, Kramer HB, Coebergh JA, Pettingill R, Maxwell S, Nibber A, et al. Antibodies to GABAA receptor alpha1 and gamma2 subunits: clinical and serologic characterization. Neurology (2015) 84:1233–41. doi: 10.1212/WNL.0000000000001326
40. Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. (2014) 13:167–77. doi: 10.1016/S1474-4422(13)70282-5
41. Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol Rev. (2017) 97:839–87. doi: 10.1152/physrev.00010.2016
42. Hoftberger R, Sabater L, Marignier R, Aboul-Enein F, Bernard-Valnet R, Rauschka H, et al. An optimized immunohistochemistry technique improves NMO-IgG detection: study comparison with cell-based assays. PLoS ONE (2013) 8:e79083. doi: 10.1371/journal.pone.0079083
43. Mayer MC, Breithaupt C, Reindl M, Schanda K, Rostasy K, Berger T, et al. Distinction and temporal stability of conformational epitopes on myelin oligodendrocyte glycoprotein recognized by patients with different inflammatory central nervous system diseases. J Immunol. (2013) 191:3594–604. doi: 10.4049/jimmunol.1301296
44. Reindl M, Di Pauli F, Rostasy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol. (2013) 9:455–61. doi: 10.1038/nrneurol.2013.118
45. Dale RC, Merheb V, Pillai S, Wang D, Cantrill L, Murphy TK, et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain (2012) 135(Pt 11):3453–68. doi: 10.1093/brain/aws256
46. Carvajal-Gonzalez A, Leite MI, Waters P, Woodhall M, Coutinho E, Balint B, et al. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain (2014) 137(Pt 8):2178–92. doi: 10.1093/brain/awu142
47. Motomura M, Lang B, Johnston I, Palace J, Vincent A, Newsom-Davis J. Incidence of serum anti-P/O-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci. (1997) 147:35–42. doi: 10.1016/S0022-510X(96)05303-8
48. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet (2004) 364:2106–12. doi: 10.1016/S0140-6736(04)17551-X
49. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
50. Pandit L, Asgari N, Apiwattanakul M, Palace J, Paul F, Leite MI, et al. Demographic and clinical features of neuromyelitis optica: a review. Mult Scler. (2015) 21:845–53. doi: 10.1177/1352458515572406
51. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. (2005) 202:473–7. doi: 10.1084/jem.20050304
52. Waters P, Reindl M, Saiz A, Schanda K, Tuller F, Kral V, et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry (2016) 87:1005–15. doi: 10.1136/jnnp-2015-312601
53. Shimizu F, Schaller KL, Owens GP, Cotleur AC, Kellner D, Takeshita Y, et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci Transl Med. (2017) 9:eaai9111. doi: 10.1126/scitranslmed.aai9111
54. Baumann M, Sahin K, Lechner C, Hennes EM, Schanda K, Mader S, et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J Neurol Neurosurg Psychiatry (2015) 86:265–72. doi: 10.1136/jnnp-2014-308346
55. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain (2017) 140:3128–38. doi: 10.1093/brain/awx276
56. Peschl P, Schanda K, Zeka B, Given K, Bohm D, Ruprecht K, et al. Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J Neuroinflamm. (2017) 14:208. doi: 10.1186/s12974-017-0984-5
57. Mader S, Gredler V, Schanda K, Rostasy K, Dujimovic I, Pfaller K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflamm. (2011) 8:184. doi: 10.1186/1742-2094-8-184
58. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflamm. (2016) 13:279. doi: 10.1186/s12974-016-0717-1
59. Jarius S, Ruprecht K, Stellmann JP, Huss A, Ayzenberg I, Willing A, et al. MOG-IgG in primary and secondary chronic progressive multiple sclerosis: a multicenter study of 200 patients and review of the literature. J Neuroinflamm. (2018) 15:88. doi: 10.1186/s12974-018-1108-6
60. Querol L, Nogales-Gadea G, Rojas-Garcia R, Martinez-Hernandez E, Diaz-Manera J, Suarez-Calvet X, et al. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. (2013) 73:370–80. doi: 10.1002/ana.23794
61. Devaux JJ, Miura Y, Fukami Y, Inoue T, Manso C, Belghazi M, et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology (2016) 86:800–7. doi: 10.1212/WNL.0000000000002418
62. Stathopoulos P, Alexopoulos H, Dalakas MC. Autoimmune antigenic targets at the node of Ranvier in demyelinating disorders. Nat Rev Neurol. (2015) 11:143–56. doi: 10.1038/nrneurol.2014.260
63. Querol L, Rojas-Garcia R, Diaz-Manera J, Barcena J, Pardo J, Ortega-Moreno A, et al. Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e149. doi: 10.1212/NXI.0000000000000149
64. Querol L, Devaux J, Rojas-Garcia R, Illa I. Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications. Nat Rev Neurol. (2017) 13:533–47. doi: 10.1038/nrneurol.2017.84
65. Querol L, Nogales-Gadea G, Rojas-Garcia R, Diaz-Manera J, Pardo J, Ortega-Moreno A, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology (2014) 82:879–86. doi: 10.1212/WNL.0000000000000205
66. Miura Y, Devaux JJ, Fukami Y, Manso C, Belghazi M, Wong AH, et al. Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain (2015) 138(Pt 6):1484–91. doi: 10.1093/brain/awv054
67. Nagelhus EA, Veruki ML, Torp R, Haug FM, Laake JH, Nielsen S, et al. Aquaporin-4 water channel protein in the rat retina and optic nerve: polarized expression in Muller cells and fibrous astrocytes. J Neurosci. (1998) 18:2506–19. doi: 10.1523/JNEUROSCI.18-07-02506.1998
68. Zeka B, Hastermann M, Kaufmann N, Schanda K, Pende M, Misu T, et al. Aquaporin 4-specific T cells and NMO-IgG cause primary retinal damage in experimental NMO/SD. Acta Neuropathol Commun. (2016) 4:82. doi: 10.1186/s40478-016-0355-y
69. Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. (2013) 12:157–65. doi: 10.1016/S1474-4422(12)70310-1
70. Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. (2014) 13:276–86. doi: 10.1016/S1474-4422(13)70299-0
71. Armangue T, Santamaria J, Dalmau J. When a serum test overrides the clinical assessment. Neurology (2015) 84:1379–81. doi: 10.1212/WNL.0000000000001425
72. Zandi MS, Paterson RW, Ellul MA, Jacobson L, Al-Diwani A, Jones Jl, et al. Clinical relevance of serum antibodies to extracellular N-methyl-D-aspartate receptor epitopes. J Neurol Neurosurg Psychiatry (2015) 86:708–13. doi: 10.1136/jnnp-2014-308736
73. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. (2016) 15:391–404. doi: 10.1016/S1474-4422(15)00401-9
74. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. (2010) 30:5866–75. doi: 10.1523/JNEUROSCI.0167-10.2010
75. Peng X, Hughes EG, Moscato EH, Parsons TD, Dalmau J, Balice-Gordon RJ. Cellular plasticity induced by anti-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann Neurol. (2015) 77:381–98. doi: 10.1002/ana.24293
76. Koneczny I. A new classification system for IgG4 autoantibodies. Front Immunol. (2018) 9:97. doi: 10.3389/fimmu.2018.00097
77. Dahm L, Ott C, Steiner J, Stepniak B, Teegen B, Saschenbrecker S, et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol. (2014) 76:82–94. doi: 10.1002/ana.24189
78. Hammer C, Stepniak B, Schneider A, Papiol S, Tantra M, Begemann M, et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry (2014) 19:1143–9. doi: 10.1038/mp.2013.110
79. Castillo-Gomez E, Oliveira B, Tapken D, Bertrand S, Klein-Schmidt C, Pan H, et al. All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol Psychiatry (2017) 22:1776–84. doi: 10.1038/mp.2016.125
80. Zerche M, Weissenborn K, Ott C, Dere E, Asif AR, Worthmann H, et al. Preexisting serum autoantibodies against the NMDAR subunit NR1 modulate evolution of lesion size in acute ischemic stroke. Stroke (2015) 46:1180–6. doi: 10.1161/STROKEAHA.114.008323
81. Doss S, Wandinger KP, Hyman BT, Panzer JA, Synofzik M, Dickerson B, et al. High prevalence of NMDA receptor IgA/IgM antibodies in different dementia types. Ann Clin Transl Neurol. (2014) 1:822–32. doi: 10.1002/acn3.120
Keywords: paraneoplastic neurological syndrome, autoimmune encephalitis, onconeuronal antibodies, test methods, cell-based assay, tissue-based assay, anti-neuronal antibodies
Citation: Ricken G, Schwaiger C, De Simoni D, Pichler V, Lang J, Glatter S, Macher S, Rommer PS, Scholze P, Kubista H, Koneczny I and Höftberger R (2018) Detection Methods for Autoantibodies in Suspected Autoimmune Encephalitis. Front. Neurol. 9:841. doi: 10.3389/fneur.2018.00841
Received: 12 July 2018; Accepted: 18 September 2018;
Published: 10 October 2018.
Edited by:
Johann Sellner, Universitätsklinikum Salzburg, AustriaReviewed by:
Andrea Harrer, Paracelsus Medizinische Privatuniversität, AustriaCopyright © 2018 Ricken, Schwaiger, De Simoni, Pichler, Lang, Glatter, Macher, Rommer, Scholze, Kubista, Koneczny and Höftberger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Romana Höftberger, cm9tYW5hLmhvZWZ0YmVyZ2VyQG1lZHVuaXdpZW4uYWMuYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.